

Abstract
NF-κB signaling regulates diverse processes such as cell death, inflammation, immunity, and cancer. The activity of NF-κB is controlled by methionine 1-linked linear polyubiquitin, which is assembled by the linear ubiquitin chain assembly complex (LUBAC) and the ubiquitin-conjugating enzyme UBE2L3. Recent studies found that the deubiquitinase OTULIN breaks the linear ubiquitin chain, thus inhibiting NF-κB signaling. Despite the essential role of OTULIN in NF-κB signaling has been established, the regulatory mechanism for OTULIN is not well elucidated. To discover the potential regulators of OTULIN, we analyzed the OTULIN protein complex by proteomics and revealed several OTULIN-binding proteins, including LUBAC and tripartite motif-containing protein 32 (TRIM32). TRIM32 is known to activate NF-κB signaling, but the mechanism is not clear. Genetic complement experiments found that TRIM32 is upstream of OTULIN and TRIM32-mediated NF-κB activation is dependent on OTULIN. Mutagenesis of the E3 ligase domain showed that the E3 ligase activity is essential for TRIM32-mediated NF-κB activation. Further experiments found that TRIM32 conjugates polyubiquitin onto OTULIN and the polyubiquitin blocks the interaction between HOIP and OTULIN, thereby activating NF-κB signaling. Taken together, we report a novel regulatory mechanism by which TRIM32-mediated non-proteolytic ubiquitination of OTULIN impedes the access of OTULIN to the LUBAC and promotes NF-κB activation.
Keywords: NF-κB, LUBAC, TNF, linear ubiquitination, TRIM, proteomics
Introduction
The Methionine 1 (Met1)-linked linear ubiquitination, a post-translational modification of the canonical NF-κB signaling pathway, is critical for NF-κΒ-dependent inflammatory signaling and immune responses (Shimizu et al., 2015). The linear ubiquitin chain is assembled by the linear ubiquitin chain assembly complex (LUBAC), a ubiquitin E3 ligase complex comprising HOIP (also known as RNF31), HOIL-1L (also known as RBCK1), and SHARPIN (Haas et al., 2009; Tokunaga et al., 2009; Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011) together with the E2 conjugating enzyme UBE2L3 (Fu et al., 2014). Linear polyubiquitin plays a critical role in regulating the canonical NF-κB pathway, such as TNFα, IL-1, CD40, and TLR signaling pathways (Haas et al., 2009; Tokunaga et al., 2009; Hostager et al., 2010; Gerlach et al., 2011; Ikeda et al., 2011; Tokunaga et al., 2011; Zak et al., 2011). Linear ubiquitin is also involved in NOD2-mediated inflammatory signaling (Damgaard et al., 2012) and genotoxic stress (Niu et al., 2011).
The OTU domain-containing deubiquitinase with linear linkage specificity (OTULIN), also known as FAM105B, specifically cleaves the Met1-linked and head-to-tail linear polyubiquitin. Genetic studies have shown a critical role of OTULIN in limiting the linear polyubiquitination. Excessive linear polyubiquitin is found in OTULIN knockout mice, which is lethal due to acute systemic inflammation (Damgaard et al., 2016). OTULIN deficiency in humans causes severe autoinflammatory syndrome (Damgaard et al., 2016; Zhou et al., 2016). Recent studies showed that OTULIN interacts with HOIP via its PUB-interacting motif (PIM), which binds to the N-terminal PUB domain of HOIP. Phosphorylation of the tyrosine 56 prevents OTULIN from binding to HOIP (Elliott et al., 2014; Schaeffer et al., 2014). However, the tyrosine kinase for the phosphorylation and whether this phosphorylation is induced by external signals are unknown. Furthermore, whether there are other post-translational modifications of OTULIN is also not clear.
Tripartite motif-containing protein 32 (TRIM32) is a member of the tripartite motif family, which possesses an N-terminal RING domain that confers E3 ubiquitin ligase activity, a B-box domain, a coiled-coil region, and a C-terminal NHL repeats domain that mediates protein interaction. TRIM32 has multi-faceted roles in diverse physiological processes, such as muscular dystrophy (Schoser et al., 2005; Guglieri et al., 2008; Saccone et al., 2008; Neri et al., 2013), tumorigenesis (Horn et al., 2004; Albor and Kulesz-Martin, 2007; Kano et al., 2008), the etiology of psoriasis (Liu et al., 2010), neuronal development and differentiation (Kudryashova et al., 2009; Schwamborn et al., 2009; Sato et al., 2011; Hillje et al., 2013; Lionel et al., 2014), innate immunity (Zhang et al., 2012; Yang et al., 2017), and host intrinsic immune defense to viral infection (Fu et al., 2015). Genetic mutations in the TRIM32 NHL repeats cause recessive hereditary muscle disorders, including limb-girdle muscular dystrophy 2H (LGMD2H) (Schoser et al., 2005; Guglieri et al., 2008; Saccone et al., 2008; Neri et al., 2013). Similar phenotypes are observed in the TRIM32 knockout mice (Kudryashova et al., 2009; Nicklas et al., 2012) and the knock-in mice that carry a disease associated TRIM32 mutation (Kudryashova et al., 2011). It has been shown that the NF-κB pathway is perturbed in limb-girdle muscular dystrophy (LGMD) (Baghdiguian et al., 1999). More interestingly, TRIM32 knockout mice also develop to an atopic dermatitis-like inflammatory skin condition due to impaired NF-κB activity (Liu et al., 2017b). Others and we have shown that overexpression of TRIM32 strongly activates NF-κB activity (Albor et al., 2006; Li et al., 2011). However, the molecular basis for the role of TRIM32 in NF-κB signaling pathway is yet to be determined.
Here, we show that TRIM32 activates NF-κB by non-proteolytic ubiquitination of OTULIN. TRIM32 interacts with OTULIN through the C-terminal NHL repeats. TRIM32 with mutations in the NHL repeat abolishes the interaction with OTULIN and NF-κB activation. More importantly, the E3 ubiquitin ligase activity is essential for TRIM32-mediated NF-κB activation. Mechanistic studies found that TRIM32 conjugates K63-linked polyubiquitin onto OTULIN, and the polyubiquitin blocks the interaction between OTULIN and HOIP, thereby activating NF-κB signaling. Thus, we report a new post-translational modification of OTULIN, which is critical for the regulation of NF-κB activity.
Results
Proteomic analysis of the OTULIN protein complex
Although the role of OTULIN in NF-κB pathway has been established, the regulatory mechanism for OTULIN is not well elucidated. To discover potential new regulators of OTULIN, we analyzed the OTULIN protein complex by affinity purification coupled with mass spectrometry (AP–MS) (Wang et al., 2017). In this regard, FLAG-tagged human OTULIN was transfected into HEK293 cells to generate stable cell lines. After we obtained the stable cell lines by hygromycin selection, the OTULIN protein complexes were affinity purified using the anti-FLAG antibody and then analyzed by mass spectrometry (Figure 1A). The AP–MS was biologically repeated. To efficiently reduce false positives in AP–MS, we adopted the well-established statistical method SAINT (Choi et al., 2011; Choi et al., 2012) and compared OTULIN proteomics dataset with a database of 200 protein complexes purified under identical conditions. Using a stringent statistical SAINT score cutoff of 0.89 (P < 0.01), we identified 10 high-confidence candidate interacting proteins (HCIPs) (Supplementary Table S1), including HOIP and HOIL-1 (Figure 1B). HOIP and HOIL-1 are known interactors of OTULIN (Keusekotten et al., 2013; Rivkin et al., 2013; Elliott et al., 2014; Fu et al., 2014; Schaeffer et al., 2014), which substantiates the high quality of our OTULIN protein interaction network.
Figure 1.
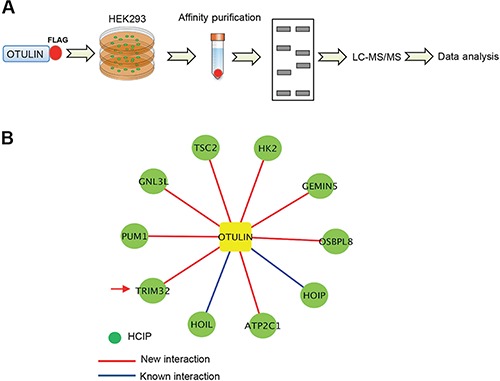
Proteomic analysis of the OTULIN protein complex. (A) Proteomic analysis pipeline. (B) The OTULIN protein interaction network. OTULIN and the HCIPs are shown as square and circles, respectively. The blue line indicates a previously known interaction and the red line indicates a new interaction. The arrow indicates TRIM32.
TRIM32 interacts and co-localizes with OTULIN
Our proteomics study found eight new OTULIN-interacting proteins, including TRIM32 (Figure 1B). We were interested in TRIM32 because others and we have shown that TRIM32 activates NF-κB reporter (Albor et al., 2006; Li et al., 2011). However, the underlying mechanism is not clear. We hypothesize that TRIM32 activates NF-κB signaling by modulation of OTULIN. To investigate this hypothesis, we first examined the interaction between TRIM32 and OTULIN by co-immunoprecipitation (co-IP). Different tagged TRIM32 and OTULIN were co-transfected into HEK293 cells. The reciprocal co-IP validated the interaction between TRIM32 and OTULIN (Figure 2A). Furthermore, we examined the interaction between endogenous OTULIN and TRIM32 in HEK293 cells treated with TNFα. After TNFα stimulation, the interaction between endogenous OTULIN and TRIM32 was increased (Figure 2B), suggesting TNFα induces OTULIN–TRIM32 interaction. We then examined the subcellular localization of FLAG-tagged TRIM32 and HA-tagged OTULIN in the lung epithelial cell line, A549 cells. Immunofluorescence assay (IFA) displayed a cytosolic co-localization between TRIM32 and OTULIN (Figure 2C). Consistently, endogenous TRIM32 also co-localized with endogenous OTULIN in the cytoplasm (Figure 2D). As both TRIM32 and OTULIN are diffusely expressed in cytoplasm, IFA cannot determine their interaction in situ. To determine the in situ interaction of OTULIN-TRIM32, we further performed proximity ligation assay (PLA). The PLA showed that TNFα increased the in situ endogenous interaction of OTULIN–TRIM32 (Figure 2E). These data demonstrate that TNFα promotes the interaction between TRIM32 and OTULIN.
Figure 2.
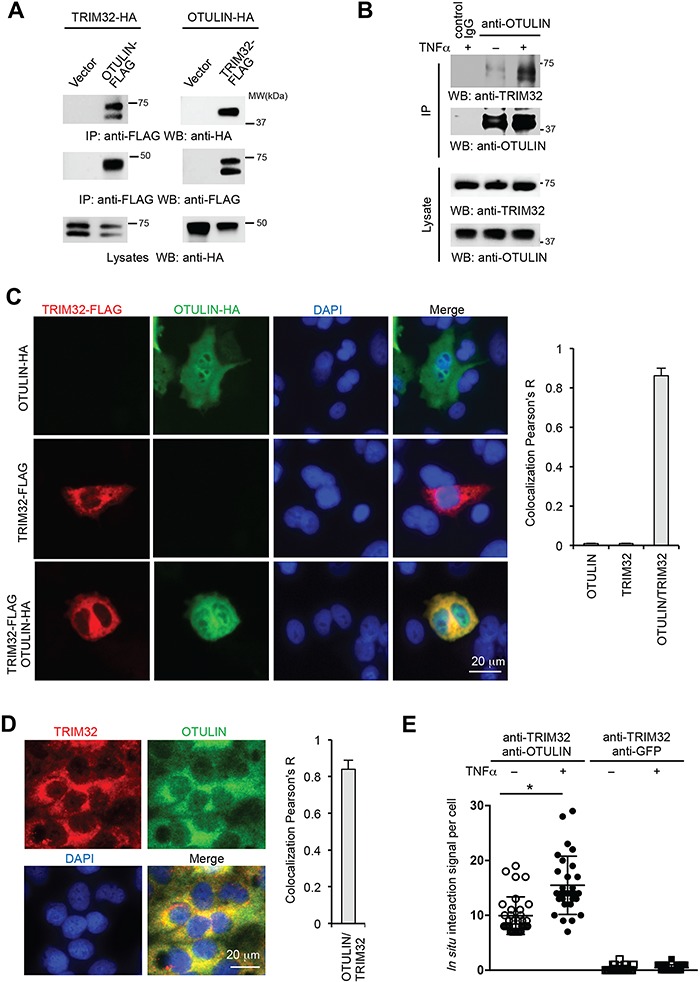
TRIM32 interacts and co-localizes with OTULIN. (A) Reciprocal immunoprecipitation between TRIM32 and OTULIN. Different tagged TRIM32 and OTULIN were co-transfected into HEK293 cells. After 48 h, cell lysates were immunoprecipitated and blotted with the indicated antibodies. (B) HEK293 cells were treated with 10 ng/ml TNFα for 30 min, and then the cell lysates were immunoprecipitated with anti-OTULIN antibody and blotted with anti-TRIM32 antibody to detect endogenous TRIM32. (C) A549 cells were transfected with FLAG-tagged TRIM32 or/and HA-tagged OTULIN. Cells were fixed and stained with the indicated antibody. Red: FLAG; green: HA; blue: DAPI, a nuclear stain. Right panel shows quantitated TRIM32–OTULIN colocalization data with three representative images. (D) A549 cells in the chamber slide were fixed with cold methanol and incubated with anti-TRIM32 and anti-OTULIN antibodies. Red: TRIM32; green: OTULIN; blue: DAPI, a nuclear stain. Right panel shows quantitated TRIM32–OTULIN colocalization data with three representative images. (E) In situ interaction between TRIM32 and OTULIN with vs. without 10 ng/ml TNFα stimulation in A549 cells determined by the PLA. The anti-GFP antibody was used as control antibody for PLA assays. *P < 0.05.
Domains required for TRIM32–OTULIN interaction
To define the domain(s) responsible for TRIM32 and OTULIN association, we first dissected the TRIM32 domains. We generated a panel of TRIM32 truncates by mutagenesis (Figure 3A) and transfected them with HA-tagged OTULIN into HEK293 cells. We found that the C-terminal five NHL repeats are sufficient to bind to OTULIN (Figure 3B). Conversely, the mutants lacking the NHL repeats lost the ability to bind to OTULIN (Figure 3B). Similarly, we made a couple of OTULIN deletion mutants (Figure 3C). These OTULIN mutants were co-transfected with TRIM32 into HEK293 cells. Co-IP showed that the OTU domain is responsible for the interaction with TRIM32 (Figure 3D). Furthermore, co-IP found that the NHL repeats (C356) interacted with the OTU domain (Figure 3E). The combined experiments indicate that the NHL repeats are necessary and sufficient for binding to OTULIN, while the OTU domain is required for TRIM32 interaction. Several mutations in the NHL repeats of TRIM32 have been found in the LGMD2H patients (Frosk et al., 2002; Saccone et al., 2008). We examined the effects of these mutations on the interaction with OTULIN. Consistent with a previous report (Locke et al., 2009), the upper band of FLAG tagged TRIM32 was mono-ubiquitinated and all three mutations (R394H, D487N, and D588 deletion) abolished the ubiquitination (Figure 3F). The monoubiquitination of TRIM32 has been found to be the consequence of self-ubiquitination of TRIM32 (Locke et al., 2009). However, the role of monoubiquitination is unknown. Co-IP further showed that these mutations impaired the interaction between TRIM32 and OTULIN (Figure 3F), suggesting a potential role of NF-κB in the LGMD2H.
Figure 3.
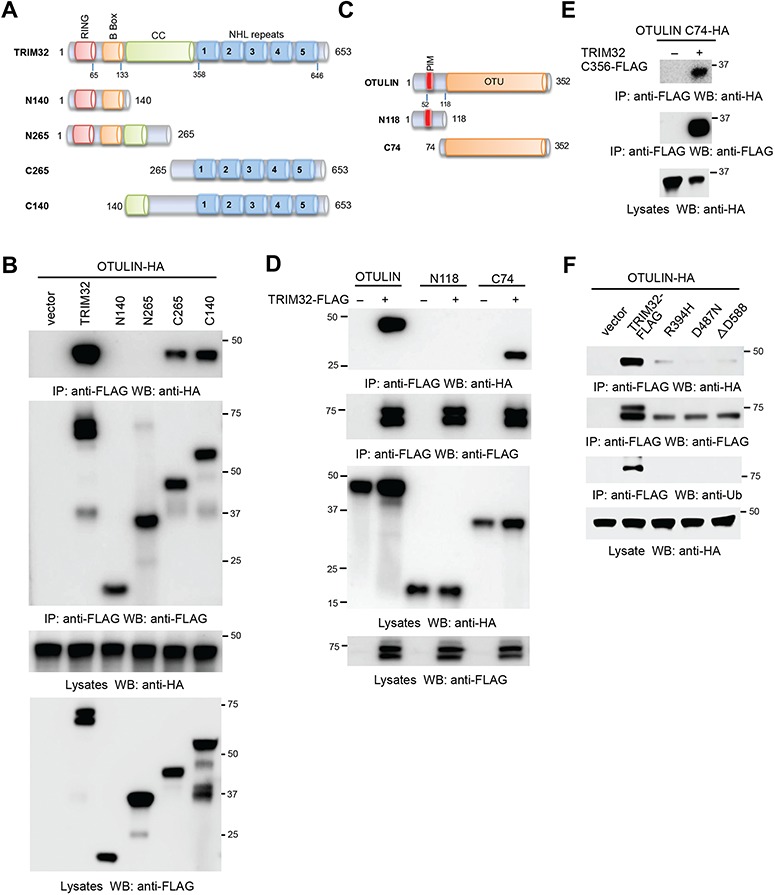
The domains required for the interaction between TRIM32 and OTULIN. (A) The schematics of TRIM32 mutants. RING, really interesting new gene; CC, coiled coil; NHL, NCL-1, HT2A, and Lin-41. (B) HA-tagged OTULIN (OTULIN-HA) was co-transfected with FLAG-tagged TRIM32 (TRIM32-FLAG) or the indicated mutant into HEK293 cells. After 48 h, cell lysates were immunoprecipitated and blotted as indicated. (C) The schematics of OTULIN mutants. (D) TRIM32-FLAG was co-transfected with OTULIN-HA or the indicated mutant into HEK293 cells. After 48 h, cell lysates were immunoprecipitated and blotted with the indicated antibodies. (E) The FLAG-tagged TRIM32 mutant (C356-FLAG) and the HA-tagged OTULIN mutant (C74-HA) were transfected into HEK293 cells. After 48 h, cell lysates were immunoprecipitated and blotted with the indicated antibodies. (F) OTULIN-HA was co-transfected with TRIM32-FLAG or the indicated point mutant into HEK293 cells. After 48 h, cell lysates were immunoprecipitated and blotted as indicated.
TRIM32 activates NF-κB via OTULIN
Our previous study found that overexpression of TRIM32 activates NF-κB activity (Li et al., 2011). To corroborate our previous findings, we examined the effects of TRIM32 deficiency on NF-κB activation. We first knocked out TRIM32 in HEK293 cells by CRISPR (Figure 4A) and treated the knockout cell lines with TNFα. The IκBα and p65 phosphorylation were reduced in the TRIM32-deficient cells (Figure 4A). Consistently, real-time PCR found that knockout of TRIM32 impaired the mRNA expression of A20 and VCAM-1 (Figures 4B and C). These data corroborate that TRIM32 positively regulates NF-κB signaling pathway.
Figure 4.
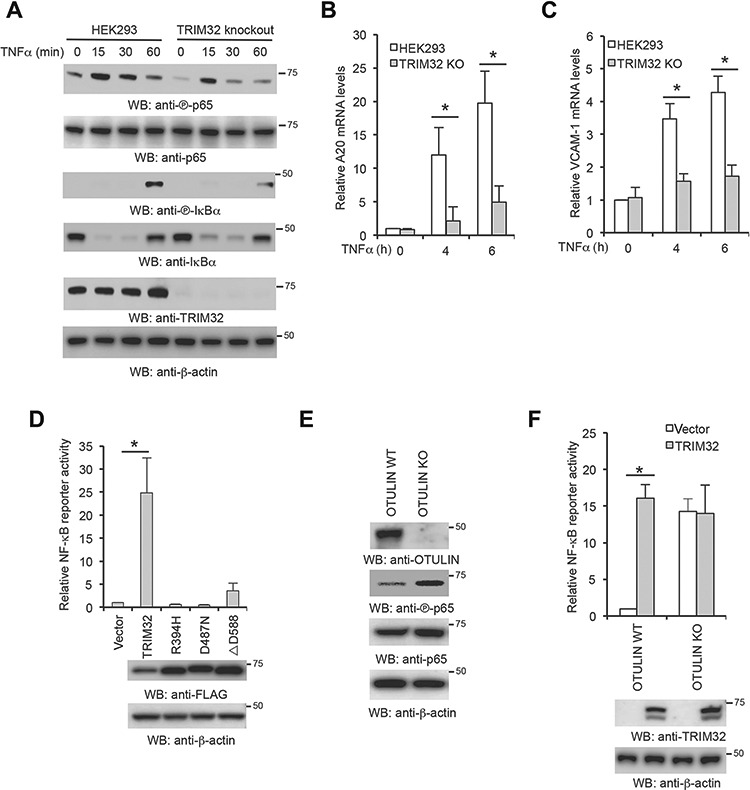
TRIM32 mediates NF-κB activation via OTULIN. (A–C) HEK293 and TRIM32 knockout cells were stimulated with 10 ng/ml TNFα for the designated times. (A) Cell lysates were blotted as indicated. (B and C) Real-time PCR was performed to determine A20 (B) and VCAM-1 (C) mRNA levels. (D) HEK293 cells were transfected with TRIM32 or the indicated mutant with the NF-κB luciferase reporter. After 48 h, luciferase activities were measured. (E) The lysates of wild-type (WT) HEK293 and the indicated OTULIN knockout (KO) generated by CRISPR were blotted with the indicated antibodies. (F) The indicated OTULIN WT and KO cells were transfected with TRIM32 together with the NF-κB luciferase reporter and pRL-SV40. After 48 h, luciferase activities were measured. Data represent mean ± SD of three independent experiments. The P-value was calculated (two-tailed Student’s t-test). *P < 0.05.
To delineate the mechanism by which TRIM32 activates NF-κB, we examined whether the interaction with OTULIN is required for TRIM32-mediated NF-κB activation. Wild-type TRIM32 or the NHL point mutants were transfected with NF-κB reporter into HEK293 cells. Interestingly, all mutants failed to activate NF-κB (Figure 4D). Since the NHL repeats are required for binding to OTULIN (Figure 3F), these data suggest that the interaction with OTULIN is critical for TRIM32 activity. To further corroborate that TRIM32-mediated NF-κB activation is dependent on OTULIN, we examined the effect of TRIM32 overexpression in OTULIN knockout cells. We first generated OTULIN knockout cell lines in HEK293 cells by CRISPR. Consistent with previous reports (Damgaard et al., 2016), knockout of OTULIN increased NF-κB activity as indicated by the phosphorylation of p65 (Figure 4E). Next, we transfected TRIM32 into OTULIN wild-type and knockout cells. TRIM32 failed to activate NF-κB in OTULIN deficient cells (Figure 4F). Taken together, these data conclude that TRIM32 is upstream of OTULIN and the interaction with OTULIN is required for TRIM32-mediated NF-κB activity.
TRIM32 ubiquitinates OTULIN in cells and in vitro
As a ubiquitin E3 ligase, the ligase activity is critical for the functions of TRIM32 in various physiological processes. However, whether the ligase activity is required for TRIM32-mediated NF-κB activation is not clear. Therefore, we made a ligase-deficient mutant by deletion of the RING domain. As shown in Figure 5A, deletion of the RING domain abolished TRIM32-induced NF-κB activity. We further made C44S/C47S mutation, which disrupts the rigid cross-braced architecture of the RING domain and destroys E3 ligase activity. Consistently, the C44S/C47S mutant failed to activate NF-κB activity (Figure 5B), suggesting the ubiquitin E3 ligase activity is required.
Figure 5.
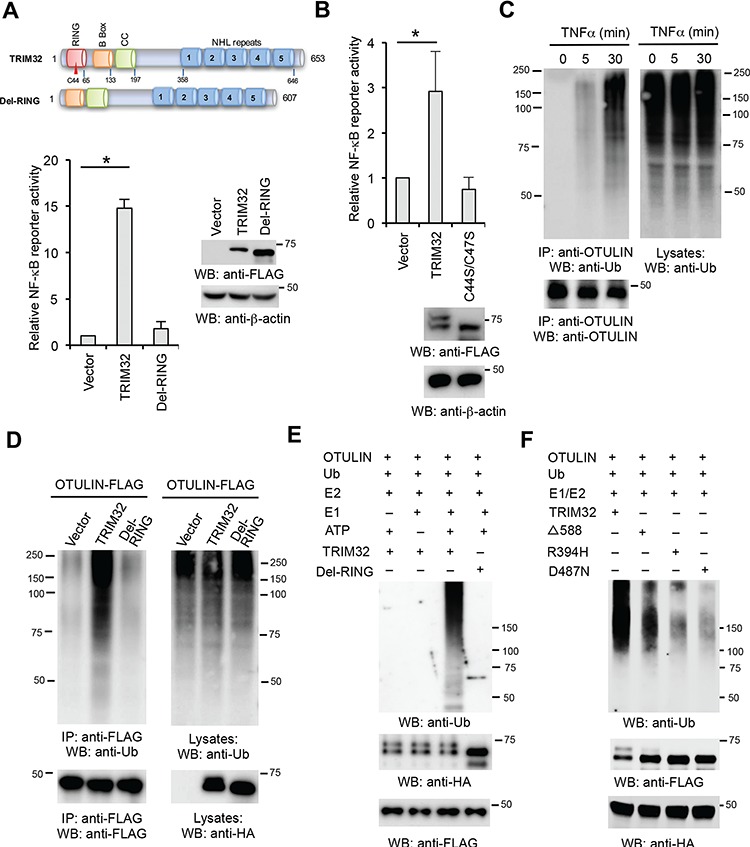
TRIM32 ubiquitinates OTULIN. (A) HEK293 cells were transfected with TRIM32 or the RING deletion mutant with the NF-κB luciferase reporter and pRL-SV40. After 48 h, luciferase activities were measured. The right panel shows protein expression levels of TRIM32 and the mutant by western blotting. (B) HEK293 cells were transfected with TRIM32 or the C44S/C47S mutant with the NF-κB luciferase reporter and pRL-SV40. After 48 h, luciferase activities were measured. Data represent mean ± SD of three independent experiments. The P-value was calculated (two-tailed Student’s t-test). *P < 0.05. The lower panel shows protein expression levels of TRIM32 and the mutant by western blotting. (C) HEK293 cells were stimulated with 10 ng/ml TNFα for the designated times. Cell lysates were immunoprecipitated with anti-OTULIN antibody and blotted as indicated. (D) OTULIN-FLAG was co-transfected with vector, TRIM32-HA, or the indicated mutant into HEK293 cells. Cell lysates were immunoprecipitated with anti-FLAG antibody and blotted with the indicated antibodies. (E) In vitro ubiquitination of OTULIN by TRIM32. FLAG-tagged OTULIN, HA-tagged TRIM32, or the indicated mutant, plus E1, E2 (UBCH5A), ATP, and ubiquitin were added as indicated and incubated at 30°C for 2 h. The reaction mixtures were immunoprecipitated with anti-FLAG antibody. After washing with 1 M urea, the OTULIN protein was eluted with FLAG peptide. The eluates were blotted with the indicated antibodies. (F) In vitro ubiquitination of OTULIN by TRIM32 and the NHL mutants. HA-tagged OTULIN, FLAG-tagged TRIM32, or the indicated mutant, plus E1, E2 (UBCH5A), ATP, and ubiquitin were added as indicated and incubated at 30°C for 2 h. The reaction mixtures were immunoprecipitated with anti-FLAG antibody. After washing with 1 M urea, the OTULIN protein was eluted with FLAG peptide. The eluates were blotted with the indicated antibodies.
The requirement of E3 ligase activity implicates that TRIM32 may ubiquitinate OTULIN. We first examined whether OTULIN ubiquitination is induced by an external signal. HEK293 cells were stimulated with TNFα followed by immunoprecipitation with anti-OTULIN antibody. OTULIN ubiquitination was barely detected in the resting cells; however, the ubiquitination of OTULIN appeared 5 min after TNFα stimulation (Figure 5C), suggesting that TNFα induces OTULIN ubiquitination. We next examined the effects of TRIM32 on the ubiquitination of OTULIN. OTULIN was transfected with TRIM32 or the RING deletion mutant into HEK293 cells. As shown in Figure 5D, TRIM32 increased the levels of OTULIN ubiquitination. However, ectopic expression of the RING deletion mutant showed little or no effect on OTULIN ubiquitination (Figure 5D). Consistently, the in vitro ubiquitination assay found that wild-type TRIM32, but not the RING deletion mutant, heavily conjugated ubiquitin onto OTULIN (Figure 5E). Similarly, the NHL point mutations, including R394H, D487N, and D588 deletion, dramatically impaired the ubiquitin conjugation activity on OTULIN in vitro (Figure 5F).
TRIM32 mediates a non-proteolytic ubiquitination of OTULIN
Proteins modified with polyubiquitin are potential targets for proteasomal degradation. To determine the role of TRIM32 in OTULIN protein turnover, we transfected HEK293 cells with different doses of TRIM32. However, TRIM32 overexpression had little effects on the protein stability of endogenous OTULIN (Figure 6A). Several ubiquitination linkages, such as K63-linked ubiquitination, have non-proteolytic roles. To determine the linkage, HEK293 cells were co-transfected with OTULIN plus wild-type ubiquitin or a ubiquitin construct in which only the indicated lysine residue is not mutated and the other six lysines are mutated to arginines. As expected, the K48-only ubiquitin was not coupled onto OTULIN while wild-type and K63-only ubiquitin were conjugated onto OTULIN (Figure 6B). We further transfected OTULIN with the K48R or K63R ubiquitin mutants into HEK293 cells. Consistently, the K48R ubiquitin, but not the K63R ubiquitin, was conjugated to OTULIN (Figure 6B), indicating that OTULIN is conjugated with K63-linked polyubiquitin.
Figure 6.
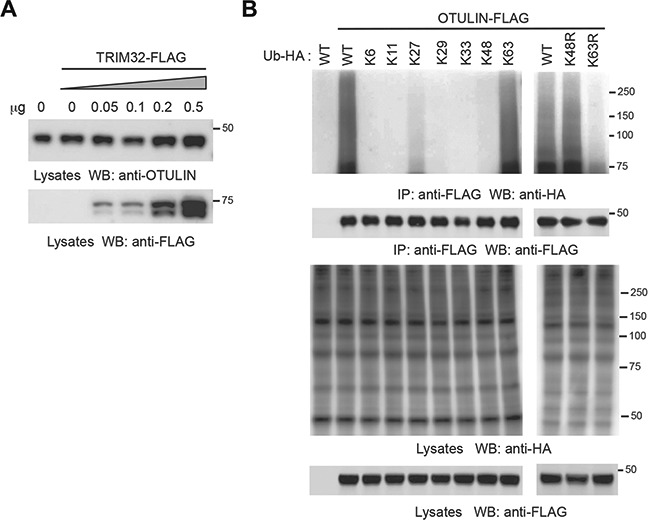
TRIM32 mediates non-proteolytic ubiquitination of OTULIN. (A) HEK293 cells were transfected with different doses of TRIM32. After 48 h, cell lysates were blotted with the indicated antibodies. (B) OTULIN-FLAG was transfected with wild-type ubiquitin or the indicated mutant into HEK293 cells. After 48 h, cell lysates were immunoprecipitated with anti-FLAG antibody and blotted as indicated.
Ubiquitination impairs OTULIN-mediated suppression of NF-κB activity
Four ubiquitination sites (K64, K66, K116, and K180) of OTULIN were detected by mass spectrometry (Kim et al., 2011; Akimov et al., 2018), but these sites have not been validated. Thus, we first mutated each of these lysines to arginine and transfected them with TRIM32 into HEK293 cells. As shown in Figure 7A, the point mutations had little effect on OTULIN ubiquitination. We further made two double-site mutations, K64R/K66R and K116R/K180R. The K64R/K66R mutant dramatically reduced TRIM32-mediated ubiquitination of OTULIN (Figure 7B), suggesting that K64 and K66 are the two major ubiquitination sites. Next, we examined the effect of K64R/K66R mutation on the function of OTULIN. Wild-type OTULIN or the mutant was transfected with the LUBAC (HOIP, HOIL-1, and SHARPIN) into HEK293 cells. Consistent with previous reports (Elliott et al., 2014; Schaeffer et al., 2014), OTULIN inhibited LUBAC-induced NF-κB activity (Figure 7C). More importantly, the K64R/K66R mutant showed much higher inhibitory activity than wild-type OTULIN (Figure 7C). These data suggest that TRIM32-mediated ubiquitination impedes OTULIN function.
Figure 7.
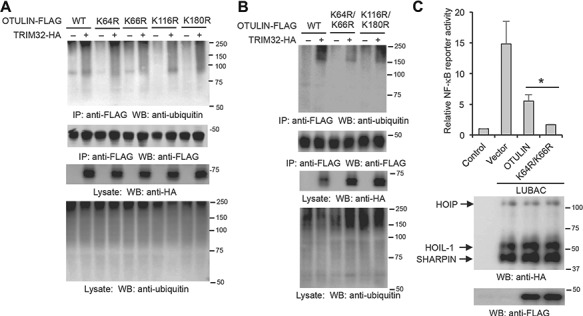
K64 and K66 are the major ubiquitination sites of OTULIN. (A and B) HEK293 cells were transfected with TRIM32 and OTULIN or the indicated OTULIN mutant. After 48 h, cell lysates were immunoprecipitated and blotted with the indicated antibodies. (C) OTULIN or the indicated mutant was transfected into HEK293 cells along with LUBAC, NF-κB luciferase reporter, and pRL-SV40. After 48 h, luciferase activities were measured. *P < 0.05. The protein expression levels of LUBAC and OTULIN were determined by western blotting.
Ubiquitination blocks the interaction between OTULIN and HOIP
As these two ubiquitination sites are proximal to the PIM motif required for HOIP interaction, we speculated that the ubiquitination disrupts the interaction between HOIP and OTULIN. To test this hypothesis, we transfected TRIM32 and HOIP together with OTULIN or the K64R/K66R mutant into HEK293 cells. Co-IP found that TRIM32 weakened the interaction between HOIP and OTULIN (Figure 8A). We next examined the effect of TRIM32 and the RING deletion mutant on the interaction between OTULIN and HOIP. As shown in Figure 8B, overexpression of TRIM32 but not the mutant impaired OTULIN–HOIP interaction. Conversely, deficiency of TRIM32 increases the association between OTULIN and HOIP (Figure 8C). We further examined the effect of TRIM32 on the OTULIN-mediated suppression of LUBAC. Consistently, wild-type TRIM32, but not the mutant, rescued LUBAC-mediated NF-κB activation (Figure 8D). Taken together, our data suggest that ubiquitination perturbs the interaction between OTULIN and HOIP, thereby blocking the access of OTULIN to the linear polyubiquitin generated by the LUBAC (Figure 8E).
Figure 8.
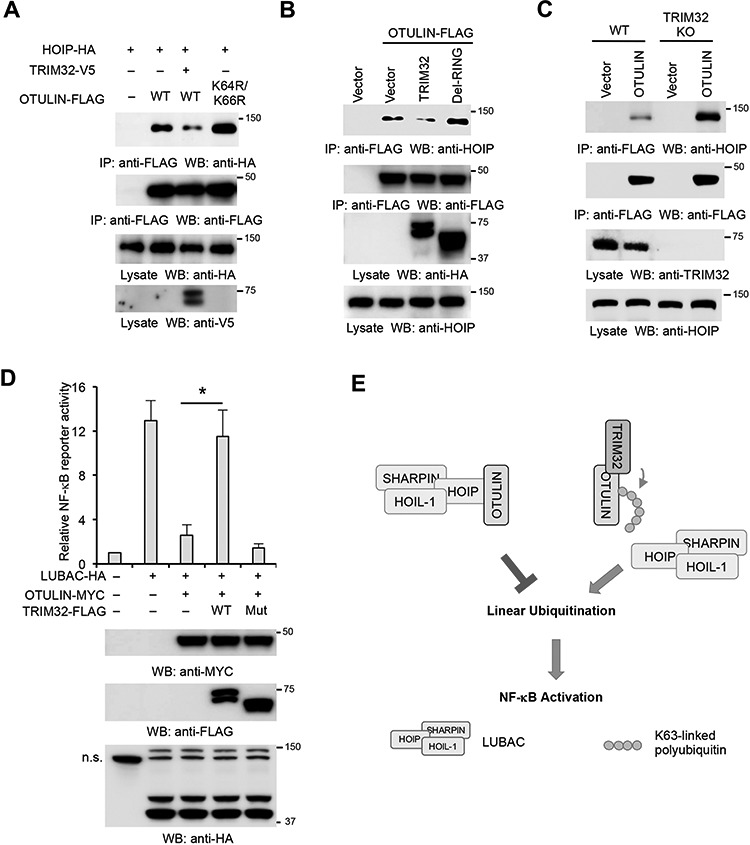
The non-proteolytic ubiquitination perturbs OTULIN–HOIP interaction. (A) HOIP, TRIM32, and OTULIN or the K64R/K66R mutant were transfected into HEK293 cells in the indicated combination. After 48 h, cell lysates were immunoprecipitated and blotted with the indicated antibodies. (B) OTULIN-FLAG was transfected with TRIM32 or the RING deletion mutant (Mut) into HEK293 cells. After 48 h, cell lysates were immunoprecipitated with anti-FLAG antibody and blotted as indicated. (C) OTULIN-FLAG was transfected into HEK293 and the TRIM32 knockout cells. After 48 h, cell lysates were immunoprecipitated with anti-FLAG antibody and blotted as indicated. (D) TRIM32 or the RING deletion mutant was transfected into HEK293 cells along with OTULIN, LUBAC, NF-κB luciferase reporter, and pRL-SV40 in the indicated combination. After 48 h, luciferase activities were measured. *P < 0.05. The protein expression levels of TRIM32, LUBAC, and OTULIN were determined by western blotting. The non-specific band (n.s.) is indicated. (E) The working model for TRIM32-mediated OTULIN ubiquitination and NF-κB activation.
Discussion
NF-κB plays an important role in the immune system and participates in cell survival, differentiation, and proliferation (Hayden and Ghosh, 2012). Upon TNFα stimulation, TNF receptor 1 (TNFR1) recruits a series of intermediary adaptors, such as TRAF2, RIP1, and LUBAC. These adaptors further recruit and activate the IKK kinase complex consisting of IKKα, IKKβ, and NEMO. The activated IKK complex phosphorylates IκBα, which leads to IκBα protein degradation and subsequent release and activation of NF-κB. In addition to phosphorylation, K63-linked ubiquitination has been shown to provide a platform for recruitment of downstream signaling molecules and stabilization of the TNFR1 signalosome. Recently, the linear polyubiquitin generated by the LUBAC has been found to be critical for NF-κB activation.
OTULIN has been shown to inhibit NF-κB signaling pathway by removal of the linear polyubiquitin (Keusekotten et al., 2013). Further studies show that OTULIN binds to the PUB domain of HOIP through its PIM motif (Elliott et al., 2014; Schaeffer et al., 2014). This interaction is regulated by tyrosine phosphorylation of OTULIN. The Y56 phosphorylation of OTULIN abolishes the interaction with HOIP. However, whether the Y56 phosphorylation is induced by TNF or other ligands is not clear. Moreover, the tyrosine kinase and phosphatase for Y56 are unknown. Here, we report another post-translational modification of OTULIN. We find that TRIM32 confers the K63-linked polyubiquitin onto OTULIN and the polyubiquitin blocks the interaction between OTULIN and LUBAC. Unlike the proteolytic polyubiquitins, K63-linked polyubiquitin mediates protein–protein interaction through the interaction between polyubiquitin and ubiquitin-associated (UBA) domain. However, our data show that the K63-linked polyubiquitin blocks the interaction even though HOIP has a UBA domain. There might be several explanations. First, the UBA domain of HOIP has been shown to interact with the ubiquitin-like domain of SHARPIN (Liu et al., 2017a). Thus, the UBA domain of HOIP is pre-occupied by SHARPIN as this interaction is critical for the integrity of the LUBAC. Secondly, as the ubiquitination sites (K64 and K66) are proximal to Y56, the polyubiquitination may cause a conformational change of PIM motif, thereby inhibiting the interaction between PIM and PUB domain. Last, the K63-linked polyubiquitin might recruit the unidentified tyrosine kinase. Further studies will examine whether TRIM32-mediated ubiquitination induces the Y56 phosphorylation of OTULIN or vice versa.
Genetic mutation of the TRIM32 B-box domain in humans is responsible for Bardet–Biedl syndrome, which has a pleiotropic phenotype often associated with retinal degeneration (Blacque and Leroux, 2006; Chiang et al., 2006). Mutations in the TRIM32 NHL domains cause recessive hereditary muscle disorders, including LGMD2H (Schoser et al., 2005; Guglieri et al., 2008; Saccone et al., 2008; Neri et al., 2013). LGMDs are characterized by progressive symmetrical atrophy and weakness of the proximal limb, scapular pelvic girdle, and trunk muscles, without affecting facial muscles (Fardeau et al., 1996). Similar phenotypes are observed in TRIM32 knockout mice (Kudryashova et al., 2009; Nicklas et al., 2012) and the knock-in mice that carry a disease-associated TRIM32 mutation (Kudryashova et al., 2011). However, the underlying mechanism is not clear. Interestingly, it has been shown that the NF-κB pathway is perturbed in the LGMD2A (Baghdiguian et al., 1999). Our study showed that the three mutations in the NHL repeats of LGMD2H patients abolished the interaction with OTULIN and NF-κB activation. Thus, it is plausible that TRIM32-mediated NF-κB activation plays a role in the LGMD2H. Future experiments will investigate the role of NF-κB in LGMD2H diseases.
TRIM32 has been reported to interact with one of the protein inhibitors of activated STAT (PIAS), PIASy (Albor et al., 2006). TRIM32 promotes PIASy ubiquitination and degradation, thereby activating NF-κB activity. Their interaction was induced by treatment with UVB/TNFα and involved redistribution of PIASy from the nucleus to the cytoplasm (Albor et al., 2006). However, NF-κB p65 is cytosolic in resting cells, how overexpression of TRIM32 activates NF-κB nuclear translocation via PIASy is not clear. Furthermore, deficiency of TRIM32 impairs p65 phosphorylation (Liu et al., 2017b); however, PIASy and PIAS1 have been shown to inhibit the DNA binding ability of p65, but not p65 activation and nuclear translocation (Liu et al., 2005; Tahk et al., 2007). These discrepancies imply that TRIM32 might also play a role at early steps of NF-κB signaling pathways. Our study now demonstrates that TRIM32 also regulates the upstream of NF-κB signaling.
In conclusion, we report a novel non-proteolytic ubiquitination of OTULIN by TRIM32. The polyubiquitin interferes with the interaction between OTULIN and HOIP, thereby preventing OTULIN from removing the linear ubiquitin generated by the LUBAC and in turn activating NF-κB signaling. This new regulatory mechanism of OTULIN may offer new opportunities for intervention in inflammatory, infectious, autoimmune diseases, and cancers.
Materials and methods
Cells
HEK293 cells (ATCC, #CRL-1573) were maintained in Dulbecco’s Modified Eagle Medium (Life Technologies) containing antibiotics (Life Technologies) and 10% fetal bovine serum (Life Technologies). A549 cells (ATCC, #CCL-185) were cultured in RPMI Medium 1640 (Life Technologies) plus 10% fetal bovine serum and 1× MEM Non-Essential Amino Acids Solution (Life Technologies).
Plasmids and antibodies
Mutants of TRIM32-FLAG and OTULIN-FLAG were constructed using a Q5® Site-Directed Mutagenesis Kit (New England Biolabs).
Primary antibodies used are as follows: anti-β-actin (Abcam, #ab8227), anti-FLAG (Sigma, #F3165), anti-ubiquitin (Cell Signaling Technology, #3933S), anti-HA (Cell Signaling Technology, #3724), anti-IκBα (Cell Signaling Technology, #4814), anti-phospho-IκBα (Ser32) (Cell Signaling Technology, #2859), anti-GFP (Abcam, #ab290), anti-NF-κB p65 (Bioss, bs-0465R), anti-phospho-NF-κB p65 (Ser536) (Cell Signaling Technology, #3033S), anti-OTULIN (Cell Signaling Technology, #14127), anti-TRIM32 (R&D Systems, AF6515), and anti-TRIM32 as described in our previous study (Hillje et al., 2011). Secondary antibodies used are as follows: goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, #sc-2055), goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, #sc-2030), Alexa Fluor 594 goat anti-mouse IgG (H + L) (Life Technologies, #A11005), and Alexa Fluor 488 goat anti-rabbit IgG (H + L) (Life Technologies, #A11034).
Sample preparation, western blotting, and immunoprecipitation
Approximately 1 × 106 cells were lysed in 500 μl of tandem affinity purification (TAP) lysis buffer [50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 0.5% Nonidet P40, 10% glycerol, complete EDTA-free protease inhibitor cocktail tablets (Roche)] for 30 min at 4°C. The lysates were then centrifuged for 30 min at 15000 × rpm. Supernatants were collected and mixed with 1× Lane Marker Reducing Sample Buffer (Thermo Fisher Scientific). Western blotting and immunoprecipitation were performed as described in a previous study (Wang et al., 2017). For ubiquitin detection, all beads were washed with 1 M urea for 15 min, three times to exclude potential binding of unanchored polyubiquitin.
Proteomics analysis of OTULIN protein complex
AP–MS experiments were performed as previously described (Li et al., 2011). For protein purification, HEK293 cells stably expressing FLAG-tagged OTULIN were collected and lysed in 10 ml of the TAP lysis buffer. Cell lysates were pre-cleared with 50 μl of protein A/G resin before the addition of 20 μl of anti-FLAG resin (Sigma) and incubated for 16 h at 4°C on a rotator. The resin was washed three times and transferred to a spin column with 40 μl of the FLAG peptide for 1 h at 4°C. The purified samples were sent for mass spectrometry analysis.
Proteins found in the control group were considered as non-specific binding proteins. The SAINT algorithm (http://sourceforge.net/projects/saint-apms) was used to evaluate the MS data. Proteins with SAINT score <0.89 are considered as non-specific binding proteins. We manually removed ribosomal proteins from the final HCIP list because these proteins are prone to associate with RNA-binding proteins.
IFA
Cells were cultured in the Lab-Tek II CC2 Chamber Slide System 4-well (Thermo Fisher Scientific). After the indicated treatment, the cells were fixed and permeabilized in cold methanol for 10 min at −20°C. Then, the slides were washed with 1× PBS for 10 min and blocked with Odyssey Blocking Buffer (LI-COR Biosciences) for 1 h. The slides were incubated in Odyssey Blocking Buffer with appropriately diluted primary antibodies at 4°C for 12 h. Images were captured and analyzed using an iRiSTM Digital Cell Imaging System (Logos Biosystems). Automated imaging software (Fiji ImageJ) with colocalization 2 application package was used to quantitate colocalization.
PLA
The PLA was performed using Duolink® In Situ Red Starter Kit Mouse/Rabbit (Sigma, #DUO92101-1KT) according to the manufacturer’s protocol.
Real-time PCR
Total RNA was prepared using the RNeasy Mini Kit (Qiagen). One microgram of RNA was reverse transcribed into cDNA using a QuantiTect reverse transcription kit (Qiagen). For one real-time reaction, 10 μl of SYBR Green PCR reaction mix (Eurogentec) including a 1/10 volume of the synthesized cDNA plus an appropriate oligonucleotide primer pair were analyzed on a 7500 Fast Real-time PCR System (Applied Biosystems). The comparative Ct method was used to determine the relative mRNA expression of genes normalized by the housekeeping gene GAPDH. The primer sequences are as follows: A20, forward primer 5′-TCACAGCTTTCCGCATATTG-3′, reverse primer 5′-GGACTTTGCGAAAGGATCG-3′; VCAM-1, forward primer 5′-GGGAAGATGGTCGTGATCCTT-3′; reverse primer 5′-TCTGGGGTGGTCTCGATTTTA-3′; GAPDH, forward primer 5′-AGGTGAAGGTCGGAGTCA-3′, reverse primer 5′-GGTCATTGATGGCAACAA-3′.
Plasmid transfection
HEK293 and A549 cells were transfected using Lipofectamine 3000 or Lipofectamine LTX Transfection Reagent (Life Technologies) according to the manufacturer’s protocol.
CRISPR/Cas9
The single guide RNA (sgRNA) targeting sequences are: human TRIM32 sgRNA, 5′-TTATGGGGGAGCTGCAGCGG-3′; OTULIN sgRNA-2, 5′-ACCTCCATACGGCGAGTCCG-3′; OTULIN sgRNA-3, 5′-CAGATCTGTGTAACCCGTCT-3′. The sgRNA was cloned into lentiCRISPR v2 vector (Sanjana et al., 2014) (Addgene). The lentiviral construct was transfected into HEK293 cells using Lipofectamine 2000. Cells were selected with 10 μg/ml puromycin for 14 days. Single clones were expanded for knockout confirmation by western blotting and DNA sequencing.
In vitro ubiquitination
In vitro ubiquitination assays were performed according to the manufacturer’s manual (Boston Biochem). Ubiquitin (3 μg), E1 (200 ng), UBCH5A (300 ng) (Boston Biochem), OTULIN-FLAG (0.5 μg) bound to the anti-FLAG resin (Sigma), and TRIM32-HA or the mutant (0.5 μg) were incubated at 30°C in the ubiquitin assay buffer (20 mM Tris-HCl, pH 7.5, plus 1× ERS buffer) for 2 h. Two hours later, the anti-FLAG resin was washed with 1 M urea for 15 min to exclude potential binding of unanchored polyubiquitin. Then the resin was incubated with 40 μl of 0.2 μg/ml FLAG peptide to elute OTULIN protein. The eluates were subsequently analyzed by SDS-PAGE followed by western blotting.
Statistical analysis
The sample size was sufficient for data analysis using paired two-tailed Student’s t-test. For all statistical analyses, differences were considered to be statistically significant at values of P < 0.05.
Supplementary Material
Funding
This work was supported by the National Institutes of Health (R15AI126360, R21AI137750, R01AI141399, and P20GM103648 to S.L.; R01AI121288 to M.E.D.), the Research Advisory Committee Fund (to L.W.), and the Oklahoma Center for the Advancement of Science and Technology (HR17-045 to S.L.).
Conflict of interest: none declared.
References
- Akimov V., Barrio-Hernandez I., Hansen S.V.F., et al. (2018). UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat. Struct. Mol. Biol. 25, 631–640. [DOI] [PubMed] [Google Scholar]
- Albor A., El-Hizawi S., Horn E.J., et al. (2006). The interaction of Piasy with Trim32, an E3-ubiquitin ligase mutated in limb-girdle muscular dystrophy type 2H, promotes Piasy degradation and regulates UVB-induced keratinocyte apoptosis through NFκB. J. Biol. Chem. 281, 25850–25866. [DOI] [PubMed] [Google Scholar]
- Albor A., and Kulesz-Martin M. (2007). Novel initiation genes in squamous cell carcinomagenesis: a role for substrate-specific ubiquitylation in the control of cell survival. Mol. Carcinog. 46, 585–590. [DOI] [PubMed] [Google Scholar]
- Baghdiguian S., Martin M., Richard I., et al. (1999). Calpain 3 deficiency is associated with myonuclear apoptosis and profound perturbation of the IκBα/NF-κB pathway in limb-girdle muscular dystrophy type 2A. Nat. Med. 5, 503–511. [DOI] [PubMed] [Google Scholar]
- Blacque O.E., and Leroux M.R. (2006). Bardet–Biedl syndrome: an emerging pathomechanism of intracellular transport. Cell. Mol. Life Sci. 63, 2145–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang A.P., Beck J.S., Yen H.J., et al. (2006). Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet–Biedl syndrome gene (BBS11). Proc. Natl Acad. Sci. USA 103, 6287–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Larsen B., Lin Z.Y., et al. (2011). SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H., Liu G., Mellacheruvu D., et al. (2012). Analyzing protein-protein interactions from affinity purification-mass spectrometry data with SAINT. Curr. Protoc. Bioinformatics Chapter 8, Unit8.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard R.B., Nachbur U., Yabal M., et al. (2012). The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol. Cell 46, 746–758. [DOI] [PubMed] [Google Scholar]
- Damgaard R.B., Walker J.A., Marco-Casanova P., et al. (2016). The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166, 1215–1230.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P.R., Nielsen S.V., Marco-Casanova P., et al. (2014). Molecular basis and regulation of OTULIN–LUBAC interaction. Mol. Cell 54, 335–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardeau M., Hillaire D., Mignard C., et al. (1996). Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 119, 295–308. [DOI] [PubMed] [Google Scholar]
- Frosk P., Weiler T., Nylen E., et al. (2002). Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 70, 663–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B., Li S., Wang L., et al. (2014). The ubiquitin conjugating enzyme UBE2L3 regulates TNFα-induced linear ubiquitination. Cell Res. 24, 376–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B., Wang L., Ding H., et al. (2015). TRIM32 senses and restricts influenza A virus by ubiquitination of PB1 polymerase. PLoS Pathog. 11, e1004960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach B., Cordier S.M., Schmukle A.C., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471, 591–596. [DOI] [PubMed] [Google Scholar]
- Guglieri M., Straub V., Bushby K., et al. (2008). Limb-girdle muscular dystrophies. Curr. Opin. Neurol. 21, 576–584. [DOI] [PubMed] [Google Scholar]
- Haas T.L., Emmerich C.H., Gerlach B., et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36, 831–844. [DOI] [PubMed] [Google Scholar]
- Hayden M.S., and Ghosh S. (2012). NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillje A.L., Pavlou M.A., Beckmann E., et al. (2013). TRIM32-dependent transcription in adult neural progenitor cells regulates neuronal differentiation. Cell Death Dis. 4, e976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillje A.L., Worlitzer M.M., Palm T., et al. (2011). Neural stem cells maintain their stemness through protein kinase Cζ-mediated inhibition of TRIM32. Stem. Cells 29, 1437–1447. [DOI] [PubMed] [Google Scholar]
- Horn E.J., Albor A., Liu Y., et al. (2004). RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis 25, 157–167. [DOI] [PubMed] [Google Scholar]
- Hostager B.S., Fox D.K., Whitten D., et al. (2010). HOIL-1L interacting protein (HOIP) as an NF-κB regulating component of the CD40 signaling complex. PLoS One 5, e11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F., Deribe Y.L., Skanland S.S., et al. (2011). SHARPIN forms a linear ubiquitin ligase complex regulating NF-κB activity and apoptosis. Nature 471, 637–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano S., Miyajima N., Fukuda S., et al. (2008). Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 68, 5572–5580. [DOI] [PubMed] [Google Scholar]
- Keusekotten K., Elliott P.R., Glockner L., et al. (2013). OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W., Bennett E.J., Huttlin E.L., et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashova E., Struyk A., Mokhonova E., et al. (2011). The common missense mutation D489N in TRIM32 causing limb girdle muscular dystrophy 2H leads to loss of the mutated protein in knock-in mice resulting in a Trim32-null phenotype. Hum. Mol. Genet. 20, 3925–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudryashova E., Wu J., Havton L.A., et al. (2009). Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 18, 1353–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wang L., Berman M., et al. (2011). Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 35, 426–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionel A.C., Tammimies K., Vaags A.K., et al. (2014). Disruption of the ASTN2/TRIM32 locus at 9q33.1 is a risk factor in males for autism spectrum disorders, ADHD and other neurodevelopmental phenotypes. Hum. Mol. Genet. 23, 2752–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Yang R., Wong K.A., et al. (2005). Negative regulation of NF-κB signaling by PIAS1. Mol. Cell. Biol. 25, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Wang Y., Gong Y., et al. (2017a). Structural insights into SHARPIN-mediated activation of HOIP for the linear ubiquitin chain assembly. Cell Rep. 21, 27–36. [DOI] [PubMed] [Google Scholar]
- Liu Y., Lagowski J.P., Gao S., et al. (2010). Regulation of the psoriatic chemokine CCL20 by E3 ligases Trim32 and Piasy in keratinocytes. J. Invest. Dermatol. 130, 1384–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wang Z., De La Torre R., et al. (2017b). Trim32 deficiency enhances Th2 immunity and predisposes to features of atopic dermatitis. J. Invest. Dermatol. 137, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke M., Tinsley C.L., Benson M.A., et al. (2009). TRIM32 is an E3 ubiquitin ligase for dysbindin. Hum. Mol. Genet. 18, 2344–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri M., Selvatici R., Scotton C., et al. (2013). A patient with limb girdle muscular dystrophy carries a TRIM32 deletion, detected by a novel CGH array, in compound heterozygosis with a nonsense mutation. Neuromuscul. Disord. 23, 478–482. [DOI] [PubMed] [Google Scholar]
- Nicklas S., Otto A., Wu X., et al. (2012). TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS One 7, e30445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J., Shi Y., Iwai K., et al. (2011). LUBAC regulates NF-NF-κB activation upon genotoxic stress by promoting linear ubiquitination of NEMO. EMBO J. 30, 3741–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkin E., Almeida S.M., Ceccarelli D.F., et al. (2013). The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature 498, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone V., Palmieri M., Passamano L., et al. (2008). Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum. Mutat. 29, 240–247. [DOI] [PubMed] [Google Scholar]
- Sanjana N.E., Shalem O., and Zhang F. (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T., Okumura F., Kano S., et al. (2011). TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 124, 3492–3502. [DOI] [PubMed] [Google Scholar]
- Schaeffer V., Akutsu M., Olma M.H., et al. (2014). Binding of OTULIN to the PUB domain of HOIP controls NF-NF-κB signaling. Mol. Cell 54, 349–361. [DOI] [PubMed] [Google Scholar]
- Schoser B.G., Frosk P., Engel A.G., et al. (2005). Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann. Neurol. 57, 591–595. [DOI] [PubMed] [Google Scholar]
- Schwamborn J.C., Berezikov E., and Knoblich J.A. (2009). The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 136, 913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y., Taraborrelli L., and Walczak H. (2015). Linear ubiquitination in immunity. Immunol. Rev. 266, 190–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahk S., Liu B., Chernishof V., et al. (2007). Control of specificity and magnitude of NF-κB and STAT1-mediated gene activation through PIASy and PIAS1 cooperation. Proc. Natl Acad. Sci. USA 104, 11643–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F., Nakagawa T., Nakahara M., et al. (2011). SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471, 633–636. [DOI] [PubMed] [Google Scholar]
- Tokunaga F., Sakata S., Saeki Y., et al. (2009). Involvement of linear polyubiquitylation of NEMO in NF-NF-κB activation. Nat. Cell Biol. 11, 123–132. [DOI] [PubMed] [Google Scholar]
- Wang L., Fu B., Li W., et al. (2017). Comparative influenza protein interactomes identify the role of plakophilin 2 in virus restriction. Nat. Commun. 8, 13876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Liu T.T., Lin H., et al. (2017). TRIM32-TAX1BP1-dependent selective autophagic degradation of TRIF negatively regulates TLR3/4-mediated innate immune responses. PLoS Pathog. 13, e1006600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak D.E., Schmitz F., Gold E.S., et al. (2011). Systems analysis identifies an essential role for SHANK-associated RH domain-interacting protein (SHARPIN) in macrophage toll-like receptor 2 (TLR2) responses. Proc. Natl Acad. Sci. USA 108, 11536–11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Hu M.M., Wang Y.Y., et al. (2012). TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J. Biol. Chem. 287, 28646–28655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Yu X., Demirkaya E., et al. (2016). Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early-onset autoinflammatory disease. Proc. Natl Acad. Sci. USA 113, 10127–10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.