

Abstract
Background and Objective
BC 007 is a substance with a novel and innovative mode of action for the first-time causal treatment of chronic heart failure, associated with the occurrence of autoantibodies against the β1-adrenoceptor, and other diseases of mostly the heart and vascular system, being accompanied by the occurrence of functionally active agonistic autoantibodies against G-protein-coupled receptors (fGPCR-AAb). The proposed mechanism of action of BC 007 is the neutralisation of these pathogenic autoantibodies which stimulate the respective receptor. To evaluate the safety, tolerability, pharmacokinetics and mode of action of BC 007, single intravenous infusions of increasing concentration were given to healthy young males and healthy elderly autoantibody-negative and autoantibody-positive participants of both sexes.
Methods
This study was subdivided into three parts. Part A was a single-centre, randomised, double-blind, placebo-controlled safety and tolerability study including healthy young male autoantibody-negative Whites (N = 23) and Asians (N = 1), testing doses of 15, 50 and 150 mg BC 007 (Cohorts 1–3) and elderly male and female Whites (N = 8), testing a dose of 150 mg BC 007 (Cohort 4), randomly assigned in a 3:1 ratio to BC 007 or placebo. Open-label Part B included fGPCR-AAb-positive subjects (50 and 150 mg BC 007, Cohorts 1 and 2, respectively). Open-label Part C included fGPCR-AAb-positive subjects for testing doses of 300, 450, 750, 1350 mg and 1900 mg BC 007. Lower doses were either given as an infusion or divided into a bolus plus infusion up to a dose of 300 mg followed by a constant bolus of 150 mg up to a dose of 750 mg, while at doses of 1350 mg and 1900 mg it was a slow infusion with a constant infusion rate. Infusion times increased with increasing dose from 20 min (15, 50 or 150 mg) to 40 min (300, 450 or 750 mg), 75 min (1350 mg) and 105 min (1900 mg).
Results
The mean observed BC 007 area under the concentration–time curve (AUC0–24) increased with increasing dose in a dose proportional manner (slope estimate of 1.039). No serious adverse events were observed. Drug-related adverse events were predominantly the expected mild-to-moderate increase in bleeding time (aPTT), beginning with a dose of 50 mg, which paralleled the infusion and returned to normal shortly after infusion. fGPCR-AAb neutralisation efficiency increased with increasing dose and was achieved for all subjects in the last cohort.
Conclusion
BC 007 is demonstrated to be safe and well tolerated. BC 007 neutralised fGPCR-AAb, showing a trend for a dose-response relationship in elderly healthy but fGPCR-AAb-positive subjects.
ClinicalTrials.gov Registration Number
Key Points
| The mean BC 007 area under the concentration–time curves (AUC(0–t) and AUCinf) increased linearly with the dose. The plasma half-life time was fast and ranged from 3.0 min at 15 mg in the young population up to 11 min at 1900 mg BC 007 in the elderly population. |
| No treatment emergent adverse events (TEAEs) leading to subject discontinuation, no severe TEAEs, no serious adverse events (SAEs) and no deaths were reported throughout the entire clinical Phase I. The most common reported adverse event was a slight-to-moderate increase of the anticoagulatory effect accompanying the infusion beginning at a dose of 50 mg which normalised quickly within minutes after infusion end. |
| BC 007, a new drug candidate for heart failure treatment, with innovative mode of action neutralised autoantibodies against G-protein-coupled receptors in elderly healthy G-protein-coupled receptor autoantibody-positive participants. The neutralisation lasted, in most cases, for the entire observation period of 1 month. |
Introduction
Despite ongoing progress in pharmacological and non-pharmacological treatment of heart failure (HF) over the last three decades [1], HF is still a growing public health problem [2]. Up-to-date identified factors which trigger HF are manifold, as summarised in a comprehensive review by Hershberger et al. [3]. The impact of autoimmunity on HF pathogenesis is also subject of current discussion [4]. In particular, with the positive outcome after the removal of agonistic acting autoantibodies which stimulate the β1-adrenoceptors (β1-AR), using immunoadsorption, a direct relationship between functionally active autoantibodies specific for the β1-adrenoceptor (fβ1-AR-AAb) and the pathogenesis of dilated cardiomyopathy (DCM) has been seen [5]. A tremendous beneficial effect of immunoadsorption (increase in survival rate by 43.9%, 5 years after treatment) was received on top of continuing conventional treatment [6, 7] and persisted over years [7]. It is very important to correctly identify the patients positive for functionally active autoantibodies, using bioassays; in patients who do not have functionally active autoantibodies, this removal therapy did not, of course, show any beneficial effect [7]. Since binding but not receptor activating β1-AR-AAbs have been described [8] and also since the quality of solid phase based assays (ELISAs) in this respect are generally in question [9, 10], a correct analytic identifying the functionally active autoantibodies is a critical parameter and prerequisite for treatment success.
Independent of this described treatment success in the clinic, animal experiments have also proven the cardiotoxic effect of fβ1-AR-AAbs when induced by immunisation with the epitope peptide of the second extracellular loop of the human β1-AR, a sequence which is fully homologous to rats. Moreover, the transfer of the generated fβ1-AR-AAb-containing serum samples to healthy control rats transferred the cardiomyopathic phenotype [11].
Immunoadsorption therapy, however, has not established itself as a routine therapy for various reasons, despite its great success.
With BC 007, a substance has now been identified capable of neutralising functionally active autoantibodies in vivo, which are directed to the G-protein coupled receptors (fGPCR-AAbs) including the fβ1-AR-AAbs [12]. This includes fβ1-AR-AAbs which target the first and also those which target the second extracellular loop of the β1-AR. In this way, all fβ1-AR-AAb-positive HF patients are included in the possible therapy spectrum, and moreover, other cases of cardiomyopathy, such as Chagas’ cardiomyopathy or peripartum cardiomyopathy, in which the fβ1-AR-AAbs show slightly different epitopes compared to HF patients [13]. This is a tremendous advantage over a drug-development strategy which is based on the use of a specific autoantibody epitope peptide sequence only, as was done with the development of COR-1 [14], which has already failed for several reasons [14, 15].
BC 007 is a short single-stranded DNA sequence (15mer) which unfolds its AAB neutralising effect ex vivo and in vivo via aptamer functionality [16].
BC 007 showed no binding on a variety of tested G-protein coupled receptors itself, including the β1-ARs (Eurofin Cerep Safety-screen 44 Panel). It binds the fGPCR-AAbs. BC 007 did not show any signs of toxicity in 14-day repeat dose studies in rats (study no. SBC007S003, data not shown) and beagle dogs (study no. SBC007S004, data not shown.). In both studies, no signs of local intolerance or systemic toxicity were noted including the absence of effects on reproductive organs and signs of immune toxicology. The No-Observed-Adverse-Effect-Level (NOAEL) was concluded to be above 90 mg BC 007/kg IV for beagle dogs and above 100 mg BC 007/kg IV for rats.
BC 007 is, therefore, an excellent drug candidate to be tested for causal treatment of autoimmune associated chronic HF.
With the present study, the safety, tolerability, pharmacokinetics and also mode of action to neutralise fGPCR-AAbs were tested at doses between 15–1900 mg BC 007, intravenously applied over a period between 20 and 105 min as a bolus plus infusion or infusion only.
Methods
Study Drug
Clinical trial samples of BC 007 (molecular mass as sodium salt 5033.83 g/mol) for infusion was manufactured by BioSpring GmbH (Frankfurt am Main, Germany) and filled by LYOCONTRACT GmbH (Ilsenburg, Germany). Shortly before application the vials containing lyophilised powder were reconstituted using commercially available sodium chloride for injection.
Study Population
This study included 73 White and one Asian subjects, aged 18–75 years. Part A included 32 subjects, 24 treated and eight placebos, Part B included 12 subjects in two dose cohorts and Part C included 30 subjects in five cohorts of six subjects each. Demographic data and an overview of baseline characteristics are shown in Table 1.
Table 1.
Baseline characteristics of participating subjects, subdivided into study parts and attributive cohorts
| Parameter | Part A | Part B | Part C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cohort 1 | Cohort 2 | Cohort 3 | Placebo 1–3 | Cohort 4 | Placebo 4 | Cohort 1 | Cohort 2 | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 | |
| N | 6 | 6 | 6 | 6 | 6 | 2 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Male, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 5 (83.3) | 2 (100) | 3 (50) | 2 (33.3) | 4 (66.7) | 3 (50) | 3 (50) | 4 (66.7) | 3 (50) |
| Female, n (%) | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 3 (50) | 4 (66.7) | 2 (33.3) | 3 (50) | 3 (50) | 2 (33.3) | 3 (50) |
| Race | |||||||||||||
| White, n (%) | 5 (83.3) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 2 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) |
| Asian, n (%) | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Age (years) | |||||||||||||
| Mean (SD) | 33.5 (9.14) | 35.8 (4.54) | 34.8 (5.71) | 36.2 (8.42) | 67.2 (1.83) | 68.0 (0.00) | 64.3 (3.88) | 61.5 (5.47) | 64.7 (5.54) | 65.0 (7.38) | 60.2 (3.54) | 62.8 (6.34) | 61.7 (3.08) |
| Range | 24–45 | 29–42 | 27–42 | 21–44 | 65–69 | 68–68 | 60–69 | 56–69 | 57–74 | 56–72 | 56–64 | 56–74 | 57–65 |
| Height (cm) | |||||||||||||
| Mean (SD) | 178.5 (7.06) | 183.7 (6.62) | 181.5 (4.51) | 175.7 (5.99) | 174.0 (5.06) | 168.0 (1.41) | 168.8 (4.17) | 171.2 (11.55) | 169.5 (5.89) | 172.7 (9.05) | 172.8 (11.05) | 172.0 (10.55) | 173.7 (9.99) |
| Range | 169–190 | 175–192 | 174–185 | 168–185 | 167–180 | 167–169 | 162–174 | 156–185 | 162–178 | 164–184 | 155–184 | 159–183 | 159–187 |
| BMI (kg/m2) | |||||||||||||
| Mean (SD) | 25.58 (2.38) | 24.92 (2.32) | 26.63 (3.01) | 24.63 (0.93) | 24.18 (1.41) | 24.15 (2.62) | 26.13 (2.79) | 25.58 (2.14) | 26.13 (2.15) | 25.27 (2.98) | 24.62 (2.79) | 26.53 (3.50) | 27.55 (1.45) |
| Range | 22–29 | 22–28 | 22–29 | 24–26 | 22–26 | 22–26 | 21–29 | 23–29 | 23–29 | 21–29 | 21–28 | 22–30 | 25–29 |
BMI body mass index, SD standard deviation
Study Design
The study was subdivided into three parts (Fig. 1). While Part A was exclusively focused on safety and tolerability, Parts B and C were dedicated to additionally testing BC 007’s mode of action (NCT02955420).
Fig. 1.

Study design. FUP follow up period, i.v. intravenous, N number of participants, PK pharmacokinetic, fGPCR-AAb functionally active agonistic autoantibodies against G-protein-coupled receptors
Part A was a single-centred randomised double-blind placebo-controlled safety and tolerability test, including healthy young males, fGPCR-AAb-negative whites (N = 23) and Asians (N = 1), testing doses of 15, 50 and 150 mg BC 007 intravenous with infusion (Cohorts 1–3) and elderly male and female whites (N = 8), testing a dose of 150 mg BC 007 (Cohort 4), all randomly assigned in a 3:1 ratio to BC 007 or placebo. Open label Part B included fGPCR-AAb-positive subjects (50 and 150 mg BC 007, Cohorts 1 and 2, respectively). Open label Part C included fGPCR-AAb-positive subjects for testing doses of 300, 450, 750, 1350 and 1900 mg BC 007. Lower doses were given either as an infusion (one per cohort, sentinel) or divided into a bolus plus infusion up to a dose of 300 mg or with a constant bolus of 150 mg up to 750 mg, three per cohort in Part C; while at doses of 1350 mg and 1900 mg it was slow infusion only. Infusion times increased with increasing dose from 20 min (50 or 150 mg) to 40 min (300, 450 or 750 mg), 75 min (1350 mg) and 105 min (1900 mg).
All three parts of this study (Parts A–C) consisted of three phases, beginning with a screening phase of up to 28 days before enrolment (day − 28 until day − 2), followed by single intravenous application assigned as described above (Day 1) and stationary follow-up for safety checks for 24 hours and outpatient follow-up safety checks at 7–10 days (Part A and B) and 8–12 days (Part C) after dosing.
For Parts B and C, these follow-up visits and an additional outpatient visit 28–32 days after dosing (Part C) were used to verify the fGPCR-AAb status.
Subject safety was carefully checked by monitoring electrocardiograms (ECG), blood pressure, injection site reactions and various laboratory safety tests checking vital signs, performing physical examinations and monitoring reports of adverse events. Blood samples were collected before the infusion began until 24 h thereafter for analyses of clinical chemistry, haematology, coagulation (including coagulation parameters at bedside during infusion of 1900 mg) and pharmacokinetics. One interim and one final follow-up visit for fGPCR-AAb status determination in subjects of Part C took place approximately one week and one month, respectively, after administration of doses of 300 mg and higher. For pharmacokinetic analysis, urine was collected.
Pharmacokinetics, Safety and Mode of Action Evaluation
Sample Collection
For safety laboratory analysis, blood samples for plasma concentration measurements were taken before dosing, during dosing, 4 h (Part A, Part B, Part C Cohorts 4–5) or 2 h (Part C Cohorts 1–3) and 24 h after dosing, as well as 7–10 or 8–12 days after dosing for Parts A and B or C, respectively.
For pharmacokinetic analysis, blood samples for plasma concentration measurements were taken pre-dose, at the end of the bolus or alternatively, 1 min after the start of the infusion for sentinel subjects (infusion only), during and immediately after the end of infusion, and also at initially shorter and later longer time intervals up to 24-h post-start bolus/infusion in Parts A and B.
In Part C, blood samples were taken pre-dose, 2 min post-end of bolus or 3 min after start of infusion (when receiving infusion only). Subsequently, blood was taken at several time points during infusion followed by immediately after end of infusion and at several timepoints up to 240 min after start of infusion.
Urine was collected before dosing, after start of dosing every 4 h for 12 h, and afterwards in one interval from 12 to 24 h. At doses of 1350 mg and 1900 mg BC 007, urine was collected at intervals of 0–2, 2–4, 4–8, 8–12 h and 12- to 24-h after start infusion.
For mode of action analysis (fGPCR-AAb neutralisation status), serum was taken before dosing, 24 h and about one week and one month after dosing beginning with the dose of 300 mg BC 007.
Pharmacokinetic Testing
BC 007 Concentration in Plasma and Urine
Hybridisation Assay for BC 007 Concentration in Plasma and Urine BC 007 plasma and urine concentrations were estimated by Accelero® Bioanalytic GmbH on the basis of a validated enzyme-linked sandwich hybridisation assay using probes adapted to BC 007 within an analytical range of 5–150 ng/mL.
Quality control samples included three different concentrations in each sample batch. To ensure precision, the CV % had to be ≤ 20% [25% at lower and upper limit of quantification (LLOQ/ULOQ)] for all samples. With respect to accuracy, the concentration had to be within ± 20% (± 25% at LLOQ/ULOQ) of the nominal level at each concentration level. At least 75% of the calibration standards and 67% of the QC samples had to meet this criterion. Furthermore, the total error of the QC sample values had to be ≤ 30% (≤ 40% at LLOQ/ULOQ).
Pharmacokinetic parameters maximum observed plasma concentration (Cmax), plasma concentration at end of infusion (Cend_infusion), time corresponding to occurrence of maximum observed plasma concentration (Tmax), area under the plasma concentration-time curve from time zero to last quantifiable concentration and from time zero to time/24-h postdose (AUC(0–inf), AUC(0–t), AUC(0–24)), terminal elimination rate constant (λz), elimination half-life (t1/2), clearance (CL) and apparent volume of distribution (Vz) were calculated on the basis of actual sampling times using WinNonlin Professional (Version 6.3). AUC(0–inf) calculation was based on a linear up/log-linear down trapezoidal method.
HPLC-Assay for the Determination of Metabolites (BC 007 n–x) in Plasma and Urine LGC Axiolabs GmbH was assigned for the explorative estimation of BC 007’s 3′-exonuclease cleavage products (n-1 up to n-3), developing a BC 007-specific anion exchange high-performance liquid chromatography (HPLC) method.
Determination of the fGPCR-AAb Status
For fGPCR-AAb immune status assessment a qualitative cell-based assay (bioassay) was exploited.
The bioassay recorded the basal beating rate and the chronotropic response of spontaneously beating cultured neonatal cardiomyocytes to beating rate-influencing agents, such as GPCR-agonists and fGPCR-AAbs.
For this study, adapted to semiautomatic cell-beat-rate recording [17], the assay was used to identify functionally active autoantibodies against the alpha1-adrenoceptor (fα1-AR AAb), the beta1-adrenoceptor (fβ1-AR AAb), the beta2-adrenoceptor (fβ2-AR AAb) and the endothelin ETA-receptor (fETA AAb). The first three caused a positive chronotropic cell response and the latter a negative chronotropic response. The exact assignment of the chronotropic effect to the specific autoantibodies was made through neutralising the antibody effect by administering 0.1 µmol/L of specific receptor blockers: prazosin, bisoprolol, ICI 118.551 and BQ 123 for fα1-AR AAb, fβ1-AR AAb, fβ2-AR AAb and fETA AAb, respectively [18]. For this purpose, the blocker was added after the AAb developed its chronotropic effect on the cells. A subsequent neutralisation of the chronotropic effect by the specific blocker assigned the fGPCR-AAb to the respective receptor [19, 20].
Statistics
All statistical analyses were performed using SAS® Version 9.2 or higher (SAS Institute Inc., Cary, North Carolina, USA). All statistical tests were two-sided and performed at the 5% level of significance [95% confidence interval (CI) was computed]. Categorical variables were described using frequencies and percentages.
Results
Pharmacokinetics of BC 007
The mean plasma BC 007 concentration–time profiles of all single cohorts of Parts A, B and C are depicted in Figs. 2, 3, 4 in a semi-logarithmic scale.
Fig. 2.

Mean plasma BC 007 concentration–time profiles of Part A, bolus with infusion (inf): solid curve; infusion only: dotted curve; subjects who received the infusion with a bolus component, 50% of the total dose was delivered as a bolus and the remainder infused over 20 min; infusion was delivered over 20 min. a Cohort 1 (15 mg BC 007, 6 young males fGPCR-AAb-negative), b Cohort 2 (50 mg BC 007, 6 young males fGPCR-AAb-negative), c Cohort 3 (150 mg BC 007, 6 young males fGPCR-AAb-negative), d Cohort 4 (150 mg BC 007, 6 elderly males and females fGPCR-AAb-negative). Error bars indicate the standard deviation. LOQ limit of quantification (5 ng/mL), fGPCR-AAb functionally active agonistic autoantibodies against G-protein-coupled receptors
Fig. 3.

Mean plasma BC 007 concentration–time profiles of Part B, bolus with infusion (inf): solid curve; infusion only: dotted curve; in subjects who received the infusion with a bolus component, 50% of the total dose was delivered as a bolus and the remainder infused over 20 min; infusion was delivered over 20 min. a Cohort 1 (50 mg BC 007, 6 elderly males and females fGPCR-AAb-positive), b Cohort 2 (150 mg BC 007, 6 elderly males and females fGPCR-AAb positive). Error bars indicate the standard deviation. LOQ limit of quantification (5 ng/mL), fGPCR-AAb functionally active agonistic autoantibodies against G-protein-coupled receptors
Fig. 4.

Mean plasma BC 007 concentration–time profiles of Part C, bolus with infusion (inf): solid curve; infusion only: dotted curve, in subjects who received the infusion with a bolus component, 150 mg BC 007 was delivered as a bolus and the remainder infused over 40 min. All cohorts comprised six elderly male and female fGPCR-AAb-positive participants. a Cohort 1 (300 mg BC 007 in total, bolus 150 mg, infusion time 40 min), b Cohort 2 (450 mg BC 007 in total, bolus 150 mg, infusion time 40 min). c Cohort 3 (750 mg BC 007 in total, bolus 150 mg, infusion time 40 min). d Cohort 4 (1350 mg BC 007 infusion only, infusion time 75 min). e Cohort 5 (1900 mg BC 007 infusion only, infusion time 105 min). Error bars indicate the standard deviation. LOQ limit of quantification (5 ng/mL), fGPCR AAb functionally active agonistic autoantibodies against G-protein-coupled receptors
The main information obtained from a visual inspection of the plasma concentration–time curves following administration of 15, 50 and 150 mg infusions over 20 min (Part A, Fig. 2) was the rapid increase in concentrations with comparable highest concentrations for the 150 mg dose level, whether receiving a bolus dose or not. For the 15 mg dose level, the highest concentration was observed for the bolus group and the reverse was observed for the 50 mg dose.
The main information from a visual inspection of the plasma concentration–time curves following administration of 50 and 150 mg infusions over 20 min in elderly male and female subjects was, comparable to the Part A results, an obvious rapid increase of concentrations with comparable highest concentrations, whether receiving a bolus dose or not at 50 mg (Fig. 3a) and highest concentrations for the subjects without a bolus at 150 mg (Fig. 3b).
Concentrations of BC 007 declined rapidly with all dose levels below the quantifiable limit at 2-h post-dose.
The main information from a visual inspection of the BC 007 plasma concentration–time curves following administration of 300, 450 and 750 mg with a bolus (150 mg each) and without a bolus (40 min infusion), and 1350 mg (75 min) and 1900 mg (105 min) infusions only showed, comparable to Parts A and B, a rapid increase in concentrations with comparable highest concentrations for the 750 mg dose level, whether receiving a bolus dose or not (Fig. 4c). For the 300 and 450 mg dose levels, the mean highest concentration for doses with a bolus were higher than without a bolus (Fig. 4a, b). This was all expected, given the different proportion of dose administered as a bolus. At doses of 300 mg and 450 mg with a bolus, the plasma concentrations of BC 007 declined rapidly below the quantifiable limit at 2-h post-dose and at 450 mg without a bolus at 4-h post-dose. For the 750 and 1350 mg doses without a bolus, the concentrations were below the quantifiable limit by 2-h and 4-h post-dose, respectively. Concentrations were measured for at least one subject through 8-h post-dose for the 750 mg with a bolus and through 4-h post-dose for the 1900 mg.
Plasma pharmacokinetic parameters for all study participants of Parts A, B and C are given in Table 2.
Table 2.
Plasma pharmacokinetic parameters for all study participants
| Study part | Cohort no. | Dose (mg) | Mode of administration bolus/infusion (b/i) (n) | AUC(0–t) (h·ng/mL) | AUC(0–inf) (h·ng/mL) | Cmax (ng/mL) | Tmax (min) | t1/2 (min) | CL (mL/min) | Vz (mL) | λz (h−1) | C0 (ng/mL) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Randomised blinded | ||||||||||||
| A | 1 | 15 | b/i (5) | 381 | 395 | 1755 | 10 | 2.9 | 677 | 2956 | 15 | 1765 |
| − /i (1) | 262 | 262 | 858 | 20 | 2.9 | 953 | 3985 | 14 | NC | |||
| 2 | 50 | b/i (5) | 727 | 729 | 2294 | 10 | 3.5 | 1182 | 5964 | 12 | 1037 | |
| − /i (1) | 976 | 976 | 3340 | 21 | 4.2 | 854 | 5135 | 10 | NC | |||
| 3 | 150 | b/i (5) | 2062 | 2067 | 7566 | 21 | 3.3 | 1274 | 5998 | 13 | 1874 | |
| − /i (1) | 1887 | 1892 | 7063 | 10 | 3.1 | 1321 | 5941 | 13 | NC | |||
| 4 | 150 | b/i (5) | 2209 | 2211 | 7433 | 21 | 4.6 | 1182 | 8171 | 10 | 2213 | |
| − /i (1) | 2698 | 2700 | 9051 | 10 | 4.3 | 926 | 5693 | 10 | NC | |||
| Open label | ||||||||||||
| B | 1 | 50 | b/i (5) | 631 | 633 | 2931 | 10 | 3.3 | 1388 | 6658 | 13 | 2297 |
| − /i (1) | 742 | 745 | 2389 | 20 | 3.4 | 1119 | 5493 | 12 | NC | |||
| 2 | 150 | b/i (5) | 1665 | 1667 | 7625 | 1 | 3.6 | 1793 | 9089 | 12 | 8078 | |
| − /i (1) | 2105 | 2106 | 8495 | 10 | 4.2 | 1187 | 7124 | 10 | NC | |||
| Open label | ||||||||||||
| C | 1 | 300 | b/i (3) | 5717 | 5727 | 19,931 | 3 | 3.7 | 904 | 4753 | 11 | 35,694 |
| − /i (3) | 5709 | 5721 | 9453 | 10 | 3.5 | 876 | 4407 | 12 | NC | |||
| 2 | 450 | b/i (3) | 7280 | 7293 | 20,370 | 3 | 3.4 | 1043 | 5162 | 12 | 38,452 | |
| − /i (3) | 6784 | 6792 | 11,350 | 20 | 5.4 | 1167 | 9835 | 10 | NC | |||
| 3 | 750 | b/i (2) | 9782 | 9363 | 16,545 | 3.5 | 4.5 | 1335 | 8630 | 9 | 20,148 | |
| − /i (3) | 8974 | 9014 | 15,415 | 20 | 4.2 | 1436 | 8755 | 10 | NC | |||
| 4 | 1350 | − /i (5) | 18,977 | 18,990 | 18,833 | 25 | 9.9 | 1226 | 17,100 | 5 | NC | |
| 5 | 1900 | − /i (6) | 28,678 | 28,692 | 22,207 | 12 | 11 | 1187 | 18,550 | 5 | NC | |
NC not calculated, AUC area under the concentration–time curve, b bolus, i infusion, Tmax time corresponding to occurrence of maximum observed plasma concentration, t1/2 elimination half-life, CL clearance, Vz apparent volume of distribution, λz terminal elimination rate constant, C0 initial concentration at t = 0
Across all study parts, including a steadily increasing dose (15–1900 mg) and infusion time (20–105 min) and also different dosing regimen (with and without bolus), Tmax covered an expected range between 1–25 min.
The mean Cmax values increased with the dose and ranged in Part A (15–150 mg) from 1755 to 7566 ng/mL (with bolus) and 858 to 9051 ng/mL (without bolus); in Part B (50 mg, 150 mg) 2931 and 7625 ng/mL (with bolus) and 2389 and 8495 ng/mL (without bolus); in Part C (300, 450, 750 mg) from 19,931 to 20,370 ng/mL (with bolus) and 9453 to 15,415 ng/mL (without bolus), and at 1350 and 1900 mg without bolus from 18,833 to 22,207 ng/mL.
The mean AUC(0–24) values increased with the dose and ranged in Part A (15–150 mg) from 381 to 2209 h·ng/mL (with bolus) and 262 to 2698 h·ng/mL (without bolus); in Part B (50 mg, 150 mg) 631 and 1665 h·ng/mL (with bolus) and 742 and 2105 h·ng/mL (without bolus); in Part C (300, 450, 750 mg) from 5717 to 9782 h·ng/mL (with bolus) and 5709 to 8974 h·ng/mL (without bolus) and at 1350 and 1900 mg without bolus from 18,977 and 28,678 h·ng/mL.
The elimination half-life was generally short and amounted to dose-dependent increasing values between 2.9 min in the young population at lower doses and infusion rates and 11 min in the elderly population at higher doses and infusion rates, neglecting the different dosing routines. The increase in half-life with dose is likely to be dependent on the possibility of a subsequent elimination phase for a longer time at higher doses while at lower doses the half-life might be more representative of the distribution phase.
BC 007 Dose Proportionality
The relationship between AUC and dose was assessed using a power model to fit the natural logarithm of the parameter to the natural logarithm of the dose for the data of Parts B and C of the study. All data of all doses with or without bolus were included; the plot for AUC(0–t) is depicted in Fig. 5.
Fig. 5.

Relationship between BC 007 dose and mean area under the concentration–time curve (0–24 h; AUC(0–24)) following single intravenous infusions
Slope estimates revealed that the parameters AUC(0-t) and AUC(0–inf) (data not shown) increased in a dose-proportional manner across the dose range 50–1900 mg, showing slope estimates of 1.0390 (95% CI 0.9578–1.1202) and 1.0388 (95% CI 0.9563–1.1214), respectively.
With respect to C0, this parameter was affected by the increase in concentration of bolus by parallel infusion. A proper assessment of dose proportionality of C0 would require a bolus dose in the absence of infusion. The more than dose-proportional increase of the slope estimate of C0 of 1.8051 across the dose range 50–750 mg administered with a bolus, has to be, therefore, considered with caution.
Additionally, because of the change of infusion time, a power model for Cmax has not been reported.
BC 007 Urine Concentration
The fraction of the dose (fe (0–24)) excreted unchanged in urine in 24 hrs after dosing was less than 0.01% for all treatments (see Table 3). Renal clearance (CLR) was estimated to range from 0.0093 to 0.098 mL/min for doses with a bolus and 0.0 to 0.2 mL/min for subjects receiving doses without a bolus (Part A), 0.062 and 0.1 mL/min for 50 and 150 mg doses with a bolus and 0.025 and 0.055 mL/min for subjects receiving the same doses without a bolus, respectively (Part B), and 0.19 to 0.82 mL/min for doses with a bolus and 0.14 to 2.8 mL/min for subjects receiving doses without a bolus (Part C).
Table 3.
Urine pharmacokinetic parameters for all study participants
| Study Part | Cohort no. | Dose (mg) | Mode of administration bolus/infusion (b/i) (n) |
Ae (µg) (SD) | fe (0–24) | CLR (mL/min) |
|---|---|---|---|---|---|---|
| A | 1 | 15 | b/i (5) | 0.00 (0) | 0.000012 | 0.0093 |
| − /i (1) | 0.00 (NC) | 0.0 | 0 | |||
| 2 | 50 | b/i (5) | 0.59 (1.3) | 0.000012 | 0.017 | |
| − /i (1) | 0.00 (NC) | 0 | 0 | |||
| 3 | 150 | b/i (5) | 12.00 (1.1) | 0.000077 | 0.098 | |
| − /i (1) | 23.00 (NC) | 0.00015 | 0.2 | |||
| 4 | 150 | b/i (5) | 10.00 (4.1) | 0.000067 | 0.085 | |
| − /i (1) | 14.00 (NC) | 0.000093 | 0.086 | |||
| B | 1 | 50 | b/i (5) | 2.00 (1.2) | 0.00004 | 0.062 |
| − /i (1) | 1.10 (NC) | 0.000023 | 0.025 | |||
| 2 | 150 | b/i (5) | 8.10 (1.8) | 0.000054 | 0.1 | |
| − /i (1) | 7.00 (NC) | 0.000046 | 0.055 | |||
| C | 1 | 300 | b/i (3) | 100.0 (74.0) | 0.00035 | 0.34 |
| − /i (3) | 48.0 (21.0) | 0.00016 | 0.14 | |||
| 2 | 450 | b/i (3) | 82.0 (20.0) | 0.00018 | 0.19 | |
| − /i (3) | 120.0 (25.0) | 0.00027 | 0.31 | |||
| 3 | 750 | b/i (2) | 540.0 (NC) | 0.00064 | 0.82 | |
| − /i (3) | 560.0 (97.0) | 0.00074 | 1.00 | |||
| 4 | 1350 | − /i (5) | 1100.0 (330) | 0.00081 | 0.97 | |
| 5 | 1900 | − /i (6) | 4700.0 (1500) | 0.0025 | 2.80 |
NC not calculated, Ae cumulative amount of unchanged drug excreted into urine, b bolus, CLR renal clearance, fe fraction of intravenous administered drug that is excreted unchanged in urine, i infusion
BC 007 Metabolites (n–x)
According to Shaw et al. who investigated an ex vivo successive 3′-exonuclease degradation in monkey plasma and serum [21], the n–x degradation was investigated as an exploratory parameter. In the current study, at higher doses of BC 007, a 3′-exonuclease degradation was detectable down to n-3 and amounted in total to about 7% of the plasma concentration of full-length BC 007, exemplarily shown for 1900 mg BC 007 in Fig. 6.
Fig. 6.
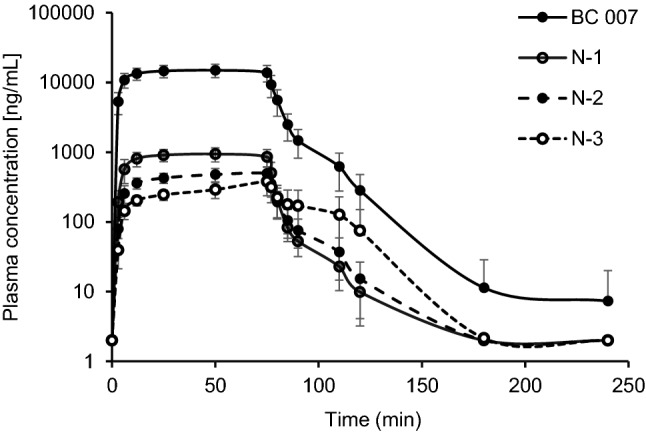
Mean plasma concentration–time profiles of 3′-exonuclease degradation products of BC 007 at the applied dose of 1900 mg
N-1 was the major metabolite of the 3′-exonulease products with an AUC0–t of 1116.3 and 1542.9 h·ng/mL at a dose of 1350 and 1900 mg BC 007, respectively. Comparable to the parent compound, the concentration declined quickly after stopping the infusion, showing a t1/2 of about 8 min.
fGPCR-AAb Status After Dosing (Mode of Action)
The fGPCR-AAb neutralisation effect showed a clear tendency of a dose–effect relationship, as shown in Table 4.
Table 4.
Dose–effect on fGPCR-AAb-neutralisation 1 day, about 1 week and 1 month after BC 007 application in healthy elderly but fGPCR-AAb-positive subjects
| Study part | Cohort no. | Dose (mg) | fGPCR-AAb status (# negative/# positive) | Effect | |||
|---|---|---|---|---|---|---|---|
| Before dosing | 1 day after dosing | 17–12 days after dosing | 28–32 days after dosing | ||||
| Part B | 1 | 50 | 0/6 | 0/6 | 0/6 | n.d. | |
| 2 | 150 | 0/6 | 1/5 | 0/6 | n.d. | (+) | |
| Part C | 1 | 300 | 0/6 | 2/4 | 1/5 | 2/4 | + |
| 2 | 450 | 0/6 | 2/4 | 2/4 | 3/3 | ++ | |
| 3 | 750 | 0/6 | 1/5 | 2/4 | 1/5 | ++ | |
| 4 | 1350 | 0/5 | 2/3 | 2/3 | 4/1 | ++ | |
| 5 | 1900 | 0/6 | 5/1 | 5/1 | 6/0 | +++ | |
(# negative/# positive) number of fGPCR-AAb-negative/number of fGPCR-AAb-positive, mg milligram, n.d. not determined, no. number, +, ++, +++ fGPCR-AAb-neutralization effect, fGPCR-AAb functionally active agonistic autoantibodies against G-protein-coupled receptors
At a dose of 1900 mg BC 007 given as an infusion over 105 min the fGPCR-AAbs were neutralised in all six treated subjects one month after dosing. At lower doses, the share of successfully fGPCR-AAb neutralised patients in the total number of treated patients was lower, as shown in Table 4. A more detailed breakdown according to the single fAAb subtypes can be taken from Müller et al. [22].
BC 007 Safety
No treatment emergent adverse event (TEAE) leading to subject discontinuation, no severe TEAEs, no serious adverse events (SAEs) and no deaths were reported throughout the entire clinical Phase I.
The most common reported adverse event was a slight-to-moderate anticoagulatory effect accompanying the infusion and seen via increased activated partial thromboplastin time (aPTT), increased prothrombin time (PT) and increased international normalised ratio (INR). This was due to the fact that the sequence of BC 007 was originally selected as a potential short-lasting thrombin inhibitor for transient anticoagulation during coronary bypass graft surgery. However, in this indication it was not successful because the lack of a persistent effect resulted in a suboptimal dosing profile; too high a dose for a therapeutic effect. This anticoagulatory effect was observed in all cohorts, beginning at a dose of 50 mg. The anticoagulatory effect normalised quickly, within minutes after the end of the infusion.
Besides this anticoagulatory effect, the following were recorded:
In Part A (24 treated subjects), no clinically significant changes in vital signs, ECG and temperature assessments were observed. Throughout all cohorts of this study part (15–150 mg BC 007), two subjects showed slightly elevated alanine aminotransferase and two subjects showed slightly elevated aspartate amino transferase. One subject reported mild palpitations and another subject a mild headache, both assessed as possibly related to investigational medical product (IMP).
In Part B (12 treated subjects, 50 and 150 mg), no clinically significant changes in vital signs, ECG and temperature assessments were observed. No adverse events with a possible relation to IMP were reported, except the above-mentioned anticoagulatory effect.
In Part C (300, 450, 750, 1350 mg and 1900 mg BC 007, Cohorts 1–5, altogether 30 treated subjects), no clinically significant changes in vital signs and temperature assessments were observed, except for one subject in Cohort 3 who showed elevated systolic and diastolic blood pressure on several measurements, which were, however, unrelated to infusion of BC 007. No clinically significant ECG changes or post-dose trends were observed in any subjects. One subject in Cohort 3 showed a QRS angle between − 15 and − 50 in all ECGs, unrelated to infusion of BC 007. ECGs with values between − 35° and − 50° were assessed as left anterior hemi block, being not clinically relevant.
Further, a first-degree atrioventricular (AV) block in one subject of Cohort 2, a first-degree AV block in another subject and prolonged QTcB/F in one subject of Cohort 4 and first degree AV block in one subject of Cohort 5, left anterior hemi block in another subject were recorded. However, all these findings were assessed as: a) not clinically relevant; and b) not related to BC 007. One subject in Cohort 1 experienced mild dizziness with a possible relation to IMP. One had mild loose stool and a third moderate nausea after the end of the bolus injection. In Cohort 4 (1350 mg), sleepiness of mild intensity in one subject was recorded. In Cohort 5 (1900 mg), four out of the six subjects experienced four AEs assessed as possibly related to IMP. However, all were the before-mentioned moderately prolonged aPTT.
In all subjects of Cohorts 4 and 5 (1350 and 1900 mg), an increase in uric acid was seen, beginning 50 min after the start of the infusion, with peak values at 120 and 110 min after the start of the infusion, respectively, with only slight elevation left on Day 2 and complete return to baseline on Day 8. This increase was within the normal physiologic range [23].
Placebo data from all cohorts were pooled within each age group (young: Cohorts 1–3 or elderly: Cohort 4) for safety analyses. One subject of the younger group experienced mild headache, an unrelated TEAE.
Discussion
Neutralisation of fGPCR-AAbs, including β1-AR-AAb, is a novel therapeutic principle, acknowledging the impact of autoantibodies on the pathogenesis of diseases of the heart and vascular system. BC 007 has shown its fGPCR-AAb-neutralising effect in vitro [12], in spontaneously hypertensive rats in vivo [16] and recently with outcome benefit in fβ1-AR-AAb positive Doberman pinschers with dilated cardiomyopathy [24]. Currently, it is under development for the causal treatment of diseases which are associated with the occurrence of fGPCR-AAbs (for review see [25]), such as HF here the β1-AR-AAb [4, 26].
Preclinical safety and pharmacology tests showed no signs of local intolerance or systemic toxicity in rats (study no. SBC007S003, data not shown) and dogs (study no. SBC007S004, data not shown).
This clinical study Phase I was designed to test the safety and tolerability of BC 007 in humans as well as the mode of action when intravenously applied. It was, therefore, also designed to find an appropriate dosing regimen for fGPCR-AAb-positive patients. With the latter, the dose was the primary focus of interest; second, was the application mode, which means infusion time and combination with or without a bolus.
Overall, the AUC increased in a dose-proportional manner across the dose range 50–1900 mg, including all data.
The plasma clearance showed values in the range 0.68–1.79 L/min, obviously not depending on the dose.
Elimination was rapid and increased with increasing dose from 2.9 min at 15 mg BC 007 in the young population up to 11 min at 1900 mg in the elderly. The increase in half-life with dose is likely to be dependent on the possibility of a subsequent elimination phase for a longer time at higher doses, while at lower doses the half-life might be more representative of the distribution phase.
The apparent volume of distribution is small but increased from about 3 L at a dose of 15 mg in the young population up to 18.5 L at 1900 mg BC 007 in the elderly, as it was derived from half-life, increasing with dose. A generally small apparent volume of distribution is typical for intravascularly confined highly water-soluble compounds when plasma-protein binding is low.
Comparing the mean values of the essential pharmacokinetic parameters between infusion with and without bolus especially in Part C (300, 450 and 750 mg BC 007; in this study part a constant bolus of 150 mg and subgroup sizes comparable), no trend for one or the other group (with or without bolus) with respect to AUC, CL and apparent distribution volume became visible, except for Tmax, of course, which was reached immediately after bolus application compared to infusion where Tmax was 10 and 20 min conforming to expectations, since Tmax is expected to be reached within the end of the infusion when a steady state is achieved based on 3–5 half-lives. The AUC(0–24) values were identical to the AUC(0–inf) for all treatments, as expected, given the short half-life of the compound.
Cmax was lower with the infusion only at the 300 and 450 mg group, (with bolus vs without bolus: 19,931 vs 9453 ng/mL and 20,370 vs 11,359 ng/mL, respectively), an effect which disappeared at 750 mg BC 007 (16,545 vs 15,415 ng/mL). Since the bolus was kept constant at 150 mg and infusion time increased, its influence diminished with the increasing total dose. In general, at the higher dose of 750 mg the differences disappeared. Restrictions with respect to the increase of the bolus were partially made as a precaution on the basis of observations by Gilbert et al. [27] who reported a: ‘hypersensitivity reaction that occurred in 1 subject at a dose…given by a rapid intravenous push’. Thereafter, the drug was diluted and ‘the rate of administration was reduced…it was given as a slow intravenous bolus…’, avoiding such incidents.
It was also decided to continue the dose escalation with infusion without bolus (1350 mg and 1900 mg) from the pharmacokinetic perspective. Doing this, the infusion time was prolonged from 40 min at 750 mg BC 007 to 75 min at 1350 mg and 105 min at 1900 mg, this way keeping the infusion rate constant at about 18 mg/min. In this way, safety and pharmacodynamic aspects were considered at the same time. From the pharmacokinetic perspective, the increased duration of BC 007 steady state was more likely to neutralise fGPCR-AAbs bound to the GPCR-receptor.
With respect to metabolism, the proportion of 7% of 3′-exonuclease cleaved compound is low, especially since Shaw et al. published an effective 3′-exonuclease decay in monkey plasma and serum in 1995 [21]. It was, therefore, also already looked for further down breakdown products, identifying the increase of nucleobase decay products immediately after the start of the infusion, which was described in detail by Davideit et al. [23]. This fast and full degradation down to nucleic base degradation products in humans was an astonishing fact and one of the first reports about the fate of DNA-drugs in humans.
Looking at the influence of age on pharmacokinetic parameters, the 150 mg groups of fGPCR-AAb-negative subjects of Part A can be compared. Following the 150 mg dose with a bolus (5 participants each), the arithmetic mean results for Cmax were slightly higher in healthy males aged 18–45 years (Cohort 3) compared to healthy males (N = 3) and females (N = 1) aged 55–70 years (Cohort 4) and the AUC parameters were slightly lower for Cohort 3. The elderly subject (N = 1) receiving 150 mg without a bolus had approximately 43% higher AUC values and a 28% higher Cmax than the young subject (N = 1) receiving 150 mg without a bolus. As expected, the clearance was approximately 43% higher for the young subject receiving 150 mg without a bolus compared to the elderly subject receiving the same treatment. However, given the limited sample size (N = 1), this difference cannot be considered statistically significant, even though lower clearance is expected in an elderly population.
The higher dose, especially of 1900 mg, showed a satisfying fGPCR-AAb-neutralising effect. In general, a trend for a dose–effect relationship became clearly visible. The fGPCR-AAb-positive participants were, however, healthy people. It is very likely that the occurrence of autoantibodies is a result of a very early autoimmune response. As previously mentioned [22], it can be speculated that, since during an early immune response, normally a multiplicity of low-avidity immunoglobulin (IgG) antibodies target a multitude of separate epitopes of a target molecule, only the subsequent clonal selection (beginning pathogenesis) increases the avidity of the antibodies and limits the number of epitopes [28]. This might determine a dosing regimen and has to be investigated in future effectivity tests when testing different indications.
Nevertheless, these existing mode of action data combined with the excellent safety profile support the application of 1350 mg BC 007 for the neutralisation of fβ1-AR-AAbs in autoantibody positive HF patients (Phase IIa, in progress, NCT04192214), compromising and combining the neutralisation of 80% of all fGPCR-AAbs (4 out of 5) one month after application and the complete neutralisation of fβ1-AR-AAbs at each tested dose beginning with 450 mg [22].
The observed good safety profile up to the highest tested amount of 1900 mg at an infusion rate of 18 mg/mL permitted the use of 1350 mg BC 007 (18 mg/mL) for testing the persistence of the fβ1-AR-AAb neutralisation in HF patients.
BC 007 proved to be safe and well tolerated. No SAEs were observed. The main drug-related adverse event was an infusion accompanying a slight-to-moderate anticoagulatory effect which normalised shortly after the end of the infusion.
Conclusion
BC 007 is a substance with a novel mode of action for the causal treatment of diseases associated with the occurrence of pathogenic functionally acting autoantibodies against G-protein coupled receptors, which mostly affect the heart and vascular system (e.g. heart failure and pulmonary hypertension). BC 007 is applied as infusion. In this Phase 1 study, BC 007 proved to be safe, well tolerated and demonstrated its ability to neutralise the autoantibodies. BC 007's half-life time was short and was between 3 and 11 min in all conditions tested. The area under the concentration–time curves (AUC) increased linearly with the dose. Based on this Phase I study results, a currently ongoing Phase IIa study has been established, testing its effect on fβ1-AR-AAb–associated heart failure patients.
Acknowledgements
This manuscript was proofread by Proof-Reading-Service, PRS.
Compliance with Ethical Standards
Funding
This study and the Open Access fee were funded by the Berlin Cures Holding AG. The authors have no other relevant affiliations or financial involvement with any organisation or entity with a financial interest in or financial conflict with the subject or materials discussed in the manuscript apart from those which are disclosed.
Conflict of interest
Niels-Peter Becker, Annekathrin Haberland, Katrin Wenzel, Peter Göttel, Gerd Wallukat, Hanna Davideit, Sarah Schulze-Rothe, Anne-Sophie Hönicke, Ingolf Schimke, Sabine Bartel, Johannes Müller, Susanne Becker are employed at Berlin Cures GmbH. Annekathrin Haberland, Peter Göttel, Gerd Wallukat, Ingolf Schimke, Johannes Müller are shareholders of Berlin Cures Holding AG, the parent company of the Berliner branch Berlin Cures GmbH. All other authors have nothing to declare.
Ethical standards
The study was conducted in accordance with the principles set forth in the Declaration of Helsinki as amended in 1996, the Guidelines of the International Council for Harmonisation (ICH) on Good Clinical Practice (GCP) (CPMP/ICH/135/95), as well as the requirements of the European Union Data Protection Directive 95/46/EC and other applicable regulatory requirements. The study was approved by the Berlin ethics commision “Landesamt für Gesundheit und Soziales Berlin, Ethikkommission des Landes Berlin”.
Informed consent
Before each subject was enrolled in this clinical study, written informed consent was obtained according to the regulatory and legal requirements of the country which hosted the study.
References
- 1.McMurray JJV, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. 2012;14:803–869. doi: 10.1093/eurjhf/hfs033. [DOI] [PubMed] [Google Scholar]
- 2.Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a Report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines developed in collaboration with the International Society for Heart And Lung Transplantation. J Am Coll Cardiol. 2009;53:e1–e90. doi: 10.1016/j.jacc.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez P, Briasoulis A. Immune modulation in heart failure: the promise of novel biologics. Curr Treat Options Cardiovasc Med. 2018;20:26. doi: 10.1007/s11936-018-0617-z. [DOI] [PubMed] [Google Scholar]
- 5.Wallukat G, Müller J, Hetzer R. Specific removal of beta1-adrenergic autoantibodies from patients with idiopathic dilated cardiomyopathy. N Engl J Med. 2002;347:1806. doi: 10.1056/NEJM200211283472220. [DOI] [PubMed] [Google Scholar]
- 6.Müller J, Wallukat G, Dandel M, Bieda H, Brandes K, Spiegelsberger S, et al. Immunoglobulin adsorption in patients with idiopathic dilated cardiomyopathy. Circulation. 2000;101:385–391. doi: 10.1161/01.CIR.101.4.385. [DOI] [PubMed] [Google Scholar]
- 7.Dandel M, Wallukat G, Englert A, Lehmkuhl HB, Knosalla C, Hetzer R. Long-term benefits of immunoadsorption in β(1)-adrenoceptor autoantibody-positive transplant candidates with dilated cardiomyopathy. Eur J Heart Fail. 2012;14:1374–1388. doi: 10.1093/eurjhf/hfs123. [DOI] [PubMed] [Google Scholar]
- 8.Bornholz B, Roggenbuck D, Jahns R, Boege F. Diagnostic and therapeutic aspects of β1-adrenergic receptor autoantibodies in human heart disease. Autoimmun Rev. 2014;13:954–962. doi: 10.1016/j.autrev.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 9.Haberland A, Muller J, Wallukat G, Wenzel K. Antigen-free control wells in an ELISA set-up for the determination of autoantibodies against G protein-coupled receptors-a requisite for correct data evaluation. Anal Bioanal Chem. 2018;410:5101–5105. doi: 10.1007/s00216-018-1172-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oaks M, Michel K, Downey FX, Thohan V. Xenoreactive antibodies and latent fibrin formation in VAD and cardiac transplant recipients can confound the detection and measurement of anti-AT1R antibodies. Am J Transplant. 2018;18:2763–2771. doi: 10.1111/ajt.14753. [DOI] [PubMed] [Google Scholar]
- 11.Jahns R, Boivin V, Hein L, Triebel S, Angermann CE, Ertl G, et al. Direct evidence for a beta 1-adrenergic receptor-directed autoimmune attack as a cause of idiopathic dilated cardiomyopathy. J Clin Invest. 2004;113:1419–1429. doi: 10.1172/JCI200420149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haberland A, Holtzhauer M, Schlichtiger A, Bartel S, Schimke I, Muller J, et al. Aptamer BC 007—a broad spectrum neutralizer of pathogenic autoantibodies against G-protein-coupled receptors. Eur J Pharmacol. 2016;789:37–45. doi: 10.1016/j.ejphar.2016.06.061. [DOI] [PubMed] [Google Scholar]
- 13.Haberland A, Wallukat G, Schimke I. The patent situation concerning the treatment of diseases associated with autoantibodies directed against G-protein-coupled receptors. Pharm Pat Anal. 2013;2:231–248. doi: 10.4155/ppa.12.88. [DOI] [PubMed] [Google Scholar]
- 14.Nnane IP, Plotnikov AH, Peters G, Johnson M, Kojak C, Vutikullird A, et al. Pharmacokinetics and safety of single intravenous doses of JNJ-54452840, an Anti-β1-adrenergic receptor antibody cyclopeptide, in healthy male Japanese and Caucasian participants. Clin Pharmacokinet. 2016;55:225–236. doi: 10.1007/s40262-015-0309-8. [DOI] [PubMed] [Google Scholar]
- 15.Stork S, Plotnikov AN, Peters G, Davies BE, Nnane I, Rivas D, et al. Efects of JNJ-54452840, an anti-β1 receptor antibody cyclopeptide in heart failure patients: a randomized, double-blind, parallel-group, phase-2 pilot study. Cardiovasc Pharmacol Open Access. 2016;5:4. doi: 10.4172/2329-6607.1000190. [DOI] [Google Scholar]
- 16.Wallukat G, Müller J, Haberland A, Berg S, Schulz A, Freyse E-J, et al. Aptamer BC007 for neutralization of pathogenic autoantibodies directed against G-protein coupled receptors: a vision of future treatment of patients with cardiomyopathies and positivity for those autoantibodies. Atherosclerosis. 2016;244:44–47. doi: 10.1016/j.atherosclerosis.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Davideit H, Haberland A, Bartel S, Schulze-Rothe S, Müller J, Wenzel K. Determination of agonistically acting autoantibodies to the adrenergic beta-1 receptor by cellular bioassay. Methods Mol Biol. 2019;1901:95–102. doi: 10.1007/978-1-4939-8949-2_8. [DOI] [PubMed] [Google Scholar]
- 18.Wallukat G, Prüss H, Müller J, Schimke I. Functional autoantibodies in patients with different forms of dementia. PLoS One. 2018;13:e0192778. doi: 10.1371/journal.pone.0192778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenzel K, Schulze-Rothe S, Haberland A, Müller J, Wallukat G, Davideit H. Performance and in-house validation of a bioassay for the determination of beta1-autoantibodies found in patients with cardiomyopathy. Heliyon. 2017;3:e00362. doi: 10.1016/j.heliyon.2017.e00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wallukat G, Wenzel K, Schimke I. Analytics of functional autoantibodies in patients with chagas disease. Methods Mol Biol. 2019;1955:247–261. doi: 10.1007/978-1-4939-9148-8_19. [DOI] [PubMed] [Google Scholar]
- 21.Shaw JP, Fishback JA, Cundy KC, Lee WA. A novel oligodeoxynucleotide inhibitor of thrombin. I. In vitro metabolic stability in plasma and serum. Pharm Res. 1995;12:1937–1942. doi: 10.1023/A:1016243923195. [DOI] [PubMed] [Google Scholar]
- 22.Muller J, Haberland A, Becker N-P, et al. The DNA-based therapeutic agent BC 007 completely neutralizes agonistic autoantibodies directed against β1-adrenoceptors: results of a phase 1 trial. J Am Coll Cardiol. 2018;71(11 Supplement):A645. doi: 10.1016/S0735-1097(18)31186-0. [DOI] [Google Scholar]
- 23.Davideit H, Becker S, Muller J, Becker N-P, Gottel P, Abay A, et al. In-vivo degradation of DNA-based therapeutic BC 007 in humans. Eur J Drug Metab Pharmacokinet. 2019;44:567–578. doi: 10.1007/s13318-019-00541-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werner S, Wallukat G, Becker N-P, et al. The aptamer BC 007 for treatment of dilated cardiomyopathy: evaluation in Doberman Pinschers of efficacy and outcomes. ESC Heart Fail. 2020 doi: 10.1002/ehf2.12628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallukat G, Schimke I. Agonistic autoantibodies directed against G-protein-coupled receptors and their relationship to cardiovascular diseases. Semin Immunopathol. 2014;36:351–363. doi: 10.1007/s00281-014-0425-9. [DOI] [PubMed] [Google Scholar]
- 26.Luft FC. Activating autoantibodies and cardiovascular disease. Physiology (Bethesda). 2013;28:254–261. doi: 10.1152/physiol.00014.2013. [DOI] [PubMed] [Google Scholar]
- 27.Gilbert JC, DeFeo-Fraulini T, Hutabarat RM, Horvath CJ, Merlino PG, Marsh HN, et al. First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation. 2007;116:2678–2686. doi: 10.1161/CIRCULATIONAHA.107.724864. [DOI] [PubMed] [Google Scholar]
- 28.DeFranco AL. The germinal center antibody response in health and disease. F1000Res. 2016 doi: 10.12688/f1000research.7717.1. [DOI] [PMC free article] [PubMed] [Google Scholar]