

Abstract
Cancer vaccines initiate antitumor responses in a subset of patients, but the lack of clinically meaningful biomarkers to predict treatment response limits their development. Here, we design multifunctional RNA-loaded magnetic liposomes to initiate potent antitumor immunity and function as an early biomarker of treatment response. These particles activate DCs more effectively than electroporation leading to superior inhibition of tumor growth in treatment models. Inclusion of iron oxide enhances DC transfection and enables tracking of DC migration with Magnetic Resonance Imaging (MRI). We show that T2*-weighted MRI intensity in lymph nodes is a strong correlate of DC trafficking and is an early predictor of antitumor response. In preclinical tumor models, MRI-predicted “responders” identified two days after vaccination had significantly smaller tumors 2-5 weeks after treatment and lived 73% longer than MRI-predicted “non-responders.” These studies therefore provide a simple, scalable nanoparticle formulation to generate robust antitumor immune responses and predict individual treatment outcome with MRI.
Keywords: iron oxide, dendritic cells, cancer immunotherapy, biomarker, liposome, RNA, RNA-lipoplex
Graphical Abstract
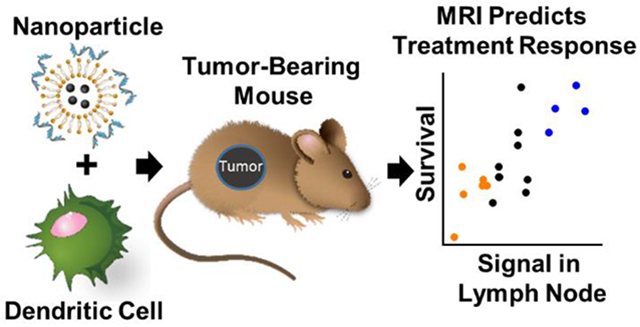
Cancer immunotherapy has produced impressive tumor regression in settings where conventional treatments yield no benefit.1 However, even the most effective immunotherapy strategies extend survival for only a subset of patients.2-6 Although multiple pretreatment biomarkers suggest susceptibility to immunotherapy, there are currently no robust markers that predict clinical response.7-9 Future development of promising cancer immunotherapies will require dynamic biomarkers to differentiate responding and nonresponding patients before tumor progression.9
We previously demonstrated that DC migration to vaccine-site draining lymph nodes (VDLNs) as assessed by SPECT/CT imaging of Indium111-labeled DCs just two days after vaccination may provide an early biomarker of overall survival in GBM patients treated with RNA-pulsed DC vaccines.10 However, radioactive cell labelling for PET and SPECT is cumbersome and not widely available in the clinical setting.11 Clinical evaluation of this biomarker will require a widely available method to sensitively track DC migration. MRI is a widely available imaging modality that has been used to qualitatively track large numbers of cells in humans, but MRI-based quantification of cell migration to lymph nodes remains challenging.12-18
Additionally, although electroporation is widely used to deliver RNA to DCs in clinical trials,10, 19-23 an immune-stimulatory replacement to electroporation could further enhance therapeutic benefit. Nanomaterials are attractive for non-viral mRNA delivery,24 but few nanoparticles reach clinical utilization due to the complexity of large-scale clinical grade manufacturing.7, 25, 26 Here, we overcome this limitation with scalable, multifunctional nanoliposomes based on previously translated materials that efficiently transfect DCs with RNA, stimulate profound DC activation, and establish MRI-detected DC migration as a biomarker of antitumor response to DC vaccines (Scheme 1).
Scheme 1: Schematic Illustration of IO-RNA-NPs.
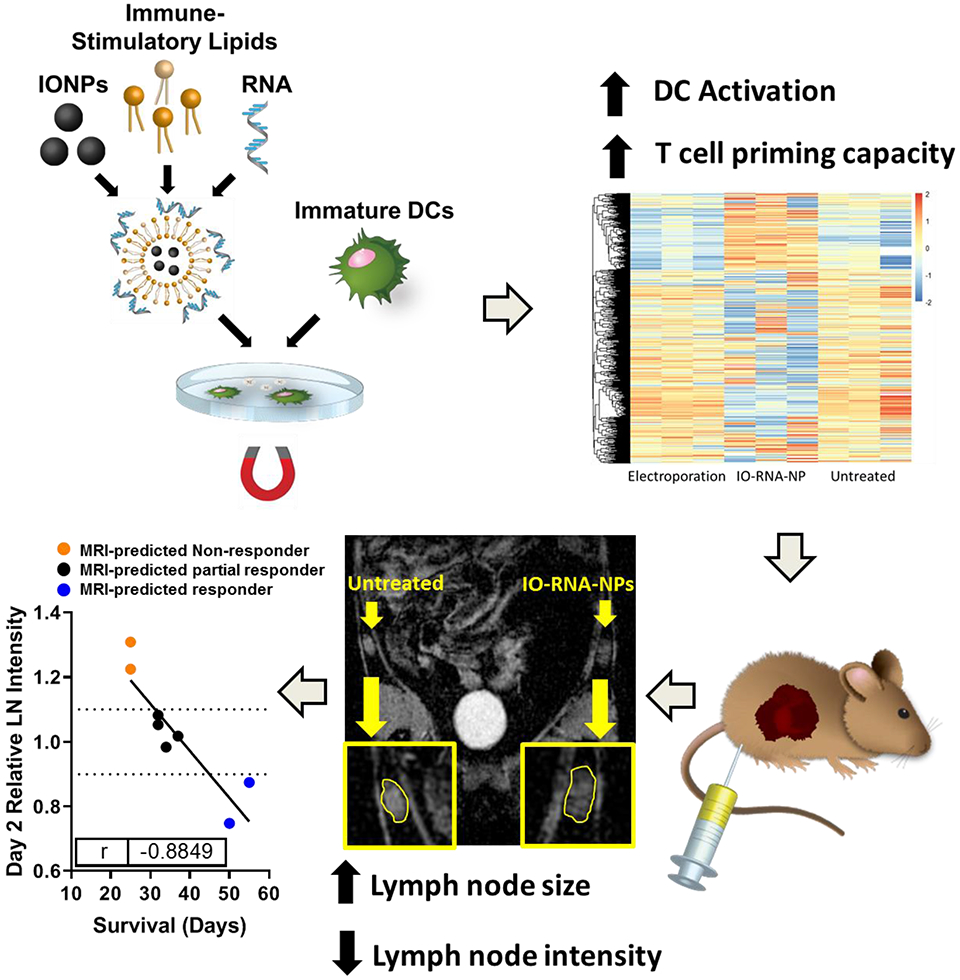
IO-RNA-NPs were generated by combining commercially available IONPs and mRNA encoding tumor antigens with a combination of previously translated lipids with exceptional capacity for mRNA delivery and DC activation. Incubation of these particles with DCs in the presence or absence of a magnetic field led to profound DC activation characterized by dramatic changes in RNA expression and enhanced capacity to stimulate antigen specific T cells. IO-RNA-NPs enabled MRI-based detection of DC migration to lymph nodes that correlated directly with survival in murine tumor models.
In this study, we first screen a library of lipid particles with proven safety profiles in humans to develop an optimized lipid nanoparticle formulation for DC activation in vitro. We then combine these immune-stimulatory RNA-loaded cationic nanoliposomes (RNA-NPs) with the T2 MRI contrast-enhancing effects of iron oxide nanoparticles (IONPs). The resulting iron oxide loaded RNA-NPs (IO-RNA-NPs) deliver RNA to DCs, activate those DCs, and enable prediction of tumor regression with MRI. We find that IO-RNA-NPs dramatically change gene expression profiles in DCs compared to electroporation, leading to increased expression of costimulatory markers, production of inflammatory cytokines (e.g. IFN-α), and enhanced migration to lymph nodes. Importantly, we also demonstrate that DCs loaded with RNA encoding tumor antigens via IO-RNA-NPs inhibit tumor growth in a treatment model in which RNA electroporated DCs yield no benefit. In contrast to previous work demonstrating qualitative MRI changes with IONP-loaded DCs,13, 27-29 we then demonstrate that MRI-detected DC trafficking predicts long-term inhibition of tumor growth and survival in murine tumor models. Substantial reduction in T2*-weighted MRI intensity in treated lymph nodes two days after vaccination correlates strongly with reduced tumor size 2-5 weeks after vaccination and predicts a 73% increase in median survival compared to treated mice without this change. Taken together, our findings demonstrate that hese DC-activating IO-RNA-NPs stimulate robust inhibition of tumor growth and enable early prediction of antitumor response to DC vaccines with a widely available imaging modality.
RESULTS AND DISCUSSION
Design of immune-stimulatory iron oxide loaded liposomes
We first sought to develop a simple, translatable method to deliver mRNA to DCs and track their movement with MRI. IONPs are attractive MRI-contrast agents due to their proven clinical utility, but present methods to optimize IONPs for RNA delivery in the preclinical setting utilize polymers without proven safety records in humans (e.g. polyethylenimine). Cationic liposomes are attractive agents for mRNA delivery in the clinical setting due to their simple, scalable synthesis and favorable safety profiles in animals and humans. However, lipid nanoparticle formulations currently in clinical evaluation are not optimized for DC activation in vitro, and previous attempts to develop cationic liposomes for DC activation in vitro limited evaluation to expression of activation markers instead of functional outcomes (e.g. capacity for transfected DCs to activate antigen specific T cells). Here, we created a library of lipid nanoparticles using commercially available materials with established safety profiles in clinical trials7 and evaluated their capacity to transfect and activate bone marrow-derived DCs (BMDCs) in vitro using both basic (i.e., transfection efficiency, viability) and functional tests (Supplementary Table 1). To do this, we developed a scoring system based on BMDC transfection, viability, expression of co-stimulatory markers (CD80, CD86, CD40), and capacity to stimulate antigen-specific T cells in vitro (Supplementary Fig 1). We found that the inclusion of cholesterol in 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) liposomes produced the most effective particles for transfection and activation of murine DCs, with Activation Scores 18 times higher than those achieved by DOTAP liposomes without cholesterol (Supplementary Fig 1).
We then developed a method to incorporate commercially available IONPs into these DC-activating cationic liposomes to enable MRI tracking without significantly increasing synthesis complexity. Since cationic liposomes have positively charged interiors, we reasoned that addition of negatively charged IONPs during particle formation could produce liposomes with solid iron oxide cores. We therefore rehydrated cationic lipids with various concentrations of carboxylated IONPs (0, 1, 10, 100, or 150ug IONPs per mg lipid) and incubated the resulting liposomes with mRNA to generate RNA-lipoplexes without IONPs (RNA-NPs) or with IONPs (IO-RNA-NPs). Inclusion of IONPs resulted in formation of 100-300nm liposomes with clear lipid bilayers and solid cores of IONPs evident by Cryo-TEM (Fig 1a). Consistent with IONP encapsulation within liposomes, particle size (0ug: 207.9±67.2nm; 100ug: 208.7±92.3nm) and charge (0ug: 44.1±4.5meV; 100ug: 40.2±7.58meV) remained consistent regardless of IONP content (Fig 1b, Supplementary Table 2). However, particles including iron oxide exhibited substantial magnetic properties (Saturation Magnetization=233938A/m) (Fig 1c).
Figure 1. Development and characterization of iron oxide loaded RNA-nanoparticles.
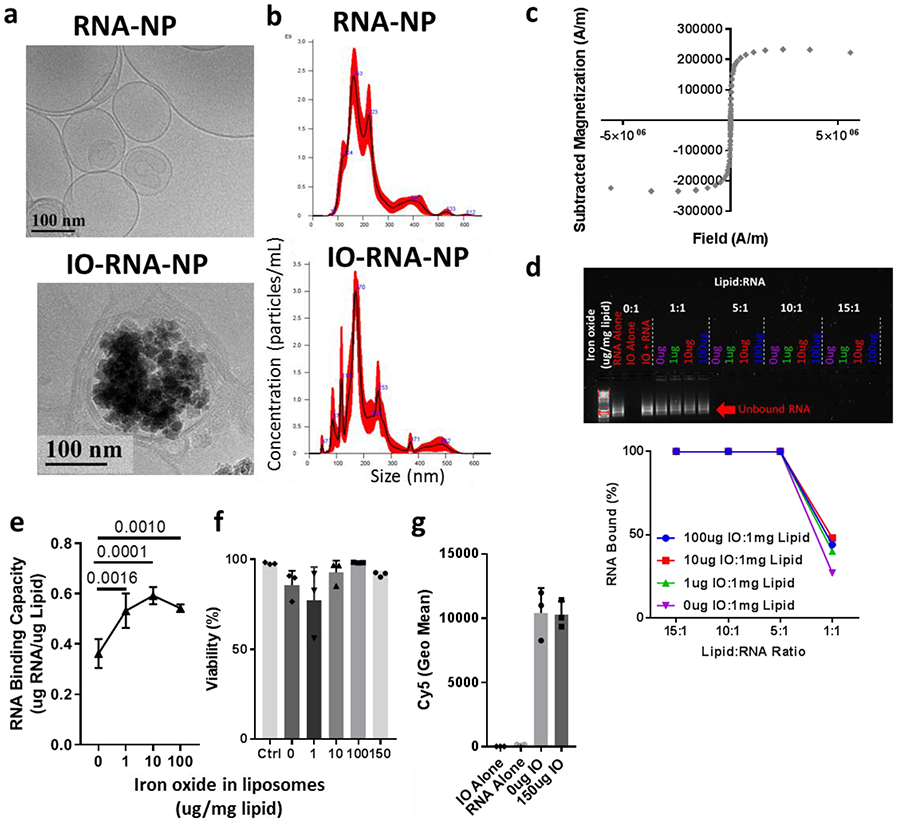
a, Representative Cryo-TEM of RNA-NPs with or without iron oxide (IO). b, Size distribution of RNA-NPs with and without IO (100ug IO:1mg lipid) assessed by Nanosight. c, Saturation magnetization of IO-RNA-NPs (100ug IO:1mg lipid) assessed with a SQUID magnetometer. d, Agarose gel electropheresis demonstrating RNA bound by different formulations of IO-RNA-NPs (labelled as the mass of IONPs in each formulation per mg lipid) after 15 minute incubation with RNA at different lipid:RNA ratios. e, RNA-binding capacity for RNA-NPs containing varying amounts of iron oxide. Numbers on graph are p values derived from one-way ANOVA and Tukey’s tests with n=4 per group. f, Viability of DC2.4s after 24 hour incubation with RNA-NPs assessed by flow cytometry (n=3). g, Uptake of Cy5-labelled RNA by DC2.4s assessed by flow cytometry after overnight incubation with RNA-NPs (n=3).
We then tested whether the inclusion of IONPs would impede RNA binding and delivery. All particles bound 100% of available RNA under experimental conditions (15:1 Lipid:RNA ratio) (Fig 1d), and particles with increased IONP content demonstrated slightly higher RNA binding capacity in the setting of excess RNA (0ug: 0.36 μg RNA/μg Lipid; 100μg: 0.54μg RNA/μg Lipid; p=0.0010) (Fig 1e). Importantly, overnight incubation of particles with DC2.4s, an immortalized cell line of DCs, demonstrated that inclusion of even high amounts of IONPs did not increase toxicity (Fig 1f) or reduce RNA-delivery (Fig 1g). We then evaluated the mechanism by which IO-RNA-NPs were taken into cells. In line with previous demonstrations of cationic liposome uptake via macropinocytosis, co-incubation of DCs with the macropinocytosis inhibitors rottlerin and cytochalasin D each significantly reduced IO-RNA-NP uptake (Supplementary Fig 2)30. Taken together, this evidence suggests that rehydration of cationic lipids in the presence of negatively charged IONPs produces IO-RNA-NPs with the magnetic properties of IONPs and the RNA-delivery capacity of cationic liposomes.
IONPs increase DC transfection and activation
Next, we sought to confirm that IONPs would not inhibit transfection efficiency and DC activation. Interestingly, fluorescent images and flow cytometry revealed a dose-dependent increase in transfection efficiency for particles with increasing iron oxide content in DC2.4s (0μg: 10.6%; 150μg: 33.0%, p=0.0037; Pearson’s r=0.9194, p=0.0272) (Fig 2a-b), and primary BMDCs (0μg: 7.0%; 100μg: 9.9%; p=0.001) (Fig 2c). However, the addition of unbound IONPs that were not previously complexed with cationic liposomes produced no benefit (RNA-NPs: 7.0%±0.96; RNA-NPs+IONPs: 5.8%±0.05; p=0.244) (Fig 2c). These results suggest that IONPs increase transfection efficiency but only when incorporated within liposomes.
Figure 2. Iron oxide enhances transfection and activation of dendritic cells.
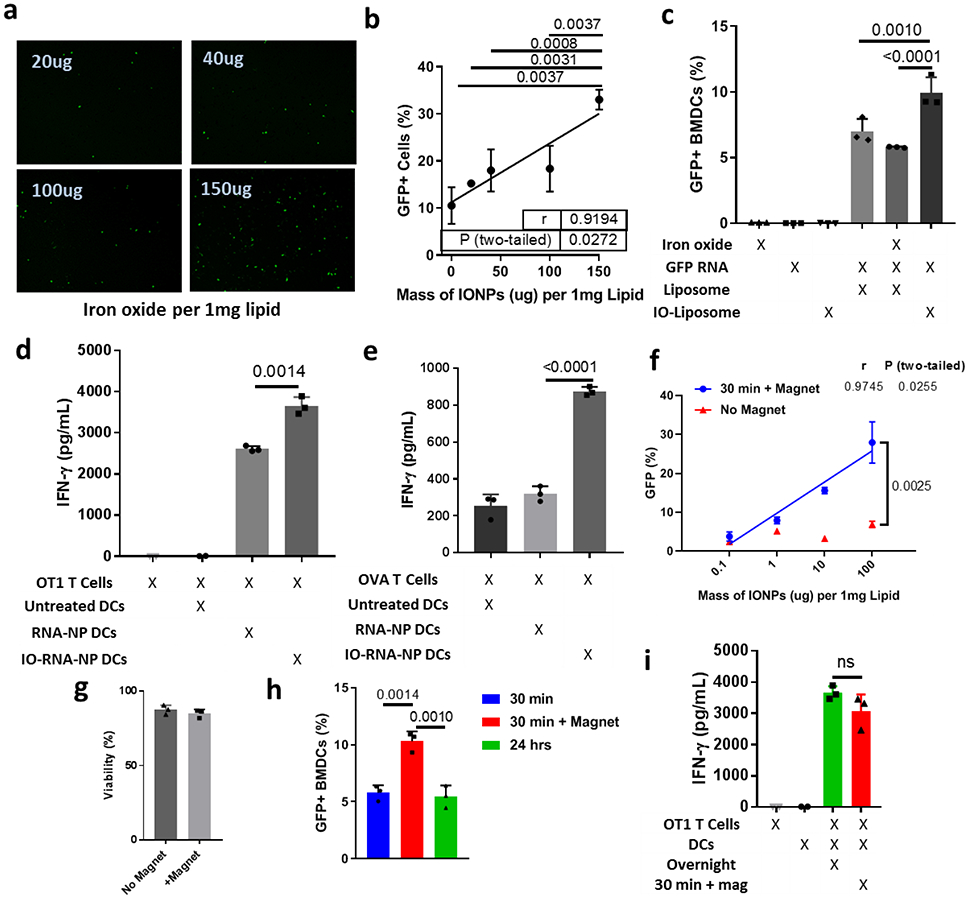
a, Representative images of GFP expression in DC2.4s after 24-hour incubation with RNA-NPs synthesized with varying amounts of iron oxide per 1mg lipid. b, Quantification of transfection efficiency from (a) via flow cytometry. Pearson’s correlation and ANOVA with Tukey’s tests were used for statistical analysis. c, Fluorescence in BMDCs after transfection with GFP-RNA-loaded liposomes with no iron oxide, iron oxide encapsulated inside the liposomes (IO-Liposome), or iron oxide added to the media outside the liposomes. An ANOVA with Tukey’s tests were used for statistical analysis. d-e, ELISA for IFN-gamma produced after a two-day co-culture combining DCs loaded with OVA RNA via RNA-NPs or IO-RNA-NPs with naïve splenocytes from OT1 mice (d) or antigen experienced OVA T-cells (e). f, Transfection efficiency in DC2.4s after a 30-minute incubation with IO-RNA-NPs in the presence or absence of a magnetic field. A Pearson’s correlation coefficient was used for statistical analysis. g, Viability of BMDCs 24 hours after a 30-minute incubation with IO-RNA-NPs in the presence or absence of a magnetic field. h, GFP expression in BMDCs after either a 30-minute incubation with IO-RNA-NPs (100ug IONP:1mg lipid) in the presence or absence of a magnetic field or an overnight incubation in the absence of a field. One-way ANOVA with Tukey’s tests were used for statistical analysis. i, ELISA for IFN-γ produced during a two-day co-culture of antigen-naïve OT1 T-cells and BMDCs treated with IO-RNA-NPs bearing OVA mRNA either overnight or for 30 minutes in the presence of a magnetic field. For all experiments, n=3, error bars represent standard deviation from the mean, and numbers are P values calculated from two-tailed unpaired two sample t tests or Tukey’s tests as appropriate. Results in a-c, and f-i are each representative of at least two replicate experiments. ns=not significant.
We then evaluated whether this increase in transfection was accompanied by enhanced DC activation and function. While RNA-NPs slightly increased expression of the costimulatory molecules CD80 and CD86 beyond that achieved with inflammatory cytokines IL-4 and GM-CSF, IO-RNA-NPs further enhanced expression of CD86 and co-expression of both molecules (Supplementary Fig 3a and 3b). Given this suggestion that IONPs contribute to a more activated DC phenotype, we evaluated the impact of IONPs on DC function. Bone marrow-derived dendritic cells (BMDCs) were treated with RNA-NPs or IO-RNA-NPs bearing mRNA encoding ovalbumin (OVA) and incubated with naïve OVA-specific OT1 T-cells or antigen-experienced OVA T-cells. While both particle constructs induced substantial T-cell activation as measured by IFN-ɣ production at 48 hours, inclusion of IONPs within RNA-NPs significantly enhanced activation of antigen-experienced T-cells (RNA-NP DCs: 2611.8±67.06pg/mL; IO-RNA-NP DCs: 3653.5±216.8pg/mL; p=0.0014) (Fig 2d) and priming of naïve T-cells (RNA-NP DCs: 319.0±41.29pg/mL; IO-RNA-NP DCs: 874.6±23.9pg/mL; p<0.0001) (Fig 2e) in an antigen-specific manner (Supplementary Figure 3c). These unexpected results demonstrate that inclusion of IONPs in liposomes enhances BMDC activation and priming of antitumor T-cell responses.
We next evaluated whether IONP-mediated DC transfection could be further enhanced with application of external magnetic fields arranged to pull particles into contact with cells. Addition of magnetic fields enhanced transfection efficiency of DCs in a manner dependent on the concentration of IONPs within liposomes (Fig 2f), resulting in a 3-fold increase in transfection efficiency at the highest tested concentration of IONPs (-Mag: 6.87+/−0.8; +Mag: 27.93+/−5.3; p=0.0025) without compromising viability (Fig 2g). Importantly, a thirty-minute incubation with IO-RNA-NPs under the influence of a magnetic field produced double the transfection efficiency compared to overnight incubation with primary BMDCs (Overnight: 5.5±0.97%; 30min+Mag: 10.3±0.85%; p=0.0010) (Fig 2h), and comparable T-cell priming capacity (Overnight: 3653.5±216.8pg/mL; 30min+Mag: 3070.4±536.1pg/mL; p=0.16) (Fig 2i). Magnetic fields can therefore be used to significantly reduce transfection time while maintaining high levels of transfection efficiency.
IO-RNA-NPs activate DCs more effectively than electroporation
Although liposome-mediated RNA delivery leads to robust DC activation in vivo, electroporation remains the preferred technique for delivering RNA to DCs ex vivo in the clinical setting due to its high transfection efficiency (60-80% of BMDCs) compared to other non-viral transfection methods. Indeed, electroporation transfected a greater percentage of BMDCs than IO-RNA-NPs (Electroporation: 81.1%; IO-RNA-NPs: 17.733%; p<0.0001) (Fig 3a). However, electroporation bypasses pattern recognition receptors that contribute to DC activation by avoiding natural antigen processing in endosomes and phagolysosomes. The clear difference in these transfection methods is visible with fluorescent microscopy of DCs after delivery of Cy3-labelled RNA (Fig 3b). RNA delivered by IO-RNA-NPs clusters into bright intracellular compartments consistent with endosomes and phagolysosomes where it interacts with the endosomally restricted toll like receptor TLR7 (Fig 3b, Supplementary Fig 2c). In contrast, RNA delivered by electroporation is diluted throughout the cytosol (Fig 3b). We hypothesized that this natural antigen uptake might allow IO-RNA-NPs to activate DCs more effectively than electroporation.
Figure 3. IO-RNA-NPs enhance DC activation and migration compared to electroporation.
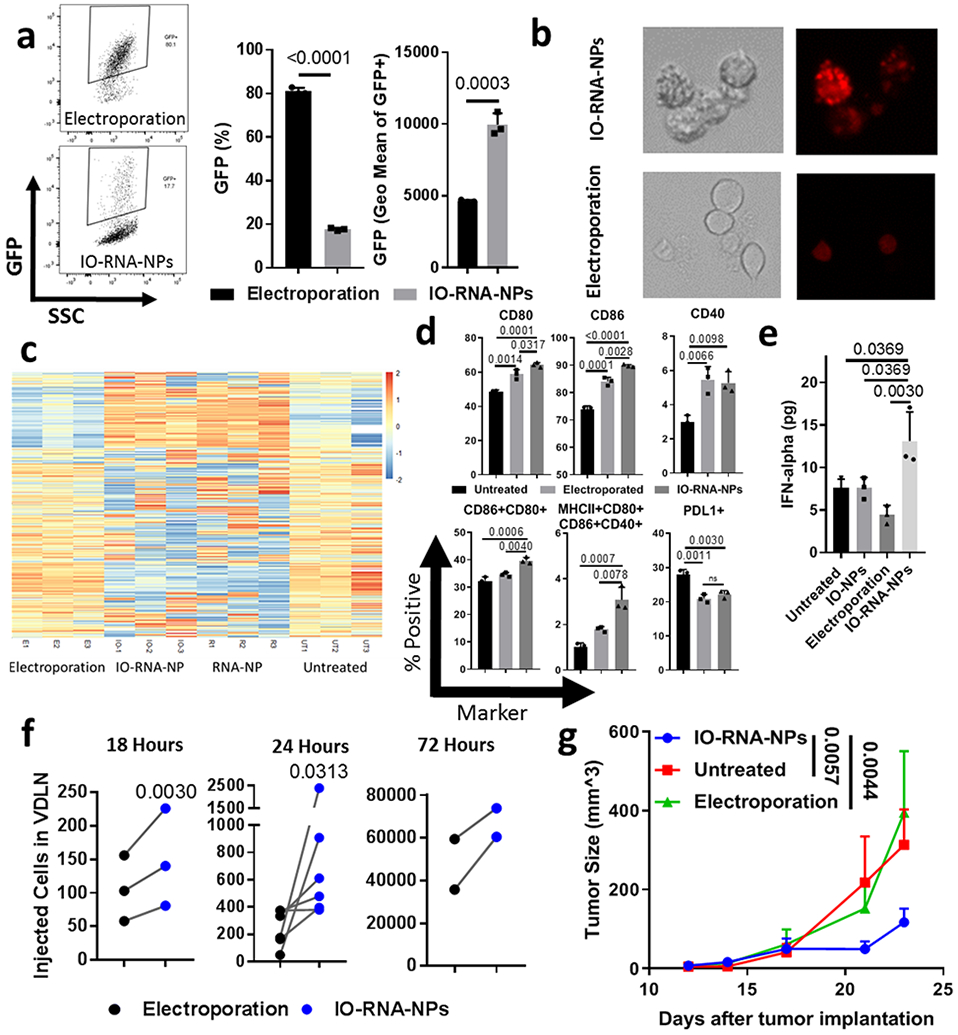
a, Representative flow cytometry plots (left), transfection efficiency (center) and geometric mean fluorescence intensity (right) 24 hours after transfection of BMDCs with GFP RNA via electroporation or IO-RNA-NPs (n=3). b, Fluorescent microscope images of DC2.4s incubated overnight with Cy3-labelled RNA-NPs. c, Heat map comparing mRNA expression in BMDCs 24 hours after treatment with GFP mRNA via RNA-NPs, IO-RNA-NPs, or Electroporation. d-e, Phenotypic markers of activation assessed by flow cytometry (d) and IFN-alpha release assessed by ELISA (e) for BMDCs 24 hours after treatment with electroporation or IO-RNA-NPs. One-way ANOVA and Tukey’s tests were used for statistical analysis. f, Migration of IO-RNA-NP-loaded BMDCs to VDLN at varying timepoints after intradermal injection. Statistical analysis was completed with Wilcoxon matched-pairs rank sum test for n>4 or student’s paired t test for n<4. g, Summary data showing tumor growth in mice (Untreated: n=6; Electroporation: n=6; IO-RNA-NPs: n=13) with subcutaneous B16F10-OVA tumors after a single vaccination with 500,000 BMDCs pulsed with OVA mRNA via electroporation or IO-RNA-NPs and 10 million naïve OT1 T-cells. Data is pooled from two independent experiments. A two-way ANOVA was used for statistical analysis. Results in a, b, and d-g are each representative of at least 2 replicate experiments. Numbers on graphs are P values.
To test this hypothesis, we evaluated the effects of transfection via electroporation and IO-RNA-NPs on DC activation, including gene expression, upregulation of costimulatory molecules, secretion of inflammatory cytokines, cell migration to lymph nodes, and antitumor immune responses.
We first evaluated the impact of each transfection method on gene expression. BMDCs treated with IO-RNA-NPs exhibited significant changes in RNA expression profiles after 24 hours compared to no treatment or electroporation (Fig 3c), including markedly enhanced expression of gene sets related to antiviral defense, Type I Interferon production, toll-like receptor (TLR) signaling, the innate immune response, and lymphocyte migration (Supplementary Fig 4, Supplementary Table 3). Further analysis of individual genes responsible for lymphocyte homing and activation revealed significant upregulation of the T cell activating cytokine IL-12 and the chemokines CCL3 and CCL4, which are both heavily implicated in attraction of T cells to DCs in lymphoid organs (Supplementary Fig 4). Interestingly, the expression of all of these gene sets was essentially equivalent in the absence of iron oxide (0 gene sets significantly upregulated with IO-RNA-NPs compared to RNA-NPs), indicating that RNA is a larger contributor to IO-RNA-NP-mediated DC activation than IONPs (Fig 3c). We therefore completed future studies using IO-RNA-NPs as our standard formulation unless otherwise specified.
These broad changes in gene expression profiles with lipid-mediated RNA delivery suggest a substantial advantage of liposome-mediated DC transfection compared to transfection via electroporation and led us to consider the functional consequences of this increased gene expression. Although both IO-RNA-NPs and electroporation increased expression of costimulatory molecules by flow cytometry, IO-RNA-NPs induced higher co-expression of these markers (Fig 3d, Supplementary Fig 5). We next evaluated whether this activation phenotype was accompanied by increased secretion of IFN-α, which we and others previously showed is required to initiate antitumor immune responses to systemic RNA-NPs.30-32 In accordance with our RNA-sequencing data, we found that IO-RNA-NPs increased production of IFN-α (IO-RNA-NP DCs: 13.1±3.4pg; Untreated DCs: 7.6±1.3pg; p=0.0369) (Fig 3e). In contrast, electroporation non-significantly decreased IFN-α production relative to controls (Electroporated DCs: 4.45±1.1pg; Untreated DCs 7.6±1.0pg; p=0.2879) (Fig 3e). This result suggests that IO-RNA-NPs stimulate an IFN-α-dependent antitumor immune response that is absent in electroporated cells.
We previously reported in a blinded and randomized pilot clinical trial that enhanced migration of RNA-loaded DC vaccines to VDLNs correlated with improved survival in patients with GBM.10 We therefore compared the migratory capacity of DCs loaded with IO-RNA-NPs or electroporation in a murine model of our clinical protocol in which DCs are prepared in the setting of GM-CSF and IL-4.10 In this experiment, each mouse received contralateral intradermal injections of DsRed+ DCs treated with RNA electroporation or IO-RNA-NPs. In three separate experiments, each evaluating different time points, DCs loaded with IO-RNA-NPs migrated to lymph nodes more efficiently than those treated with electroporation (18 hours: p=0.003, n=3; 24 hours: p=0.0313, n=6; and 72 hours: p=0.16, n=2) (Fig 3f).
We then evaluated the impact of IO-RNA-NPs and electroporation on inhibition of tumor growth in an established tumor model. We found that a single vaccination with DCs loaded with OVA mRNA via IO-RNA-NPs 5 days after tumor inoculation significantly inhibited growth of subcutaneous B16F10-OVA tumors (IO-RNA-NP vs Untreated: p=0.0057; IO-RNA-NP vs Electroporation: p=0.0044) (Fig 3g). In contrast, DCs treated with OVA RNA electroporation provided no treatment benefit (Electroporation vs Untreated: p=0.8) (Fig 3g).
Superior DC activation in the setting of increased type I IFN production and upregulation of gene sets denoting toll-like receptors compared to electroporation suggested a major role for endosomally restricted pattern recognition receptors in IO-RNA-NP-mediated DC activation. To confirm this effect, we generated DCs from either C57Bl6 or TLR7−/− mice and treated them with IO-RNA-NPs. As expected, DCs from TLR7−/− mice exhibited significantly diminished upregulation of costimulatory markers in response to IO-RNA-NP stimulation, indicating a role for TLR7 in IO-RNA-NP-mediated DC activation (Supplementary Fig 6). We then evaluated the therapeutic importance of TLR7 signaling in treatment of mice with subcutaneous B16F10-OVA tumors. While IO-RNA-NP-loaded C57Bl6 DCs produced marked inhibition of tumor growth and extended survival compared to either no treatment (p=0.0252) or IO-RNA-NP-loaded TLR7−/− DCs (p=0.0138), IO-RNA-NP-loaded TLR7−/− DCs failed to produce any therapeutic benefit (Supplementary Fig 6). Taken together, this evidence indicates that IO-RNA-NPs induce robust DC activation characterized by immune-related gene signatures, expression of costimulatory molecules, secretion of inflammatory cytokines (IFN-α), and enhanced migration to lymph nodes. This DC activation is dependent on TLR7-signaling and sufficient to inhibit tumor growth in a treatment model in which a clinically utilized RNA delivery technique (RNA electroporation) yields no benefit. These results suggest that IO-RNA-NPs are a promising alternative to electroporation for DC vaccines.
Cell tracking with MRI
We next sought to evaluate the utility of IO-RNA-NPs for DC tracking with MRI. DsRed+ BMDCs were treated with IO-RNA-NPs and injected intradermally into the inguinal areas of naïve mice. After two days, MRI images revealed a visible increase in volume in treated lymph nodes across multiple imaging parameters (Fig 4a). Researchers blinded to treatment group quantified these results by manually drawing regions of interest around inguinal LNs in every slice in which they appeared. Relative size was calculated as the total volume of each LN compared to the contralateral, untreated LN. Relative intensity was calculated as the average T2*-weighted MRI intensity of the treated LN compared to the contralateral, untreated LN. Flow cytometry (Fig 4b) revealed that the relative increase in size of the VDLN compared to the contralateral untreated lymph node correlated strongly with absolute counts of DsRed+ cells in treated lymph nodes (r=0.9015; p=0.0002) (Fig 4c). Furthermore, optimized T2*-weighted MRI sequences detected reductions in intensity in VDLN compared to contralateral untreated lymph nodes (Relative Intensity of VDLN) consistent with high concentrations of iron oxide. These reductions in MRI intensity correlated strongly with both iron oxide content (r=−0.6392; p=0.0006) (Fig 4d) and absolute counts of injected cells in VDLNs (r=−0.7727; p=0.0074) (Fig 4e). In order to confirm that the changes in T2*-weighted MRI intensity in the treated lymph node were due to the presence of iron oxide, we duplicated this observation in mice treated with bare IONPs without cells (r=−0.6425; p=0.0451) (Supplementary Fig 7). We also repeated the same experiment in the absence of IONPs to demonstrate that correlation of MRI intensity with cell count is dependent on IONP uptake in DCs (Supplementary Fig 8). Together, these results demonstrate the capacity for MRI to distinguish lymph nodes with high and low concentrations of IONP-loaded DCs.
Figure 4. IO-RNA-NPs enable quantitative cell tracking with MRI.
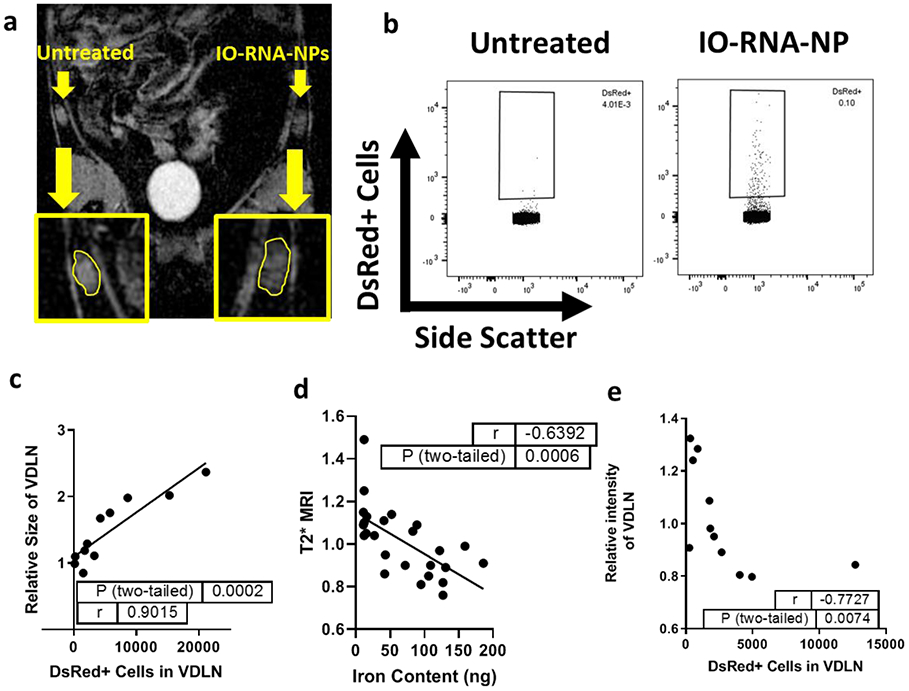
a, T2*-weighted MRI image 48 hours after vaccination with IO-RNA-NP-loaded DsRed+ DCs in the left inguinal area. Yellow borders indicate lymph nodes on treated (right) and untreated (left) sides. b, Exemplary flow cytometry plots demonstrating gating on DsRed+ cells in lymph nodes. c, Correlation of relative lymph node size between treated and untreated lymph nodes and the absolute count of DCs in that lymph node. Data is combined from 2 independent experiments. d-e, Correlation of relative T2*-weighted MRI intensity in treated and untreated lymph nodes with iron oxide content (d) and absolute counts of labelled cells (e). Data is representative of two replicate experiments. p values and r values are derived from a Pearson correlation for parametric data with no outliers (c, d) and Spearman’s correlation for data with significant outliers (e).
MRI as an early biomarker of antitumor response in early and established tumor models
Having shown that IO-RNA-NPs stimulate robust DC activation and produce consistent changes to MRI intensity in lymph nodes, we evaluated the utility of MRI-detected DC migration as a biomarker to predict antitumor immune response. We first developed a model of early cancer in which vaccination produces variable antitumor responses. Intradermal injection of DCs loaded with IO-RNA-NPs bearing OVA mRNA on the same day of tumor implantation significantly inhibited growth of subcutaneous B16F10-OVA tumors compared to untreated controls (Day 19: p=0.0376; Day 23: p=0.0351; Day 25: p=0.0328) (Fig 5a). However, large cohorts of treated mice demonstrated heterogeneous inhibition of tumor growth (Fig 5b). These differences became pronounced on Day 27, at which a subset of mice had substantial tumors while others had none (Fig 5b). Comparison of Day 2 MRI images revealed that MRI-detected LN size did not correlate with treatment outcome (Supplementary Fig 9). However, tumor size on Day 27 did correlate well with reduction in T2*-weighted MRI intensities in treated LNs compared to the contralateral, untreated LNs in the same mice (r=0.5045; p=0.0166) (Fig 5c). The predictive value of this biomarker was further confirmed upon following these mice to endpoint, as reduction in T2*-weighted MRI intensity two days after treatment also correlated directly with survival (r=−0.6168; p=0.0022) (Fig 5d).
Figure 5. MRI-detected DC migration predicts response to DC vaccines.
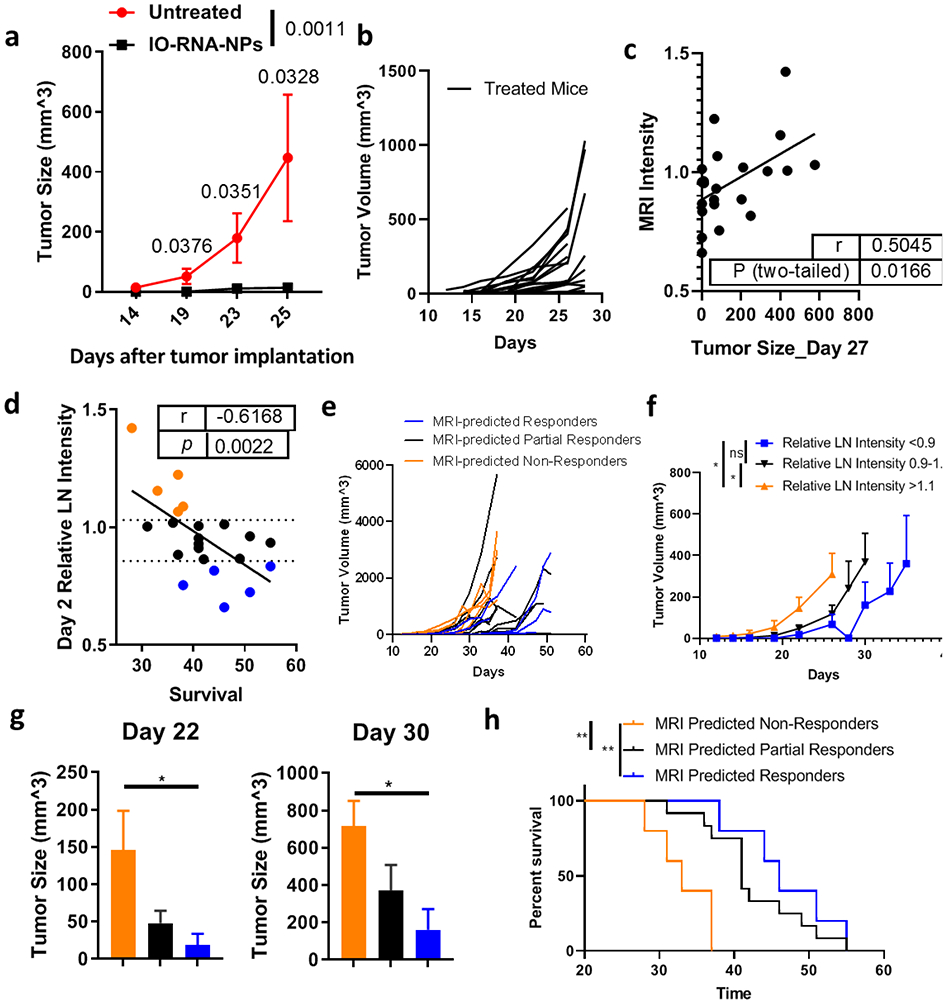
Mice with subcutaneous B16F10-OVA tumors were treated with BMDCs loaded with IO-RNA-NPs bearing ovalbumin mRNA. a, Tumor growth over time between treated mice (n=7) and untreated mice (n=5). Numbers on graph are P values calculated by unpaired student’s t tests. b, Growth of individual treated tumors from two independent experiments (n=20). c-d, Correlation of the relative change in MRI-detected lymph node intensity in treated compared to untreated lymph nodes (Relative LN Intensity) on Day 2 with Day 27 tumor size (c) and survival (d). Dotted lines demarcate the 25th and 75th percentiles of relative MRI intensity in lymph nodes. Datapoints from mice with substantial MRI-predicted DC migration indicated by relative VDLN intensity in the bottom 25th percentile are blue, those from mice with moderate VDLN intensity in the middle 50th percentile are black, and those from mice with high VDLN intensity in the top 75th percentile are orange. Data are combined from two independent experiments which each resulted in statistically significant correlations (see Supplementary Fig 10-11). Numbers on graphs c-d are derived from a Pearson correlation. e-h, Individual tumor growth curves (e), summary data (f), tumor sizes at multiple timepoints (g), and survival (h) separated by MRI intensity on Day 2 after vaccination. Numbers on graphs are p values calculated from ANOVA (f), Mann-Whitney (g), or Log-Rank tests (h).
We then evaluated whether T2*-weighted MRI-intensity in treated lymph nodes could be used to separate treated mice into meaningful groups to predict treatment outcome. We therefore separated mice into groups based on the Day 2 T2*-weighted MRI intensity in their treated LNs compared to the contralateral, untreated LN. We found that mice with substantial DC migration indicated by MRI-intensities in the bottom 25th percentile (MRI-predicted Responders) had significantly smaller tumors than mice with minimal DC migration indicated by MRI intensities in the top 75th percentile (MRI-predicted Non-Responders) throughout the analysis (p=0.0202) (Fig 5e-f) and on individual days including Day 22 (p=0.0473) and Day 30 (p=0.0317) (Fig 5h), leading to a 39% increase in median survival (MRI-predicted Responders: 46 days; MRI-predicted Non-Responders: 33 Days; p=0.0004). Furthermore, mice with intermediate DC migration exhibited intermediate antitumor activity (Fig 5f-h). These results provide strong evidence that MRI-detected DC migration two days after treatment is an early, dynamic biomarker of antitumor immune responses.
Having established utility in an early tumor model, we examined the predictive capacity of MRI-detected DC migration in the setting of established tumors. In this experiment, all mice had palpable subcutaneous tumors before treatment with IO-RNA-NP-loaded DCs (Fig 6a). Here, we again observed significant inhibition of tumor growth with treatment (p<0.0001) (Fig 6b) despite much faster tumor growth and heterogeneous efficacy among treated mice (Fig 6c). Analysis of tumor growth curves with MRI taken two days after vaccination again revealed that early decreases in intensity in treated lymph nodes correlated strongly with future inhibition of tumor growth (Day 2 MRI Intensity vs Day 14 Tumor Size: r=0.8052, p<0.0001; Day 2 MRI Intensity vs Day 17 Tumor Size: r=0.7246; p=0.0013) (Fig 6d, Fig 6e). Mice with high T2*-weighted MRI intensity in treated lymph nodes had substantial tumors, while those with low intensity had none. As expected, this correlation was absent in a cohort of animals treated with DCs that did not contain IONPs (Supplementary Figure 14). This evidence suggests that MRI imaging just two days after vaccination is sufficiently sensitive to predict subsequent antitumor immune responses to IONP-loaded DC vaccines.
Fig 6. MRI-detected DC migration at two days post-vaccine predicts antitumor efficacy of therapeutic DC vaccine.
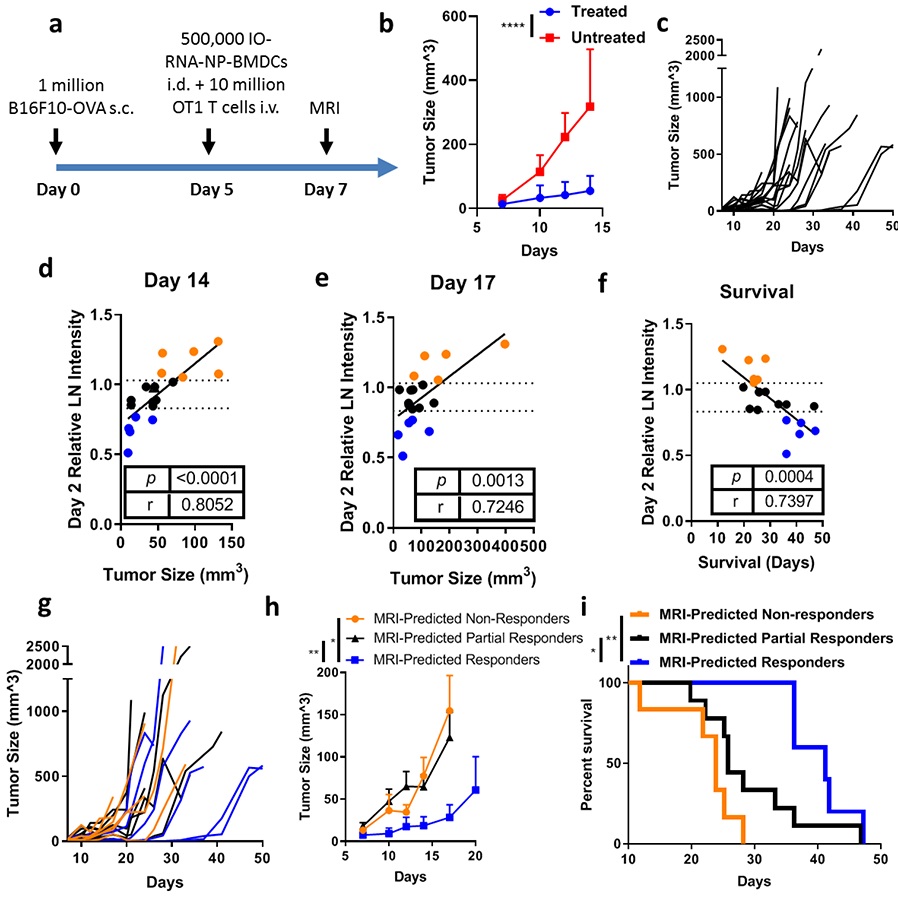
a, Schematic of treatment schedule. Mice received subcutaneous injection of 1 million B16F10-OVA cells in the lateral flank. On Day 5 mice received intradermal (i.d.) injection of 500,000 BMDCs loaded with IO-RNA-NPs bearing OVA RNA and intravenous (i.v.) injection of 10 million OT1 T-cells. Mice were imaged with MRI after two days and followed for tumor growth and survival. b, Summary data for treated and untreated mice. Statistical analysis was completed with ANOVA. c, Individual tumor growth curves for all treated mice before deaths (n=20). d-e, Correlation of T2*-weighted MRI intensity on Day 2 with tumor size on Day 14 (d) and Day 17 (e). Dotted lines demarcate the 25th and 75th percentiles of relative MRI intensity in lymph nodes. f, Correlation of T2*-weighted MRI intensity on Day 2 with survival. g-h, Individual tumor growth curves (g) and summary data (h) through deaths of all treated mice. Numbers on the graph are p values calculated using a mixed effects model. i, Survival curves for all treated mice. Numbers on graph are p values calculated using a Log-Rank test. Data are combined from two independent experiments (Supplementary Fig 12-13). P and r values in d, e, and f are derived from Mixed Effects Linear Regression Model with batch included as a random effect.
We then continued to observe these animals to determine whether MRI-detected DC migration predicts the more clinically relevant outcome of survival. Importantly, Day 2 MRI intensity in treated lymph nodes correlated strongly with survival (r=−0.7397; p=0.0004) (Fig 6f).
To evaluate the predictive capacity of this method, we divided these mice into groups based on Day 2 MRI intensity as in Fig 5. We found that mice with significant reductions in MRI intensity in treated LNs two days after vaccination revealed by relative MRI intensity below the 25th percentile on Day 2 (MRI-Predicted Responders) had significantly smaller tumors throughout the analysis than those with no significant change to LN intensity indicated by MRI intensities in the middle 50th percentile (MRI-Predicted Partial Responders) or those with MRI intensities in the top 75th percentile (MRI-Predicted Non-Responders) (MRI-Predicted Partial Responders vs MRI-Predicted Responders: p=0.0011; MRI-Predicted Non-Responders vs MRI-Predicted Responders: p=0.0058) (Fig 6g-h). Additionally, these MRI-Predicted Responders with substantial DC migration to lymph nodes lived significantly longer than MRI-Predicted Partial Responders (p=0.0443; log rank analysis) and almost twice as long as MRI-Predicted Non-Responders (p=0.0014; log rank analysis) (MRI-Predicted Non-Responders: 23.8 days; MRI-Predicted Partial Responders: 25.8 days; MRI-Predicted Responders: 41.2 days) (Fig 6g). These results indicate that MRI-imaging of DC trafficking can be used as a highly correlative biomarker to distinguish long-term antitumor responses to IO-RNA-NP-loaded DC vaccines just two days after vaccination.
Clinical implementation of immunotherapy will require readily accessible biomarkers to predict which patients will respond to treatment before tumor progression.9 Although our previous work suggests that migration of radio-labelled DCs to lymph nodes may correlate with antitumor activity,10 the requirement for radioactive cell labelling prohibits widespread evaluation of this technique as a biomarker in the clinical setting. Here, we show that MRI-detected DC migration just two days after vaccination with an RNA-based DC vaccine correlates strongly with long-term inhibition of tumor growth and survival in a fast-growing murine tumor model. In contrast to previous descriptions of qualitative MRI imaging of DCs loaded with IONPs, we show that IO-RNA-NPs enable MRI-based differentiation of “responders” and “non-responders”. This early biomarker of antitumor response measured with a widely accessible imaging modality promises significant benefits both for researchers evaluating improvements to existing therapies and clinicians deciding whether to continue a given cellular immunotherapy. Furthermore, this proof-of-principle in DC vaccines supports the use of MRI-detected cell migration as a dynamic biomarker for other migration-dependent immunotherapies (e.g. CAR T-cells, Adoptive cellular therapy, NK cells, or whole tumor cell vaccines).
IONPs garnered substantial attention as alternatives to radioactive imaging techniques after demonstrating superiority over SPECT imaging for fine spatial localization of DCs in LNs.12 Since that time, IONPs have been used to track DC migration to LNs in a variety of applications. Most work in this area has involved DCs that were loaded with IONPs after exposure to peptides encoding tumor antigen. Recently, more sophisticated approaches include the co-delivery of IONPs with tumor peptides. However, these approaches are limited by the low immunogenicity of peptide-based DC vaccines and the belief that MRI lacks sufficient quantitative sensitivity to provide prognostic function. Here, we develop a theranostic approach in which IO-RNA-NPs induce robust DC activation and sufficient changes in MRI intensity to predict tumor regression. In contrast to previous reports of qualitative cell tracking with MRI, we demonstrate that comparison of MRI intensity in treated and untreated LNs provides sufficient quantitation to differentiate responders, partial responders, and non-responders. Our observations suggest that enhanced DC migration to LNs detected by MRI correlates strongly with antitumor efficacy.
These findings hold direct relevance for the translation of this approach to imaging DC vaccines in human patients and are being developed for incorporation into our ongoing clinical trials. Although the number of DCs injected and the size of lymph nodes varies between species, the strength of this correlation across a wide range of MRI intensities in multiple tumor models in mice clearly supports further evaluation of this technique in humans. However, the sensitivity, specificity, and dynamic range of MRI intensities achievable in human DC studies are important variables that will impact the utility of MRI-based imaging of RNA-IONP loaded DCs as a biomarker for clinical outcomes in cancer patients. Future work should also consider whether we could increase the quantitative sensitivity of this technique with use of semi- or fully automated segmentation to determine regions of interest.
Although other groups have utilized lipids for DC transfection in vitro, we describe a simple approach to select a particle construct for both transfection and DC activation. Additionally, our lipid nanoparticle vaccine forgoes the use of peptides as antigen in exchange for mRNA, which we and others have shown to have many advantages including rapid clinical grade manufacturing and robust immunostimulatory effects. This exchange results in creation of a nanoparticle construct with the MRI-imaging capability of IONPs and potent immune stimulatory effects of mRNA.
We demonstrate that simple, translatable IO-RNA-NPs based on an immune-activating lipid formulation stimulate DCs to produce IFN-α, migrate to lymph nodes, and express activation markers to a greater extent than electroporation, leading to improved antitumor immune responses compared to electroporation despite inferior transfection efficiency. This seemingly contradictory result builds on early observations that delivery of bare RNA stimulates robust T-cell activation despite very low transfection efficiency, and suggests that RNA uptake and processing in intracellular vesicles is an important step in DC activation. Although it is likely that the injected DCs migrate to lymph nodes and stimulate T cells directly, it is also possible that the injected DCs transfer antigen to other LN-resident cells. Future work could further explore the precise mechanism by which DC vaccines initiate antitumor T cell responses. Regardless, our results support the use of IO-RNA-NPs as an immune-stimulatory alternative to electroporation for DC vaccines.
Our data also suggests a role for IONPs in lipid-mediated DC transfection. Although IONPs have been shown to increase production of inflammatory cytokines in Th1 T-cells and induce an antitumor M1 phenotype in macrophages, the role of iron oxide in DC transfection and activation remains unknown. Here, we show that IONPs enhance lipid-mediated DC transfection, activation and T-cell priming, but only when incorporated inside of liposomes. The presence of these advantages without substantial changes to DC activation phenotype or gene expression profiles suggest that IONPs may have a role in enhancing lipid-mediated DC transfection that complements the primary DC activating capacity of mRNA.
Although many multifunctional nanoparticles have been developed in the preclinical setting, the high costs and complexity of mass production in clinical grade manufacturing (cGMP) conditions often preclude their translation into clinical use. The nanoparticles that are used in humans tend to be liposomes with simple, scalable synthesis methods. Here, we sought to overcome this barrier by incorporating commercially available IONPs into immune-stimulating liposomes using only materials with proven safety records in humans and a synthesis method that is already proven scalable in the clinical setting. These dendritic cell-activating IO-RNA-NPs generate superior antitumor immune responses compared to electroporation and establish MRI-detected DC migration as a dynamic biomarker of response to immune therapy. Our use of a proven synthesis technique to generate multifunctional particles that can be imaged with a widely available modality should serve as a suggested roadmap for development of other simple, translatable nanomaterials to improve cancer therapy.
CONCLUSIONS
In this study, we describe multifunctional IO-RNA-NPs that produce potent antitumor immune responses and enable early prediction of survival benefit from IO-RNA-NP-loaded DC vaccines with MRI. We applied a quantitative scoring system to identify a formulation of immune stimulatory lipids that strongly activates DCs. Remarkably, these DCs secrete IFN-alpha and migrate to lymph nodes even more effectively than DCs loaded with RNA by conventional methods (i.e. electroporation), leading to marked inhibition of tumor growth in murine models. Moreover, the inclusion of IONPs in these particles enables rapid, enhanced DC transfection in the presence of magnetic fields and high-resolution imaging of cell migration with MRI. We found that IONP signal in the vaccination-site draining LN two days post-vaccination correlates strongly with long-term inhibition of tumor growth and survival outcome in multiple treatment models. This platform therefore provides a powerful tool to activate DCs and predict individual treatment responses to DC vaccines well before physical signs of tumor progression.
METHODS
Particle characterization
Size: IO-RNA-NPs were diluted 2000 times with cold PBS and measured with a NanoSight NS300 (Malvern). Particle size was calculated from over 1400 frames using 5 acquisitions per sample and 60 s per acquisition. Data was processed using NTA 3.3 Dev Build 3.3.104 (Camera Type: sCMOS). Selected plots and data are representative of four independent batches for each particle construct. Charge: Zeta potential was evaluated with a Nicomp ZLS Z3000. Reported measurements are averages of 5 cycles for each particle that are representative of 3 independent batches. Magnetism: Measurements of magnetism were made using a Quantum Design MPMS-3 Superconducting Quantum Interference Device (SQUID) magnetometer. Particles were prepared and analyzed in PBS at 2.5mg/mL. Magnetization curves were obtained by applying a 10 Oe (0.8 kA/m) field at varying temperatures from 4K to 345K. RNA Binding: Liposomes were loaded with RNA at Liposome:RNA ratios of 15:1, 10:1, 5:1, 1:1, or 0:1 and incubated for 15 minutes to allow liposome formation before staining with RNA-loading buffer. 20uL of IO-RNA-NPs were then loaded into each well of a 1% agarose gel and electrophoresed at 80V for 20 minutes. Free RNA was assessed with a ChemiDoc imaging system (Bio-Rad) and Image Lab software (Bio-Rad). The relative RNA binding capacity for each particle was calculated as: Bound RNA (%) * Total RNA (ug), where “Bound RNA” is calculated as 1- (SampleBand intensity/RNA Alone Band intensity). Cryogenic Electron Microscopy: Sample preparation for cryogenic transmission electron microscopy (Cryo-TEM) was performed in the Electron Microscopy Core of the University of Florida’s Interdisciplinary Center for Biotechnology Research. Three microliter aliquots of suspended liposomes were applied to C-flat holey carbon grids (Protochips, Inc.) and vitrified using a Vitrobot™ Mark IV (FEI Co.) operated at 4°C with ~90% humidity in the control chamber. The vitrified sample was stored under liquid nitrogen and transferred into a Gatan cryo-holder (Model 626/70) for imaging. The sample was examined using a 4k x 4k CCD camera (Gatan, Inc.) on a Tecnai (FEI Co.) G2 F20-TWIN Transmission Electron Microscope operated at a voltage of 200 kV using low dose conditions (~20 e/Å2).
RNA Preparation and Labeling
Green Fluorescent Protein (GFP) and OVA RNA were generated as previously described31. Isolated RNA was labeled with Cy3 and Cy5 dye using commercially available Arcturus Turbo Labeling kits (ThermoFisher Scientific) according to manufacturer instructions.
Cell culture
DC2.4s are an immortalized dendritic cell line that were a kind gift from John Sampson, Duke University33. B16F10-OVA is a murine melanoma cell line expressing the chicken ovalbumin gene (OVA) that was received as a kind gift from Dr. Richard G. Vile, PhD, at Mayo Clinic. Both cell types were cultured at 37°C with 5% CO2 in high glucose DMEM with pyruvate supplemented with 10% heat inactivated fetal bovine serum (FBS) and 1% Penicillin/Streptomycin (LifeTechnologies).
Synthesis of IO-RNA-NPs
Liposome synthesis: Liposomes were prepared as previously described.31 Briefly, the cationic liposome DOTAP was acquired from Avanti, Polar Lipids Inc. (Alabaster, AL, USA) in the dehydrated form. 25-100mg of the dehydrated liposome was mixed with Cholesterol 700000P (Avanti Polar Lipids) at a ratio of 3:1 DOTAP:Cholesterol in chloroform. The chloroform was evaporated in a nitrogen gas chamber, resulting in a thin layer of lipid. This lipid was then rehydrated to 2.5mg/mL with PBS. For IO-RNA-NPs, the lipid cake was instead rehydrated with a dense solution of 120-nm carboxylated IONPs (NanoMag-D, Micromod) at a concentration of 10 mg/mL before being brought to 2.5mg lipid/mL with PBS. The rehydrated liposomes were then placed in a 50°C water bath and vortexed every 10 minutes for 1 hour. Liposomes were then stored at room temperature overnight before being vortexed, placed in a bath sonicator for 5 minutes, and filtered through a 0.45 μm syringe filter (Whatman Puradisc) and afterwards a 0.20 μm syringe filter (PALL Acrodisc syringe filter with Supor membrane). IO-RNA-NP complex formation: IO-RNA-NPs were prepared as described previously for RNA-NPs.31 10μg mRNA were added to 150μg IO-RNA-NPs (per 2 million cells) in PBS buffer. The mixture was incubated at room temperature for 15 minutes to ensure complex formation before addition to DCs.
IO-RNA-NP transfection of DCs
Overnight Transfection: IO-RNA-NPs were added to DCs in culture at 160ug IO-RNA-NPs:2 million DCs overnight. 30 Minute Transfection: DCs were pulled to the bottom of 24-well plates by centrifugation for 1 minute at 100rcf before addition of IO-RNA-NPs. Particles were left in media for 30 minutes in the presence or absence of a magnetic field created by neodymium iron boron (Nd2Fe14B) permanent magnetic disks. After 30 minutes, plates were again centrifuged at 100rcf for 1 minute before IO-RNA-NPs were removed and replaced with fresh media.
Mice
C57Bl/6, OT1 Transgenic (C57Bl/6-Tg(TcraTcrb)1100Mjb/J),DsRed (B6.Cg-Tg(CAG-DsRed*MST)1Nagy/J), and TLR7−/− (B6.129S1-Tlr7tm1Flv/J) mice were purchased from Jackson Laboratories. Animal procedures were approved by the University of Florida Institutional Animal Care and Use Committee (UFIACUC201607966).
Dendritic cell generation
DCs were isolated from murine bone marrow based on previously established methods. Briefly, tibias and femurs were harvested from C57Bl/6, DsRed, or TLR7−/− mice and bone marrow was flushed using 25-gauge syringe with serum-containing media. Red blood cells were lysed with 10 mL Pharmlyse (BD Bioscience) before suspending mononuclear cells in complete DC media (RPMI-1640, 5% FBS, 1 M HEPES (LifeTechnologies), 55 mM b-mercaptoethanol (LifeTechnologies), 100 mM Sodium pyruvate (LifeTechnologies), 10 mM nonessential amino acids (LifeTechnologies), 200 mM L-glutamine (LifeTechnologies), 10 mg GM-CSF (R&D Systems), 10 mg IL4 (R&D Systems), 1% Penicillin/Streptomycin (LifeTechnologies). Cells were then cultured in six-well plates at a concentration of 8 x 105 cells/mL in a total volume of 3mL/well. Non-adherent cells were discarded, and media was replaced at day 3. At day 7, non-adherent cells were collected and re-plated into 100 mm culture dishes at a density of 106 cells/mL in a total volume of 5 mL/dish. Twenty-four hours later, non-adherent cells were collected, transfected with mRNA via electroporation, RNA Alone, RNA-NPs, or IO-RNA-NPs, and left overnight.
T Cell Generation
Naïve OVA-specific T cells: T cells specific for OVA peptide epitopes 257-264 were generated from spleens of OT1 transgenic mice (C57Bl/6-Tg(TcraTcrb)1100Mjb/J). OT1 splenocytes were isolated by RBC lysis and suspended in PBS for immediate use in co-culture or treatment. Antigen-experienced T Cells: Antigen-experienced T cells were prepared as previously described.4 Briefly, C57Bl6 mice received intradermal vaccination with OVA-pulsed DCs. Splenocytes were isolated from these mice 1 week after vaccination and cultured for 5 days with OVA-pulsed BMDCs in T cell media with IL-2 at a splenocyte:DC ratio of 4,000,000:400,000. Activated T cells were split into new wells as they reached confluence.
Co-culture assays
DCs were transfected with OVA or GFP mRNA via electroporation, co-culture (RNA-Alone), RNA-NPs, or IO-RNA-NPs. After 24 hours, treated DCs were co-cultured with naïve OT1 splenocytes or antigen-experienced OVA T cells in a 96 well plate at a T cell:DC ratio of 400,000:40,000. Supernatants were collected after 48 hours and evaluated with ELISA for interferon-γ (ebioscience).
Flow cytometric analysis
Flow cytometry was performed using the BD Biosciences FACS Canto-II using antibodies from BD Biosciences, Biolegend, and Invitrogen. For in vitro experiments, DCs were harvested, washed with PBS, and stained for 20 minutes. Samples were then washed twice with PBS and suspended in FACS buffer. Cell counts and viability were assessed with Vi-Cell XR Cell Viability Analyzer (Beckman Coulter). For in vivo experiments, lymph nodes were harvested into cold PBS, diced with razor blade and digested in papain for 20 minutes at 37C before filtering through a 70μm cell strainer, washing with PBS, and staining for 20 minutes with appropriate antibodies. Counting beads were added to each tube immediately before flow analysis.
Analysis of iron oxide content
Iron oxide content in LNs was measured using the Iron Assay Kit (Sigma Aldrich) according to package instructions.
MRI imaging and analysis
Image Acquisition: MRI imaging was performed on a 11T MRI magnet (Magnex Scientific, 11.1 T/40cm bore) equipped with a Bruker AV3 HD console and Paravision 6.01 software using a custom built 30mm ID quadrature birdcage transmit-receiver volume coil at the UF AMRIS facility 18-72 hours after intradermal injection of IO-RNA-NP-loaded DCs in the inguinal area. Mice were imaged under isoflurane anesthesia and monitored via continuous measurements of body temperature and respirations according to UFIACUC201607966. Circulating warm water from a temperature-controlled water heater was used to maintain body temperature. Two-dimensional MRI sequences with an image size of 192x192, field of view of 25.08mm x 25.921mm, section thickness of 1.0 mm and 6 slices were collected with respiratory triggering for a variety of image parameters. T2*-weighted images were collected using the following parameters to evaluate IONP-dependent changes in hypointensity in lymph nodes: repetition time (TR) = 90 ms, echo time (TE) = 5 ms, flip angle=10° with fat saturation. A set of five other imaging sequences was taken to evaluate lymph node size. Sequence 1 (T2*): TR= 90 ms, TE=3 ms, flip angle=10°. Sequence 2 (T2* with fat sat): TR= 90 ms, TE=3 ms, flip angle=10° with fat saturation. Sequence 3 (T2_407/17): TR=407.204ms, TE=17 ms, echo spacing=4.25, RARE factor=4, with fat saturation. Sequence 4 (T2 with fat sat): TR= 500ms, TE= 14ms, echo spacing=7ms, RARE factor=4. Sequence 5 (T2): TR=3000 ms, TE= 28ms, echo spacing = 3ms, RARE factor=8, with fat saturation.
Image analysis: Image analysis was completed using ImageJ (NIH). Researchers blinded to treatment group manually created regions of interest around each inguinal lymph node for each slice in which it appeared. MRI intensity in each lymph node was calculated as the average of lymph node intensities across all slices in which that lymph node appeared for T2*-weighted MRI sequences. Relative intensity was calculated as the MRI intensity of the treated lymph node divided by the MRI intensity of the untreated lymph node for T2*-weighted MRI images. Lymph node volume was calculated as the product of the slice thickness (1mm) and the summed areas of each lymph node for each slice (e.g. V= Slice Thickness * (AreaSlice 1 + AreaSlice 2 + AreaSlice 3 … + AreaSlice n)). Relative size was calculated for each imaging sequence as the volume of the treated lymph node divided by the volume of the untreated lymph node on the contralateral side. Relative volume was calculated for all above imaging sequences and reported as the average relative volume across imaging sequences.
Treatment models
Tumor implantations: B16F10-OVA cells were harvested with 0.05% trypsin (Gibco), washed once in serum-containing medium, and washed once in Dulbecco’s phosphate-buffered saline (DPBS). Cell pellets were resuspended in DPBS at a concentration of 107 cells/mL. 1 million B16F10-OVA cells were subcutaneously injected with a 25-gauge syringe into the left flank of C57Bl/6 mice anesthetized with isoflurane. Mice that failed to establish tumors were excluded. Subcutaneous tumors were measured every 2-4 days with WESTWARD Digital Caliper. Animals bearing subcutaneous tumors were euthanized upon reaching humane endpoints of tumor size or tumor ulceration. Adoptive Cellular Therapy: Naïve or antigen experienced T cells were generated as described above, suspended in PBS at 100 million/mL, and injected into tumor-bearing mice at 100uL per mouse. DC Vaccines: RNA-pulsed DCs prepared as described above were collected and suspended in PBS at a final concentration of 1 x 107 cells/mL. 50 uL was administered intradermally in the inguinal area for each treated mouse. Sample size: Sample sizes were the smallest that would provide reasonable sensitivity to compare two groups. Additional mice were included in IO-RNA-NP groups to allow for subset analysis.
Immunofluorescence
DC2.4s were imaged in PBS 24 hours after incubation with Cy3-labelled mRNA. Cells were stained with DAPI and anti-TLR7 antibody (Invitrogen). Images were taken with an Olympus IX70 Inverted Fluorescent Microscope or Nikon A1RMP Confocal Microscope.
Gene expression analysis
BMDCs were harvested from three independent samples per treatment group 24 hours after transfection with GFP mRNA via electroporation, RNA-NPs, or IO-RNA-NPs at 10μg mRNA per 2 million cells. RNA was then isolated from each sample using commercially available RNeasy mini kits (Quiagen, cat#74104) as per the manufacturer instructions and analyzed for purity using a NanoDrop 2000 spectrophotometer (ThermoFisher Scientific) and Agilent 2100 BioAnalyzer. RNA was prepared for directional sequencing using Illumina RNA-Sequencing libraries (Poly A) at the UF Interdisciplinary Center for Biotechnology Research (ICBR). Paired end RNA sequencing with 100 cycles in 2 lanes were completed with an Illumina HiSeq 3000. Gene set enrichment was evaluated using the gene set enrichment analysis (GSEA) software available from the Broad Institute (genepattern.broadinstitute.org). Nine gene sets were selected from the C7 Immunologic Signatures gene set collection based on the hypothesis that RNA uptake in endosomes initiates a toll-like receptor-dependent DC activation phenotype. The gene set for lymphocyte migration and individual analysis of IL-12, CCL3, and CCL4 were completed based on reviewer suggestion.
Statistical analysis
Data are summarized as the mean ± standard deviation for in vitro experiments and mean ± SEM for tumor-growth experiments. Unpaired data is analyzed with two-tailed unpaired student’s t tests or ANOVA with Tukey’s tests for multiple comparisons. Paired data is analyzed with Wilcoxon matched-pairs rank sum test for experiments with n>3 and two-tailed paired student’s t tests when n≤3. Tumor growth over time is measured with two-way ANOVA for individual experiments. For Fig 5 two independent experiments were combined and evaluated with a mixed effects linear regression with experiment modeled as a random effect. Raw data for each experiment can be found in Supplementary Figures. Survival is measured with a log-rank test. Statistical analysis was conducted using GraphPad Prism version 8 and SAS. Statistical significance was defined as p<0.05. Linear correlations were evaluated with Pearson’s correlation coefficient for data without significant outliers. Spearman’s correlation was utilized in datasets with significant outliers to minimize their impact.
Supplementary Material
Supplementary Fig. 1: Translatable nanoparticles transfect and activate DCs.
Supplementary Fig. 2: IO-RNA-NPs are taken up by micropinocytosis.
Supplementary Fig. 3: IO-RNA-NPs induce antigen specific DC activation.
Supplementary Fig. 4: IO-RNA-NPs induce expression of antiviral gene sets.
Supplementary Fig. 5: IO-RNA-NPs enhance activation of BMDCs.
Supplementary Fig. 6: Activating capacity of RNA-NPs is dependent on TLR7 activation.
Supplementary Fig. 7: T2*-weighted MRI intensity correlates with iron oxide content in LNs
Supplementary Fig. 8: T2*-weighted MRI intensity does not correlate with cell count in the absence of IONPs.
Supplementary Fig. 9: LN size does not correlate strongly with efficacy.
Supplementary Fig. 10: MRI-detected DC migration predicts response to DC vaccines_Experiment 1.
Supplementary Fig. 11: MRI-detected DC migration predicts response to DC vaccines_Experiment 2.
Supplementary Fig. 12: MRI-detected DC migration at two days post-vaccine predicts antitumor efficacy of therapeutic DC vaccine_Experiment 1.
Supplementary Fig. 13: MRI-detected DC migration at two days post-vaccine predicts antitumor efficacy of therapeutic DC vaccine_Experiment 2.
Supplementary Fig. 14: LN intensity does not predict outcome in absence of IONPs.
Supplementary Table 1: Summary data for lipid library.
Supplementary Table 2: Characterization of iron oxide loaded RNA-nanoparticles.
Supplementary Table 3: LN size does not correlate strongly with efficacy.
ACKNOWLEDGEMENT
We thank P. Castillo for assistance with co-culture experiments. We also thank M. Unni for assistance with SQUID magnetometer and D. Modlin for assistance with figure preparation. We also thank H. Zeng and J. Long from the University of Florida AMRIS facility, and R. Alvarado from the University of Florida Interdisciplinary Center for Biotechnology Research Electron Microscopy Core. This work was supported by the Circle of Hope Foundation (AG), the University of Florida Health Cancer Center Dissertation Award (AG), and NIH/NCI awards 1R01CA175517 (DM), R01CA195563 (DM), K08CA199224 (ES), TL1TR001428 (AG), and 1F30CA228280-01 (AG). A portion of this work was performed in the McKnight Brain Institute at the National High Magnetic Field Laboratory’s AMRIS Facility, which is supported by National Science Foundation Cooperative Agreement No. DMR-1157490 and DMR-1644779 and the State of Florida. The content is solely the responsibility of the authors and does not necessarily represent the views of the funding sources.
Footnotes
SUPPORTING INFORMATION
Additional supporting results including liposome selection, IO-RNA-NP characterization, effects of IO-RNA-NPs on DC activation, role of TLR7 in antitumor response to RNA-NP-based DC vaccines, correlation of MRI intensity with DC migration, and each individual repeat of large efficacy experiments are included in Supplementary Figures 1-14 and Supplementary Tables 1-3. This supporting information is available free of charge at http://pubs.acs.org.
Competing Interests
D.A.M. has patented immunotherapy related technologies that have been licensed by Annias Immunotherapeutics, Inc., Immunomic Therapeutics, Inc., Celldex Therapeutics, Inc., and iOncologi, Inc. D.A.M. receives research funding from Immunomic Therapeutics, serves as a consultant/advisor to Bristol-Myers Squibb, Inc., Tocagen, Inc., and Oncorus, Inc., and is co-founder of iOncologi, Inc., a biotechnology company specializing in immuno-oncology. The other authors declare no conflicts.
References
- 1.Rosenberg SA; Yang JC; Sherry RM; Kammula US; Hughes MS; Phan GQ; Citrin DE; Restifo NP; Robbins PF; Wunderlich JR; Morton KE; Laurencot CM; Steinberg SM; White DE; Dudley ME, Durable Complete Responses in Heavily Pretreated Patients with Metastatic Melanoma Using T-Cell Transfer Immunotherapy. Clin Cancer Res 2011, 17, 4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schadendorf D; Hodi FS; Robert C; Weber JS; Margolin K; Hamid O; Patt D; Chen TT; Berman DM; Wolchok JD, Pooled Analysis of Long-Term Survival Data from Phase Ii and Phase Iii Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 2015, 33, 1889–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Burg SH; Arens R; Ossendorp F; van Hall T; Melief CJ, Vaccines for Established Cancer: Overcoming the Challenges Posed by Immune Evasion. Nat Rev Cancer 2016, 16, 219–233. [DOI] [PubMed] [Google Scholar]
- 4.Garon EB; Rizvi NA; Hui R; Leighl N; Balmanoukian AS; Eder JP; Patnaik A; Aggarwal C; Gubens M; Horn L; Carcereny E; Ahn MJ; Felip E; Lee JS; Hellmann MD; Hamid O; Goldman JW; Soria JC; Dolled-Filhart M; Rutledge RZ, et al. , Pembrolizumab for the Treatment of Non-Small-Cell Lung Cancer. N Engl J Med 2015, 372, 2018–2028. [DOI] [PubMed] [Google Scholar]
- 5.Larkin J; Chiarion-Sileni V; Gonzalez R; Grob JJ; Cowey CL; Lao CD; Schadendorf D; Dummer R; Smylie M; Rutkowski P; Ferrucci PF; Hill A; Wagstaff J; Carlino MS; Haanen JB; Maio M; Marquez-Rodas I; McArthur GA; Ascierto PA; Long GV, et al. , Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015, 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellmann MD; Rizvi NA; Goldman JW; Gettinger SN; Borghaei H; Brahmer JR; Ready NE; Gerber DE; Chow LQ; Juergens RA; Shepherd FA; Laurie SA; Geese WJ; Agrawal S; Young TC; Li X; Antonia SJ, Nivolumab Plus Ipilimumab as First-Line Treatment for Advanced Non-Small-Cell Lung Cancer (Checkmate 012): Results of an Open-Label, Phase 1, Multicohort Study. Lancet Oncol 2017, 18, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grippin AJ; Sayour EJ; Mitchell DA, Translational Nanoparticle Engineering for Cancer Vaccines. OncoImmunology 2017, 6, e1290036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishino M; Ramaiya NH; Hatabu H; Hodi FS, Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat Rev Clin Oncol 2017, 14, 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lesterhuis WJ; Bosco A; Millward MJ; Small M; Nowak AK; Lake RA, Dynamic versus Static Biomarkers in Cancer Immune Checkpoint Blockade: Unravelling Complexity. Nat Rev Drug Discov 2017, 16, 264–272. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell DA; Batich KA; Gunn MD; Huang M-N; Sanchez-Perez L; Nair SK; Congdon KL; Reap EA; Archer GE; Desjardins A; Friedman AH; Friedman HS; Herndon Ii JE; Coan A; McLendon RE; Reardon DA; Vredenburgh JJ; Bigner DD; Sampson JH, Tetanus Toxoid and Ccl3 Improve Dendritic Cell Vaccines in Mice and Glioblastoma Patients. Nature 2015, 519, 366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Srinivas M; Aarntzen EHJG; Bulte JWM; Oyen WJ; Heerschap A; de Vries IJM; Figdor CG, Imaging of Cellular Therapies. Adv. Drug Deliv. Rev. 2010, 62, 1080–1093. [DOI] [PubMed] [Google Scholar]
- 12.de Vries IJ; Lesterhuis WJ; Barentsz JO; Verdijk P; van Krieken JH; Boerman OC; Oyen WJ; Bonenkamp JJ; Boezeman JB; Adema GJ; Bulte JW; Scheenen TW; Punt CJ; Heerschap A; Figdor CG, Magnetic Resonance Tracking of Dendritic Cells in Melanoma Patients for Monitoring of Cellular Therapy. Nat Biotechnol 2005, 23, 1407–1413. [DOI] [PubMed] [Google Scholar]
- 13.Verdijk P; Scheenen TW; Lesterhuis WJ; Gambarota G; Veltien AA; Walczak P; Scharenborg NM; Bulte JW; Punt CJ; Heerschap A; Figdor CG; de Vries IJ, Sensitivity of Magnetic Resonance Imaging of Dendritic Cells for In Vivo Tracking of Cellular Cancer Vaccines. Int J Cancer 2007, 120, 978–984. [DOI] [PubMed] [Google Scholar]
- 14.Noh YW; Jang YS; Ahn KJ; Lim YT; Chung BH, Simultaneous In Vivo Tracking of Dendritic Cells and Priming of an Antigen-Specific Immune Response. Biomaterials 2011, 32, 6254–6263. [DOI] [PubMed] [Google Scholar]
- 15.Cho NH; Cheong TC; Min JH; Wu JH; Lee SJ; Kim D; Yang JS; Kim S; Kim YK; Seong SY, A Multifunctional Core-Shell Nanoparticle for Dendritic Cell-Based Cancer Immunotherapy. Nat Nanotechnol 2011, 6, 675–682. [DOI] [PubMed] [Google Scholar]
- 16.Mou Y; Hou Y; Chen B; Hua Z; Zhang Y; Xie H; Xia G; Wang Z; Huang X; Han W; Ni Y; Hu Q, In Vivo Migration of Dendritic Cells Labeled with Synthetic Superparamagnetic Iron Oxide. Int J Nanomedicine 2011, 6, 2633–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Chickera S; Willert C; Mallet C; Foley R; Foster P; Dekaban GA, Cellular Mri as a Suitable, Sensitive Non-Invasive Modality for Correlating In Vivo Migratory Efficiencies of Different Dendritic Cell Populations with Subsequent Immunological Outcomes. Int Immunol 2012, 24, 29–41. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Z; Li W; Procissi D; Li K; Sheu AY; Gordon AC; Guo Y; Khazaie K; Huan Y; Han G; Larson AC, Antigen-Loaded Dendritic Cell Migration: Mr Imaging in a Pancreatic Carcinoma Model. Radiology 2015, 274, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Batich KA; Reap EA; Archer GE; Sanchez-Perez L; Nair SK; Schmittling RJ; Norberg P; Xie W; Herndon JE 2nd; Healy P; McLendon RE; Friedman AH; Friedman HS; Bigner D; Vlahovic G; Mitchell DA; Sampson JH, Long-Term Survival in Glioblastoma with Cytomegalovirus Pp65-Targeted Vaccination. Clin Cancer Res 2017, 23, 1898–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anguille S; Van de Velde AL; Smits EL; Van Tendeloo VF; Juliusson G; Cools N; Nijs G; Stein B; Lion E; Van Driessche A; Vandenbosch I; Verlinden A; Gadisseur AP; Schroyens WA; Muylle L; Vermeulen K; Maes MB; Deiteren K; Malfait R; Gostick E, et al. , Dendritic Cell Vaccination as Postremission Treatment to Prevent or Delay Relapse in Acute Myeloid Leukemia. Blood 2017, 130, 1713–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilgenhof S; Corthals J; Heirman C; van Baren N; Lucas S; Kvistborg P; Thielemans K; Neyns B, Phase Ii Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (Trimixdc-Mel) Plus Ipilimumab in Patients with Pretreated Advanced Melanoma. J Clin Oncol 2016, 34, 1330–1338. [DOI] [PubMed] [Google Scholar]
- 22.Bol KF; Figdor CG; Aarntzen EH; Welzen ME; van Rossum MM; Blokx WA; van de Rakt MW; Scharenborg NM; de Boer AJ; Pots JM; Olde Nordkamp MA; van Oorschot TG; Mus RD; Croockewit SA; Jacobs JF; Schuler G; Neyns B; Austyn JM; Punt CJ; Schreibelt G, et al. , Intranodal Vaccination with mRNA-Optimized Dendritic Cells in Metastatic Melanoma Patients. Oncoimmunology 2015, 4, e1019197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aarntzen EH; Schreibelt G; Bol K; Lesterhuis WJ; Croockewit AJ; de Wilt JH; van Rossum MM; Blokx WA; Jacobs JF; Duiveman-de Boer T; Schuurhuis DH; Mus R; Thielemans K; de Vries IJ; Figdor CG; Punt CJ; Adema GJ, Vaccination with mRNA-Electroporated Dendritic Cells Induces Robust Tumor Antigen-Specific Cd4+ and Cd8+ T Cells Responses in Stage Iii and Iv Melanoma Patients. Clin Cancer Res 2012, 18, 5460–5470. [DOI] [PubMed] [Google Scholar]
- 24.Phua KKL; Nair SK; Leong KW, Messenger RNA (mRNA) Nanoparticle Tumour Vaccination. Nanoscale 2014, 6, 7715–7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anselmo AC; Mitragotri S, Nanoparticles in the Clinic. Bioeng Transl Med 2016, 1, 10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi J; Kantoff PW; Wooster R; Farokhzad OC, Cancer Nanomedicine: Progress, Challenges and Opportunities. Nat Rev Cancer 2017, 17, 20–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Vries IJM; Lesterhuis WJ; Barentsz JO; Verdijk P; van Krieken JH; Boerman OC; Oyen WJG; Bonenkamp JJ; Boezeman JB; Adema GJ; Bulte JWM; Scheenen TWJ; Punt CJA; Heerschap A; Figdor CG, Magnetic Resonance Tracking of Dendritic Cells in Melanoma Patients for Monitoring of Cellular Therapy. Nat Biotech 2005, 23, 1407–1413. [DOI] [PubMed] [Google Scholar]
- 28.Ahrens ET; Bulte JWM, Tracking Immune Cells In Vivo Using Magnetic Resonance Imaging. Nat Rev Immunol 2013, 13, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulte JWM, In Vivo Mri Cell Tracking: Clinical Studies. Am J Roentgenol 2009, 193, 314–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kranz LM; Diken M; Haas H; Kreiter S; Loquai C; Reuter KC; Meng M; Fritz D; Vascotto F; Hefesha H; Grunwitz C; Vormehr M; Husemann Y; Selmi A; Kuhn AN; Buck J; Derhovanessian E; Rae R; Attig S; Diekmann J, et al. , Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [DOI] [PubMed] [Google Scholar]
- 31.Sayour EJ; De Leon G; Pham C; Grippin A; Kemeny H; Chua J; Huang J; Sampson JH; Sanchez-Perez L; Flores C; Mitchell DA, Systemic Activation of Antigen-Presenting Cells via RNA-Loaded Nanoparticles. Oncoimmunology 2017, 6, e1256527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sayour EJ; Grippin A; De Leon G; Stover B; Rahman M; Karachi A; Wummer B; Moore G; Castillo-Caro P; Fredenburg K; Sarkisian MR; Huang J; Deleyrolle LP; Sahay B; Carrera-Justiz S; Mendez-Gomez HR; Mitchell DA, Personalized Tumor RNA Loaded Lipid-Nanoparticles Prime the Systemic and Intratumoral Milieu for Response to Cancer Immunotherapy. Nano Lett 2018, 18, 6195–6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Z; Reznikoff G; Dranoff G; Rock KL, Cloned Dendritic Cells Can Present Exogenous Antigens on Both Mhc Class I and Class Ii Molecules. J Immunol 1997, 158, 2723–2730. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1: Translatable nanoparticles transfect and activate DCs.
Supplementary Fig. 2: IO-RNA-NPs are taken up by micropinocytosis.
Supplementary Fig. 3: IO-RNA-NPs induce antigen specific DC activation.
Supplementary Fig. 4: IO-RNA-NPs induce expression of antiviral gene sets.
Supplementary Fig. 5: IO-RNA-NPs enhance activation of BMDCs.
Supplementary Fig. 6: Activating capacity of RNA-NPs is dependent on TLR7 activation.
Supplementary Fig. 7: T2*-weighted MRI intensity correlates with iron oxide content in LNs
Supplementary Fig. 8: T2*-weighted MRI intensity does not correlate with cell count in the absence of IONPs.
Supplementary Fig. 9: LN size does not correlate strongly with efficacy.
Supplementary Fig. 10: MRI-detected DC migration predicts response to DC vaccines_Experiment 1.
Supplementary Fig. 11: MRI-detected DC migration predicts response to DC vaccines_Experiment 2.
Supplementary Fig. 12: MRI-detected DC migration at two days post-vaccine predicts antitumor efficacy of therapeutic DC vaccine_Experiment 1.
Supplementary Fig. 13: MRI-detected DC migration at two days post-vaccine predicts antitumor efficacy of therapeutic DC vaccine_Experiment 2.
Supplementary Fig. 14: LN intensity does not predict outcome in absence of IONPs.
Supplementary Table 1: Summary data for lipid library.
Supplementary Table 2: Characterization of iron oxide loaded RNA-nanoparticles.
Supplementary Table 3: LN size does not correlate strongly with efficacy.