

SUMMARY
Following deployment of an augmented validation system by the Worldwide Protein Data Bank (wwPDB) partnership, the quality of crystal structures entering the PDB has improved. Of significance are improvements in quality measures now prominently displayed in the wwPDB Validation Report. Comparisons of PDB depositions made before and after introduction of the new reporting system show improvements in quality measures relating to pairwise atom-atom clashes, sidechain torsion angle rotamers, and local agreement between the atomic coordinate structure model and experimental electron density data. These improvements are largely independent of resolution limit and sample molecular weight. No significant improvement in the quality of associated ligands was observed. Principal component analysis revealed that structure quality could be summarized with three measures (Rfree, Real Space R-factor Z-score, and a combined molecular geometry quality metric), which can in turn be reduced to a single overall quality metric readily interpretable by all PDB archive users.
Keywords: structure validation, protein crystal structure, Protein Data Bank, structure quality, multivariate analysis, principal component analysis, PDB, wwPDB, RCSB, OneDep
Graphical Abstract
eTOC Blurb
Two years after deployment of wwPDB OneDep Deposition/Annotation/Validation system and official wwPDB Validation Report, Shao et al. analyzed the individual and condensed structure quality measures, revealing quality improvements in protein crystal structures deposited to the PDB, but little improvement in the quality of bound ligands.
INTRODUCTION
Quality improvement is central to management of the Protein Data Bank (PDB), the single open-access global repository for experimentally determined, three-dimensional (3D) atomic-level structures of biological macromolecules. High quality structure data are critical for much of biomedical research and drug discovery. The PDB archive is managed by the Worldwide Protein Data Bank partnership (wwPDB; http://wwpdb.org) (Berman et al., 2003), which currently includes three founding regional data centers, located in the US (RCSB Protein Data Bank or RCSB PDB; http://rcsb.org), Japan (Protein Data Bank Japan or PDBj; http://pdbj.org), and Europe (Protein Data Bank in Europe or PDBe; http://pdbe.org), plus a global NMR specialist data repository BioMagResBank, made up of deposition sites in the US (BMRB; http://www.bmrb.wisc.edu) and Japan (PDBj-BMRB; http://bmrbdep.pdbj.org). Together, wwPDB partners collect, biocurate, validate, and disseminate standardized PDB data to the public without any limitations on usage. During the deposition and biocuration processes, wwPDB partners also examine data quality and present a validation report to Depositors for quality control and improvement.
As of November 2016, the PDB archive numbered more than 124,000 experimentally determined 3D structures of biological macromolecules and their complexes with various ligands. Approximately 90% of structures in the PDB have been determined using crystallography (primarily X-ray), with the remaining structures determined using nuclear magnetic resonance spectroscopy (~9%, NMR) and electron microscopy (<1%, 3DEM). Since 2008, experimental data have been required to accompany PDB depositions of atomic coordinates derived from X-ray or NMR (http://www.wwpdb.org/news/news?year=2007#29-November-2007). Availability of these data allows the quality of a given PDB entry be assessed from various perspectives, including experimental data, the geometry of the atomic coordinates and agreement with known stereochemistry, and goodness-of-fit between atomic coordinates and experimental data. For more than a decade, the RCSB PDB has been tracking >130 Depositor-reported and RCSB PDB-calculated data quality measures for X-ray structures including molecular features such as Molecular Weight and sequence, data quality statistics such as R-merge and I/σ(I) versus resolution, structural solution and refinement statistics such as Rwork and Rfree, plus model quality measures such as Ramachandran outliers and stereochemistry violations.
Method-specific validation can be performed at various stages throughout the structure determination pipeline. For example, in the steps of finalizing X-ray crystal structure model, an increasing number of validation tools can be used with structure refinement programs such as PHENIX (Adams et al., 2010), REFMAC (Murshudov et al., 1997), BUSTER (Smart et al., 2008), SHELX (Sheldrick et al., 2008), and CNS (Brünger et al., 1998). These provisions have been augmented by review and modification programs such as COOT (Emsley et al., 2010), components in the comprehensive CCP4 package (Winn et al., 2011), and services such as PDB_REDO (Joosten et al., 2014).
To better understand and improve the quality of data released into the PDB archive, the wwPDB partnership formed a series of expert Validation Task Forces (VTF) to make recommendations as to which experimental data/metadata should be archived and how they should be validated from X-ray crystallography (Read et al., 2011), NMR (Montelione et al., 2013), and 3DEM (Henderson et al., 2012). Task force recommendations have been implemented in the form of a software pipeline which produces an official wwPDB Validation Report (Gore et al., 2012). This validation pipeline was integrated into the wwPDB OneDep Deposition/Biocuration/Validation system (OneDep) first deployed in January 2014 for crystallography, and then in January 2016 for NMR and 3DEM (Young et al., submitted). The OneDep system allows Depositors to view validation assessments during the deposition process. Pre-deposition validation is strongly encouraged using a separate anonymous wwPDB Validation Server (http://validate.wwpdb.org) or a Web Service API (http://wwpdb.org/validation/onedep-validation-web-service-interface). At the conclusion of the biocuration process, a final official wwPDB Validation Report is provided to the Depositor by the OneDep system. The wwPDB strongly encourages use of these final wwPDB Validation Reports (http://wwpdb.org/validation/validation-reports) during manuscript review. wwPDB Validation Reports are now required for submitting structure determination manuscripts to an increasing number of journals, including Structure (http://crosstalk.cell.com/blog/show-us-your-pdb-validation-reports) and Nature Publishing Group publications (Editorial, 2016). The final wwPDB Validation Reports are made public in concert with release of each PDB entry (typically at the time of publication of the associated primary citation). Provision of validation information prior to and during structure deposition, is intended to help Depositors make any necessary corrections before their PDB entries are made public. Annually, the wwPDB updates data quality statistics for the entire PDB archive and publishes updated Validation Reports for all previously released entries.
The new wwPDB Validation Report (Gore et al., 2012) provides comprehensive quality assessments calculated using community-standard software tools, including DCC (Yang et al., 2016), EDS (Kleywegt et al., 2004), Mogul (Bruno et al., 2004), MolProbity (Chen et al., 2010), and Xtriage (Adams et al., 2010). Presentation of quality metrics in the wwPDB Validation Reports is accompanied by summary illustrations of these measures in the form of five graphical sliders for the free R-factor (Rfree) (Brünger, 1992), non-bonded atom-atom clashes assessed by a scaled Clashscore (Chen et al., 2010), % Ramachandran Outliers (Ramachandran et al., 1963), % Sidechain Rotamer Outliers (Chen et al., 2010), and % Real Space R-factor Z-Score (RSRZ) Outliers (Kleywegt et al., 2004). These sliders provide percentile scores, showing how the quality of a given entry compares to all structures archived in the PDB and to the subset of PDB structures determined at similar resolution.
Figure 1 displays examples of slider images for structures of higher quality (PDB: 4DI8) (Hobbs et al., 2012), intermediate quality (PDB: 2HYU) (Shao et al., 2006), and lower quality (PDB: 2GUW) (http://dx.doi.org/10.2210/pdb2guw/pdb). The right side blue color zone denotes better quality values and the left side red color zone denotes lower quality values. These five metrics were introduced into the new wwPDB Validation Report as primary quality measures, following recommendations from X-ray crystallography experts convened as the wwPDB X-ray VTF (Read et al., 2011).
Figure 1.
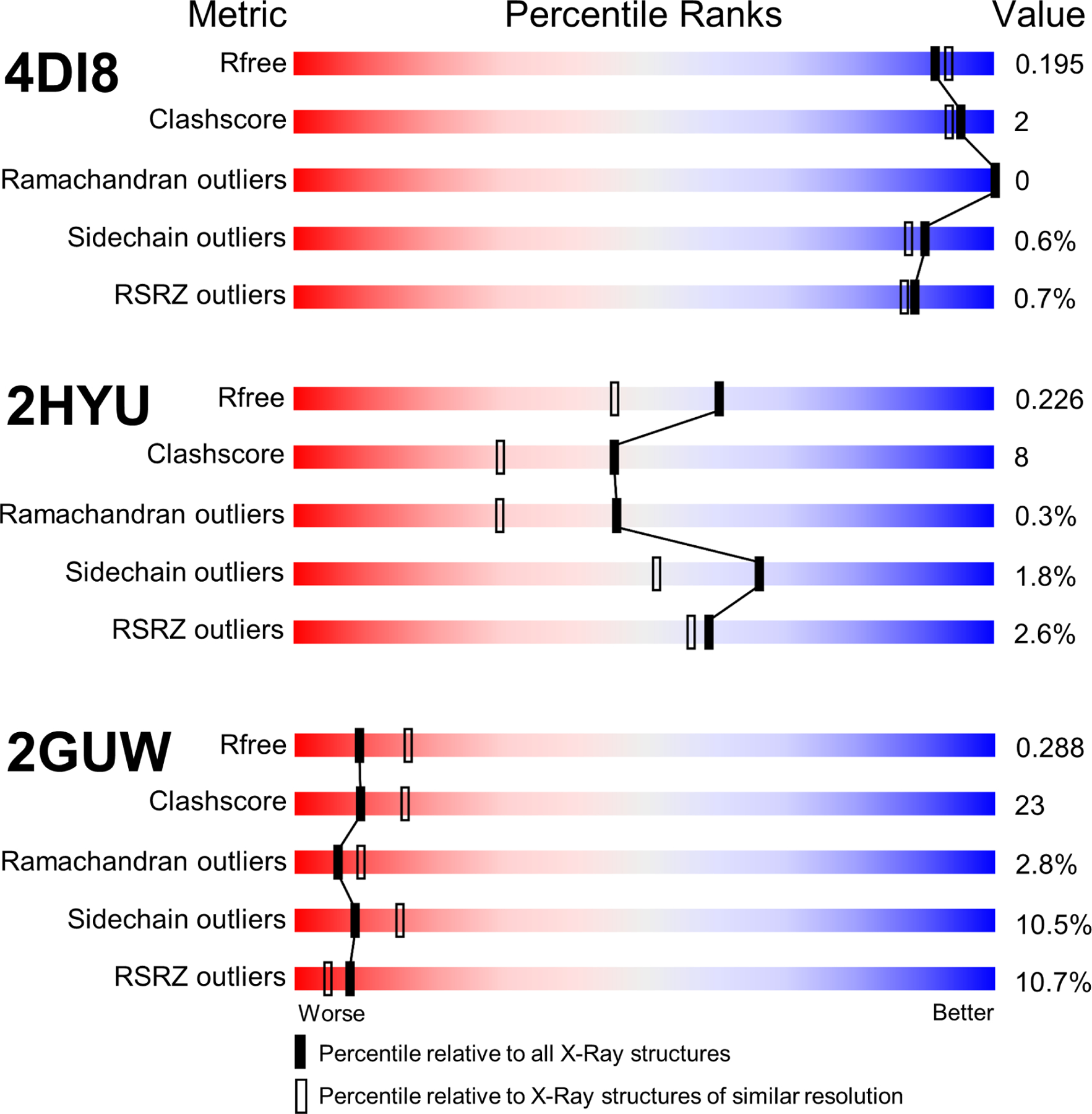
Slider images of five structure quality measures for entries of higher quality (PDB: 4DI8), intermediate quality (PDB: 2HYU), and lower quality (PDB: 2GUW).
To assess the data quality for co-crystal structure determinations of proteins or nucleic acids with one or more bound ligands, the wwPDB Validation Report provides the results of Mogul (Bruno et al., 2004) validation against the small molecule X-ray crystal structure data in the Cambridge Structural Database (Groom et al., 2016). The fit of ligand atomic coordinates to corresponding experimental electron density is also assessed. Figure 2 illustrates electron density maps for a well-known ligand co-factor (NADP) found in two archival entries, PDB: 1ZK4 (Schlieben et al., 2005) and PDB: 2FZD (Steuber et al., 2006). The 2FZD co-crystal structure was determined at 1.08Å resolution, and the fit of the atomic coordinates of the NADP co-factor to the experimental electron density is excellent (Figure 2A) with clean difference map (Figure 2C). In contrast, co-crystal structure 1ZK4, determined at 1.0Å resolution, was identified by the TWILIGHT program (Weichenberger et al., 2013) as an example of poor fit of the atomic coordinates of NADP to the experimental electron density (Figure 2B) with noisy difference map (Figure 2D). wwPDB Validation Reports for 2FZD and 1ZK4 provide quantitative assessments of the quality of the common NADP ligand, including Real space R factor, RSR (Jones et al., 1991); real space correlation coefficient, RSCC (Brändén and Jones, 1990); Occupancy Weighted Average B factor, OWAB; and Bond Length Root-Mean-Square deviation (RMS) Z-score and Bond Angle RMSZ provided by Mogul (Bruno et al., 2004). The NADP ligand quality metrics for 2FZD (RSR=0.05; RSCC=0.99; OWAB=5.5Å2; Bond Length RMSZ=1.0Å; Bond Angle RMSZ=1.1°) are significantly superior to those of 1ZK4 (RSR=0.67; RSCC=−0.06; OWAB=94.8Å2; Bond Length RMSZ=1.6Å; Bond Angle RMSZ=2.0°). The values of these five ligand quality metrics provided in the wwPDB Validation Report correlate well with the goodness-of-fit readily apparent in the electron density maps illustrated in Figure 2.
Figure 2.
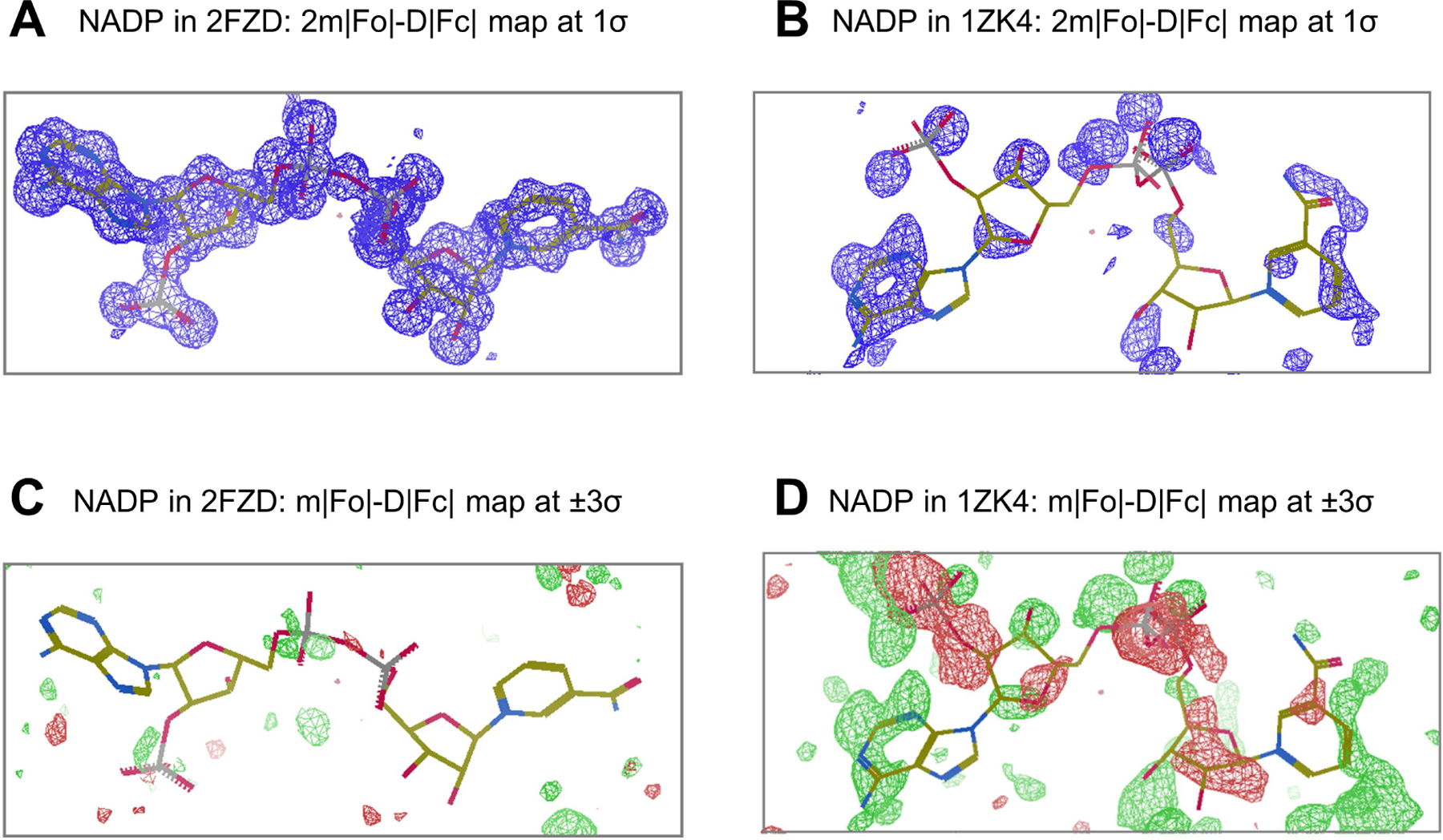
Bound NADP ligands (stick model) and the corresponding electron density maps well resolved in co-crystal structure PDB: 2FZD and poorly resolved in PDB: 1ZK4. (A) and (B): 2m|Fo|-D|Fc| map contoured at +1σ (blue) for 2FZD and 1ZK4, respectively; (C) and (D): m|Fo|-D|Fc| difference map contoured at +3σ (green)/−3σ (red) for 2FZD and 1ZK4, respectively.
Prior to introduction of the OneDep system, presentation of validation data at the time of PDB deposition varied owing to differences in software tools used at each wwPDB data deposition site. At the RCSB PDB, the legacy validation report included a report of data consistency, geometrical and stereochemical issues, real space R factor, and real space correlation coefficient. This legacy report was accompanied by the output from the Molprobity and DCC software tools. Prior to 2014, validation reports were not incorporated into the PDB archive, and data quality information therein was restricted to enumeration of exceptional geometrical outliers and stereochemical errors reported in PDB format REMARKs and CAVEAT records.
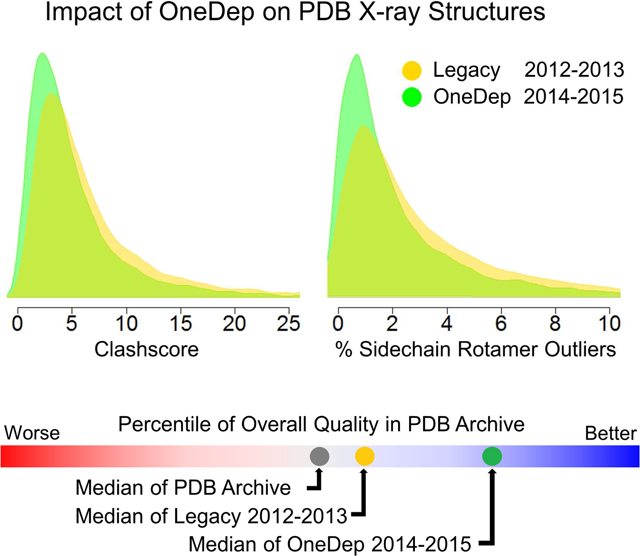
Since its initial deployment in January 2014, more than 21,000 X-ray crystallographic entries have been processed by the OneDep system. To assess the impact of the new validation report on the overall quality of structure data added to the PDB repository, we have compared entries deposited in 2014–2015 via the OneDep system with a collection of entries deposited in 2012–2013 via legacy systems. Both single and multivariate analyses have been used to compare quality measures for structure entries in the Legacy (2012–2013) and New (2014–2015) groups. We document that the overall quality of the deposited entries has improved since deployment of the OneDep system and introduction of the wwPDB validation report. Herein, we report the outcomes of our analyses and discuss how these results could guide future improvements in crystal structure quality assessment and reporting.
RESULTS AND DISCUSSION
Comparison of primary structure quality measures between X-ray entries deposited via the New OneDep system versus Legacy systems.
(A) Quality comparisons between New and Legacy entries
Our analyses initially focused on the five quality measures depicted in the wwPDB Validation Report slider graphic: Rfree, Clashscore, % Ramachandran Outliers, % Sidechain Rotamer Outliers, and % RSR Z-score Outliers. Figure 3A illustrates comparisons of primary quality measures, before and after OneDep deployment for 17538 X-ray structures deposited in 2012–2013 via legacy deposition systems (Legacy group) and 10387 structures deposited in 2014–2015 via OneDep system (New group). Each quality measure is displayed using both side-by-side box plots and overlaid probability density plots for Legacy (yellow) and New (green) groups. For each box plot, the median value is shown with black horizontal line and the Inter-Quartile Range (IQR; 25% to 75%) is represented by the height of the box. Table 1 provides quantitative details of each comparison.
Figure 3.

Comparison of structure quality measures for PDB X-ray structure data deposited via the new OneDep system (New) vs. the legacy systems (Legacy). (A) Upper: Boxplot comparisons of five structure quality measures for 2012–2013 Legacy group (yellow) vs. 2014–2015 New group (green); Lower: Probability density plot overlay between Legacy group (solid yellow) and New group (green line depicts the outline and solid green highlights the difference of distribution from that of Legacy group). (B) Comparisons between Legacy and New groups stratified by diffraction resolution limit. (C) Comparisons stratified by asymmetric unit molecular weight (MW).
Table 1:
Quantitative comparisons of median values and IQRs computed for structure.and ligand quality measures for Legacy vs. New. “Median Change” = “Median New” minus “Median Legacy”, and “Median Change by %” = “Median Change” divided by “Median Legacy”.
| Measures | Median Legacy |
Median New |
Median Change |
Median Change by % |
IQR Legacy |
IQR New |
|
|---|---|---|---|---|---|---|---|
| Overall | Rfree | 0.231 | 0.232 | 0.001 | 0.5% | 0.049 | 0.051 |
| Clashscore | 5.00 | 3.80 | −1.20 | −24.0% | 5.65 | 4.41 | |
| %Rama Outliers | 0.00 | 0.00 | 0.00 | 0.0% | 0.40 | 0.31 | |
| %Rotamer Outliers | 2.11 | 1.35 | −0.76 | −36.0% | 3.49 | 2.48 | |
| %RSRZ Outliers | 3.09 | 2.80 | −0.29 | −9.4% | 4.04 | 3.92 | |
| Ligand | RSR | 0.17 | 0.17 | 0 | 0% | 0.12 | 0.12 |
| RSCC | 0.92 | 0.92 | 0 | 0% | 0.11 | 0.11 | |
| Bond Length RMSZ | 0.6 | 0.59 | −0.01 | −1.7% | 0.76 | 0.76 | |
| Bond Angle RMSZ | 0.79 | 0.75 | −0.04 | −5.1% | 1.23 | 1.18 | |
| OWAB | 45.1 | 45.5 | 0.4 | 0.8% | 38.8 | 37.9 | |
| RSZD+ | 0.9 | 0.9 | 0 | 0% | 1.7 | 1.6 | |
| RSZD- | −0.6 | −0.5 | 0.1 | 16.7% | 1.4 | 1.3 | |
| Resolution Limit (A) | 2.05 | 2.1 | 0.05 | 2.4% | 0.74 | 0.83 | |
| MW (Da) | 56377 | 58288 | 1911 | 3.4% | 68042 | 73492 |
For the quality measures of the New and Legacy groups, median values of Rfree and % Ramachandran Outliers showed little improvement; median values of Clashscore, % Sidechain Rotamer Outliers, and % RSRZ Outliers all fell (improved); the IQRs for Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers all declined (improved); the IQRs for Rfree and % RSRZ Outliers did not change significantly. The overlaid probability density plots between Legacy and New groups show shift of density (difference of probability distribution highlighted in green) from higher values (poor) to lower values (better) for Clashscore and % Sidechain Rotamer Outliers, and the reduced peak width that corresponds to reduced IQR. Slight shifts of probability densities to better quality values are also seen for % Ramachandran Outliers and % RSRZ Outliers, but not for Rfree.
Recognizing that Ramachandran Outliers and Sidechain Rotamer Outliers are quality measures specific to proteins, we also compared Legacy vs. New depositions after excluding PDB entries containing nucleic acid polymers. Analysis results obtained absent nucleic acid polymers were very similar to those observed for all structures (data not shown). Further analyses described below were performed with all structures regardless whether or not nucleic acid was present.
We separately analyzed the data deposited via PDB legacy systems in year 2014, an overlap period during which both legacy and OneDep deposition systems were operated in parallel, to determine whether or not timely improvement or retrogress could be confounding our comparisons. Comparison of the primary quality measures between 5381 Legacy entries and 3395 New entries deposited in 2014 alone (data not shown) shows similar trends to those seen in Legacy 2012–2013 vs. New 2014–2015 comparison displayed in Figure 3A.
Some of our findings can be more fully appreciated with a historical perspective. The Rfree cross-validation method was introduced in 1992 (Brünger, 1992) and subsequently implemented in all major crystal structure refinement software packages. Following its introduction, Rfree rapidly became the most visible quality metric used during refinement of crystal structures. It is, therefore, not surprising that we saw no improvement in the Rfree quality metric when comparing New vs. Legacy (Figure 3A). Indeed, median values of Rfree computed annually for PDB X-ray depositions have not changed significantly over the past decade (data not shown). In our comparisons, both the median and the IQR of the Rfree metric changed little for New vs. Legacy.
G.N. Ramachandran first analyzed (Ф,Ψ) polypeptide backbone torsion angles for protein structures in the early 1960s (Ramachandran et al., 1963), revealing preferred regions within the (Ф,Ψ) plot corresponding to β–strands, right-handed α–helices, and left-handed α-helices. PROCHECK (Laskowski et al., 1992), arguably the first widely used computer programs for protein structure quality assessment, enabled automated detection of (Ф,Ψ) values falling outside preferred regions. Ramachandran Outliers have been assessed routinely during X-ray refinement of protein structures for many years, and again it comes as no surprise that there has been little improvement in terms of the median of % Ramachandran Outliers when comparing New vs. Legacy (Figure 3A). We do, however, see a reduction in IQRs for % Ramachandran Outliers for New vs. Legacy. Because more than 50% of entries have no Ramachandran outliers, any newly-determined structure with even one Ramachandran outlier (e.g., PDB: 2HYU) falls into the red unfavorable percentile section of that particular Validation Report slider (Figure 1), which should perforce encourage PDB Depositors using the OneDep system to either verify that the experimental data supports the apparent outlier, or to correct errors.
Clashscore and Sidechain Rotamer Outlier quality metrics are calculated using Molprobity (Chen et al., 2010). With introduction of the new wwPDB Validation Report in 2014, both of these quality measures were prominently presented to PDB Depositors. Clashscore and % Rotamer Outliers both showed improvement for New vs. Legacy, as judged by reduced median values (Figure 3A). Reductions in IQRs for Clashscore and % Rotamer Outliers also show improvements for New vs. Legacy. The visibility of both Clashscore and Rotamer Outliers in the wwPDB Validation Report (Figure 1) appears to have sensitized PDB Depositors to these quality metrics.
Software tools used to calculate Real Space R-factor or RSR (Jones et al., 1991) and the Real Space R-factor Z-score or RSRZ (Kleywegt et al., 2004) have been widely used for more than a decade. Unlike Rfree, a measure of overall agreement between atomic coordinates and experimental data (observed structure factors), scaled RSRZ Outliers (% RSRZ Outliers) assesses local agreement between atomic coordinates and experimental electron density. In the new wwPDB Validation Report, RSRZ Outliers are highlighted in both graphical and tabular forms. % RSRZ Outliers showed a modest improvement for New vs. Legacy, as judged by reduction in median value (Figure 3A). Again, the wwPDB Validation Report slider for RSRZ Outliers (Figure 1) appears to be sensitizing PDB Depositors to the quality metric.
(B) Impact of diffraction data resolution limit and sample molecular weight
In general, 3D structure quality and diffraction data resolution are related, because the number of experimental observations available for structure refinement changes with resolution limit. Table 1 shows that average diffraction data resolution did not change significantly for 2012–2013 vs. 2014–2015. In fact, the median resolution limit of the Legacy group (2.05Å) is slightly better than that of the New group (2.10Å), and the resolution limit IQR of the Legacy group is slightly narrower than that of the New group.
To further explore the impact of resolution limit, we repeated our New vs. Legacy quality comparisons as a function of diffraction data resolution limit by dividing both New and Legacy groups into the following bins: High Resolution (~25% of the population, <1.76Å); Medium Resolution (~50%, 1.76–2.50Å); and Low Resolution (~25%, >2.50Å). For each bin, we performed the same analyses as for all data. Figure 3B illustrates box plots for Rfree, Clashscore, % Ramachandran Outliers, % Sidechain Rotamer Outliers, and % RSRZ Outliers, stratified by diffraction data resolution limit. Comparison of median values of structure quality measures between the Legacy and New groups for each resolution range is shown in Table 2.
Table 2:
Quantitative comparisons of median values computed for structure and ligand quality measures for Legacy vs. New grouped by diffraction data resolution limit.
| High (<1.76 Å) | Medium (1.76–2.50 Å) | Low (>2.50 Å) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Measures | Median Change |
Median Change |
Median Change |
|||||||
| Overall | Rfree | −0.003 | 0 | 0.001 | ||||||
| Clashscore | −1.135 | −1.18 | −2.2 | |||||||
| %Rama Outliers | 0 | 0 | −0.14 | |||||||
| %Rotamer Outliers | −0.29 | −0.91 | −2.24 | |||||||
| %RSRZ Outliers | −0.3 | −0.42 | 0.05 | |||||||
| Ligand | RSR | −0.005 | −0.004 | −0.001 | ||||||
| RSCC | 0 | 0.001 | 0.002 | |||||||
| Bond Length RMSZ | −0.03 | −0.02 | 0.04 | |||||||
| Bond Angle RMSZ | −0.05 | −0.07 | −0.02 | |||||||
| OWAB | 0.285 | 0.09 | −3.475 | |||||||
| RSZD+ | 0.1 | 0 | −0.1 | |||||||
| RSZD- | 0.1 | 0 | 0 | |||||||
Median values for Rfree increase as resolution limit goes from High to Medium to Low, as expected (Dodson et al., 1996), with little variation in IQRs. Median values for Clashscore, % Ramachandran Outliers, and % Rotamer Outliers and their respective IQRs increase (worsen) as resolution limit goes from High to Medium to Low. For % RSRZ Outliers, neither median value nor IQR changed significantly as a function of resolution.
Comparing quality measures between New and Legacy group, molecular geometry quality improved for every resolution bin of the New group, with decreased median values and/or IQRs of Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers. In terms of Rfree, the median value decreased slightly (improved) at High resolution, but was unchanged at Medium and Low resolution for New vs. Legacy, whereas IQR increased (worsened) for New vs. Legacy only at Low resolution. Finally, both median and IQRs for the local fitting measure % RSRZ Outliers decreased (improved) slightly at High and Medium resolution entries. The opposite is true at Low resolution.
To summarize, at High resolution the New group showed improvements in all five quality metrics analyzed vs. Legacy. At Medium resolution, four out of five quality metrics improved for New vs. Legacy, except for Rfree (unchanged). At Low resolution, only the three molecular geometry quality measures (Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers) showed improvement for New vs. Legacy.
Sample molecular weight (MW), or more precisely the MW of the crystallographic asymmetric unit (ASU), represents another variable that could confound assessment of quality metrics. We repeated our New vs. Legacy quality comparisons as a function of ASU MW by dividing the New and Legacy groups into three subsets: High MW (~25%, >104kDa); Medium MW (~50%, 34–104kDa); and Low MW (25%, <34kDa). For each subset, we performed the same analysis as that carried out for all data. Figure 3C illustrates box plots for Rfree, Clashscore, % Ramachandran Outliers, % Sidechain Rotamer Outliers, and % RSRZ Outliers, stratified by ASU MW. Examination of the columns in Figure 3C, reveals that for each quality measure, the New vs. Legacy comparison of either median or IQR has no significant dependence on ASU MW. We conclude, within our benchmark data sets, that the molecular weight of the asymmetric unit is not influencing structure quality improvement as assessed by the five primary quality measures.
Comparison of bound ligand model quality measures between entries deposited via the New OneDep system versus Legacy systems
(A) Comparison between New and Legacy systems
Ligand quality measures were compared between New vs. Legacy groups as described earlier. The results of this comparison are depicted in Figure 4A. No significant improvements in ligand quality metrics are discernible, except for a slight decreased in the median value of Bond Angle RMSZ, which may reflect the use of more appropriate geometric restraints during structure refinement. Table 2 also provides quantitative details of the comparison. Figure 4B summarizes the results of the same analyses performed with the benchmark data subdivided by resolution limit as for Figure 3B. Again, there are no significant differences in ligand quality metrics when analyzed as a function of diffraction data resolution limit. There is slight improvement in median values for Bond Length RMSZ and Bond Angle RMSZ at High and Medium resolution, and OWAB improved slightly at Low resolution for New vs. Legacy. Beyond these very modest improvements in quality metrics, there is little discernable change in the ligand quality measures between New vs. Legacy PDB depositions.
Figure 4.

Comparison of ligand quality measures for PDB X-ray structure data deposited via the new OneDep system (New) vs. the legacy systems (Legacy). (A) Boxplot comparisons of between Legacy and New groups. (B) Comparisons between Legacy and New groups stratified by diffraction resolution limit.
(B) RSZD ligand quality measure
Given the lack of improvement in ligand data quality in the New vs. Legacy groups, we conducted further analysis of the ligand density fitting using the real-space difference density Z-score (RSZD) described by Tickle (Tickle, 2012). Because RSZD values are calculated using the results of an m|Fo|-D|Fc| difference Fourier synthesis (|Fo| and |Fc| are observed and calculated amplitudes, respectively), they are sensitive to local discrepancies between ligand atomic coordinates (reflected in |Fc|) and the corresponding experimental electron density feature (reflected in |Fo|). When computed for the NADP ligands in 2FZD (maximum value of RSZD-Plus=+1.1; minimum value of RSZD-Minus=−3.5) and 1ZK4 (maximum value of RSZD-Plus=+22.5; minimum value of RSZD-Minus=−0.0), the differences in quality of the two instances of NADP are readily apparent from the RSZD metrics.
Using Tickle’s approach, we retrospectively computed RSZD values for all ligand instances in the PDB archive with deposited structure factors, keeping RSZD-Plus (positive features, green in Figure 2C and 2D) and RSZD-Minus (negative features, red in Figure 2C and 2D) separate throughout the calculation. For New vs. Legacy, the median value for RSZD-Plus is unchanged, with a modest decrease in IQR (Figure 4A). For New vs. Legacy group, the absolute value of the median for RSZD-Minus slightly decreased, as did IQR (Figure 4A). When analyzed as a function of diffraction data resolution, differences between New vs. Legacy in both median values and IQRs of RSZD-Plus and RSZD-Minus are observed in some cases, but they do not appear significant (Figure 4B and Table 2), confirming the lack of any significant improvement in the ligand quality measures, as described above.
Multivariate analyses of primary quality measures
(A) Principal Component Analyses
We applied Principal Component Analysis (PCA) to the five quality measures depicted in Figure 1 to explore possible interrelationships and determine whether or not assessments of overall quality can be reduced in dimensionality (i.e., <5). An initial PCA analysis was performed for all 86644 PDB archival X-ray entries with supporting structure factor data. Figure 5A shows the proportion of variance explained by each principal component, documenting that the three leading components collectively explain ~85% of total variance. The fractional contribution (loading) of each of the original quality measures is also tabulated in Figure 5A. The first Principal Component (PC1; accounting for ~50% of variance) is dominated in approximately equal proportion by the three molecular geometry quality measures (Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers). The second Principal Component (PC2; accounting for an additional ~21% of variance) is dominated by % RSRZ Outliers. The third Principal Component (PC3; accounting for an additional ~14% of variance) is dominated by Rfree. The orthogonality between PC2 and PC3 is consistent with the relatively low pairwise correlation coefficient (0.210) between Rfree and % RSRZ Outliers, since Rfree is a global measure that cannot effectively detect local errors (Dodson et al., 1996).
Figure 5.
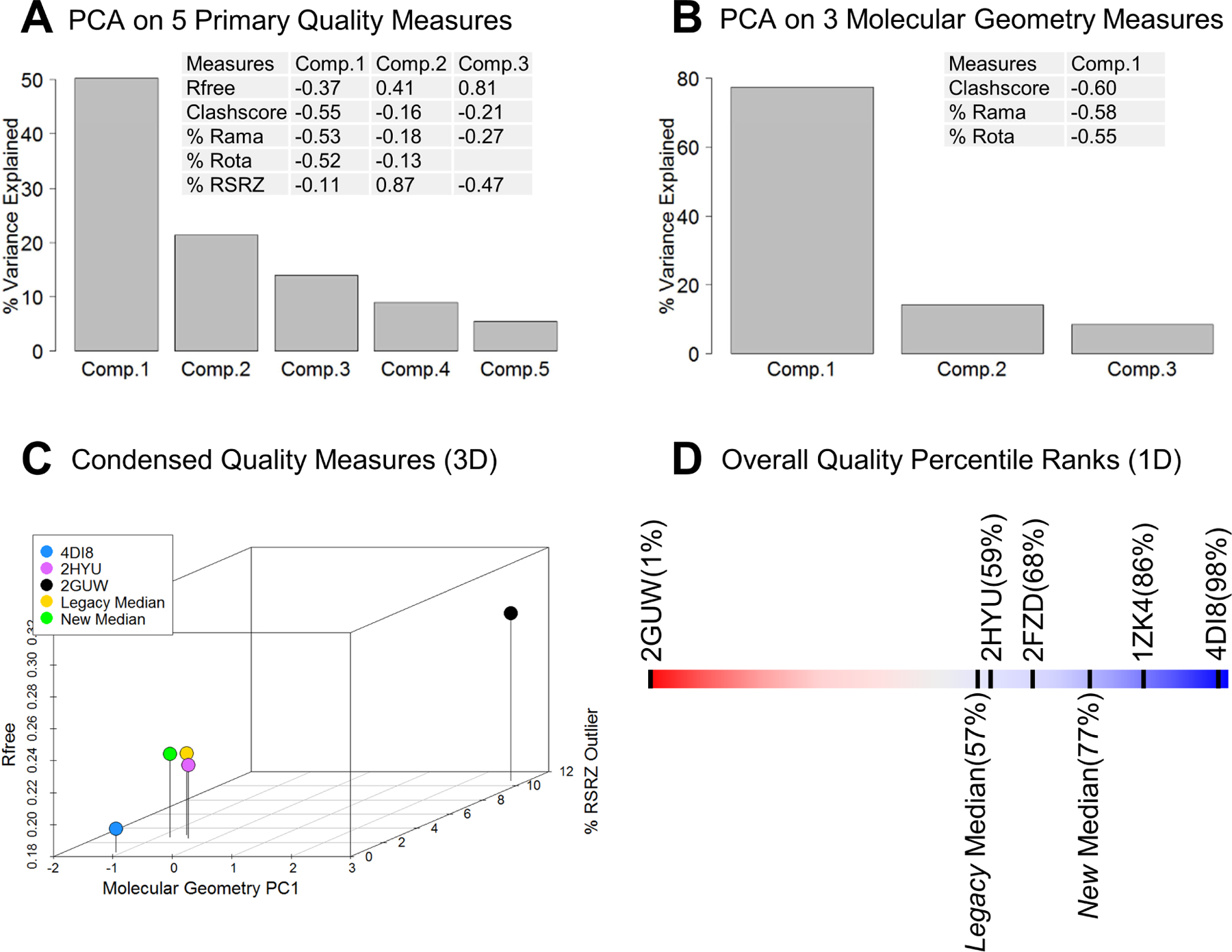
Principal Component Analyses (PCA) and reduced dimensionality quality measures. (A) PCA on five primary structure quality measures (Rfree, Clashscore, % Ramachandran Outliers, % Sidechain Rotamer Outliers, and % RSRZ Outliers). The height of gray bar represents the percentage of overall variance explained by each principal component, and embedded table lists the composition of top three principal components. (B) PCA on three molecular geometry quality measures (Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers). (C) 3D scatterplot of dimension-reduced quality measures, with X-axis for the molecular geometry 1st principal component, Y-axis for % RSRZ Outliers, and Z-axis for Rfree. Three individual PDB structures and median values for Legacy and New groups were represented by solid circles of different colors. (D) Graphical slider depicting an 1D overall structure quality ranking metric for individual structures, Legacy Median, and New Median.
Because Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers together dominate PC1, these three quality metrics can be usefully regarded as a collective molecular geometry quality metric with its combined measure being nearly orthogonal to PC2 (dominated by % RSRZ Outliers) and PC3 (dominated by Rfree), which can be easily understood as quality metrics assessing atomic structure geometry vs. local electron density map fitting vs. global fit to the diffraction data.
A second PCA analysis was performed using only Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers (Figure 5B). The first Principal Component (PC1) is made up of approximately equal contributions from each of the three standardized molecular geometry quality measures, accounting for ~77% of the overall variance. We, therefore, use this PC1 from 3-variable PCA as a composite molecular geometry metric to not only simplify the representation of three correlated individual measures, but also to overcome the difficulty of ranking individual variables with many zero values (e.g., % Ramachandran Outliers).
To assess whether or not our PCA results are strongly influenced by the timing of PDB depositions, diffraction data resolution, or ASU MW, PCA analyses on the three molecular geometry quality measures were run multiple times for different subsets of the PDB archive. The composition of the first Principal Component (PC1) is largely independent of deposition date, diffraction data resolution, or ASU MW (data not shown).
Having established that PCA outcomes are robust with respect to deposition date, diffraction data resolution, and asymmetric unit molecular weight, structures can be compared using just three approximately orthogonal quality metrics, Rfree, % RSRZ Outliers, and molecular geometry PC1 (combining Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers). Figure 5C illustrates a three-dimensional scatterplot that we can use to assess median quality of New vs. Legacy in our benchmark dataset together with the three PDB entries exemplified in Figure 1. PC1 running along the X-axis was constructed such that smaller values indicate entries with lower Clashscore, lower % Ramachandran Outliers, and lower % Sidechain Rotamer Outliers. Thus, entries falling closer to the origin in Figure 5C have better molecular geometry, Rfree, and % RSRZ Outliers quality measures. Comparison of New vs. Legacy in Figure 5C reveals marked improvement in the molecular geometry quality measure PC1, some improvement in the % RSRZ Outliers quality measure, and no significant change in Rfree.
(B) One-dimensional combined overall quality measure
Among the five primary quality measures, the three molecular geometry measures of Clashscore, % Ramachandran Outliers, and % Sidechain Rotamer Outliers are correlated with one another (pairwise correlation coefficients > 0.5), and were therefore best combined into the molecular geometry PC1 (Figure 5C). The other two approximately orthogonal measures of Rfree and % RSRZ Outliers are neither strongly correlated with any of the three molecular geometry measures nor with their combined measure of molecular geometry PC1 (correlation coefficients of 0.386 between PC1 and Rfree, and 0.063 between PC1 and % RSRZ Outliers). Further dimensionality reduction to a one-dimensional (1D) quality measure can be accomplished by averaging the scaled contributions from Rfree, % RSRZ Outliers, and the molecular geometry PC1. We used the robust ranking statistic as the scaled contribution for each quality measure. For every X-ray entry the ranking percentile of Rfree (PRfree), % RSRZ
Outliers (P%RSRZ), and the molecular geometry PC1 (PGeometry_PC1) are calculated against the entire PDB X-ray archive, with the lowest quality at 0% and the best quality at 100%, and their arithmetic mean Q1 (1D quality measure) calculated according to Equation 1.
| Equation 1 |
After the average percentile is calculated, each X-ray entry is then ranked within the population to obtain its final ranking percentile PQ1, with lowest Q1 at 0% and highest at 100%. Figure 5D exemplifies the utility of the 1D overall structure quality metric by plotting PQ1 with the familiar red/blue slider graphic (running from 0%-worst to 100%-best) for PDB: 2GUW, 2HYU, 4DI8, 1ZK4, 2FZD (Figures 1 and 2). PDB entry 2GUW has a very low overall quality percentile PQ1 (only better than 1% of the archive), while PDB entry 4DI8 has very high overall quality percentile (better than 98% of the archive). As expected, PDB entry 2HYU has an intermediate overall quality percentile of ~59%. Thus, our one-dimensional measure PQ1 can provide a simple measure with which to assess the overall quality of a given PDB X-ray entry relative to the entire archive. Using this approach, PQ1 of the median quality structure of the Legacy group is ~57%, whereas PQ1 of the median quality structure of the New group is ~77%. This ~20% improvement in the median overall quality percentile for New vs. Legacy entries provides a compelling view of structure quality improvement since introduction of the wwPDB Validation Report with the OneDep system.
Care must be taken, however, to avoid over interpretation of the 1D overall quality metric. Each of the original primary quality indicators uniquely measures an aspect of the model quality. The further the reduction of the data dimension, the more information is lost. The 1D quality measure provides a straightforward single comparison metric with the minimal loss of information and simple interpretation, and may be of value for non-structural biologists in recognizing structure quality without deeper understanding the precise meaning of each individual measure. This measure is also not sensitive to ligand quality in co-crystal structures. The overall quality metrics for 1ZK4 and 2FZD both lie within the upper 1/3 of all archived X-ray structures (~86% and ~68%, respectively), but we know that the NADP ligands common to the two co-crystal structures are markedly different in quality as judged by both Tickle’s RSZD metric and comparisons of ligand atomic coordinates to experimental electron density maps (Figure 2).
Conclusions
To assess the impact of the structure validation pipeline implemented in the new OneDep system, the five primary quality measures have been compared between a group of entries deposited during 2014–2015 using the new OneDep system and another group of entries deposited during 2012–2013 using older deposition tools. Improvements in structure quality are reflected in four of these five quality metrics (Clashscore, % Ramachandran Outliers, % Rotamer Outliers, and % RSRZ Outliers) in terms of reduced (improved) median values and/or reduced (improved) IQRs. These improvements were observed across data resolution limits and asymmetric unit molecular weights. They can be attributed, at least in part, to the visibility of the five graphic sliders in the new wwPDB Validation Report produced by the OneDep system. Explicit review of the validation report is required for assignment of the PDB accession code at the time of data deposition, thereby ensuing Depositors awareness of quality issues. None of these quality improvements could have been possible without the method and software development in structural biology and the ongoing advice from wwPDB X-ray VTF, which also contribute functionality to the wwPDB validation pipeline.
Quality improvements were restricted to the macromolecular components of PDB entries. No significant improvements were discernible in the quality of ligands present in PDB entries deposited in 2014–2015 through the new OneDep system vs. 2012–2013 through legacy systems. Efforts are currently underway to improve presentation of ligand quality metrics in the wwPDB Validation Report. The wwPDB partners, together with the Cambridge Crystallographic Data Center (CCDC; http://www.ccdc.cam.ac.uk) and the Drug Design Data Repository (D3R; https://drugdesigndata.org/), convened a joint wwPDB/CCDC/D3R Ligand Validation Workshop at Rutgers University in July 2015 to address issues of ligand quality in the PDB archive. Workshop recommendations aimed at improving validation of ligands at the time of structure deposition have been published in this journal (Adams et al., 2016).
Finally, our PCA results provide a statistical framework that can be used to understand how various structure quality measures are correlated with one another, and whether or not these measures can be reduced in dimensionality to simplify assessments of PDB structure quality. With PCA, we detected associations among the three quality measures pertaining to molecular geometry, and using their first principal component we were able to reduce primary quality assessment to three dimensions for easier graphical display. From that vantage point, we could assess structure quality with an even simpler overall quality measure that may be more accessible to the majority of PDB Users who are not structural biologists.
PROCEDURES
Benchmark Datasets
To assess the impact of the new wwPDB Validation Report, we assembled structures deposited through the new OneDep system, and structures deposited through the legacy RCSB PDB, PDBj, and PDBe deposition systems. To enable meaningful comparisons, we assembled Benchmark Datasets for 2-year intervals before and after deployment of OneDep in January 2014, including 17538 Legacy X-ray entries deposited via legacy systems in 2012–2013 and 10387 New entries deposited via OneDep in 2014–2105. All of these entries were publicly accessible at the time of writing (August 2016). Structures not included in the New group were those on hold for release and structures submitted via Legacy deposition systems that ran parallel to the new OneDep system in 2014–2015.
In addition to the statistics described in Table 1 and 2, the mean resolution limit is 2.16 Å for the Legacy group, and 2.19 Å for the New group, both skewed to the right with long tail at lower resolution; the mean molecular weight in ASU is 105kDa for the Legacy group, and 139kDa for the New group, both skewed to the right with long tail at higher molecular weight. In terms of diffraction sources, for both Legacy and New groups, ~90% of structures were determined from data collected at synchrotron facilities and ~10% at home sources. Free Electron Laser sources were used for nine structures in the Legacy group and 51 structures in the New group.
Within the Benchmark Datasets, there are 7966 unique small-molecule components and 147708 instances with occupancy >=10% (multiple instances of a ligand can occur in a given PDB entry). Because we analyze bond lengths and bond angles, ligands containing only one non-hydrogen atom (e.g., metal ions and water molecules) were excluded, as were any ligands marked as Unknown.
Comparisons of Data Quality Metrics
Conventional box plots were displayed for five structure quality measures, including Rfree, Clashscore, % Ramachandran Outliers, % Sidechain Rotamer Outliers, and % RSRZ Outliers. For each box plot, the dark horizontal line within the box represents the median value. The bottom and top of each box represent the first (25%) and third (75%) quartiles, thus the height of a box represents the Inter-Quartile Range (IQR). The vertical dashed line extended from each box’ bottom and top represent the data range below 1st quartile and above 3rd quartile. Outliers were excluded for the sake of clarity.
The probability density distribution was calculated using kernel density estimate. The density plot for both Legacy and New groups were drawn in the same scale and overlaid, thus the difference represents the shift of probability density distribution.
Benchmark Datasets were stratified by diffraction data resolution into three bins: High Resolution (~25% of the population, <1.76Å), Medium Resolution (~50%, 1.76–2.50Å), and Low Resolution (25%, >2.50Å) for comparisons of Rfree, Clashscore, Ramachandran Outliers, Rotamer Outliers, and RSRZ Outliers. Benchmark Datasets were also stratified by asymmetric unit molecular weight (MW) into the three subsets: High MW (~25%, >104kDa), Medium MW (~50%, 34–104kDa), and Low MW (25%, <34kDa) for comparisons of structure quality measures.
To assess ligand model quality, conventional box plots were displayed for five ligand quality measures provided in the wwPDB Validation Report, including RSR or real space R factor, RSCC or real space correlation coefficient, Bond lengths RMSZ and Bond angles RMSZ computed with Mogul, and OWAB or occupancy weighted average B factor. Additional box plots were displayed for RSZD-Plus and RSZD-Minus. Again Benchmark Datasets were stratified by diffraction data resolution, as for structure quality metrics.
Principal Component Analysis (PCA)
PCA was carried out on the structure quality metrics by computing a correlation matrix from which ranked eigenvalues and eigenvectors were extracted. Each variable was scaled during the analysis. To ensure no loss of information in calculating the correlation, we examined the entire public X-ray archive that includes 106237 entries deposited between 1972 and 2015 and publicly released at the time of writing. After removing entries deposited earlier without associated structure factor for which Rfree and % RZRZ Outliers cannot be calculated, we performed PCA on a total of 86644 X-ray entries. To study the potential impact of various factors, the entire data was also separated into groups by resolution limit, deposition date, or molecular weight in ASU, with PCA performed independently on each group for comparison. The weight or loading parameters calculated from PCA of the entire archive were subsequently applied to the Benchmark Datasets for comparison between the Legacy and the New groups and for making Figures 5C and 5D.
Computation
Data were extracted from both PDBx/mmCIF files and wwPDB Validation Reports for each entry in the public PDB archive and loaded into a MySQL Database (https://www.mysql.com/). Subsequent search, tabulation, and statistical calculation were performed primarily with Python (https://www.python.org/) and R (https://www.R-project.org/) programs.
Highlights.
Improved structure quality from the wwPDB OneDep system versus legacy PDB systems
Little change in ligand quality from the OneDep system versus legacy systems
Principal component analyses allow streamlining of OneDep quality metrics
ACKNOWLEDGEMENTS
This work was funded by the National Science Foundation, the National Institutes of Health, and the US Department of Energy (NSF DBI-1338415). We thank the entire wwPDB partnership for collaborative development of the OneDep system, and Dr. Shubing Wang for discussions on conducting statistical analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Adams PD, Aertgeerts K, Bauer C, Bell JA, Berman HM, Bhat TN, Blaney JM, Bolton E, Bricogne G, Brown D, et al. (2016). Outcome of the First wwPDB/CCDC/D3R Ligand Validation Workshop. Structure 24, 502–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Henrick K, and Nakamura H (2003). Announcing the worldwide Protein Data Bank. Nat. Struct. Biol 10, 980. [DOI] [PubMed] [Google Scholar]
- Brändén C, and Jones T (1990). Between objectivity and subjectivity. Nature 343, 687–689. [Google Scholar]
- Brünger AT (1992). Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, et al. (1998). Crystallographic and NMR system: a new software suite for macromolecular structure determination. Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WD, Purkis LH, Smith BR, Taylor R, Cooper RI, Harris SE, et al. (2004). Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci 44, 2133–2144. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodson E, Kleywegt GJ, and Wilson K (1996). Report of a workshop on the use of statistical validators in protein X-ray crystallography. Acta Cryst. D52, 228–234. [DOI] [PubMed] [Google Scholar]
- Editorial. (2016). Where are the data? Nat. Struct. Mol. Biol 23, 871–871. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gore S, Velankar S, and Kleywegt GJ (2012). Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Cryst. D68, 478–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom CR, Bruno IJ, Lightfoot MP, and Ward SC (2016). The Cambridge Structural Database. Acta Cryst. B72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N, et al. (2012). Outcome of the first electron microscopy validation task force meeting. Structure 20, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs ME, Malashkevich V, Williams HJ, Xu C, Sauder JM, Burley SK, Almo SC, and Raushel FM (2012). Structure and catalytic mechanism of LigI: insight into the amidohydrolase enzymes of cog3618 and lignin degradation. Biochemistry 51, 3497–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, and Kjeldgaard M (1991). Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst.A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Joosten RP, Long F, Murshudov GN, and Perrakis A (2014). The PDB_REDO server for macromolecular structure model optimization. IUCrJ 1, 213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleywegt GJ, Harris MR, Zou JY, Taylor TC, Wahlby A, and Jones TA (2004). The Uppsala Electron-Density Server . Acta Cryst D60, 2240–2249. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, and Thornton JM (1993). PROCHECK - a programs to check the stereochemical quality of protein structures. J. Appl. Cryst 26, 283–291. [Google Scholar]
- Montelione GT, Nilges M, Bax A, Guntert P, Herrmann T, Richardson JS, Schwieters CD, Vranken WF, Vuister GW, Wishart DS, et al. (2013). Recommendations of the wwPDB NMR Validation Task Force. Structure 21, 1563–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Ramachandran GN, Ramakrishnan C, and Sasisekharan V (1963). Stereochemistry of polypeptide chain configurations. J. Mol. Biol 7, 95–99. [DOI] [PubMed] [Google Scholar]
- Read RJ, Adams PD, Arendall WB 3rd, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lutteke T, Otwinowski Z, et al. (2011). A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlieben NH, Niefind K, Muller J, Riebel B, Hummel W, and Schomburg D (2005). Atomic resolution structures of R-specific alcohol dehydrogenase from Lactobacillus brevis provide the structural bases of its substrate and cosubstrate specificity. J. Mol. Biol 349, 801–813. [DOI] [PubMed] [Google Scholar]
- Shao C, Zhang F, Kemp MM, Linhardt RJ, Waisman DM, Head JF, and Seaton BA (2006). Crystallographic analysis of calcium-dependent heparin binding to annexin A2. J. Biol. Chem 281, 31689–31695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM, (2008) A short history of SHELX. Acta Cryst. A64, 112–122. [DOI] [PubMed] [Google Scholar]
- Smart OS, Brandl M, Flensburg C, Keller P, Paciorek W, Vonrhein C, Womack TO & Bricogne G (2008). Refinement with Local Structure Similarity Restraints (LSSR) Enables Exploitation of Information from Related Structures and Facilitates use of NCS. Abstr. Annu. Meet. Am. Crystallogr. Assoc. Abstract TP139, 117. [Google Scholar]
- Steuber H, Zentgraf M, Gerlach C, Sotriffer CA, Heine A, and Klebe G (2006). Expect the unexpected or caveat for drug designers: multiple structure determinations using aldose reductase crystals treated under varying soaking and co-crystallisation conditions. J. Mol. Biol 363, 174–187. [DOI] [PubMed] [Google Scholar]
- Tickle IJ (2012). Statistical quality indicators for electron-density maps. Acta Cryst. D68, 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichenberger CX, Pozharski E, and Rupp B (2013). Visualizing ligand molecules in Twilight electron density. Acta Cryst. F69, 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Cryst. D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Peisach E, Westbrook JD, Young J, Berman HM, and Burley SK (2016). DCC: a Swiss army knife for structure factor analysis and validation. J. Appl. Cryst 49, 1081–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JJ, Westbrook JD, Feng Z, Sala R, Peisach E, Oldfield TJ, Sen S, Gutmanas A, Armstrong DR, Berrisford JM, et al. (2017). OneDep: Unified wwPDB System for Deposition, Biocuration, and Validation of Macromolecular Structures in the Protein Data Bank (PDB) Archive . Structure, in press, DOI: 10.1016/j.str.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]