

Abstract
Semisynthetic artemisinins and other bioactive peroxides are best known for their powerful antimalarial activities, and they also show substantial activity against schistosomes—another hemoglobin-degrading pathogen. Building on this discovery, we now describe the initial structure—activity relationship (SAR) of antischistosomal ozonide carboxylic acids OZ418 (2) and OZ165 (3). Irrespective of lipophilicity, these ozonide weak acids have relatively low aqueous solubilities and high protein binding values. Ozonides with para-substituted carboxymethoxy and N-benzylglycine substituents had high antischistosomal efficacies. It was possible to increase solubility, decrease protein binding, and maintain the high antischistosomal activity in mice infected with juvenile and adult Schistosoma mansoni by incorporating a weak base functional group in these compounds. In some cases, adding polar functional groups and heteroatoms to the spiroadamantane substructure increased the solubility and metabolic stability, but in all cases decreased the antischistosomal activity.
Graphical Abstract
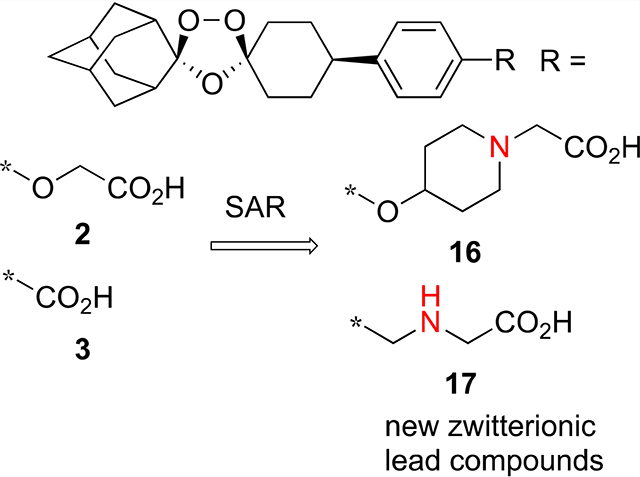
INTRODUCTION
Artemisinin and its semisynthetic derivatives dihydroartemisin, artemether, and artesunate (Figure 1) are best known for their powerful antimalarial activities,1,2 and they also show substantial activity against schistosomes—another hemoglobin-degrading pathogen.3–7 With their high activity against juvenile stage schistosomes, the semisynthetic artemisinins have promise for the chemoprophylaxis and prevention of schistosomiasis.4,6,8 In contrast, praziquantel, the only drug available for the treatment of this disease, shows little activity against the young developmental stages of the parasite and is rarely curative.4,7,8 Abundant data indicate that the pharmacophoric peroxide bond9 of semisynthetic artemisinins and other bioactive peroxides undergoes one-electron reduction by heme released during parasite hemoglobin digestion10,11 to produce carbon-centered radicals that alkylate heme and parasite proteins.10,12–21
Figure 1.

Artemisinin (ART) and its semisynthetic derivatives dihydroartemisinin (DHA), artemether (AM), and artesunate (AS).
We have shown that one class of synthetic peroxides–ozonides (1,2,4-trioxolanes)–shows promising antischistosomal activity.5 The most active of these were OZ418 (2) and OZ165 (3),22–24 carboxylic acid analogues of next-generation antimalarial ozonide OZ439 (1) (artefenomel).25 For example, single 400 mg/kg oral doses of 2 administered to Schistosoma mansoni-, Schistosoma japonicum-, or Schistosoma haematobium-infected mice reduced adult worm burden by 80, 69, and 86%, respectively.23,24 Like the semisynthetic artemisinins, ozonides 2 and 3 are even more effective against the juvenile form of the parasite; single 200 mg/kg oral doses of 2 and 3 administered to S. mansoni-infected mice reduced juvenile worm burden by 84 and 100%, respectively.24 However, with IC50 values of 44 and 39 μM against ex vivo S. mansoni and S. japonicum, 2 shows very weak in vitro activity.23,26 The structure—activity relationship (SAR) of antischistosomal ozonides has demonstrated that: (1) the spiroadamantane ring system and peroxide bond are essential for activity; (2) the core 1,2,4-trioxolane is superior to the corresponding 1,2,4-trioxane or 1,2,4,5-tetraoxane isosteres; and (3) ozonides with carboxylic acid, but not neutral or basic, functional groups show the highest antischistosomal activity. For example, 1 shows no antischistosomal activity.24 In this paper, we profile ozonides 4–25 to expand the structure–activity relationship (SAR) of 2 and 3 (Figure 2).
Figure 2.

Ozonides OZ439 (1), OZ418 (2), and OZ165 (3).
CHEMISTRY
Ozonides 4-25 were prepared as described in Schemes 1–12. Ozonide carboxylic acids 9 and 10 were prepared in 77 and 72% yields by one-pot alkylation of ozonide phenol 2627 with the corresponding bromoalkyl esters followed by hydrolysis (Scheme 1). Ozonide acylsulfonamide 13 was prepared in 62% yield by a dicyclohexylcarbodiimide (DCC)-mediated coupling of 2 with methanesulfonamide (Scheme 2). Hydroxybenzotriazole/1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (HOBt/EDC)-mediated condensation of 2 with the ethyl ester of glycine followed by ester hydrolysis afforded ozonide carboxyamide 14 in 91% yield (Scheme 2).
Scheme 1.

aReagents and conditions: 4-bromobutanoate (a) or 5-bromopentanoate (b), powdered NaOH, tetrabutylammonium hydrogensulfate, dimethoxyethane, room temperature (rt) to 60 °C, 12 h, then aq. AcOH.
Scheme 12.

aReagents and conditions: (a) O3, 10:1 cyclohexane/DCM, 0 °C, 0.25 h; (b) glycine ethyl ester hydrochloride, DIPEA, DMA, 50 °C, 5 h; (c) 1 M aq. NaOH, tetrahydrofuran (THF), rt, 12 h, then aq. AcOH; (d) methanesulfonic acid, THF, rt, 12 h.
Scheme 2.

aReagents and conditions: (a) methanesulfonamide, dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), DCM, rt, 24 h; (b) glycine ethyl ester hydrochloride, HOBt, EDC, N,N-diisopropylethylamine (DIPEA), N,N-dimethylformamide (DMF), rt, 12 h; (c) 2% aq. NaOH, EtOH, rt, 12 h, then aq. AcOH.
The reaction of ozonide acetate 2727 with N-chloroethylpi-peridine 28 according to the method of Charman et al.25 furnished ozonide ester 29 in 45% yield; hydrolysis of 29 gave ozonide piperidine carboxylic acid 15 as its sodium salt in 86% yield (Scheme 3). Alkylation of ozonide piperidine 3025 with ethyl bromoacetate produced ozonide ester 31 in 69% yield (Scheme 4). Hydrolysis of 31 afforded ozonide piperidine carboxylic acid 16 in 84% yield.
Scheme 3.

aReagents and conditions: (a) powdered NaOH, tetrabutylammonium hydrogensulfate, CH3CN, rt to 60 °C, 12 h; (b) powdered NaOH, EtOH, rt, 24 h.
Scheme 4.

aReagents and conditions: (a) ethyl bromoacetate, K2CO3, 12:1 THF/H2O, rt, 12 h; (b) 1 M aq. NaOH, EtOH, 50 °C, 20 h, then aq. AcOH.
Ozonide esters 33 and 34 were formed in 99–100% yield by alkylation of ozonide benzyl chloride 3228 with the ethyl esters of glycine and azetidine-3-carboxylic acid, respectively (Scheme 5). Ester hydrolysis of 33 and 34 followed by treatment with methanesulfonic acid afforded the aza ozonide carboxylic acids 17 and 18 as their mesylate salts in 77 and 84% yield, respectively.
Scheme 5.

aReagents and conditions: glycine ethyl ester hydrochloride (a) or ethyl azetidine-3-carboxylate hydrochloride (b), DIPEA, DMA, 50 °C, 5 h; (c) 1 M NaOH, aq. THF, rt, 12 h, then aq. AcOH; (d) methanesulfonic acid, Et2O, rt, 1 h; (e) methanesulfonic acid, EA, rt, 1 h.
Chlorination of ozonide phenol 2627 with one and nine molar equivalents of N-chlorosuccinimide (NCS) furnished the chlorinated phenol intermediates 35 (77%) and 36 (63%), respectively (Scheme 6). Alkylation of 35 and 36 with ethyl bromoacetate afforded a quantitative yield of ozonide esters 37 and 38; ester hydrolysis of the latter produced the target ozonide carboxylic acids 7 and 8 in 97 and 89% yield, respectively.
Scheme 6.

aReagents and conditions: (a) 1 equiv NCS, DMF rt, 24 h; (b) 9 equiv NCS, DMF rt, 24 h; (c) ethyl bromoacetate, K2CO3, acetone, 60 °C, 12 h; (d) 1 M aq. NaOH, THF, rt, 12 h, then aq. AcOH.
Ozonide salicylic acid 5 was synthesized in a multistep sequence starting from phenol ketal 3929 (Scheme 7). Formylation of 39 led to a quantitative yield of salicylaldehyde 40, which then underwent successive silver oxide oxidation and ketal deprotection to afford keto salicylate 41 in 96% yield. Successive Fisher esterification and acetylation of 41 furnished the keto diester 42 in 99% yield. Griesbaum coozonolysis30 of 42 and oxime ether 4331 formed ozonide diester 44 in 70% yield. Ester hydrolysis of 44 afforded the desired ozonide salicylate 5 in quantitative yield. Ozonide dicarboxylic acid 6 was obtained in 84% yield by alkylation of 5 with ethyl bromoacetate followed by hydrolysis.
Scheme 7.

aReagents and conditions: (a) paraformaldehyde, MgCl2, Et3N, THF, rt to reflux, 5 h; (b) Ag2O, H2O, 100 °C, 12 h; (c) 6 N HCl, THF, rt, 4 h; (d) acetyl chloride, MeOH, 0 °C to reflux, 4 h; (e) Ac2O, pyridine, DCM, 0 °C to rt, 5 h; (f) O3, 4:1 cyclohexane/DCM, 0 °C, 0.25 h; (g) KOH, 4:4:1 H2O/MeOH/THF, rt to 50 °C, 4 h; (h) ethyl bromoacetate, K2CO3, acetone, 60 °C, 12 h; (i) 1 M aq. NaOH, 5:1 THF/H2O, rt, 12 h, then aq. AcOH.
Coozonolysis30 of oxime ether 4728 and keto ester 46, formed by alkylation of keto phenol 45 with ethyl bromoacetate, furnished ozonide ester 48 in 15% yield (Scheme 8). Ester hydrolysis of 48 afforded the desired ozonide carboxylic acid 19 in 93% yield.
Scheme 8.

aReagents and conditions: (a) ethyl bromoacetate, K2CO3, acetone, 60 °C, 12 h; (b) O3, 5:2 cyclohexane/DCM, 0 °C, 0.25 h; (c) 15% aq. KOH, THF, rt, 12 h, then aq. AcOH.
As shown in Scheme 9, ozonide carboxylic acids 20 and 21 were synthesized from 53, a common ozonide keto phenol precursor. The synthesis of 53 began with the conversion of the mono-ketal of adamantane-2,6-dione (49)32 to its oxime ether 50 in high yield. This was followed by coozonolysis30 of 50 and keto ester 5127 to form ozonide ester ketal 52 in 43% yield. Successive ester and ketal deprotection of 52 furnished 53 in an overall yield of 89%. Alkylation of 53 with ethyl bromoacetate to form ozonide ester 54 followed by hydrolysis afforded ozonide carboxylic acid 20 in 95% overall yield. Reduction of 53 with sodium borohydride afforded the corresponding secondary alcohol 55 in 99% yield. Alkylation of 55 with ethyl bromoacetate to form ozonide ester 56 followed by hydrolysis afforded carboxylic acid 21 in 95% overall yield.
Scheme 9.

aReagents and conditions: (a) methoxyamine HCl, pyridine, EtOH, rt, 12 h; (b) O3, 3:1 cyclohexane/DCM, 0 °C, 0.25 h; (c) 1 M aq. NaOH, THF, rt, 12 h; (d) 1 M MsOH, 5:1 acetone/H2O, rt, 12 h; (e) ethyl bromoacetate, K2CO3, acetone, 55 °C, 12 h; (f) 5:1 1 M aq. NaOH/THF, rt, 12 h, then aq. AcOH; (g) NaBH4, MeOH, 0 °C, 2 h.
Reductive amination of ozonide ester 54 followed by acetylation afforded ozonide ester 57 in 80% yield (Scheme 10). Ester hydrolysis of 57 furnished ozonide acetamide carboxylic acid 22 in 94% yield. Similarly, Boc protection of the crude amino ester ozonide formed by reductive amination of 54 afforded 58 in 69% yield. Ester hydrolysis of 58 followed by treatment with methanesulfonic acid afforded ozonide amino acid 23 as its mesylate salt in 98% yield.
Scheme 10.

aReagents and conditions: (a) NaBH3CN, NH4OAc, AcOH, MeOH, rt, 12 h, then 1 M aq. NaOH; (b) acetyl chloride, pyridine, DCM, rt, 6 h; (c) 1 M aq. KOH, THF, rt, 12 h, then aq. AcOH; (d) (Boc)2O, DIPEA, DCM, 0 °C to rt, 12 h; (e) 1 M methanesulfonic acid in EtOAc, rt, 12 h.
The synthesis of ozonide 24 began with the formation of oxime ether 60 from the Boc-protected azaadamantanone 59,33 which then underwent coozonolysis30 with keto ester 51 to form ozonide ester 61 in 36% yield (Scheme 11). Ester hydrolysis of 61 furnished ozonide phenol 62 in 98% yield, which was then alkylated with ethyl bromoacetate to form ozonide ester 63 in quantitative yield. Ester hydrolysis of 63 produced ozonide carboxylic acid 64 in 90% yield. Boc deprotection of 64 with methanesulfonic acid afforded the mesylate salt of ozonide carboxylic acid 24 in quantitative yield.
Scheme 11.

aReagents and conditions: (a) methoxyamine hydrochloride, pyridine, EtOH, rt, 12 h; (b) O3, 10:1 cyclohexane/DCM, 0 °C, 0.25 h; (c) 15% aq. KOH, THF, rt, 12 h, then aq. AcOH; (d) ethyl bromoacetate, K2CO3, acetone, 60 °C, 12 h; (e) methanesulfonic acid, THF, rt, 12 h.
The synthesis of target ozonide 25 began with coozonolysis30 of oxime ether 60 and ketone 65 to form ozonide 66 in 26% yield (Scheme 12). Alkylation of glycine ethyl ester with 66 afforded ozonide ester 67 in 97% yield. Intermediate 67 then underwent successive ester hydrolysis and Boc deprotection with methanesulfonic acid to afford the dimesylate salt of ozonide carboxylic acid 25 in 82% yield. Ozonides 2–4, 11, and 12 were obtained as previously described.34–36
PHYSICOCHEMICAL, IN VITRO ABSORPTION, DISTRIBUTION, METABOLISM, AND EXCRETION (ADME), AND ANTISCHISTOSOMAL PROPERTIES
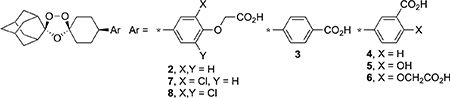
When considering the physicochemical and in vitro ADME properties of these ozonides (Tables 1–3), we note several overarching trends. First, the calculated polar surface area (PSA) values of between 68 and 117 Å2 indicate that the polarity of these compounds is unlikely to be a rate-limiting factor for membrane permeability and oral bioavailability.37 Second, most of the compounds have low aqueous solubilities even those with log D74 values ≤3. Third, these compounds had high estimated protein binding values ranging from 94.2 to >99.5%, an unsurprising result for these ozonide weak acids.
Table 1.
Physicochemical, In Vitro ADME, and Antischistosomal Data for Ozonides 2–8
 | ||||||
|---|---|---|---|---|---|---|
| compd | g log D7.4a | PSA (Å2)b | sol2.0/sol6.5 (μg/mL)c | h/m CLint (μL/(min mg) protein)d | cPPB (%)e | S. mansoni WBR (%) 1 × 400 mg/kg pof |
| 2 | 2.9 | 77.1 | <1.6/6.3–12.5 | NDg | NDh | 80i,j |
| 3 | 3.0 | 67.8 | <3.1/<1.6 | NDg | 98.8 | 74i,j |
| 4 | 3.0 | 67.8 | <1.6/<1.6 | 12/30 | 98.9 | 63 |
| 5 | 3.0 | 88.1 | <1.6/>100 | 108/20 | 99.0 | 36 |
| 6 | 1.9 | 117.2 | <3.1/>100 | <7/8 | 99.1 | 0 |
| 7 | 3.2 | 77.1 | <3.1/3.1–6.3 | <7/8 | >99.5 | 84 |
| 8 | 3.3 | 77.1 | <3.1/3.1–6.3 | NDg | ND | 32 |
log D7.4 values were estimated by chromatography.38
Calculated PSA values were generated using JChem for Excel.
Kinetic solubilities at pH 6.5 phosphate buffer and 0.01 M HCl (approx. pH 2.0).39
In vitro intrinsic clearance (CLint) values in human and mouse liver microsomes.
Protein-binding values were estimated by chromatography.40
Groups of five S. mansoni-infected NMRI mice were treated on day 49 post-infection with ozonides dissolved or suspended in 7% v/v Tween 80, 3% v/v ethanol. At 28 days post-treatment, animals were sacrificed and dissected to assess total worm burden reduction (WBR).
Analytical difficulties precluded measurement.
Poor chromatography precluded determination.
Data from Keiser et al.24
p < 0.02 from the Kruskal–Wallis test comparing the medians of the responses between the treatment and control groups.
ND = not determined.
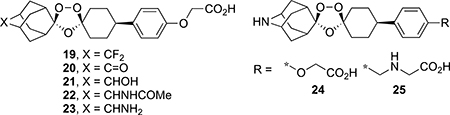
Table 3.
Physicochemical, In Vitro ADME, and Antischistosomal Data for Ozonides 19–25
 | ||||||
|---|---|---|---|---|---|---|
| compd | g log D7.4 | PSA (Å2) | sol2.0/sol6.5 (μg/mL) | h/m CLint (μL/(min mg) protein) | cPPB (%) | S. mansoni WBR (%) 1 × 400 mg/kg po |
| 19 | 2.9 | 77.1 | <1.6/12.5–25 | 23/130 | 99.4 | 57 |
| 20 | 1.9 | 94.1 | <1.6/50–100 | NDa | 97.8 | 24 |
| 21 | 1.7 | 97.3 | <1.6/25–50 | <7/<7 | 98.5 | 14 |
| 22 | 1.8 | 106.2 | 3.1–6.3/12.5–25 | 10/<7 | 98.2 | 0 |
| 23 | 1.4 | 104.7 | 6.3–12.5/<1.6 | 13/8 | 94.2 | 30 |
| 24 | 1.3 | 93.7 | 3.1–6.3/<1.6 | 35/27 | NDb | 29 |
| 25 | 1.6 | 101.0 | >100/50–100 | <7/<7 | NDb | 41 |
Apparent non-NADPH-mediated degradation.
Poor chromatography precluded determination.
The data in Table 1 show the SAR of the phenyl substructure of 2 and 3. The only compounds with high aq. solubilities at pH 6.5 were salicylate 5 and dicarboxylic acid 6; the latter was also metabolically stable. Ozonide 7, the more lipophilic chloro analogue of 2, was also metabolically stable. Ozonide 4, the meta isomer of 3, was somewhat less stable to metabolism; interestingly, addition of a phenol functional group in 5 improved the metabolic stability in mouse microsomes but substantially decreased metabolic stability in human microsomes. Of these analogues (Table 1), only 7 showed in vivo activity against chronic S. mansoni infections equal or superior to that of the two prototype ozonides 2 and 3. For comparison, at this same 400 mg/kg oral dose, dihydroartemisinin5 and praziquantel41 reduce worm burden by 66 and 96%, respectively.
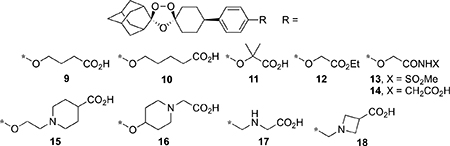
The data in Table 2 show the SAR of the carboxymethoxy substructure of 2. Extending the length of the side chain to carboxypropoxy (9) or carboxybutoxy (10) decreased aq. solubility, increased protein binding, and decreased the antischistosomal activity compared to 2. Compound 11, the gem-dimethyl analogue of 2, also had very high protein binding and was less active than 2. With the exception of ethyl ester 12 and glycine conjugate 14, the other compounds were metabolically stable; 12 was rapidly converted to 2 in both human and mouse liver microsomes, whereas 14 was converted to 2 in human but not mouse liver microsomes. Of these two compounds, only 14 showed significant antischistosomal activity. Interestingly, 14 was more and less soluble than 2 at pH 2.0 and 6.5, respectively. Acylsulfonamide 13 had the same solubility profile as 2, but showed only a modest antischistosomal activity. The amphoteric analogs 15–18 show the effects of adding a weak base functional group to the overall profiles of these ozonides. Compound 17, an aza isostere of 9, had a much superior profile than the latter. For example, 17 was less lipophilic, showed lower protein binding, and was much more active than 9 against S. mansoni in vivo. However, for the two piperidine carboxylic acids 15 and 16, only the former showed significant antischistosomal activity. Like 15, azetidine carboxylic acid 18 was less lipophilic than 2, but showed only weak antischistosomal activity.
Table 2.
Physicochemical, In Vitro ADME, and Antischistosomal Data for Ozonides 9–18
 | ||||||
|---|---|---|---|---|---|---|
| compdg | g log D7.4 | PSA (Å2) | sol2.0/sol6.5 (μg/mL) | h/m CLint (μL/(min mg) protein) | cPPB (%) | S. mansoni WBR (%) 1 × 400 mg/kg po |
| 9 | 3.6 | 77.1 | <3.1/<1.6 | <7/8 | >99.5 | 55 |
| 10 | 4.0 | 77.1 | <1.6/<1.6 | <7/13 | >99.5 | 51 |
| 11 | 3.1 | 77.1 | <1.6/3.1–6.3 | <7/7 | >99.5 | 48 |
| 12 | >5.3 | 63.2 | <1.6/<0.78 | NDa | 98.9 | 60 |
| 13 | 3.0 | 97.4 | <1.6/6.3–12.5 | <7/12 | 99.0 | 53 |
| 14 | 3.0 | 106.2 | 12.5–25/<3.1 | <7/CNDa,b | 98.5 | 76e |
| 15 | 3.8 | 81.5 | 3.1–6.3/<1.6 | <7/9 | 98.0 | 0 |
| 16 | NDc | 81.5 | NDd | <7/<7 | ND | 75e |
| 17 | 3.3 | 84.4 | <12.5/<6.3 | <7/10 | 97.6 | 90e |
| 18 | NDc | 72.3 | 6.3–12.5/<1.6 | 7/10 | ND | 43 |
Apparent non-NADPH-mediated degradation.
Putative amide hydrolysis product 2 was detected in controls and NADPH samples.
Poor chromatography precluded determination.
Poor solubility in DMSO (<1 mg/mL) precluded determination of solubility.
p < 0.02 from the Kruskal–Wallis test comparing the medians of the responses between the treatment and control groups.
CND = could not determine.
ND = not determined.
The data in Table 3 summarize the SAR of the spiroadamantane substructure of 2. Replacing the distal methylene carbon of 2 with a difluoromethylene (19) or a ketone (20) increased aq. solubility, decreased protein binding, and decreased both metabolic stability and antischistosomal activity. Decreasing lipophilicity by adding a secondary alcohol (21), acetamide (22), or primary amine (23) to the distal methylene carbon of 2 increased aq. solubility at pH 6.5 for 21 and 22, but abolished the antischistosomal activity for all three ozonides. Interestingly, the amphoteric 23 showed the lowest protein binding (94.2%) for this series of ozonides. Compounds 24 and 25, azaadamantane analogues of 2 and 17, show the consequences of replacing the methylene carbon of 2 with a nitrogen atom. Unfortunately, 24 showed low aq. solubility, intermediate metabolic stability, and low antischistosomal activity. Even though 25 was metabolically stable and was the sole ozonide in this series with high aq. solubility at both pH 2.0 and 6.5, it also showed weak antischistosomal activity.
As the data in Tables 1–3 reveal, we identified 7, 14, 16, and 17—four new ozonides with high activity against adult S. mansoni in a mouse model. As a measure of their antischistosomal selectivity, these ozonides were tested for cytotoxicity against four human cell lines: foreskin fibroblast (HFF), osteosarcoma (U-2 OS), kidney (HEK 293T), and hepatocyte (HC-04). None of the compounds inhibited these cell lines at concentrations of up to 50 μM (data not shown). Although our primary objective was to identify ozonides with high activity against the adult stage of S. mansoni, we wanted to find compounds with high activities against both adult and juvenile stages. Therefore, the most active ozonides were also tested for activity against juvenile S. mansoni in infected mice (Table 4). At a single 200 mg/kg dose, all of the compounds reduced worm burden by more than 80% in mice with juvenile stage S. mansoni infections (Table 4); of these, 2, 14, and 17 were the most effective with WBR values of 97–100%.
Table 4.
In Vivo Activity of Selected Ozonides against the Juvenile Stage of S. mansoni
Data from Keiser et al.24
p < 0.05 from the Kruskal–Wallis test comparing the medians of the responses between the treatment and control groups.
SUMMARY
In summary, these ozonide weak acids showed relatively low aqueous solubilities and high protein binding values that were independent of lipophilicity. It was possible to increase the solubility, decrease the protein binding, and maintain the high antischistosomal activity in mice infected with juvenile and adult S. mansoni by incorporating a weak base functional group in these compounds. Ozonides with para-substituted carboxymethoxy and N-benzylglycine substituents showed high antischistosomal activities. These are exemplified by 7, 14, 16, and 17—four new ozonides with log D7.4 values ranging from 3.0 to 3.3, similar to that of prototype ozonide 2 (log D7.4 of 2.9). Adding polar functional groups and heteroatoms to the spiroadamantane substructure could increase the solubility and metabolic stability, but in all cases, decreased the antischistosomal activity.
EXPERIMENTAL SECTION
General
Melting points are uncorrected. Unless otherwise noted, one-dimensional (1D) 1H and 13C NMR spectra were recorded on a 500 MHz spectrometer using CDCl3 or DMSO-d6 as solvents. All chemical shifts are reported in parts per million (ppm) and are relative to internal (CH3)4Si (0 ppm) for and CDCl3 (77.2 ppm) or DMSO-d6 (39.5 ppm) for 13C NMR. Silica gel (sg) particle size of 32–63 μm was used for all flash column chromatography experiments. Reported reaction temperatures are those of the oil bath. Gas chromatography–mass spectrometry (GCMS) data were generated with an Agilent 6890–5973N GC-MS system using a DB-5 column and helium flow rate of 1 mL/min. Combustion analysis confirmed that all target compounds have a purity of at least 95%. X-ray crystallographic data for all ozonides with 8′-aryl substituents (like 2–25) demonstrate that they have cis configurations.27,42
5-(Dispiro[adamantane-2,3′ -[1,2,4]trioxolane-5′,1″-cyclohexan]-4″-yl)-2-hydroxybenzoic acid (5)
Step 1. To a mixture of anhydrous magnesium chloride (12.21 g, 128.2 mmol), paraformaldehyde (5.78 g, 192.3 mmol), and triethylamine (17.9 mL, 128.2 mmol) in anhydrous THF (150 mL) at rt was added dropwise a solution of 4-(1,4-dioxaspiro[4.5]decan-8- yl)phenol (39)29 (15.0 g, 64.1 mmol) in THF (100 mL) under Ar. The reaction mixture was then refluxed for 5 h. After completion of the reaction, an insoluble solid was filtered and washed with THF (2 × 25 mL). After solvent removal in vacuo, the residue was dissolved in DCM (150 mL), washed with 10% HCl (3 × 25 mL) and water (3 × 25 mL), and dried over MgSO4. After filtration, the solvent was removed in vacuo to afford 2-hydroxy-5-(1,4-dioxaspiro[4.5]decan-8-yl)benzaldehyde (40) as a viscous oil (16.8 g, 100%). GCMS: m/z: 262 [M+], 1H NMR (CDCl3) δ 1.66–1.90 (m, 8H), 2.50–2.60 (m, 1H), 3.98 (s, 4H), 6.90–6.92 (m, 1H), 7.40–7.42 (m, 2H), 9.85 (s, 1H), 10.86 (s, 1H). 13C NMR (CDCl3) δ 31.4, 34.8, 41.8, 64.1, 108.1, 117.3, 120.2, 131.1, 135.9, 137.99, 159.79, 196.5. Step 2. To a stirred solution of silver nitrate (7.13 g, 41.98 mmol) in water (25 mL) was added dropwise a solution of NaOH (3.35 g, 83.96 mmol) in water (25 mL) at 0 °C. After stirring for 5 min, silver oxide was obtained as a brown semisolid. To this mixture, a solution of 40 (5.5 g, 20.99 mmol) in water (25 mL) was added and the reaction mixture was stirred at 100 °C for 12 h. After filtration of the black silver suspension, the aq. solution was neutralized (pH = ~7) with 10% HCl at 0 °C to obtain a solid that was filtered, washed with water, and dried to afford 2-hydroxy-5-(1,4-dioxaspiro[4.5]decan-8-yl)benzoic acid as a colorless solid (5.6 g, 96%). To a stirred solution of hydroxy-5-(1,4-dioxaspiro[4.5]decan-8-yl)benzoic acid (5.6 g, 20.1 mmol) in THF (75 mL) was added 6 N HCl (20 mL) dropwise at rt. After completion of the reaction, the solvent was removed in vacuo and the residue was dissolved in DCM (100 mL), washed with water (3 × 25 mL), and dried over anhydrous MgSO4. After filtration, the solvent was removed in vacuo to afford 2-hydroxy-5-(4-oxocyclohexyl)-benzoic acid (41) as a colorless solid (4.69 g, 100%). 1H NMR (DMSO-d6) δ 1.78–1.90 (m, 2H), 2.02–2.08 (m, 2H), 2.22–2.30 (m, 2H), 2.52–2.62 (m, 2H), 3.01–3.08 (m, 1H), 6.92 (d, J = 8.42 Hz, 1H), 7.47 (dd, J = 1.47, 8.42 Hz, 1H), 7.71 (d, J = 1.47 Hz, 1H), 11.2 (bs, 1H), 14.0 (bs, 1H). 13C NMR (DMSO-d6) δ 33.6, 40.8, 40.9, 112.8, 117.4, 128.1, 134.5, 136.2, 159.9, 172.2, 210.4. Step 3. To a solution of 41 (6.54 g, 27.9 mmol) in MeOH (200 mL) was added acetyl chloride (2 mL) dropwise at 0 °C. Then, the reaction mixture was heated to reflux for 4 h. Solvents were then removed in vacuo, and the residue was dissolved in DCM (100 mL), washed with water (3 × 25 mL), and dried over anhydrous MgSO4. After filtration, the solvent was removed in vacuo to afford methyl 2-hydroxy-5-(4-oxocyclohexyl)benzoate as a viscous oil (6.98 g, 100%). GCMS: m/z: 248 [M+]. Successive dropwise additions ofpyridine (11.28 mL, 139.6 mmol) and acetic anhydride (13.2 mL, 139.6 mmol) were made to a solution of methyl 2-hydroxy-5-(4-oxocyclohexyl)benzoate (6.98 g, 28.2 mmol) in DCM (50 mL) at 0 °C. The reaction mixture was stirred at rt for 5 h before solvent removal in vacuo. The residue was cooled to 0 °C to obtain a solid that was filtered, washed with water (3 × 25 mL), and dried to afford methyl 2-acetoxy-5-(4-oxocyclohexyl)benzoate (42) as a colorless solid (8.0 g, 99%). GCMS: m/z: 290 [M+], 1H NMR (CDCl3) δ 1.80–1.90 (m, 2H), 2.02–2.08 (m, 2H), 2.26 (s, 3H), 2.38–2.46 (m, 4H), 2.96–3.04 (m, 1H), 3.79 (s, 3H), 6.98 (d, J = 8.30 Hz, 1H), 7.35 (dd, J = 1.95, 8.30 Hz, 1H), 7.81 (d, J = 1.95 Hz, 1H). Step 4. A solution of 42 (5.89 g, 20.29 mmol) and O-methyl-2-adamantanone oxime (43)31 (5.45 g, 30.44 mmol) in cyclohexane (320 mL) and DCM (80 mL) at 0 °C was treated with ozone according to the method of Dong et al.42 Solvents were removed in vacuo, and the residue was triturated with 95% aq. EtOH to obtain a solid that was filtered, washed with 95% aq. EtOH, and dried to afford methyl 2-acetoxy-5-(dispiro[adamantane- 2,3/-[1,2,4]trioxolane-5/,1″-cyclohexan]-4″-yl)benzoate (44) as a colorless solid (6.5 g, 70%). Mp 132–134 °C. 1H NMR (CDCl3) δ 1.68–2.08 (m, 22H), 2.34 (s, 3H), 2.56–2.62 (m, 1H), 3.86 (s, 3H), 7.01 (d, J = 8.30 Hz, 1H), 7.38 (dd, J = 2.44 & 8.30 Hz, 1H), 7.85 (d, J = 1.95 Hz, 1H). Step 5. To a solution of 44 (2.0 g, 4.39 mmol) in 4:4:1 H2O/MeOH/THF (45 mL) was added solid KOH (2.46 g, 43.9 mmol) at rt. After the addition, the reaction mixture was stirred at 50 °C for 4 h. After cooling to rt, the reaction mixture was concentrated to 10 mL in vacuo, diluted with water (50 mL), and neutralized (pH = ~7) with 10% aq. HCl at 0 °C to obtain a solid that was filtered, washed with water, and dried to afford 5 (1.8 g, 100%) as a colorless solid. Mp 159–161 °C. 1H NMR (DMSO-d6) δ 1.46–1.56 (m, 2H), 1.64–1.96 (m, 20H), 2.43–2.52 (m, 1H), 6.55 (d, J = 8.30 Hz, 1H), 6.99 (dd, J = 2.44, 8.30 Hz, 1H), 7.52 (d, J = 1.95 Hz, 1H). 13C NMR (DMSO-d6) δ 26.0, 26.5, 31.7, 34.4, 34.5, 36.0, 36.3, 41.1, 108.5, 110.7, 115.8, 120.1, 127.8, 129.2, 133.4, 161.2, 171.8. Anal. calcd for C23H27NaO6 H2CO3: C, 59.50; H, 6.03. Found: C, 60.09; H, 5.77.
cis-Adamantane-2-spiro-3′−8′-[3-carboxy-4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (6)
Step 1. A mixture of 5 (200 mg, 0.50 mmol), ethyl bromoacetate (167 mg, 1.00 mmol), and potassium carbonate (208 mg, 1.50 mmol) in acetone (20 mL) was stirred at 60 °C for 12 h. A solid was filtered and washed with acetone, and the filtrate was concentrated in vacuo to afford cis-adamantane-2-spiro-3′−8′-[3-chloro-4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane as a colorless oil (250 mg, 87%). 1H NMR (CDCl3) δ 1.28 (m, 6H), 1.77–1.65 (m, 8H), 1.87–1.77 (m, 6H), 1.93 (d, J = 12.9 Hz, 2H), 2.07–1.97 (m, 6H), 2.54 (t, J = 12.3, 1H), 4.25 (m, 4H), 4.69 (s, 2H), 4.81 (s, 2H), 6.85 (d, J = 8.6 Hz, 1H), 7.30 (d, J = 8.4, 1H), 7.78 (s, 1H). 13C NMR (CDCl3) δ 14.1, 26.5, 26.9, 31.4, 34.6, 34.8, 36.4, 36.8, 41.88, 61.0, 61.2, 61.3, 67.0, 108.2, 111.4, 114.9, 119.9, 130.6, 131.9, 139.5, 156.4, 164.8, 167.8, 168.6. Step 2. A mixture of cis-adamantane-2-spiro-3′−8′-[3-chloro-4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (250 mg, 0. 44 mmol) and 1 M aq. NaOH (2 mL) in THF (10 mL) was stirred at rt for 12 h. The solvent was removed in vacuo, and the residue was diluted with H2O (10 mL) and acidified with AcOH (0.2 mL). The resultant precipitate was collected by filtration, washed with cold water, and dried in vacuo at 50 °C to afford 6 as a white solid (190 mg, 95%). Mp 164–165 °C. 1H NMR (DMSO-d6) δ 1.51 (q, J = 13.0, 2H), 1.98–1.62 (m, 20H), 2.59 (t, J = 12.3, 1H), 4.61 (s, 2H), 7.4 (d, J = 8.6 Hz, 1H), 7.28 (d, J = 8.2 Hz, 1H), 7.39 (s, 1H). 13C NMR (DMSO-d6) δ 26.3, 26.7, 31.6, 34.5, 34.7, 36.2, 36.6, 40.9, 68.4, 108.5, 111.1, 116.2, 124.2, 128.9, 131.0, 139.4, 155.5, 168.3, 171.7. Anal. calcd for C25H30O8 H2O: C, 63.01; H, 6.77. Found: C, 63.38; H, 6.58. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C25H30O8Na 481.1838; found 481.1838.
cis-Adamantane-2-spiro-3 ′−8 ′-[3-chloro-4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decan (7)
Step 1. A solution of N-chlorosuccinimide (1.25 g, 9.36 mmol) in dry DMF (10 mL) was added to a solution of 2627 (3.32 g, 9.31 mmol) in dry DMF (30 mL) and stirred under nitrogen at rt for 24 h protected from light. The mixture was poured into ice water and then extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with water (100 mL) and brine (25 mL), dried with MgSO4, and concentrated in vacuo to give a semisolid that was purified by chromatography (sg, 9:1 hexane:ethyl acetate) to afford 2-chloro-(4-dispiro[adamantane-2,3′-[1,2,4]trioxolane-5′,1″-cyclohexan]-4″-yl)phenol (35) (2.80 g, 77%) as a white solid. Mp 119–121 °C. 1H NMR (CDCl3) δ 1.62–2.01 (m, 22H), 2.44–2.50 (m, 1H), 5.41 (s, 1H), 6.92 (d, J= 8.0 Hz, 1H), 7.1 (dd, J = 8.0, 2.0 Hz, 1H), 7.15 (d, J = 2.0 Hz, 1H). 13C NMR (CDCl3) δ 26.7, 27.1, 31.5, 31.7, 34.7, 34.8, 34.99, 35.00, 36.6, 37.0, 42.1, 108.4, 111.7, 116.2, 119.8, 126.8, 126.9, 127.3, 139.8, 149.7. Step 2. A mixture of 35 (210 mg, 0.54 mmol), ethyl bromoacetate (180 mg, 1.08 mmol), and potassium carbonate (224 mg, 1.62 mmol) in acetone (20 mL) was stirred at 60 °C for 12 h. The solid was filtered and washed with acetone, and the filtrate was concentrated in vacuo to afford cis-adamantane-2-spiro-3′−8′-[3-chloro-4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′ -trioxaspiro[4.5]decane (37) as a colorless oil (0.257 g, 100%). 1H NMR (CDCl3) δ 7.22 (d, J = 2.1 Hz, 1H), 7.01 (dd, J = 8.5, 2.1 Hz, 1H), 6.77 (d, J = 8.5 Hz, 1H), 4.66 (s, 2H), 4.25 (q, J = 7.1 Hz, 2H), 2.48 (t, J = 12.1 Hz, 2H), 2.07–1.97 (m, 6H), 1.93 (d, J = 12.8 Hz, 2H), 1.87–1.59 (m, 15H), 1.28 (t, J = 7.2 Hz, 3H). 13C NMR (CDCl3) δ 14.1, 26.5, 26.9, 31.4, 34.6, 34.8, 36.4, 36.8, 41.8, 61.4, 66.5, 108.2, 111.4, 114.2, 123.1, 125.5, 129.0, 140.8, 151.9, 168.5. Step 3. A mixture of 37 (110 mg, 0.23 mmol) and 1 M aq. NaOH (1 mL) in THF (10 mL) was stirred at rt for 12 h. The solvent was removed in vacuo, and the residue was diluted with H2O (10 mL) and acidified with AcOH (0.2 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 7 as a white solid (100 mg, 97%). Mp 145–146 °C. 1H NMR (CDCl3) δ 2.12–1.54 (m, 22H), 2.48 (t, J =12.4 Hz, 1H), 4.67 (s, 2H), 6.79 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 7.23 (s, 1H), 8.07 (brs, 1H). 13C NMR (CDCl3) δ 26.5, 26.9, 31.4, 34.6, 34.8, 36.4, 36.8, 41.9, 66.2, 108.2, 111.5, 114.4, 123.2, 125.9, 129.1, 141.3, 151.4, 173.2. Anal. calcd for C24H29ClO6 H2O: C, 61.73; H, 6.69. Found: C, 62.00; H, 6.40.
cis-Adamantane-2-spiro-3′ −8′-[3,5-dichloro-4-(carboxymethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decan (8)
Step 1. To a solution of 26 27 (410 mg, 1.0 mmol) in dry DMF (5 mL) was added dropwise a solution of N-chlorosuccinimide (1.25 g, 9.36 mmol) in dry DMF (3 mL). The reaction mixture was stirred under nitrogen at rt for 24 h. The mixture was poured on ice and then extracted with EtOAc (2 × 20 mL). The combined organic extracts were washed with water (30 mL) and brine (20 mL), dried over anhydrous MgSO4, and concentrated in vacuo. The crude product was purified by chromatography (sg, hexane/EA, 25:1) to afford cis-adamantane-2-spiro-3′−8′-(3,5-dichloro-4-hydroxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane (36) (270 mg, 63%) as a white solid. Mp 143–145 °C. 1H NMR (400 MHz, CDCl3) 8 1.61–1.87 (m, 14H), 1.90–2.08 (m, 8H), 2.45 (tt, J = 12.5, 3.2 Hz, 1H), 5.70 (s, 1H), 7.10 (s, 2H); 13C NMR (100 MHz, CDCl3) 8 26.5, 26.9, 31.4, 34.5, 34.8, 36.4, 36.8, 41.8, 108.0, 111.6, 120.8, 126.6, 139.6, 146.0. Step 2. A mixture of 36 (270 mg, 0.63 mmol), ethyl bromoacetate (159 mg, 0.95 mmol), and potassium carbonate (175 mg, 1.26 mmol) in acetone (20 mL) was stirred at 60 °C for 12 h. The solid was removed by filtration and washed with acetone, and the filtrate was concentrated in vacuo to afford cis-adamantane-2-spiro-3′−8′-[3,5-dichloro-4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]-decane (38) (322 mg, 100%) as a white solid. Mp 99–100 °C. 1H NMR (CDCl3) δ 1.30 (t, J = 7.1 Hz, 3H), 1.58–1.69 (m, 6H), 1.691.84 (m, 8H), 1.87–1.93 (m, 2H), 1.94–2.05 (m, 6H), 2.46 (tt, J = 12.3, 3.3 Hz, 1H), 4.28 (q, J = 7.1 Hz, 2H), 4.56 (s, 2H), 7.11 (s, 2H); 13C NMR (CDCl3) δ 14.1, 26.4, 26.8, 31.1, 34.4, 34.8, 36.4, 36.7, 41.9, 61.3, 69.3, 107.8, 111.5, 127.3, 128.8, 144.3, 148.4, 167.9. Step 3. A mixture of 38 (322 mg, 0.63 mmol) and 1 N aq. NaOH (2 mL) in THF (20 mL) was stirred at rt for 12 h. The removal of the solvents in vacuo gave a white residue, which was suspended in H2O (10 mL) and acidified with AcOH to pH 3. The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 8 (270 mg, 89%) as a white solid. Mp 142–143 °C. 1H NMR (CDCl3) δ 1.61–1.87 (m, 14H), 1.90–2.09 (m, 8H), 2.49 (tt, J = 12.3, 3.4 Hz, 1H), 4.64 (s, 2H), 7.15 (s, 2H); 13C NMR (CDCl3) 8 26.5, 26.9, 31.2, 34.4, 34.8, 36.4, 36.8, 42.0, 68.9, 107.9, 111.6, 127.4, 128.7, 144.8, 148.0, 171.2. Anal. calcd for C24H28Cl2O6: C, 59.63; H, 5.84. Found: C, 59.89; H, 6.00.
cis-Adamantane-2-spiro-3′−8′-[4-(carboxypropoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (9)
To a solution of 2627 (250 mg, 0.70 mmol) in dry dimethoxyethane (25 mL) were added powdered NaOH (168 mg, 4.20 mmol) and Bu4NHSO4 (48 mg, 0.15 mmol), and the resulting mixture was stirred at rt for 30 min before addition of ethyl 4-bromobutanoate (205 mg, 1.05 mmol). The reaction mixture was stirred at 60 °C for 12 h, then concentrated in vacuo. The residue was suspended in H2O (20 mL) and acidified with AcOH to pH 3. The precipitate was filtered, washed with H2O, and dried in vacuo at 50 °C to afford 9 (240 mg, 77%) as a white solid. Mp 148–149 °C. 1H NMR (CDCl3) δ 1.87–1.57 (m, 14H), 1.94 (d, J = 12.8 Hz, 2H), 2.06–1.97 (m, 6H), 2.09 (p, J = 6.7 Hz, 2H), 2.49 (tt, J = 12.1, 3.3 Hz, 1H), 2.57 (t, J = 7.3 Hz, 2H), 3.98 (t, J = 6.1 Hz, 2H), 6.81 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3) 8 24.5, 26.5, 26.9, 30.6, 31.7, 34.77, 34.83, 36.4, 36.8, 42.1, 66.5, 108.5, 111.4, 114.4, 127.6, 138.5, 157.1, 179.2. Anal. calcd for C26H34O6: C, 70.56; H, 7.74. Found: C, 70.70; H, 7.75.
cis-Adamantane-2-spiro-3′−8′-[4-(carboxybutoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decan (10)
To a solution of 2627 (250 mg, 0.70 mmol) in dry dimethyoxyethane (25 mL) were added powdered NaOH (168 mg, 4.20 mmol) and Bu4NHSO4 (48 mg, 0.15 mmol), and the resulting mixture was stirred at rt for 30 min before the addition of methyl 5-bromopentanoate (220 mg, 1.05 mmol). The reaction mixture was stirred at 60 °C for 12 h, then concentrated in vacuo. The residue was suspended in H2O (20 mL) and acidified with AcOH to pH 3. The precipitate was filtered, washed with H2O, and dried in vacuo at 50 °C to afford 10 (230 mg, 72%) as a white solid. Mp 144–145 °C. 1H NMR (CDCl3) δ 1.87–1.61 (m, 18H), 1.94 (d, J = 12.8 Hz, 2H), 2.06–1.97 (m, 6H), 2.43 (d, J = 6.9 Hz, 2H), 2.48 (tt, J = 15.7, 5.0 Hz, 1H), 3.93 (t, J = 5.3 Hz, 2H), 6.80 (d, J = 8.4 Hz, 2H), 7.10 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3) 8 21.5, 26.5, 26.9, 28.7, 31.7, 33.8, 34.77, 34.83, 36.4, 36.8, 42.1, 67.3, 108.5, 111.4, 114.4, 127.6, 138.3, 157.3, 179.3. Anal. calcd for C27H36O6: C, 71.03; H, 7.95. Found: C, 70.84; H, 7.78.
cis-Adamantane-2-spiro-3′−8′-[4-[2-[methylsulfonamino]-2-oxoethoxy]phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (13)
To a mixture of 2 (150 mg, 0.36 mmol), methanesulfonamide (47 mg, 0.40 mmol), and DMAP (10 mg, 0.07 mmol) in DCM (20 mL) was added DCC (82 mg, 0.40 mmol). The resulting mixture was stirred at rt for 24 h. The solid was filtered, and the filtrate was concentrated in vacuo to produce a residue that was purified by chromatography (sg, DCM) to afford 13 (110 mg, 62%) as a white solid. Mp 148–149 °C; 1H NMR (CDCl3) δ 2.08–1.68 (m, 22H), 2.55 (td, J1 = 12 Hz, J2 = 3 Hz), 3.38 (s, 3H), 4.85 (s, 2H), 6.88 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.5 Hz, 2H), 8.84 (br, 1H); 13C NMR (CDCl3) δ 26.5, 26.9, 31.6, 34.7, 41.7, 42.1, 67.4, 108.3, 111.5, 114.7, 128.2, 140.9, 154.8, 167.7. Anal. calcd for C25H33NO7S 0.25 H2O: C, 60.53; H, 6.81; N, 2.82. Found: C, 60.87; H, 6.86; N, 2.84.
cis-Adamantane-2-spiro-3′−8′-[4-[2-[(carboxymethyl)amino]-2-oxoethoxy]phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (14)
Step 1. To a mixture of 2 (415 mg, 1.0 mmol) and HOBt (162 mg, 1.2 mmol) in DMF (10 mL) was added EDC (230 mg, 1.2 mmol) followed by the addition of glycine ethyl ester hydrochloride (154 mg, 1.1 mmol) and DIPEA (155 mg, 1.2 mmol). The resulting mixture was stirred at rt for 12 h; diluted with EA (50 0 mL); washed with brine (2 × 50 mL), 1 N HCl (20 mL), 1 N NaOH (20 mL), and brine (20 mL); dried over anhydrous MgSO4; filtered; and concentrated in vacuo to give a colorless residue that was dissolved in a mixture of ethanol (40 mL) and 2% aq. NaOH (4 mL). The resulting mixture was stirred at rt for 12 h and then concentrated in vacuo to afford a white residue that was suspended in H2O (50 mL), acidified with AcOH to pH 3, filtered, washed with H2O, and dried in vacuo to afford 14 (430 mg, 91%) as a white solid. Mp 147–148 °C; 1H NMR (DMSO-d6) δ 1.50–1.58 (m, 2H), 1.66–1.93 (m, 19H), 2.59 (t, J =15 Hz, 1H), 3.81 (d, J = 7.2 Hz, 2H), 4.49 (s, 2H), 6.91 (d, J = 11.2 Hz, 2H),7.15 (d, J = 11.2 Hz, 2H), 8.35 (t, J = 7.2 Hz, 1H), 12.63 (bs, 1H); 13C NMR (DMSO-d6) 8 26.3, 26.7, 31.7, 34.6, 34.7, 36.3, 36.6, 40.9, 41.2, 67.4, 108.6, 111.0, 115.2, 127.9, 139.2, 156.5, 168.7, 171.4. Anal. calcd for C26H33NO7: C, 66.23; H, 7.05; N, 2.97. Found: C, 66.40; H, 6.96; N, 2.60.
Sodium 1-(2-(4-Dispiro[adamantane-2,3′-[ 1,2,4]trioxolane-5′,1″-cyclohexan]-4″-yl)phenoxy)ethyl)piperidine-4-carboxylate (15)
Step 1. To a solution of cis-adamantane-2-spiro-3′−8′-(4′-acetoxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane (27)27 (430 mg, 1.1 mmol) in dry acetonitrile (20 mL) were added powdered NaOH (259 mg, 6.5 mmol) and tetrabutylammonium hydrogensulfate (110 mg, 0.3 mmol) according to the method of Charman et al.25 The mixture was stirred at rt for 0.5 h. Then, ethyl 1-(2-chloroethyl)piperidine-4-carboxylate (28)43 (475 mg, 2.2 mmol) was added and the reaction stirred at 60 °C for 12 h. The inorganic solid was filtered off and washed with CH2O2. After removal of the solvents in vacuo, the residue was dissolved in EtOAc (50 mL). The organic layer was washed with water and brine and dried over MgSO4. The removal of the solvent in vacuo afforded ethyl 1-(2-(4-dispiro[adamantane-2,3′-[1,2,4]trioxolane-5′,1″-cyclohexan]-4″-yl)phenoxy) ethyl)piperidine-4-carboxylate (29) (260 mg, 45%) as a white solid. Mp 70–72 °C; 1H NMR (CDCl3) δ 1.25 (t, J = 7.3 Hz, 3H), 1.69–2.04 (m, 26H), 2.15–2.19 (m, 2H), 2.26–2.30 (m, 1H), 2.46–2.51 (m, 1H), 2.77 (t, J = 5.9 Hz, 3H), 2.95 (brs, 2H), 4.07 (t, J = 5.9 Hz, 2H), 4.13 (q, J = 7.3 Hz, J = 14.2 Hz, 2H), 6.83 (d, J = 8.3 Hz, 2H), 7.10 (d, J = 8.3 Hz, 2H); 13C NMR (CDCl3) 8 14.2, 26.5, 26.9, 28.2, 31.6, 34.7, 34.8, 36.4, 36.8, 41.0, 42.0, 53.4, 57.4, 60.3, 66.0. 108.4, 111.3, 114.4, 127.6, 138.5, 157.1, 175.0. Step 2. To a solution of 29 (223 mg, 0.41 mmol) in EtOH (5 mL) was added powdered NaOH (50 mg, 1.2 mmol), and the reaction mixture was stirred for 24 h at rt. Filtration afforded 15 (190 mg, 86%) as a white solid. Mp 165–167 °C; 1H NMR (CD3OD) 8 1.66–2.23 (m, 29H), 2.50–2.55 (m, 1H), 2.77 (t, J = 5.5 Hz, 3H), 3.01 (brs, 2H), 4.09 (t, J = 5.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 7.21 (d, J = 8.5 Hz, 2H); 13C NMR (CD3OD) 8 28.2, 28.6, 30.4, 33.0, 36.0, 38.0, 38.1, 43.4, 45.6, 55.3, 58.7, 66.7, 109.8, 112.4, 115.7, 128.8, 140.0, 158.7, 183.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C30H42NO6 512.3012; found 512.3026.
2-(4-(4-dispiro[adamantane-2,3′-[ 1,2,4]trioxoiane-5′,1″-cyclohexan]-4″-yl)phenoxy)piperidin-1-yl)acetic acid (16)
Step 1. To a solution of cis-adamantane-2-spiro-3–8-[4-(4-piperidinyloxy)phenyl]- 1,2,4-trioxaspiro[4.5]decane (30)25 (0.30 g, 0.68 mmol) and ethyl 2-bromoacetate (0.14 g, 0.84 mmol) in THF (12 mL) was added a solution of potassium carbonate (0.09 g, 0.65 mmol) in water (1 mL). The reaction mixture was stirred at rt for 12 h. After solvent removal in vacuo, the crude product was purified by crystallization from 4:1 EtOH/H2O to afford ethyl 2-(4-(4-(dispiro[adamantane-2,3′-[1,2,4]-trioxolane-5′,1″-cyclohexan]-4″-yl)phenoxy)piperidin-1-yl)acetate (31) as a colorless solid (0.25 g, 69%). Mp 106–107 °C; 1H NMR (CDCl3) δ 1.28 (t, J = 7.0 Hz, 3H), 1.64–2.08 (m, 26H), 2.44–2.56 (m, 3H), 2.76–2.85 (m, 2H), 3.24 (s, 2H), 4.19 (q, J = 7.0 Hz, 2H), 4.26–4.34 (m, 1H), 6.82 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 26.5, 26.9, 30.6, 31.6, 34.7, 34.8, 36.4, 36.8, 42.0, 50.3, 59.6, 60.6, 71.9, 108.4, 111.4, 116.0, 127.7, 138.5, 155.7, 170.5. Step 2. To a solution of 31 (0.53 g, 1.0 mmol) in EtOH (20 mL) was added 1 N aq. NaOH (2 mL). The resulting mixture was stirred at 50 °C for 20 h. The reaction mixture was then diluted with water (5 mL) and AcOH (5 mL). The precipitate was filtered, washed with water, and dried in vacuo at 40 °C to afford 16 (0.42 g, 84%) as a colorless solid. Mp 171–172 °C. 1H NMR (DMSO-d6) δ 1.42–1.60 (m, 2H), 1.61–2.10 (m, 24H), 2.44–2.56 (m, 1H), 2.68–2.82 (m, 2H), 2.90–3.08 (m, 2H), 3.22 (s, 2H), 4.32–4.46 (m, 1H), 6.88 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6) δ 26.3, 26.7, 30.7, 31.7, 34.6, 34.7, 36.3, 36.6, 41.2, 48.3, 55.4, 71.0, 108.6, 111.0, 116.4, 128.1, 139.2, 155.2, 167.7, HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H40NO6 498.2856; found 498.2854.
cis-Adamantane-2-spiro-3′−8′-[4-[[(carboxymethyi)amino]-methyl]phenyi]-1′,2′,4′-trioxaspiro[4.5]decane mesyiate (17)
Step 1. A mixture of cis-adamantane-2-spiro-3′−8′-[4-(chloromethyl)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (32)28 (0.30 g, 0.77 mmol), glycine ethyl ester hydrochloride (1.30 g, 9.32 mmol), and DIPEA (1.50 g, 11.65 mmol) in DMA (20 mL) was stirred at 50 °C for 5 h. The reaction mixture was then cooled to rt, diluted with EA (100 mL), and washed successively with H2O (50 mL), 1 M HCl (15 mL), and brine (2 × 50 mL). The organic layer was dried over MgSO4, filtered, concentrated, and dried in vacuo at 50 °C to afford cis-adamantane-2-spiro-3′−8′-[4-[[(2-ethoxy-2-oxoethyl)amino]methyl]-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (33) (340 mg, 97%) as a light yellow solid. Mp 77–78 °C. 1H NMR (CDCl3) δ 1.27 (t, J = 7.0 Hz, 3H), 1.69–2.05 (m, 22H), 2.53 (t, J = 12.0 Hz, 1H), 1.89 (s, 2H), 3.40 (s, 2H), 4.18 (q, J = 7.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.0 Hz); 13C NMR (CDCl3) δ 14.3, 26.5, 26.9, 31.5, 34.7, 42.6, 50.2, 53.0, 60.7, 108.4, 111.4, 126.9, 128.4, 137.4, 145.1, 172.5. Step 2. A mixture of 33 (340 mg, 0.75 mmol) and 1 M aq. NaOH (1 mL) in THF (20 mL) was stirred at rt for 12 h. After removal of the solvents in vacuo, the residue was suspended in H2O (20 mL) and acidified with AcOH (0.2 mL). The resultant precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford cis-adamantane-2-spiro-3′−8′-[4-[[(carboxymethyl)amino]methyl]-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (290 mg, 90%) as a white solid. To a suspension of cis-adamantane-2-spiro-3′−8′-[4-[[(carboxymethyl)amino]methyl]phenyl]-1′,2′,4′-trioxaspiro[4.5]-decane (230 mg, 0.54 mmol) in Et2O (10 mL) was added methanesulfonic acid (150 mg, 1.56 mmol) in Et2O (2 mL), and the resulting mixture was stirred at rt for 1 h. The precipitate was then filtered, washed with Et2O, and dried in vacuo at 50 °C to afford 17 as a white solid (240 mg, 85%). Mp 142–143 °C. 1H NMR (DMSO-d6) δ 1.55–1.93 (m, 22H), 2.30 (s, 3H), 2.65 (t, J = 12.0 Hz, 1H), 3.85 (s, 2H), 4.12 (s, 2H), 7.29 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 2H), 9.24 (br, 2H), 13.79 (br, 1H); 13C NMR (DMSO-d6) δ 26.3, 26.7, 31.4, 34.5, 34.7, 36.3, 36.6, 41.7, 46.9, 50.2, 108.5, 111.1, 127.4, 129.7, 130.7, 147.5, 168.4. Anal. calcd for C26H37NO8S: C, 59.64; H, 7.12; N, 2.67. Found: C, 59.26; H, 6.90; N, 2.41.
cis-Adamantane-2-spiro-3′−8′-[4-((3-(carbonyi)azetidin-1-yl)-methyi)phenyi]-1′,2′,4′-trioxaspiro[4.5]decane mesyiate (18)
Step 1. A mixture of 32 (0.22 g, 0.57 mmol), ethyl azetidine-3-carboxylate hydrochloride (1.02 g, 6.17 mmol), and DIPEA (0.99 g, 7.65 mmol) in DMA (15 mL) was stirred at 50 °C for 5 h. Then, the reaction mixture was cooled to rt, diluted with EA (100 mL), and washed successively with H2O (50 mL), 1 M HCl (15 mL), and brine (2 × 50 mL). The organic layer was dried over MgSO4, filtered, and concentrated. The obtained residue was dried in vacuo at 50 °C to afford cis-adamantane-2-spiro-3′−8′-[4-((3-(ethoxycarbonyl)azetidin-1-yl)methyl)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (34) (270 mg, 99%) as a white solid. Mp 144–145 °C. 1H NMR (CDCl3) δ 1.64–1.76 (m, 8H), 1.76–1.86 (m, 6H), 1.92 (d, J = 12.9 Hz, 2H), 1.96–2.5 (m, 6H), 2.51 (tt, J = 12.0, 3.2 Hz, 1H), 3.34 (d, J = 3.8 Hz, 2H), 3.60 (d, J =11.2 Hz, 4H), 4.14 (qd, J = 7.1, 1.8 Hz, 2H), 7.14 (t, J = 4.7 Hz, 2H), 7.19 (d, J = 7.6 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 26.5, 26.9, 31.4, 33.9, 34.7, 34.8, 36.4, 36.8, 42.6, 56.6, 60.9, 62.7, 108.4, 111.4, 126.9, 128.7, 134.6, 145.4, 172.9. Step 2. A mixture of 34 (270 mg, 0.56 mmol) and 1 M aq. NaOH (2 mL) in THF (15 mL) was stirred at rt for 12 h and then concentrated in vacuo. The residue was diluted with H2O (20 mL) and acidified with AcOH to pH 5. The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford cis-adamantane-2-spiro-3′−8′-[4-((3-(carbonyl)azetidin-1-yl)methyl)phenyl]-1′,2′,4′-trioxaspiro[4.5]-decane (250 mg, 98%) as a white solid. To a stirred solution of cis-adamantane-2-spiro-3′−8′-[4-((3-(carbonyl)azetidin-1-yl)methyl)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (250 mg, 0.55 mmol) in EA (10 as added dropwise a solution of methanesulfonic acid (106 mg, 1.10 mmol) in EA (2 mL). The resulting mixture was stirred at rt for 1 h. The precipitate was filtered, washed with Et2O, and dried in vacuo at 50 °C to afford 18 as a white solid (262 mg, 86%). Mp 151–152 °C. 1H NMR (DMSO-d6) δ 1.57 (qd, J = 13.0, 4.1 Hz, 2H), 1.62–1.96 (m, 20H), 2.34 (s, 3H), 2.64 (tt, J = 12.2, 3.5 Hz, 1H), 3.62 (q, J = 9.1 Hz, 1H), 4.08–4.28 (m, 4H), 4.34 (d, J = 17.8 Hz, 2H), 7.29 (d, J = 7.8 Hz, 2H), 7.39 (d, J = 7.9 Hz, 2H), 10.16 (s, 1H), 13.12 (brs, 1H); 13C NMR (DMSO-d6) δ 26.3, 26.7, 31.4, 32.5, 34.5, 34.7, 36.3, 36.6, 41.7, 55.2, 55.5, 57.3, 108.5, 111.1, 127.7, 128.6, 130.6, 147.7, 171.8. Anal. calcd for C28H39NO8S: C, 61.18; H, 7.15; N, 2.55. Found: C, 61.00; H, 7.30; N, 2.37.
cis-6,6-Difluoroadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)-pheny l]-1′,2′,4′ -trioxaspiro[4.5]decane (19)
Step 1. A mixture of4-(4-hydroxyphenyl)cyclohexanone (45) (5.71 g, 30 mmol), ethyl bromoacetate (6.01 g, 36 mmol), and K2CO3 (8.29 g, 60 mmol) in acetone (150 mL) was stirred at reflux for 12 h. The solid was removed by filtration, and the filtrate was concentrated in vacuo to afford ethyl 2-(4-(4-oxocyclehexyl)phenoxyl)acetate (46) as a light yellow oil (8.29 g, 100%). 1H NMR (CDCl3) δ 1.30 (t, J = 7.0 Hz, 3H), 1.90 (dt, J1 = 12.5 Hz, J2 = 6.0 Hz, 2H), 2.19–2.22 (m, 2H), 2.48–2.53 (m, 4H), 2.98 (tt, J1 = 9.0 Hz, J2 = 3.0 Hz, 1H), 4.27 (q, J = 7.0 Hz, 2H), 4.60 (s, 2H), 6.87 (d, J = 8.5 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 34.1, 41.4, 41.9, 61.4, 65.6, 114.8, 127.7, 138.0, 156.5, 169.0, 211.1. Step 2. Amixture of 46 (0.51 g, 1.86 mmol) and 6,6-difluoroadamantan-2-one O-methyl oxime (47)28 (0.40 g, 1.86 mmol) in cyclohexane (100 mL) and DCM (40 mL) was treated with ozone according to the method of Dong et al.41 After removal of the solvents in vacuo, the residue was recrystallized from MeOH to afford cis-6,6-difluoroadamantane-2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (48) (0.13 g, 15%) as a white solid. Mp 112–113 °C. 1H NMR (CDCl3) δ 1.28 (t, J = 7.0 Hz, 3H), 1.68 (m, 2H), 1.79–2.06 (m, 18H), 2.50 (t, J = 12.5 Hz, 1H), 4.27 (q, J = 7.0 Hz, 3H), 4.59 (s, 2H), 6.83 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 30.7 (t, 3J = 3.5 Hz), 30.9 (t, 3J = 3.5 Hz), 34.2 (t, 2J = 34.3 Hz), 34.4, 34.7, 42.2, 61.3, 65.6, 108.9, 109.5, 114.6, 124.0 (1J = 121.7 Hz), 127.8, 139.2, 156.3, 169.0. Step 3. A mixture of 48 (120 mg, 0.25 mmol) and 15% aq. KOH (0.5 mL) in THF (10 mL) was stirred at rt for 12 h. The removal of the solvents in vacuo gave a white residue which was suspended in H2O (10 mL) and acidified with AcOH (0.5 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 19 (105 mg, 93%) as a white solid. Mp 155–156 °C. 1H NMR (CD3OD) δ 1.65 (qd, J1 = 13.0 Hz, J2 = 3.0 Hz, 2H), 1.81–2.10 (m, 18H), 2.56 (t, J = 12.0 Hz, 1H), 4.48 (s, 2H), 6.86 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H); 13C NMR (CD3OD) δ 30.36 (t, 3J = 3.9 Hz), 30.42 (t, 3J = 3.9 Hz), 31.4, 34.2, 34.3 (t, 2J = 21.9 Hz), 34.7 (t, 3J = 21.9 Hz), 34.8, 41.8, 53.4, 66.0, 108.77, 108.80, 114.2, 123.7 (t, 1J = 150.8 Hz), 127.1, 138.6, 156.9, 173.6, Anal. calcd for C24H28F2O6: C, 63.99; H, 6.27. Found: C, 63.81; H, 6.07.
cis-6-Oxoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (20)
Step 1. To a solution of adamantane-2,6-dione mono ethylene ketal (49)32 (3.90 g, 18.72 mmol) in dry EtOH (100 mL) was added pyridine (2.45 g, 30.90 mmol) followed by methoxyamine hydrochloride (2.40 g, 28.09 mmol). The reaction mixture was stirred at rt for 12 h, concentrated in vacuo, and partitioned between DCM (100 mL) and H2O (50 mL). The organic phase was separated, and the aq. layer was extracted with DCM (3 × 30 mL). The organic layers were combined and washed with 1 M HCl (30 mL), sat. aq. NaHCO3 (30 mL) and brine (30 mL), dried over MgSO4, and concentrated to afford adamantane-2,6-dione mono ethylene ketal O-methyl oxime (50) (4.27 g, 96%) as a white solid. Mp 90–91 °C. 1H NMR (CDCl3) δ 1.74 (d, J = 12.5 Hz, 2H), 1.80 (d, J = 12.5 Hz, 2H), 1.91 (s, 2H), 2.47 (s, 2H), 3.41 (s, 2H), 3.82 (s, 3H), 3.99 (s, 4H); 13C NMR (CDCl3): δ 27.9, 34.5, 34.7, 35.8, 36.3, 61.0, 64.4, 109.8, 165.2. Step 2. A mixture of 50 (4.24 g, 17.91 mmol) and 4-(4-oxocyclohexyl)phenyl acetate (51) (4.16 g, 17.91 mmol) in cyclohexane (450 mL) and DCM (150 mL) was treated with ozone according to the method of Dong et al.42 After solvent removal in vacuo, the residue was recrystallized from EtOH to afford cis-6-oxoadamantane-2-spiro-3′−8′-(4-acetoxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane ethylene ketal (52) (3.35 g, 43%) as a white solid. Mp 156–157 °C. 1H NMR (CDCl3) δ 1.66–2.05 (m, 20H), 2.28 (s, 3H), 2.54 (t, J = 12.0 Hz, 1H), 3.94 (s, 4), 7.21 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3): δ 21.1, 31.5, 31.6, 34.6, 34.8, 35.1, 35.3, 42.4, 64.3, 108.5, 109.9, 110.3, 121.4, 127.7, 143.6, 148.9, 169.7. Step 3. A mixture of 52 (3.35 g, 7.34 mmol), 1 M NaOH (10 mL) and THF (100 mL) was stirred at rt overnight, then concentrated in vacuo. The residue was suspended in H2O (30 mL) and acidified with AcOH (2 mL). The resulting precipitate was filtered, washed with water, and dried in vacuo to afford cis-6-oxoadamantane-2-spiro-3′−8′-(4-hydroxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane ethylene ketal (2.97 g, 98%) as a white solid. Mp 164–165 °C. 1H NMR (500 MHz, CDCl3) δ 1.64–2.04 (m, 20H), 2.48 (t, J = 12.0 Hz, 1H), 3.95 (s, 4H), 4.98 (brs, 1H), 6.79 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 31.6, 34.7, 34.8, 35.1, 35.3, 42.0, 64.3, 108.6, 110.0, 110.2, 115.2, 127.8, 138.4, 153.9. Step 4. A suspension of cis-6-oxoadamantane-2-spiro-3′−8′-(4-hydroxyphenyl)-1′,2′,4′-trioxaspiro[4.5] decane ethylene ketal (3.10 g, 7.48 mmol) in 1 M methanesulfonic acid in 5:1 acetone/H2O (42 mL) was stirred at rt for 12 h. The resulting solid was filtered, washed with water, and dried in vacuo to afford cis-6-oxoadamantane-2-spiro-3′−8′-(4-hydroxyphen-yl)-1′,2′,4′-trioxaspiro[4.5]decane (53) (2.52 g, 91%) as a white solid. Mp 187–188 °C. 1H NMR (DMSO-d6) δ 1.52 (q, J = 12.0 Hz, 2H), 1.75 (d, J =11.5 Hz, 2H), 1.86 (d, J = 12.0 Hz, 2H), 1.95 (s, 4H), 2.7 (s, 4H), 2.18 (d, J = 12.0 Hz, 2H), 2.33 (d, J = 16.0 Hz, 2H), 2.51 (s, 1H), 6.66 (d, J = 8.5 Hz, 2H), 6.99 (d, J = 8.5 Hz, 2H); 13C NMR (DMSO-d6) δ 31.8, 34.5, 35.50, 35.53, 35.7, 41.2, 44.5, 45.0, 109.2, 109.4, 115.5, 127.8, 136.5, 156.0, 214.6. Step 5. A mixture of 53 (600 mg, 1.62 mmol), ethyl bromoacetate (325 mg, 1.94 mmol), and potassium carbonate (447 mg, 3.24 mmol) in acetone (30 mL) was stirred at 55 °C for 12 h. After filtering and washing with acetone, the filtrate was concentrated in vacuo to afford cis-6-oxoadamantane- 2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5] decane (54) as a white solid (740 mg, 100%). Mp 138–139 °C. 1H NMR (CDCl3) δ 1.30 (t, J = 7.0 Hz, 3H), 1.68–1.75 (m, 2H), 1.84–1.90 (m, 4H), 1.92 (d, J = 13.0 Hz, 2H), 1.99 (d, J = 13.0 Hz, 2H), 2.07 (d, J = 13.0 Hz, 2H), 2.15 (s, 2H), 2.27 (d, J = 13.0 Hz, 2H), 2.35 (d, J = 12.5 Hz, 2H), 2.48 (d, J = 16.0 Hz, 2H), 2.52 (t, J = 12.0 Hz, 2H), 4.27 (q, J = 7.0 Hz, 2H), 4.60 (s, 2H), 6.84 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 31.5, 34.6, 35.68, 35.74, 35.9, 42.0, 44.7, 45.2, 61.3, 65.6, 109.0, 109.3, 114.7, 127.7, 139.2, 156.3, 169.1,215.9. Step 6. A mixture of 54 (230 mg, 0.50 mmol) and 1 M aq. NaOH (2 mL) in THF (15 mL) was stirred at rt for 12 h. Removal of the solvents in vacuo gave a white residue, which was suspended in H2O (10 mL) and acidified with AcOH (0.5 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 20 (205 mg, 95%) as a white solid. Mp 171–172 °C. 1H NMR (CD3OD) δ 1.67–1.75 (m, 2H), 1.85–1.90 (m, 4H), 1.94 (d, J = 13.0 Hz, 2H), 1.99 (d, J = 13.0 Hz, 2H), 2.7 (d, J = 12.5 Hz, 2H), 2.15 (s, 2H), 2.27 (d, J = 13.0 Hz, 2H), 2.35 (d, J = 12.0 Hz, 2H), 2.49 (d, J = 16.5 Hz, 2H), 2.53 (t, J = 12.5 Hz, 1H), 4.66 (s, 2H), 6.86 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 31.5, 34.6, 35.7, 35.8, 35.9, 42.0, 45.1, 65.0, 109.1, 109.2, 114.7, 127.9, 139.7, 155.8, 172.8, 216.3. Anal. calcd for C24H28O7: C, 67.28; H, 6.59. Found: C, 67.54; H, 6.36.
cis-6-Hydroxyadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (21)
Step 1. To a solution of 53 (100 mg, 0.27 mmol) in methanol (10 mL) at 0 °C was added NaBH4 (17 mg, 0.68 mmol). The resulting mixture was stirred at 0 °C for 2 h and then quenched with H2O (1 mL). After solvent removal in vacuo, the residue was extracted in EA (20 mL) and washed with saturated NaHCO3 (10 brine (10 mL). The organic layer was dried over MgSO4, filtered, and concentrated to afford cis-6-hydroxyadamantane-2-spiro-3′−8′-(4-hydroxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane (55) (99 mg, 99%) as a white solid. Mp 159–160 °C. 1H NMR (DMSO-d6) δ 1.46–2.10 (m, 20H), 2.48 (t, J = 12.0 Hz, 1H), 5.59 (d, J = 2.0 Hz, 1H), 4.67 (d, J = 3.0 Hz, 1H), 6.66 (d, J = 8.5 Hz, 2H), 6.98 (d, J = 8.5 Hz, 2H), 9.13 (s, 1H); 13C NMR (DMSO-d6) δ 28.5, 31.8, 32.9, 33.3, 34.61, 34.63, 35.2, 35.9, 41.2, 71.5, 108.6, 111.0, 115.5, 127.8, 136.6, 156.0. Step 2. A mixture of 55 (90 mg, 0.24 mmol), ethyl bromoacetate (48 mg, 0.29 mmol), and potassium carbonate (67 mg, 0.48 mmol) in acetone (10 mL) was stirred at 55 °C for 12 h. The solid was filtered and washed with acetone, and the filtrate was concentrated in vacuo to afford cis-6-hydroxyadamantane-2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (56) as a white solid (110 mg, 100%). Mp 138–139 °C. 1H NMR (CDCl3) δ 1.29 (t, J = 7.0 Hz, 3H), 1.62–2.14 (m, 20H), 2.49 (t, J = 12.0 Hz, 2H), 3.81 (s, 1H), 4.26 (q, J =7.0 Hz, 2H), 4.58 (s, 2H), 6.82 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 28.2, 31.6, 32.9, 33.29, 33.32, 34.7, 35.1, 35.8, 42.0, 61.3, 65.6, 73.2, 108.6, 110.8, 114.6, 127.7, 139.4, 156.3, 169.1. Step 3. A mixture of 56 (190 mg, 0.41 mmol) and 1 M aq. NaOH (2 mL) in THF (15 mL) was stirred at rt for 12 h. The removal of the solvents in vacuo gave a white residue, which was suspended in H2O (10 mL) and acidified with AcOH (0.5 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 21 (170 mg, 95%) as a white solid. Mp 153–154 °C. 1H NMR (DMSO-d6) δ 1.46–1.92 (m, 18H), 2.02 (d, J = 11.5 Hz, 1H), 2.8 (d, J = 11.0 Hz, 1H), 2.56 (t, J = 12.0 Hz, 1H), 2.58 (s, 1H), 4.60 (s, 2H), 4.66 (br, 1H), 6.79 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 12.96 (br, 1H); 13C NMR (DMSO-d6) δ 28.5, 31.8, 32.9, 33.3, 34.6, 35.2, 35.9, 41.2, 65.0, 71.5, 108.6, 111.0, 114.8, 127.89, 138.9, 156.5, 170.8. Anal. calcd for C24H30O7: C, 66.96; H, 7.02. Found: C, 66.96; H, 6.89.
cis-6-Acetamidoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (22)
Step 1. To a solution of 54 (1.02 g, 2.23 mmol) in MeOH (50 mL) were added ammonium acetate (2.27 g, 26.81 mmol) and acetic acid (0.5 mL). The resulting mixture was stirred at rt for 0.5 h before addition of sodium cyanoborohydride (0.58 g, 8.94 mmol). The reaction mixture was stirred at rt for 12 h and then quenched with 1 M aq. NaOH (10 mL). After solvent removal in vacuo, the residue was extracted with EA (100 mL) and washed with water (30 mL) and brine (30 mL). The organic layer was dried over MgSO4, filtered, and concentrated to afford cis-6-aminoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane as a gray residue (1.02 g, 100%), which was used as starting material for next step without purification. Step 2. cis-6-Aminoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4-trioxaspiro[4.5]decane as a gray residue (1.02 g, 100%), which was used as starting material for next step without purification. Step 2. cis-6-Aminoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (220 mg, 0.48 mmol) was treated with acetyl chloride (57 mg, 0.72 mmol) and pyridine (76 mg, 0.96 mmol) in DCM (10 mL) at 0 °C. After stirring at rt for 6 h, the reaction mixture was quenched with H2O (1 mL); diluted with DCM (10 mL); washed successively with 1 M HCl (5 mL), H2O (5 mL), and brine (5 min); and then dried over MgSO4. After filtration and solvent removal in vacuo, the residue was purified by chromatography (sg, 10:1 DCM/MeOH) to afford cis-6-acetamidoadamantane-2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (57) (200 mg, 83%) as a white solid. Mp 89–90 °C. lH NMR (CDCl3) δ 1.29 (t, J = 7.0 Hz, 2H), 1.69–2.10 (m, 23H), 2.49 (t, J = 12.0 Hz, 1H), 3.97 (d, J = 7.0 Hz, 1H), 4.26 (q, J = 7.0 Hz, 2H), 4.58 (s, 2H), 5.77 (d, J = 7.0 Hz, 1H), 6.83 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 23.7, 29.1, 29.2, 30.2, 30.5, 31.5, 31.6, 34.0, 34.6, 34.7, 35.3, 35.5, 42.0, 52.4, 61.3, 65.6, 108.8, 110.5, 114.6, 127.7, 139.3, 156.3, 169.1, 169.5. Step 3. A mixture of 57 (170 mg, 0.34 mmol) and 1 M aq. NaOH (1 mL) in THF (10 mL) was stirred at rt for 12 h. After solvent removal in vacuo, the resulting residue was suspended in H2O (10 mL) and acidified with AcOH (0.5 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford 22 (150 mg, 94%) as a white solid. Mp 163–164 °C. 1H NMR (DMSO-d6) δ 1.51–2.02 (m, 26H), 2.55 (t, J = 12.0 Hz, 1H), 3.75 (d, J = 6.0 Hz, 1H), 4.56 (s, 2H), 6.79 (d, J = 8.5 Hz, 2H), 7.10 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 7.0 Hz, 1H); 13C NMR (DMSO-d6) δ 23.2, 28.8, 28.9, 30.3, 30.7, 31.7, 34.2, 34.5, 34.6, 35.4, 35.5, 41.2, 52.4, 65.4, 108.7, 110.7, 114.7, 127.8, 138.7, 156.7, 169.2, 170.9. Anal. calcd for C26H33NO7: C, 66.23; H, 7.05; N, 2.97. Found: C, 66.44; H, 6.90; N, 2.73.
cis-6-Aminoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane mesylate (23)
Step 1. cis-6-Aminoadamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (700 mg, 1.53 mmol) was treated with (Boc)2O (667 mg, 3.07 mmol) and DIPEA (793 mg, 6.13 mmol) in DCM (20 mL) at 0 °C. The reaction mixture was then stirred at rt overnight, diluted with DCM (30 mL), washed successively with 1 M HCl (10 mL), saturated NaHCO3 (10 mL) and brine (20 min), dried over MgSO4, filtered, and concentrated. The residue was purified by chromatography (sg, hexane/EA 5:1) to afford cis-6-((tert-butoxycarbonyl)amino)adamantane-2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′-trioxaspiro[4.5]decane (58) (590 mg, 69%) as a white solid. Mp 89–90 °C. 1H NMR (CDCl3) δ 1.29 (t, J = 7.0 Hz, 3H), 1.45 (s, 9H), 1.63–2.09 (m, 20H), 2.49 (t, J = 12.0 Hz, 1H), 3.67 (s, 1H), 4.26 (q, J = 7.0 Hz, 2H), 4.56 (s, 2H), 4.86 (s, 1H), 6.82 (d, J = 8.5 Hz, 2H), 7.11 (d, J = 8.5 Hz, 2H); 13C NMR (CDCl3) δ 14.1, 28.4, 28.9, 29.0, 30.6, 30.9, 31.49, 31.53, 34.0, 34.1, 34.6, 35.3, 35.5, 42.0, 53.5, 61.3, 65.6, 79.2, 108.6, 110.6, 114.6, 127.7, 139.3, 155.2, 156.2, 169.0. Step 2. A mixture of 58 (250 mg, 0.75 mmol) and 1 M aq. NaOH (1 mL) in THF (10 mL) was stirred at rt for 12 h. After solvent removal in vacuo, the residue was suspended in H2O (20 mL) and acidified with AcOH (0.2 mL). The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford cis-6-((tert-butoxycarbonyl)amino) adamantane-2-spiro-3′−8′-[4-(carboxymethoxy)phenyl]-1′,2′,4′ -trioxaspiro[4.5]decane as a white solid, which was then treated with 1 M methanesulfonic acid in EtOAc (6 mL) at rt for 12 h. The precipitate was filtered, washed with EA, and dried in vacuo at 50 °C to afford 23 as a white solid (232 mg, 98% over two steps). Mp 182–183 °C. 1H NMR (DMSO-d6) δ 1.52 (q, J = 12.5 Hz, 2H), 1.65 (d, J = 13.5 Hz, 1H), 1.75–2.04 (m, 17H), 2.34 (s, 3H), 2.55 (t, J = 12.0 Hz, 1H), 3.30 (s, 1H), 4.61 (s, 2H), 6.81 (d, J = 8.5 Hz, 2H), 7.11 (d, J =8.5 Hz, 2H), 7.95 (s, 3H), 12.92 (br, 1H); 13C NMR (DMSO-d6) δ 27.28, 27.33, 28.8, 29.2, 31.7, 33.57, 33.61, 34.5, 34.9, 35.1, 40.2, 41.1, 53.9, 64.9, 109.0, 110.0, 114.8, 127.9, 138.8, 156.5, 170.8. Anal. calcd for C25H35NO9S: C, 57.13; H, 6.71; N, 2.66. Found: C, 56.95; H, 6.59; N, 2.57.
cis-6-Azaadamantane-2-spiro-3′−8 ′-[4-(carboxymethoxy)-phenyl]-1′,2′,4′-trioxaspiro[4.5]decane mesylate (24)
Step 1. To a solution of 2-Boc-2-azaadamantan-6-one (59)33 (4.00 g, 15.91 mmol) in dry EtOH (100 mL) was added pyridine (2.08 g, 26.25 mmol) followed by methoxyamine hydrochloride (1.99 g, 23.87 mmol). The resulting mixture was stirred at rt for 12 h and then concentrated in vacuo. The residue was partitioned between DCM (100 mL) and H2O (50 mL). The organic phase was separated, and the aqueous layer was extracted with DCM (3 × 30 mL). The combined organic layers were successively washed with 1 M HCl (30 mL), saturated NaHCO3 (30 mL), and brine (30 mL). After drying over MgSO4, the solvent was removed in vacuo to afford 2-Boc-2-azaadamantan-6-one O-methyl oxime (60) (4.41 g, 99%) as a white solid. Mp 90–91 °C. 1H NMR (CDCl3) δ 1.48 (s, 10H), 1.78 (t, J = 10.2 Hz, 1H), 1.86 (t, J = 9.6 Hz, 1H), 1.96 (t, J = 13.5 Hz, 1H), 2.05 (t, J = 13.5 Hz, 1H), 2.69 (s, 1H), 3.61 (s, 1H), 3.83 (s, 3H), 4.30 (s, 1H), 4.42 (s, 1H). 13C NMR (CDCl3) δ 27.7, 28.5, 34.6, 35.7, 36.0, 37.0, 37.4, 45.6, 47.1, 61.1, 79.5, 154.2, 163.8. Step 2. A mixture of 60 (841 mg, 3.0 mmol) and ethyl 4-(4-oxocyclohexyl)phenyl acetate (51) (697 mg, 3.0 mmol) in cyclohexane (120 mL) and DCM (20 mL) was treated with ozone according to the method of Dong et al.42 After solvent removal in vacuo, the residue was recrystallized from MeOH to afford cis-6-Boc-6-azaadamantane-2-spiro-3′−8′-(4-acetoxyphenyl)-1′,2′,4′-trioxaspiro[4.5]decane (61) as a white solid (541 mg, 36%). Mp 162–163 °C. 1H NMR (CDCl3) δ 1.47 (s, 9H), 2.09–1.66 (m, 16H), 2.15 (s, 2H), 2.28 (s, 3H), 2.56 (t, J = 12.3 Hz, 1H),4.14 (d, J = 11.2 Hz, 1H), 4.27 (d, J= 21.8 Hz, 1H), 7.00 (d, J = 8.5 Hz, 2H), 7.20 (d, J = 8.3 Hz, 2H); 13C NMR (CDCl3) δ 21.1, 28.5, 31.4, 33.2, 33.5, 34.6, 35.0, 42.3, 44.4, 44.8, 45.8, 46.2, 79.3, 108.9, 110.0, 121.4, 127.7, 143.5, 148.9, 154.2, 169.6. Step 3. A mixture of 61 (250 mg, 0.50 mmol) and 15% aq. KOH (1 mL) in THF (10 mL) was stirred at rt for 12 h and then concentrated in vacuo. The residue was suspended in H2O (20 mL) and acidified with AcOH to pH 4. The precipitate was filtered, washed with water, and dried in vacuo to afford cis-6-Boc-6-azaadamantane-2-spiro-3′−8′-(4-hydroxyphenyl)-1′,2′,4′-trioxaspiro[4.5] decane (62) (225 mg, 98%) as a white solid. Mp 176–177 °C. 1H NMR (CDCl3) δ 1.47 (s, 9H), 1.73–1.66 (m, 2H), 1.75–2.08 (m, 14H), 2.15 (s, 2H), 2.49 (t, J = 11.4 Hz, 1H), 4.15 (d, J = 11.5 Hz, 1H), 4.27 (d, J = 21.7 Hz, 1H), 5.56 (brs, 1H), 6.77 (d, J = 8.3 Hz, 2H), 7.06 (d, J = 8.2 Hz, 2H); 13C NMR (CDCl3) δ 28.5, 31.6, 33.2, 33.5, 34.7, 34.9, 42.0, 44.5, 44.9, 45.9, 46.3, 79.6, 109.1, 109.9, 115.2, 127.7, 138.0, 154.2, 154.3. Step 4. A mixture of 62 (225 mg, 0.49 mmol), ethyl bromoacetate (100 mg, 0.60 mmol), and potassium carbonate (104 mg, 0.74 mmol) in acetone (20 mL) was stirred at 60 °C for 12 h. A solid was filtered and washed with acetone before concentrating the filtrate in vacuo to afford cis-6-Boc-6-azaadaman- tane-2-spiro-3′−8′-[4-(2-ethoxy-2-oxoethoxy)phenyl]-1′,2′,4′- trioxaspiro[4.5]decane (63) (266 mg, 100%) as a white solid. Mp 139–140 °C. 1H NMR (CDCl3) δ 1.30 (t, J = 7.1 Hz, 2H), 1.47 (s, 9H), 1.61–2.08 (m, 16H), 2.15 (s, 2H), 2.51 (t, J =11.8 Hz, 1H), 4.14 (d, J = 10.9 Hz, 1H), 4.25–4.30 (m, 3H), 4.59 (s, 2H), 6.84 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 8.4 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 28.5, 31.5, 33.2, 33.5, 34.6, 35.0, 42.0, 44.4, 44.8, 45.8, 46.2, 61.3, 65.6, 79.3, 109.0, 110.0, 114.6, 127.7, 139.3, 154.2, 156.3, 169.1. Step 5. A mixture of 63 (265 mg, 0.49 mmol) and 15% aq. KOH (1 mL) in THF (10 mL) was stirred at rt for 12 h. The removal of the solvent in vacuo gave a white residue, which was suspended in H2O (10 mL) and acidified with AcOH to pH 3. The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford cis-6-Boc-6-azadamantane-2-spiro-3′−8′-carboxymethyl-1′,2′,4′-trioxaspiro[4.5]-decane (64) (232 mg, 90%) as a white solid. Mp 158–160 °C. 1H NMR (CDCl3) δ 1.46 (d, J = 1.7 Hz, 9H), 1.65 (q, J = 12.4 Hz, 2H), 1.73–2.06 (m, 14H), 2.13 (s, 2H), 2.47 (t, J = 12.2 Hz, 1H), 4.14 (d, J =11.7 Hz, 1H), 4.26 (d, J = 28.8 Hz, 1H), 4.48 (s, 2H), 6.78 (d, J = 8.2 Hz, 2H), 7.06 (d, J = 8.2 Hz, 2H); 13C NMR (CDCl3) δ 28.5, 31.5, 33.2, 33.5, 34.6, 35.0, 41.9, 44.4, 44.8, 45.8, 46.2, 65.4, 79.5, 108.9, 109.9, 114.6, 127.8, 154.3, 155.9. Step 6. A mixture of 64 (232 mg, 0.45 mmol) and methanesulfonic acid (480 mg, 5.00 mmol) in THF (5 mL) was stirred at rt for 12 h. The precipitate was filtered and washed with Et2O to afford 24 (230 mg, 100%) as a white solid. Mp 183–184 °C. 1H NMR (DMSO-d6) δ 1.53 (q, J = 13.3, 2H), 1.71–2.18 (m, 16H), 2.37 (s, 3H), 2.57 (t, J = 12.1 Hz, 1H), 3.55 (s, 1H), 3.59 (s, 1H), 4.62 (s, 2H), 6.82 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 8.7 Hz, 2H), 8.73 (brs, 2H), 12.95 (brs, 1H); 13C NMR (DMSO-d6) δ 30.8, 31.7, 33.2, 34.3, 41.0, 45.5, 45.9, 65.0, 108.7, 109.7, 114.8, 127.9, 138.7, 156.6, 170.8. Anal. calcd for C24^3NO9S: C, 56.35; H, 6.50; N, 2.74. Found: C, 56.01; H, 6.70; N, 2.59.
cis-6-Azaadamantane-2-spiro-3′−8′-[4-[[(carboxymethyl)-amino]methyl]phenyl]-1′,2′,4′-trioxaspiro[4.5]decane mesylate (25)
Step 1. A mixture of 60 (400 mg, 1.43 mmol) and 4-(4-(chloromethyl)phenyl)cyclohexan-1-one (65) (318 mg, 1.43 mmol) in cyclohexane (120 mL) and DCM (20 mL) was treated with ozone according to the method of Dong et al.42 After solvent removal in vacuo, the residue was recrystallized from MeOH to afford cis-6-Boc- 6-azaadamantane-2-spiro-3′−8′-[4-(chloromethyl)phenyl]-1′,2′,4′--trioxaspiro[4.5]decane (66) as a white solid (182 mg, 26%). Mp 157–158 °C. 1H NMR (CDCl3) δ 1.47 (d, J = 2.1 Hz, 9H); 1.66–2.9 (m, 20H), 2.15 (s, 2H), 2.56 (t, J = 11.5 Hz, 1H), 4.15 (d, J = 10.8 Hz, 1H), 4.27 (d, J= 21.9 Hz, 1H), 4.56 (s, 2H), 7.20 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 7.9 Hz, 2H); 13C NMR (CDCl3) δ 28.5, 31.3, 33.2, 33.5, 34.9, 34.5, 42.5, 42.6, 44.3, 44.7, 45.8, 46.1, 46.2, 79.3, 108.8, 110.0, 127.1, 128.7, 135.4, 146.3, 154.2. Step 2. A mixture of 66 (182 mg, 0.37 mmol), glycine ethyl ester hydrochloride (622 mg, 9.32 mmol), and DIPEA (1.50, 11.65 mmol) in DMA (20 mL) was stirred at 50 °C for 5 h. Then, the reaction mixture was cooled to rt, diluted with EA (100 mL), and washed successively with H2O (3 × 50 mL) and brine (50 mL). The organic layer was separated and dried over MgSO4, filtered, concentrated, and dried in vacuo at 50 °C to afford cis-6-Boc-6-azaadamantane-2-spiro-3′−8′-[4-[[(2-ethoxy-2-oxoethyl)amino]methyl]phenyl]-1,2,4-trioxaspiro[4.5]decane (67) (200 mg, 97%) as a white solid. 1H NMR (CDCl3) δ 1.27 (t, J = 7.0 Hz, 3H); 1.47 (d, J = 2.2 Hz, 9H), 1.66–2.07 (m, 16H), 2.16 (s, 2H), 2.55 (t, J = 11.8 Hz, 1H), 3.41 (s, 2H), 3.77 (s, 2H), 4.15 (d, J = 10.8 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 4.28 (d, J = 21.2 Hz, 1H), 7.17 (d, J = 8.2 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H); 13C NMR (CDCl3) δ 14.2, 28.5, 31.4, 33.2, 33.5, 34.6, 35.0, 42.5, 44.4, 44.8, 45.8, 46.2, 50.1, 53.0, 60.8, 79.3, 109.0, 110.0, 126.8, 128.4, 137.3, 144.9, 154.2, 172.3. Step 3. A mixture of 67 (200 mg, 0.36 mmol) and 1 M aq. NaOH (1 mL) in THF (10 mL) was stirred at rt for 12 h and then concentrated in vacuo. The residue was suspended in H2O (10 mL) and acidified with AcOH to pH 3. The precipitate was filtered, washed with water, and dried in vacuo at 50 °C to afford cis-6-Boc-6-azaadamantane-2-spiro-3′−8′-[4-[[(carboxymethyl)amino]methyl]phenyl]-1′,2′,4′-trioxaspiro[4.5]decane, which was treated with methanesulfonic acid (480 mg, 5 mmol) in THF (5 mL). The resulting mixture was stirred at rt for 12 h before the addition of Et2O (10 mL). The supernatant was removed and to the oily residue was added Et2O (20 mL). The resulting mixture was stirred at rt for 12 h. The precipitate was filtered and washed with Et2O to afford 25 (183 mg, 82%) as a white solid. Mp 174–175 °C. 1H NMR (DMSO-d6) δ 1.58 (qd, J = 13.1, 3.9 Hz, 2H); 1.76–2.18 (m, 16H), 2.34 (s, 6H), 2.67 (tt, J = 12.4, 3.2 Hz, 1H), 3.55 (s, 1H), 3.59 (s, 1H), 3.85 (s, 2H), 4.13 (s, 2H), 7.30 (d, J = 7.9 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 8.73 (s, 2H), 9.26 (s, 2H), 13.77 (brs, 1H); 13C NMR (DMSO-d6) δ 30.77, 30.84, 31.4, 33.2, 34.2, 41.6, 45.5, 45.9, 46.9, 50.2, 108.7, 109.6, 127.4, 129.8, 130.7, 147.3, 168.4. Anal. calcd for C26H40N2O11S: C, 50.31; H, 6.50; N, 4.51. Found: C, 50.66; H, 6.58; N, 4.34. HRMS (ESI-TOf) m/z: [M + H]+ calcd for C24H33N2O5 429.2389; found 429.2397.
Polar Surface Area (PSA)
PSA values were calculated using ChemAxon Instant JChem (ver 16.4).
Partition Coefficient
Partition coefficient values (log D) of the test compounds were estimated by correlation of their chromatographic retention properties against the characteristics of a series of standard compounds with known partition coefficient values using gradient HPLC (modification of a method reported by Lombardo et al.).38
Protein Binding
Protein binding values of the test compounds were estimated by correlation of their chromatographic retention properties on a human albumin column against the characteristics of a series of standard compounds with known protein binding values. The method employed is a gradient HPLC-based derivation of the method developed by Valko et al.40
Kinetic Solubility
Compounds in DMSO (10 mg/mL) were diluted into either pH 6.5 phosphate buffer or 0.01 M HCl (approx. pH 2.0) with the final DMSO concentration being 1%. After 30 min, samples were then analyzed via nephelometry to determine a solubility range.39
In Vitro Metabolic Stability
As fully described in Coteron et al.,44 metabolic stability assays were performed by incubating test compounds in liver microsomes at 37 °C and 0.4 mg/mL protein concentration. The metabolic reaction was initiated by the addition of an NADPH-regenerating system and quenched at various time points over a 60 min incubation period by the addition of acetonitrile containing diazepam as internal standard. Control samples (contain- ing no NADPH) were included (and quenched at 2, 30, and 60 min) to monitor for potential degradation in the absence of cofactor.
Antischistosomal Screen
All animal experiments were per- formed in line with national guidelines (permit 2070). All animals were kept in polycarbonate cages under environmentally controlled conditions (temperature: 25 °C, humidity: 70%, 12:12 h light/dark photocycle), had free access to tap water and rodent food, and acclimatized for 1 week prior to infection. Cercariae of S. mansoni were obtained from infected Biomphalaria glabrata. As described by Lombardo et al.,45 4-week-old female NMRI mice (Charles Rivers, Sulzfeld, Germany) were infected subcutaneously with approximately 100 S. mansoni cercariae. At 21 or 49 days after infection, groups of four mice were treated with single oral doses of compounds in a 7% (v/v) Tween 80% and 3% (v/v) ethanol vehicle (10 mL/kg). Untreated mice (n = 8) served as controls. At 21 days post-treatment, the animals were killed by the CO2 method and dissected. Worms were removed by picking, then sexed and counted.
Cytotoxicity Screen
As fully described in Sanford et al.,46 cell lines were maintained in D10 media. Once confluent, increasing concentrations of test compound (0–100 μM) were added and incubated for 24 h. Alamar blue (10 mM) was then added to each well and incubated for 4 h. A BioTek Synergy HT plate reader was then used to determine fluorescence.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the U.S. National Institutes of Health (AI116723-01) and the European Research Council (ERC-2013-CoG 614739-A HERO) for financial support.
ABBREVIATIONS USED
- CLint
in vitro intrinsic clearance
- DCC
dicyclohexylcarbodiimide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- HOBt
hydroxybenzotriazole
- WBR
worm burden reduction
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00069.
1H and 13C{1H} NMR spectra (500 MHz) of 5–10 and 13–25 (PDF)
SMILES string computer readable identifiers for 2–25 (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00069
Contributor Information
Jianbo Wu, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 986125, United States.
Xiaofang Wang, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 986125, United States.
Francis C. K. Chiu, Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia
Ćecile Häberli, Swiss Tropical and Public Health Institute, CH-4002 Basel, Switzerland; University of Basel, CH-4003 Basel, Switzerland.
David M. Shackleford, Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia
Eileen Ryan, Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia.
Sriraghavan Kamaraj, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 986125, United States.
Vivek J. Bulbule, College of Pharmacy, University of Nebraska Medicai Center, Omaha, Nebraska 986125, United States
Alexander I. Wallick, Department of Biology, University of Nebraska at Omaha, Omaha, Nebraska 68182, United States
Yuxiang Dong, College of Pharmacy, University of Nebraska Medicai Center, Omaha, Nebraska 986125, United States.
Karen L. White, Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia
Paul H. Davis, Department of Biology, University of Nebraska at Omaha, Omaha, Nebraska 68182, United States.
Susan A. Charman, Centre for Drug Candidate Optimisation, Monash Institute of Pharmaceutical Sciences, Monash University (Parkville Campus), Parkville, Victoria 3052, Australia
Jennifer Keiser, Swiss Tropical and Public Health Institute, CH-4002 Basel, Switzerland; University of Basel, CH-4003 Basel, Switzerland.
Jonathan L. Vennerstrom, College of Pharmacy, University of Nebraska Medical Center, Omaha, Nebraska 986125, United States.
REFERENCES
- (1).Eastman RT; Fidock DA Artemisinin-based combination therapies: A vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009, 7, 864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tu Y The discovery of artemisinin (qinghaosu) and gifts from Chinese medicine. Nat. Med. 2011, 17, 1217–1220. [DOI] [PubMed] [Google Scholar]
- (3).Klayman DL Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985, 228, 1049–1055. [DOI] [PubMed] [Google Scholar]
- (4).Keiser J; Utzinger J Antimalarials in the treatment of schistosomiasis. Curr. Pharm. Des. 2012, 18, 3531–3538. [PubMed] [Google Scholar]
- (5).Zhang X-G; Z. Li, G-X; Zhao S-S; Xu F-L; Wang Y-H; Wang W. A review of dihydroartemisinin as another gift from traditional Chinese medicine not only for malaria control but also for schistosomiasis control. Parasitol Res. 2014, 113, 1769–1773. [DOI] [PubMed] [Google Scholar]
- (6).Saeed MEM; Krishna S; Greten HJ; Kremsner PG; Efferth T Antischistosomal activity of artemisinin derivatives in vivo and in patients. Pharmacol. Res. 2016, 110, 216–226. [DOI] [PubMed] [Google Scholar]
- (7).Caffrey CR; El-Sakkary N; Mader P; Krieg R; Becker K; Schlitzer M; Drewry DH; Vennerstrom JL; Grevelding CG Drug discovery and development for schistosomiasis In Neglected Tropical Diseases: Drug Discovery and Development; Swinney D; Pollastri M, Eds.; Wiley-VCH: Weinheim, Germany, 2019; pp 187–225. [Google Scholar]
- (8).Bergquist R; Utzinger J; Keiser J Controlling schistosomiasis with praziquantel: How much longer without a viable alternative? Infect. Dis. Poveriy 2017, 6, No. 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kaiser M; Wittlin S; Nehrbass-Stuedli A; Dong Y; Wang X; Hemphill A; Matile H; Brun R; Vennerstrom JL Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160). Antimicrob. Agents Chemother. 2007, 51, 2991–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Meshnick SR Artemisinin: mechanisms of action, resistance and toxicity. Int. J. Parasitol. 2002, 32, 1655–1660. [DOI] [PubMed] [Google Scholar]
- (11).Klonis N; Creek DJ; Tilley L Iron and heme metabolism in Plasmodium falciparum and the mechanism of action of artemisinins. Curr. Opin. Microbiol. 2013, 16, 722–727. [DOI] [PubMed] [Google Scholar]
- (12).Creek DJ; Charman WN; Chiu FCK; Prankerd RJ; Dong Y; Vennerstrom JL; Charman SA Relationship between antimalarial activity and haem alkylation for spiro- and dispiro-1,2,4-trioxolane antimalarials. Antimicrob. Agents Chemother. 2008, 52, 1291–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Meunier B; Robert A Heme as trigger and target for trioxane-containing antimalarial drugs. Acc. Chem. Res. 2010, 43, 1444–1451. [DOI] [PubMed] [Google Scholar]
- (14).Tilley L; Charman SA; Vennerstrom JL Semisynthetic artemisinin and synthetic peroxide antimalarials In Neglected Diseases and Drug Discovery; Palmer MJ; Wells TNC, Eds.; Royal Society of Chemistry: Cambridge, U.K, 2012; pp 33–64. [Google Scholar]
- (15).Portela J; Boissier J; Gourbal B; Pradines V; Colliere V; Cosledan F; Meunier B; Robert A Antischistosomal activity of trioxaquines: In vivo efficacy and mechanism of action on Schistosoma mansoni. PLoS Neglected Trop. Dis. 2012, 6, No. e1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Thetiot-Laurent SA-L; Boissier J; Robert A; Meunier B Schistosomiasis chemotherapy. Angew. Chem., Int. Ed. 2013, 52, 7936–7956. [DOI] [PubMed] [Google Scholar]
- (17).Wang J; Zhang CJ; Chia WN; Loh CC; Li Z; Lee YM; He Y; Yuan LX; Lim TK; Liu M; Liew CX; Lee YQ; Zhang J; Lu N; Lim CT; Hua Z-C; Liu B; Shen HM; Tan KS; Lin Q Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, No. 10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ismail HM; Barton VE; Panchana M; Charoensutthivarakul S; Biagini GA; Ward SA; O’Neill PM A click chemistry-based proteomic approach reveals that 1,2,4-trioxolane and artemisinin antimalarials share a common protein alkylation profile. Angew. Chem., Int. Ed. 2016, 55, 6401–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jourdan J; Matile H; Reift E; Biehlmaier O; Dong Y; Wang X; Maser P; Vennerstrom JL; Wittlin S Monoclonal antibodies that recognize the alkylation signature of antimalarial ozonides OZ277 (arterolane) and OZ439 (artefenomel). ACS Infect. Dis. 2016, 2, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jourdan J; Walz A; Matile H; Schmidt A; Wu J; Wang X; Dong Y; Vennerstrom JL; Schmidt RS; Wittlin S; Maser P Stochastic protein alkylation by antimalarial peroxides. ACS Infect. Dis. 2019, 5, 2067–2075. [DOI] [PubMed] [Google Scholar]
- (21).Giannangelo C; Anderson D; Wang X; Vennerstrom JL; Charman SA; Creek DJ Ozonide antimalarials alkylate heme in the malaria parasite Plasmodium falciparum. ACS Infect. Dis. 2019, 5, 2076–2086. [DOI] [PubMed] [Google Scholar]
- (22).Xiao S-H; Keiser J; Chollet J; Utzinger J; Dong Y; Vennerstrom JL; Endriss Y; Tanner M In vitro and in vivo activities of synthetic trioxolanes against major human schistosome species. Antimicrob. Agents Chemother. 2007, 51, 1440–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Xue J; Wang X; Dong Y; Vennerstrom JL; Xiao S-H Effect of ozonide OZ418 against Schistosoma japonicum harbored in mice. Parasitol. Res. 2014, 113, 3259–3266. [DOI] [PubMed] [Google Scholar]
- (24).Keiser J; Ingram K; Vargas M; Chollet J; Wang X; Dong Y; Vennerstrom JL In vivo activity of aryl ozonides against Schistosoma species. Antimicrob. Agents Chemother. 2012, 56, 1090–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Charman SA; Arbe-Barnes S; Bathurst IC; Brun R; Campbell M; Charman WN; Chiu FCK; Chollet J; Craft JC; Creek DJ; Dong Y; Matile H; Maurer M; Morizzi J; Nguyen T; Papastogiannidis P; Scheurer C; Shackleford DM; Sriraghavan K; Stingelin L; Tang Y; Urwyler H; Wang X; White KL; Wittlin S; Zhou L; Vennerstrom JL Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc. Natl. Acad. Sci. U.SA. 2011, 108, 4400–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leonidova A; Vargas M; Huwyler J; Keiser J Pharmacokinetics of the antischistosomal lead ozonide OZ418 in uninfected mice determined by liquid chromatography-tandem mass spectrometry. Antimicrob. Agents Chemother. 2016, 60, 7364–7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Tang Y; Dong Y; Karle JM; DiTusa CA; Vennerstrom JL Synthesis of tetrasubstituted ozonides by the Griesbaum coozonolysis reaction: Diastereoselectivity and functional group transformations by post-ozonolysis reactions. J. Org. Chem. 2004, 69, 6470–6473. [DOI] [PubMed] [Google Scholar]
- (28).Dong Y; Wang X; Kamaraj S; Bulbule VJ; Chiu FCK; Chollet J; Dhanasekaran M; Hein CD; Papastogiannidis P; Morizzi J; Shackleford DM; Barker H; Ryan E; Scheurer C; Tang Y; Zhao Q; Zhou L; White KL; Urwyler H; Charman WN; Matile H; Wittlin S; Charman SA; Vennerstrom JL SAR of the antimalarial ozonide artefenomel (OZ439). J. Med. Chem. 2017, 60, 2654–2668. [DOI] [PubMed] [Google Scholar]
- (29).Chu X-J; Bartkovitz D; Danho W; Swistok J; Cheung AW-H; Kurylko G; Rowan K; Yeon M; Franco L; Qi L; Chen L; Yagaloff K Discovery of 1-amino-4-phenylcyclohexane-1-carboxylic acid and its influence on agonist selectivity between human melanocortin-4 and −1 receptors in linear pentapeptides. Bioorg. Med. Chem. Lett. 2005, 15, 4910–4914. [DOI] [PubMed] [Google Scholar]
- (30).Griesbaum K; Liu X; Kassiaris A; Scherer M Ozonolyses of O-alkylated ketoximes in the presence of carbonyl groups: A facile access to ozonides. Liebigs Ann./Recl 1997, 1381–1390. [Google Scholar]
- (31).Dong Y; Vennerstrom JL Dispiro-1,2,4,5-tetraoxanes via ozonolysis of cycloalkanone O-methyl oximes: A comparison with the peroxidation of cycloalkanones in acetonitrile sulfuric acid media. J. Org. Chem. 1998, 63, 8582–8585. [Google Scholar]
- (32).Ayres FD; Khan SI; Chapman OL; Kaganove SN Improvements in the synthesis of adamantane-2,6-dione and preparation of the novel adamantane-2,6-dione mono-ketal. Tetrahedron Lett. 1994, 35, 7151–7154. [Google Scholar]
- (33).Wu J; Leas DA; Dong Y; Wang X; Ezell EL; Stack DE; Vennerstrom JL Synthesis of 2-azaadamantan-6-one: A missing isomer. ACS Omega 2018, 3, 11362–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Dong Y; Tang Y; Chollet J; Matile H; Wittlin S; Charman SA; Charman WN; Santo Tomas J; Scheurer C; Snyder C; Scorneaux B; Bajpai S; Alexander SA; Wang X; Padmanilayam M; Cheruku SR; Brun R; Vennerstrom JL Effect of functional group polarity on the antimalarial activity of spiro and dispiro-1,2,4-trioxolanes. Bioorg. Med. Chem. 2006, 14, 6368–6382. [DOI] [PubMed] [Google Scholar]
- (35).Vennerstrom JL; Dong Y; Chollet J; Matile H; Padmanilayam M; Tang Y; Charman WN Spiro and dispiro 1,2,4–trioxolane antimalarials. U. S. Patent US6,825,230, November 30, 2004.
- (36).Vennerstrom JL; Dong Y; Charman SA; Wittlin S; Chollet J; Wang X; Sriraghavan K; Zhou L; Matile H; Charman WN Spiro and dispiro 1,2,4-trioxolane antimalarials. PCT WO2009/091433A2, July 23, 2009. [Google Scholar]
- (37).Palm K; Stenberg P; Luthman K; Artursson P Polar molecular surface properties predict the intestinal absorption of drugs in humans. Pharm. Res 1997, 14, 568–571. [DOI] [PubMed] [Google Scholar]
- (38).Lombardo F; Shalaeva MY; Tupper KA; Gao F ElogDoct: A tool for lipophilicity determination in drug discovery. 2. basic and neutral compounds. J. Med. Chem. 2001, 44, 2490–2497. [DOI] [PubMed] [Google Scholar]
- (39).Bevan CD; Lloyd RS A high-throughput screening method for the determination of aqueous drug solubility using laser nephelometry in microtiter plates. Anal. Chem. 2000, 72, 1781–1787. [DOI] [PubMed] [Google Scholar]
- (40).Valko K; Nunhuck S; Bevan C; Abraham MH; Reynolds DP Fast gradient HPLC method to determine compounds binding to human serum albumin. Relationships with octanol/water and immobilized artificial membrane lipophilicity. J. Pharm. Sci. 2003, 92, 2236–2248. [DOI] [PubMed] [Google Scholar]
- (41).Dong Y; Chollet J; Vargas M; Mansour NR; Bickle Q; Alnouti Y; Huang J; Keiser J; Vennerstrom JL Praziquantel analogs with activity against juvenile Schistosoma mansoni Bioorg. Med. Chem. Lett. 2010, 20, 2481–2484. [DOI] [PubMed] [Google Scholar]
- (42).Dong Y; Chollet J; Matile H; Charman SA; Chiu FCK; Charman WN; Scorneaux B; Urwyler H; Santo Tomas J; Scheurer C; Snyder C; Dorn A; Wang X; Karle JM; Tang Y; Wittlin S; Brun R; Vennerstrom JL Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: Charting a workable SAR using simple prototypes. J. Med. Chem. 2005, 48, 4953–4961. [DOI] [PubMed] [Google Scholar]
- (43).Dumele O; Schreib B; Warzok U; Trapp N; Schalley CA; Diederich F Halogen-bonded supramolecular capsules in the solid state, in solution, and in the gas phase. Angew. ChemInt. Ed. 2017, 56, 1152–1157. [DOI] [PubMed] [Google Scholar]
- (44).Coteron JM; Marco M; Esquivias J; Deng X; White KL; White J; Koltun M; El Mazouni F; Kokkonda S; Katneni K; Bhamidipati R; Shackleford DM; Angulo-Barturen I; Ferrer SB; Jimenez-Diaz MB; Gamo FJ; Goldsmith EJ; Charman WN; Bathurst I; Floyd D; Matthews D; Burrows JN; Rathod PK; Charman SA; Phillips MA Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Lombardo FC; Pasche V; Panic G; Endriss Y; Keiser J Life cycle maintenance and drug-sensitivity assays for early drug discovery in Schistosoma mansoni. Nat. Protoc. 2019, 14, 461–481. [DOI] [PubMed] [Google Scholar]
- (46).Sanford AG; Schulze TT; Potluri LP; Hemsley RM; Larson JJ; Judge AK; Zach SJ; Wang X; Charman SA; Vennerstrom JL; Davis PH Novel Toxoplasma gondii inhibitor chemotypes. Parasitol. Int. 2018, 67, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.