

Abstract
Hemostatic clot formation is the result of a balance between the procoagulant system responding to tissue trauma and the anticoagulant system, which restricts clot formation to the injury site. Imbalances in coagulation lead to a tendency towards either thrombosis or bleeding. Over the past two years, studies published in Arteriosclerosis, Thrombosis, and Vascular Biology have provided insights into the regulation of this crucial system. Here, we highlight recent discoveries concerning the two pathways of thrombin formation, the extrinsic tissue factor (TF) pathway and the intrinsic contact pathway, and the contributions of platelet activation to the thrombotic process.
Regulation of Tissue Factor Expression and Activity
Tissue factor is a transmembrane glycoprotein cofactor constitutively expressed by subendothelial cells that serves as a high affinity receptor for, and promotes the catalytic activity of factor VIIa (FVIIa).1, 2 Upon vascular injury, TF initiates the extrinsic coagulation pathway by binding FVIIa and promoting the activation of factor X (FX) and factor IX (FIX). The importance of TF activity was explored in a recent microfluidic study by Zhu et al,3 which showed that a single molecule of TF can generate up to 92,000 molecules of thrombin and more than 200,000 fibrin monomers during a 500-second clotting window. They also calculated that the produced thrombin only has a 70 msec half-life. Thus, a single molecule of thrombin is only active long enough to produce three fibrin monomers, and robust thrombin generation is required to produce a stable clot.
Many intravascular cells, including neutrophils and monocytes, can be stimulated to express detectable levels of TF.4 These cells might also release TF into the circulation, either as the soluble extracellular form or as full-length protein incorporated into microvesicles.5 Recent studies have identified additional mechanisms regulating TF expression. Neutrophils can release neutrophil extracellular traps (NETs), which consist of DNA, histones, and granular enzymes, and Folco et al.6 showed that NET-associated effectors induce endothelial cell expression of TF through an interleukin 1α and cathepsin G-dependent pathway. According to a recent study by Liang and colleagues,7 the transcription factor Krüppel-like factor-11 (KLF11) binds to the F3 promoter to inhibit TF transcription. Under basal condition, endogenous KLF11 is sufficient to maintain low levels of TF in vascular smooth muscle and endothelial cells. Knockout of TF specifically in vascular smooth muscle cells inhibits thrombus formation in the ferric chloride injury model, while vascular smooth muscle-specific Klf11−/− mice have increased TF and a prothrombotic phenotype. Conversely, overexpression of Klf11 potently inhibited TNF-α-induced TF expression in human aortic smooth muscle cells at both the mRNA and protein levels. Thus, through its regulation of TF, KLF11 is a key controller of vascular smooth muscle cell procoagulant activity.
While procoagulant TF is clearly expressed in the vascular subendothelium, it has also been proposed that many different cell types express an inactive, encrypted form of TF that requires de-encryption.8 A recent article by Baker et al.9 has provided insight into the regulation of TF de-encryption. Hepatocytes have direct exposure to the plasma due to the fenestrated endothelium of the liver vasculature. Consequently, hepatocytes express an encrypted form of the TF/FVIIa complex. Baker et al. showed that bile acids stimulate the de-encryption of this TF/FVIIa complex in a murine cholestasis model, possibly explaining the hypercoagulability seen in patients with liver disease. The contribution of microvesicle TF activity has also been explored. Stark et al.10 found a clear correlation between cancer-derived microvesicle TF activity and the risk of deep vein thrombosis (DVT) in prostate cancer, one of the most prothrombotic type of cancers. They also showed that tumor-derived microvesicles can induce thrombosis in animal models. However, microvesicle TF was not sufficient by itself to cause DVT. Clot formation also required synergistic endothelial cell TF and surface exposure of the phospholipid phosphatidylethanolamine, but not phosphatidylserine.
Once activated, TF/FVIIa is inhibited by the anticoagulant tissue factor pathway inhibitor α (TFPIα), which is down-regulated in women taking hormonal contraception.11 Recently, Tanratana et al.12 showed that reduced TFPIα in premenopausal women on hormonal contraception results in a 2–3-fold increase in the rate of FIX activation and an increase in the amount of circulating FIXa. They also observed an inverse correlation between circulating FIXa and plasma concentrations of TFPIα and the cofactor protein S (PS). PS promotes TF/FVIIa inhibition by TFPIα, and also directly inhibits FIXa activity.11, 13 Plautz et al.14 defined the mechanism of this latter function, showing that PS interacts with the heparin-binding exosite of FIXa and that infusion of the FIXa K132A/R170A mutant protein, which cannot bind PS, significantly increases clot formation in Hemophilia B mice.
Contributions of the Contact Pathway
FIXa is also generated through the contact pathway of coagulation, a proteolytic cascade initiated by factor XII (FXII), which activates both factor XI (FXI) and the proinflammatory kallikrein‐kinin system.15 Upon exposure to negatively charged surfaces (e.g. DNA) or pathogens (e.g. long-chain polyphosphates), FXII undergoes a conformational change and produces activated FXII (FXIIa). Subsequently FXIIa activates the intrinsic coagulation system by factor XI (FXI) cleavage to form activated FXI (FXIa). FXIa then proteolytically activates FIX. FXIIa also digests prekallikrein to release plasma kallikrein (PK), which reversely activates FXII to produce an enhanced positive feedback activation loop.15
Multiple lines of evidence have shown that factor XII is dispensable for hemostasis, but promotes thrombotic processes, such as venous thromboembolism (VTE), suggesting this pathway as a viable therapeutic target.15 Lorentz et al.16 showed that AB023, an antibody which selectively blocks FXI activation by FXIIa and demonstrates a dose-dependent duration of limited anticoagulation in mice, has antithrombotic activity in a baboon model but does not increase bleeding time. They then performed a first-in-human study of the antibody. They reported a prolonged activated partial thromboplastin time in patients treated with the antibody, demonstrating its efficacy, and confirmed that the antibody does not impair either thrombin-mediated FXII activation or FXIa enzymatic activity. Most importantly, they reported no severe adverse events were observed, with only one patient experiencing a bruise at the injection site.
Similar to the TF pathway, the contact pathway is tightly downregulated in vivo, by C1 esterase inhibitor, which targets both FXIIa and PK.15 Puy et al.17 showed that the endothelial serine protease inhibitor plasminogen activator inhibitor-1 (PAI-1) also downregulates the contact pathway. Specifically, PAI-1 forms an inhibitory complex with FXIa, which is then either shed into circulation or endocytosed by endothelial cells, trafficked through endosomes and lysosomes, and degraded. Promoting these natural suppressive mechanisms represents a potential therapeutic strategy to prevent thrombosis through contact pathway inhibition.
In addition, the contact pathway influences the host response to a variety of pathological processes besides thrombosis, including atherosclerosis, stroke, and sepsis.15 Jevgenia et al.43 demonstrated that long-chain polyphosphates released by bacterial pathogens induce platelet activation and fibrin generation in vivo through a FXII-dependent mechanism and that pre-treatment with FXIa antibody suppresses platelet and fibrin consumption in a bacterial sepsis model.18, 19 Meanwhile, data also suggest cross-talk between the proinflammatory and procoagulant processes.20 Mayken et al.21 found that PK, the major proinflammatory downstream mediator of FXIIa, also contributes to FIXa activation by FXIIa, a process which operates parallel to the FXIa pathway. The ability of PK to activate FIXa could be explained by the high sequence homology between the active sites of PK and FXIa, a well-described FIX activator.22
Platelet Activation
Whether generated through the TF or contact pathway, the final substrate thrombin amplifies its own production, in part through the activation of protease-activated receptors 1 and 4 (PAR1, PAR4) on platelets, and generation of a platelet surface capable of supporting prothrombinase assembly.23 PAR1 activity is inhibited by the antiplatelet agent vorapaxar,24 and a recent study by Tourdot et al.25 has highlighted the importance of PAR4 activation in thrombosis. This group previously identified a variant in PAR4 (PAR4-Thr120), which is prevalent in African Americans and accounts for ~50% of the platelet hyperactivity observed in this group.26 They have now shown that platelets expressing PAR4-Thr120 have increased Gq and G13 activation in response to thrombin, undergo a more dramatic shape change, and produce larger clots under flow. These data suggest that PAR receptors, such as PAR4, may be valuable targets for antithrombotic therapy, particularly in individuals with the Thr120 variant.
Two recent publications have assessed the therapeutic potential of anti-PAR4 agents. First, Wilson et al.27 studied the effects of BMS-986120, a reversible PAR4 antagonist, on ex vivo platelet thrombus formation in a Phase I clinical trial. They found that platelets from BMS-986120-treated patients had dramatically reduced responses to PAR4 stimulation and formed smaller clots under high shear flow conditions, which were similar in size to those formed by platelets from patients treated with aspirin or a combination of aspirin and clopidogrel. Second, Lin et al.28 showed that the non-anticoagulant heparin SCH-28 specifically blocks thrombin-mediated PAR4 activation and reduces platelet thrombus formation under flow, and suggest that SCH-28 may be a safer alternative to traditional heparin therapy, as it does not promote FXa or thrombin inhibition by antithrombin.
Platelets are also activated through the collagen receptor glycoprotein VI (GP-VI).29 This interaction is often described as the initial activator of platelets, as collagen is exposed at the subendothelial site of injury. Recent work by Lehmann et al.30 has described an important role for GP-VI in clot progression, in a model for venous thromboembolism. They developed a microfluidic system to mimic the low-flow conditions of a venous valve. In this system, they found that TF activity promotes fibrin deposition, platelets adhere to the fibrin and expose phosphatidylserine, and additional platelets are recruited to promote thrombus growth. This final step was dependent on GP-VI, as it could be inhibited by the anti-GP-VI antibody Fab fragment ACT017 or by the fibrin fragment d-dimer, which binds GP-VI. GP-VI is also being actively pursued as a therapeutic target. Voors-Pette and colleagues31 have reported the results of a first-in-human study of ACT017. This was a safety study in healthy volunteers, and the authors reported no serious adverse events, no change in platelet count, no change in GP-VI expression on platelets, and no change in the concentration of shed, soluble GP-VI in plasma.
New studies have also elucidated the signaling pathways that lead to platelet activation in response to either thrombin or collagen. Adam et al.32 knocked out the motor protein kinesin-1 heavy chain in mice and observed a re-bleeding phenotype following tail clip and thrombus instability in the ferric chloride injury model. They subsequently showed that the platelets responded less to low-dose thrombin or collagen stimulation, defined by reduced aggregation, ATP and platelet factor-4 (PF4) secretion, and P-selectin exposure. These deficiencies could be overcome by stimulation with high concentrations of thrombin. Thus, kinesin-1 is a component of the thrombin signaling pathway in platelets. In addition, work by Wersall et al.33 identified a specific role for the Ral family of small GTPases. They knocked out either RalA or RalB or both in a mouse model and observed reduced P-selectin exposure in response to either thrombin or collagen, though total P-selectin was actually slightly increased. In contrast, they saw no change in activation of integrin αIIbβ3, secretion of α-granule cargos PF4 and TGFβ, or ex vivo thrombus formation. Though both RalA and RalB contributed to P-selectin exposure, the most dramatic phenotype was observed in double-knockout mice. However, they did observe decreased formation of platelet-leukocyte aggregates, a P-selectin-dependent process. This work shows that P-selectin exposure is regulated differently than α-granule release and is not essential for thrombus formation. Finally, Laurent and colleagues34 used knockout mice and inhibitors to show that phosphoinositide 3-kinase alpha (PI3Kα) is involved in platelet activation in response to low doses of collagen. Unlike kinesin-1 and Ral GTPases, PI3Kα was not involved in thrombin signaling. In response to collagen, knockout or inhibited platelets had reduced aggregation, dense granule secretion, and adhesion to immobilized von Willebrand factor. The mice also had reduced thrombus formation in a laser injury model, but did not show any impairment in the tail clip bleeding assay.
Platelet activation through GP-VI, PARs, and other receptors requires release of intracellular calcium stores into the cytosol.35 Recent work by Gotru and co-workers showed that these stores are regulated by transient receptor potential cation channel, subfamily M, member 7 (TRPM7), a protein which they had previously identified as a regulator of magnesium in megakaryocytes.36, 37 Platelets from TRPM7−/− mice exhibit decreased calcium release, aggregation, P-selectin exposure, and integrin activation in response to either collagen or thrombin. In addition, the authors observed reduced thrombus formation ex vivo in a whole blood flow system and showed that the mice were protected from ferric chloride-induced thrombosis and from cerebral ischemia, while there was no change in the tail bleed assay. Lopez et al.38 added to these observations by showing that calcium signaling is regulated by the calcium sensor stromal interaction molecule 1 (STIM1) and by the phosphorylation of Ser2152 on the cytoskeletal protein filamin A, which promotes its interaction with STIM1. They showed that inhibition of the STIM1-filamin A interaction enhances calcium release and platelet aggregation. Collectively, these studies have identified several new potential downstream therapeutic targets, which could selectively inhibit specific aspects of platelet function to prevent thrombosis.
Other Regulators of Platelet Function
Platelet function is also influenced by several other factors, including the adhesive protein von Willebrand Factor (vWF) and flow forces. Platelets bind to vWF through the glycoprotein IB-IX-V receptor complex.39 Chen and colleagues40 showed that by blocking vWF binding with the aptamer ARC1779, they can improve the ex vivo stability of stored platelets. They found that refrigerated murine and human platelets incubated with ARC1779 had increased recovery and half-life post-transfusion. They also had increased hemostatic function, evidenced by a reduced clot time in the tail bleed model, suggesting that vWF binding is at least partially responsible for the loss of activity and lifetime that has been observed with cold-stored platelets. Abdelgawwad et al.41 used a different approach to show the potential therapeutic benefit of blocking the platelet/vWF interaction. Thrombotic-thrombocytopenic purpura (TTP) is caused when congenital or acquired loss of the enzyme ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats 13) results in an increase in circulating high molecular weight vWF multimers.42 Abdelgawwad used the natural endocytic machinery to load platelets with ADAMTS13 and showed that these platelets have antithrombotic properties when transfused into ADAMTS13−/− mice or added into whole blood from TTP patients, supporting this process as a novel treatment approach.41 Adili and Holinstat43 similarly assessed the efficacy of introducing ADAMTS13 to treat TTP. They showed that ADAMTS13−/− mice are prone to vWF-dependent microvascular thrombi in the brain, which are independent of fibrin, and that infusion of BAX930, a recombinant form of ADAMTS13, was able to resolve these thrombi.
vWF is stored in Weibel-Palade bodies of endothelial cells and secreted into plasma.44 Recent work from Biswas et al.45 showed that vWF secretion is promoted by high mobility group box 1, and this process is dramatically enhanced by the presence of polyphosphate chains in the same size range as those secreted by activated platelets. The released vWF then supports platelet adhesion and formation of platelet/vWF “strings” on the endothelial surface, suggesting a positive feedback loop between platelets and vWF in this process. Intracellularly, Schillemans et al.46 identified syntaxin-3 as a key mediator of the membrane fusion process that allows for vWF secretion. Syntaxin-3 functions with vesicle-associated membrane protein-8, a well described exocytosis regulator, to control this process, but has no impact on Weibel-Palade body formation. vWF is also found in platelet α-granules, but interestingly, Doddapattar et al.47 found that only endothelial vWF promotes platelet adhesion and subsequent inflammation and leukocyte adhesion in the ApoE−/− atherosclerosis model, suggesting that vWF may serve different functions depending on its source.
It has long been understood that shear forces regulate vWF activity and platelet aggregation, and this appears to be particularly true for turbulent flow.48 According to the work of Bortot and colleagues,49 vWF activity is specifically regulated by turbulent flow patterns. Bortot compared laminar, transitional, and turbulent flow patterns, and observed that turbulent flow resulted in the greatest loss of vWF multimers (indicating the highest activity of ADAMTS13) and the lowest vWF activity, as measured by its ability to bind platelets and collagen.49 These data offer a possible mitigating factor to prevent spontaneous thrombosis in venous valves, where turbulent flow patterns are prevalent.34 Anyanwu et al.50 described another potential mitigating factor, the ectonucleotidase CD39. They showed that CD39−/− mice have increased thrombus size in a stasis injury model, increased P-selectin exposure and vWF levels, and increased platelet-leukocyte aggregates. Thus, the ability of CD39 to hydrolyze ATP and/or ADP appears to protect from venous thrombosis.
The pathological environment also has the power to shape platelet phenotype and function, and can impact the efficacy of antiplatelet therapies, such as aspirin and clopidogrel.51 Cameron et al.52 demonstrated that platelets isolated from patients with vascular and metabolic diseases, including patent ductus arteriosus, diabetes mellitus, and hypertension, have higher activity and are resistant to antiplatelet agents, when compared to platelets isolated from healthy volunteers. The mechanism underlying this change in platelet phenotype is not clearly understood, but is partly mediated by extracellular regulated protein kinase 5.51, 52
Summary
Over the past two years, studies published in Arteriosclerosis, Thrombosis, and Vascular Biology have improved our understanding of the regulation of thrombin generation, through both the TF and contact pathway, and of platelet activation and function (Figure 1). These studies have identified new pathways and targets, which may lead to improved anticoagulant and antithrombotic agents in the years to come.
Figure 1.
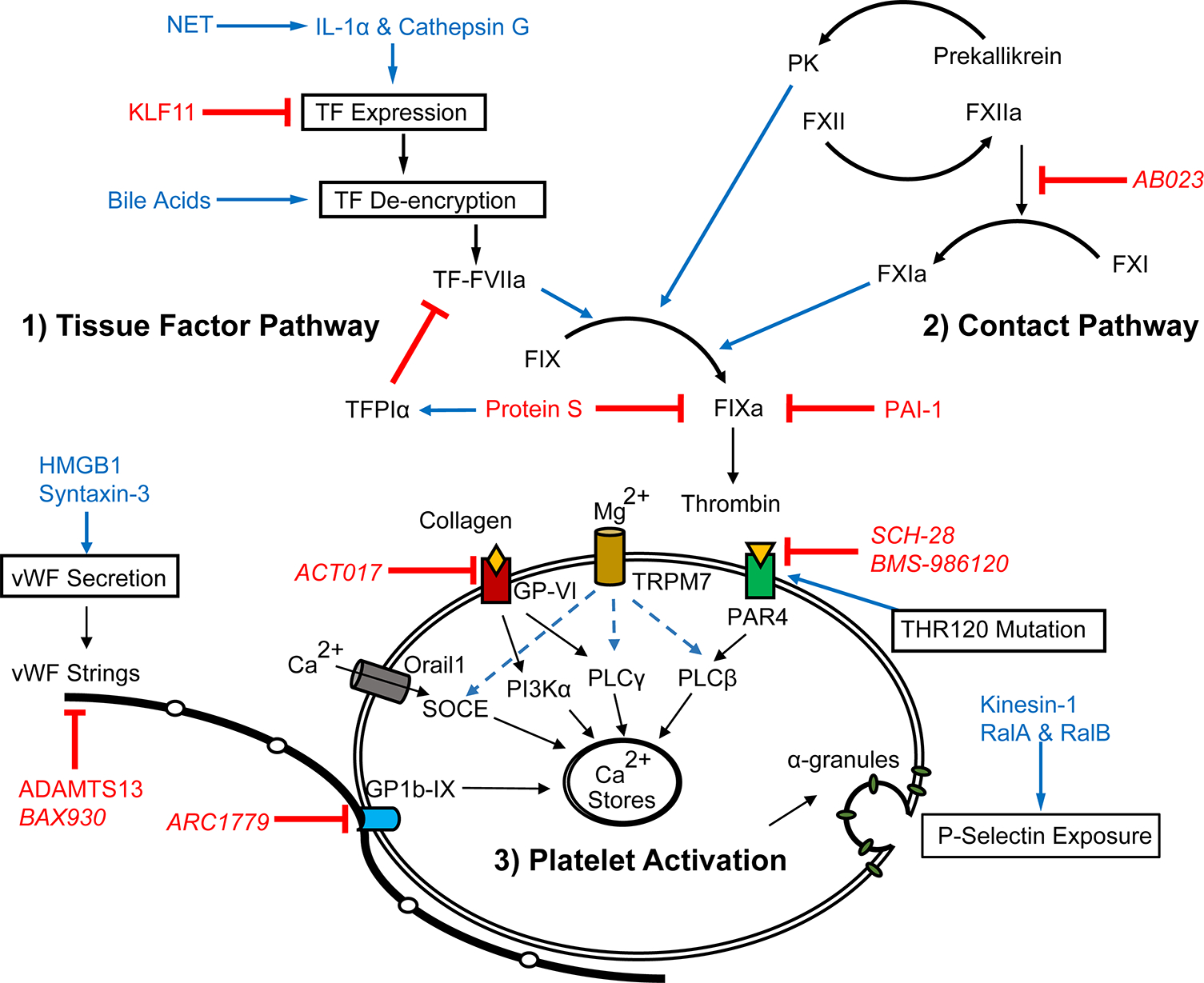
Recent advances have been made in our understanding of 1) TF expression, de-encryption, and downstream substrate FIXa activation; 2) regulation of the contact pathway and its crosstalk with inflammation; and 3) receptors, signaling and extracellular components that regulate platelet activation. Blue text indicates promoters, red indicates inhibitors, and italics indicate therapeutic agents.
Sources of Funding
This work was supported by National Institutes of Health grant HL129193.
Abbreviations:
- TF
tissue factor
- FVIIa
factor VIIa
- FX
factor X
- FIX
factor IX
- KLF11
Krüppel-like factor 11
- NET
neutrophil extracellular trap
- DVT
deep vein thrombosis
- TFPIα
tissue factor pathway inhibitor α
- PS
protein S
- FXII
factor XII
- FXI
factor XI
- PK
plasma kallikrein
- VTE
venous thromboembolism
- PAI-1
plasminogen activator inhibitor-1
- PAR4
protease activated receptor 4
- GP-VI
glycoprotein VI
- PF4
platelet factor-4
- PI3Kα
phosphoinositide 3-kinase alpha
- TRPM7
transient receptor potential cation channel, subfamily M, member 7
- STIM1
stromal interaction molecule-1
- vWF
von Willebrand factor
- ADAMTS13
a disintegrin and metalloprotease with thrombospondin type 1 repeats 13
Footnotes
Disclosures
The authors have nothing to disclose.
References
- 1.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 2.Komiyama Y, Pedersen AH, Kisiel W. Proteolytic activation of human factors ix and x by recombinant human factor viia: Effects of calcium, phospholipids, and tissue factor. Biochemistry. 1990;29:9418–9425. [DOI] [PubMed] [Google Scholar]
- 3.Zhu S, Chen J, Diamond SL. Establishing the transient mass balance of thrombosis: From tissue factor to thrombin to fibrin under venous flow. Arterioscler Thromb Vasc Biol. 2018;38:1528–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giesen PL, Rauch U, Bohrmann B, Kling D, Roque M, Fallon JT, Badimon JJ, Himber J, Riederer MA, Nemerson Y. Blood-borne tissue factor: Another view of thrombosis. Proc Natl Acad Sci U S A. 1999;96:2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Del Conde I, Shrimpton CN, Thiagarajan P, Lopez JA. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood. 2005;106:1604–1611. [DOI] [PubMed] [Google Scholar]
- 6.Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW, Libby P. Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1alpha and cathepsin g. Arterioscler Thromb Vasc Biol. 2018;38:1901–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liang W, Fan Y, Lu H, et al. Klf11 (kruppel-like factor 11) inhibits arterial thrombosis via suppression of tissue factor in the vascular wall. Arterioscler Thromb Vasc Biol. 2019;39:402–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Butenas S, Krudysz-Amblo J. Decryption of tissue factor. Thromb Res. 2012;129 Suppl 2: S18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker KS, Kopec AK, Pant A, Poole LG, Cline-Fedewa H, Ivkovich D, Olyaee M, Woolbright BL, Miszta A, Jaeschke H, Wolberg AS, Luyendyk JP. Direct amplification of tissue factor:Factor viia procoagulant activity by bile acids drives intrahepatic coagulation. Arterioscler Thromb Vasc Biol. 2019;39:2038–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stark K, Schubert I, Joshi U, et al. Distinct pathogenesis of pancreatic cancer microvesicle-associated venous thrombosis identifies new antithrombotic targets in vivo. Arterioscler Thromb Vasc Biol. 2018;38:772–786. [DOI] [PubMed] [Google Scholar]
- 11.Wood JP, Ellery PE, Maroney SA, Mast AE. Biology of tissue factor pathway inhibitor. Blood. 2014;123:2934–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanratana P, Ellery P, Westmark P, Mast AE, Sheehan JP. Elevated plasma factor ixa activity in premenopausal women on hormonal contraception. Arterioscler Thromb Vasc Biol. 2018;38:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattopadhyay R, Sengupta T, Majumder R. Inhibition of intrinsic xase by protein s: A novel regulatory role of protein s independent of activated protein c. Arterioscler Thromb Vasc Biol. 2012;32:2387–2393. [DOI] [PubMed] [Google Scholar]
- 14.Plautz WE, Sekhar Pilli VS, Cooley BC, Chattopadhyay R, Westmark PR, Getz T, Paul D, Bergmeier W, Sheehan JP, Majumder R. Anticoagulant protein s targets the factor ixa heparin-binding exosite to prevent thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:816–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Long AT, Kenne E, Jung R, Fuchs TA, Renne T. Contact system revisited: An interface between inflammation, coagulation, and innate immunity. J Thromb Haemost. 2016;14:427–437. [DOI] [PubMed] [Google Scholar]
- 16.Lorentz CU, Verbout NG, Wallisch M, Hagen MW, Shatzel JJ, Olson SR, Puy C, Hinds MT, McCarty OJT, Gailani D, Gruber A, Tucker EI. Contact activation inhibitor and factor xi antibody, ab023, produces safe, dose-dependent anticoagulation in a phase 1 first-in-human trial. Arterioscler Thromb Vasc Biol. 2019;39:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puy C, Ngo ATP, Pang J, Keshari RS, Hagen MW, Hinds MT, Gailani D, Gruber A, Lupu F, McCarty OJT. Endothelial pai-1 (plasminogen activator inhibitor-1) blocks the intrinsic pathway of coagulation, inducing the clearance and degradation of fxia (activated factor xi). Arterioscler Thromb Vasc Biol. 2019;39:1390–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zilberman-Rudenko J, Reitsma SE, Puy C, et al. Factor xii activation promotes platelet consumption in the presence of bacterial-type long-chain polyphosphate in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2018;38:1748–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silasi R, Keshari RS, Lupu C, et al. Inhibition of contact-mediated activation of factor xi protects baboons against s aureus-induced organ damage and death. Blood Adv. 2019;3:658–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Osterud B, Bouma BN, Griffin JH. Human blood coagulation factor ix. Purification, properties, and mechanism of activation by activated factor xi. J Biol Chem. 1978;253:5946–5951. [PubMed] [Google Scholar]
- 21.Visser M, van Oerle R, Ten Cate H, Laux V, Mackman N, Heitmeier S, Spronk HMH. Plasma kallikrein contributes to coagulation in the absence of factor xi by activating factor ix. Arterioscler Thromb Vasc Biol. 2020;40:103–111. [DOI] [PubMed] [Google Scholar]
- 22.Fujikawa K, Chung DW, Hendrickson LE, Davie EW. Amino acid sequence of human factor xi, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry. 1986;25:2417–2424. [DOI] [PubMed] [Google Scholar]
- 23.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chackalamannil S, Wang Y, Greenlee WJ, Hu Z, Xia Y, Ahn HS, Boykow G, Hsieh Y, Palamanda J, Agans-Fantuzzi J, Kurowski S, Graziano M, Chintala M. Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (sch 530348) with potent antiplatelet activity. J Med Chem. 2008;51:3061–3064. [DOI] [PubMed] [Google Scholar]
- 25.Tourdot BE, Stoveken H, Trumbo D, Yeung J, Kanthi Y, Edelstein LC, Bray PF, Tall GG, Holinstat M. Genetic variant in human par (protease-activated receptor) 4 enhances thrombus formation resulting in resistance to antiplatelet therapeutics. Arterioscler Thromb Vasc Biol. 2018;38:1632–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edelstein LC, Simon LM, Lindsay CR, Kong X, Teruel-Montoya R, Tourdot BE, Chen ES, Ma L, Coughlin S, Nieman M, Holinstat M, Shaw CA, Bray PF. Common variants in the human platelet par4 thrombin receptor alter platelet function and differ by race. Blood. 2014;124:3450–3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson SJ, Ismat FA, Wang Z, Cerra M, Narayan H, Raftis J, Gray TJ, Connell S, Garonzik S, Ma X, Yang J, Newby DE. Par4 (protease-activated receptor 4) antagonism with bms-986120 inhibits human ex vivo thrombus formation. Arterioscler Thromb Vasc Biol. 2018;38:448–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin YC, Ko YC, Hung SC, Lin YT, Lee JH, Tsai JY, Kung PH, Tsai MC, Chen YF, Wu CC. Selective inhibition of par4 (protease-activated receptor 4)-mediated platelet activation by a synthetic nonanticoagulant heparin analog. Arterioscler Thromb Vasc Biol. 2019;39:694–703. [DOI] [PubMed] [Google Scholar]
- 29.Kehrel B, Wierwille S, Clemetson KJ, Anders O, Steiner M, Knight CG, Farndale RW, Okuma M, Barnes MJ. Glycoprotein vi is a major collagen receptor for platelet activation: It recognizes the platelet-activating quaternary structure of collagen, whereas cd36, glycoprotein iib/iiia, and von willebrand factor do not. Blood. 1998;91:491–499. [PubMed] [Google Scholar]
- 30.Lehmann M, Schoeman RM, Krohl PJ, Wallbank AM, Samaniuk JR, Jandrot-Perrus M, Neeves KB. Platelets drive thrombus propagation in a hematocrit and glycoprotein vi-dependent manner in an in vitro venous thrombosis model. Arterioscler Thromb Vasc Biol. 2018;38:1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voors-Pette C, Lebozec K, Dogterom P, Jullien L, Billiald P, Ferlan P, Renaud L, Favre-Bulle O, Avenard G, Machacek M, Pletan Y, Jandrot-Perrus M. Safety and tolerability, pharmacokinetics, and pharmacodynamics of act017, an antiplatelet gpvi (glycoprotein vi) fab. Arterioscler Thromb Vasc Biol. 2019;39:956–964. [DOI] [PubMed] [Google Scholar]
- 32.Adam F, Kauskot A, Kurowska M, Goudin N, Munoz I, Bordet JC, Huang JD, Bryckaert M, Fischer A, Borgel D, de Saint Basile G, Christophe OD, Menasche G. Kinesin-1 is a new actor involved in platelet secretion and thrombus stability. Arterioscler Thromb Vasc Biol. 2018;38:1037–1051. [DOI] [PubMed] [Google Scholar]
- 33.Wersall A, Williams CM, Brown E, Iannitti T, Williams N, Poole AW. Mouse platelet ral gtpases control p-selectin surface expression, regulating platelet-leukocyte interaction. Arterioscler Thromb Vasc Biol. 2018;38:787–800. [DOI] [PubMed] [Google Scholar]
- 34.Laurent PA, Hechler B, Solinhac R, Ragab A, Cabou C, Anquetil T, Severin S, Denis CV, Mangin PH, Vanhaesebroeck B, Payrastre B, Gratacap MP. Impact of pi3kalpha (phosphoinositide 3-kinase alpha) inhibition on hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:2041–2053. [DOI] [PubMed] [Google Scholar]
- 35.Charo IF, Feinman RD, Detwiler TC. Inhibition of platelet secretion by an antagonist of intracellular calcium. Biochem Biophys Res Commun. 1976;72:1462–1467. [DOI] [PubMed] [Google Scholar]
- 36.Gotru SK, Chen W, Kraft P, et al. Trpm7 kinase controls calcium responses in arterial thrombosis and stroke in mice. Arterioscler Thromb Vasc Biol. 2018;38:344–352. [DOI] [PubMed] [Google Scholar]
- 37.Stritt S, Nurden P, Favier R, et al. Defects in trpm7 channel function deregulate thrombopoiesis through altered cellular mg(2+) homeostasis and cytoskeletal architecture. Nat Commun. 2016;7:11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lopez JJ, Albarran L, Jardin I, Sanchez-Collado J, Redondo PC, Bermejo N, Bobe R, Smani T, Rosado JA. Filamin a modulates store-operated ca(2+) entry by regulating stim1 (stromal interaction molecule 1)-orai1 association in human platelets. Arterioscler Thromb Vasc Biol. 2018;38:386–397. [DOI] [PubMed] [Google Scholar]
- 39.Shen Y, Romo GM, Dong JF, Schade A, McIntire LV, Kenny D, Whisstock JC, Berndt MC, Lopez JA, Andrews RK. Requirement of leucine-rich repeats of glycoprotein (gp) ibalpha for shear-dependent and static binding of von willebrand factor to the platelet membrane gp ib-ix-v complex. Blood. 2000;95:903–910. [PubMed] [Google Scholar]
- 40.Chen W, Voos KM, Josephson CD, Li R. Short-acting anti-vwf (von willebrand factor) aptamer improves the recovery, survival, and hemostatic functions of refrigerated platelets. Arterioscler Thromb Vasc Biol. 2019;39:2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdelgawwad MS, Cao W, Zheng L, Kocher NK, Williams LA, Zheng XL. Transfusion of platelets loaded with recombinant adamts13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats-13) is efficacious for inhibiting arterial thrombosis associated with thrombotic thrombocytopenic purpura. Arterioscler Thromb Vasc Biol. 2018;38:2731–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng XL, Sadler JE. Pathogenesis of thrombotic microangiopathies. Annu Rev Pathol. 2008;3:249–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adili R, Holinstat M. Formation and resolution of pial microvascular thrombosis in a mouse model of thrombotic thrombocytopenic purpura. Arterioscler Thromb Vasc Biol. 2019;39:1817–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagner DD, Olmsted JB, Marder VJ. Immunolocalization of von willebrand protein in weibel-palade bodies of human endothelial cells. J Cell Biol. 1982;95:355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Biswas I, Panicker SR, Cai X, Mehta-D’souza P, Rezaie AR. Inorganic polyphosphate amplifies high mobility group box 1-mediated von willebrand factor release and platelet string formation on endothelial cells. Arterioscler Thromb Vasc Biol. 2018;38:1868–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schillemans M, Karampini E, van den Eshof BL, et al. Weibel-palade body localized syntaxin-3 modulates von willebrand factor secretion from endothelial cells. Arterioscler Thromb Vasc Biol. 2018;38:1549–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doddapattar P, Dhanesha N, Chorawala MR, Tinsman C, Jain M, Nayak MK, Staber JM, Chauhan AK. Endothelial cell-derived von willebrand factor, but not platelet-derived, promotes atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. 2018;38:520–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peterson DM, Stathopoulos NA, Giorgio TD, Hellums JD, Moake JL. Shear-induced platelet aggregation requires von willebrand factor and platelet membrane glycoproteins ib and iib-iiia. Blood. 1987;69:625–628. [PubMed] [Google Scholar]
- 49.Bortot M, Ashworth K, Sharifi A, Walker F, Crawford NC, Neeves KB, Bark D Jr., Di Paola J. Turbulent flow promotes cleavage of vwf (von willebrand factor) by adamts13 (a disintegrin and metalloproteinase with a thrombospondin type-1 motif, member 13). Arterioscler Thromb Vasc Biol. 2019;39:1831–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Anyanwu AC, Kanthi Y, Fukase K, et al. Tuning the thromboinflammatory response to venous flow interruption by the ectonucleotidase cd39. Arterioscler Thromb Vasc Biol. 2019;39:e118–e129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cameron SJ, Ture SK, Mickelsen D, Chakrabarti E, Modjeski KL, McNitt S, Seaberry M, Field DJ, Le NT, Abe J, Morrell CN. Platelet extracellular regulated protein kinase 5 is a redox switch and triggers maladaptive platelet responses and myocardial infarct expansion. Circulation. 2015;132:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cameron SJ, Mix DS, Ture SK, Schmidt RA, Mohan A, Pariser D, Stoner MC, Shah P, Chen L, Zhang H, Field DJ, Modjeski KL, Toth S, Morrell CN. Hypoxia and ischemia promote a maladaptive platelet phenotype. Arterioscler Thromb Vasc Biol. 2018;38:1594–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]