

Summary
The etiology of aortic aneurysms is poorly understood, but it is associated with atherosclerosis, hypercholesterolemia, and abnormal TGFβ signaling in smooth muscle. Here we investigated the interactions between these different factors in aortic aneurysm development and identified a key role for smooth muscle cell (SMC) reprogramming into a mesenchymal stem cell (MSC)-like state. SMC-specific ablation of TGFβ signaling in Apoe−/− mice on a hypercholesterolemic diet led to development of aortic aneurysms exhibiting all the features of human disease, which was associated with transdifferentiation of a subset of contractile SMCs into an MSC-like intermediate state that generated osteoblasts, chondrocytes, adipocytes, and macrophages. This combination of medial SMC loss with marked increases in non-SMC aortic cell mass induced exuberant growth and dilation of the aorta, calcification and ossification of the aortic wall, and inflammation, resulting in aneurysm development.
Graphical Abstract
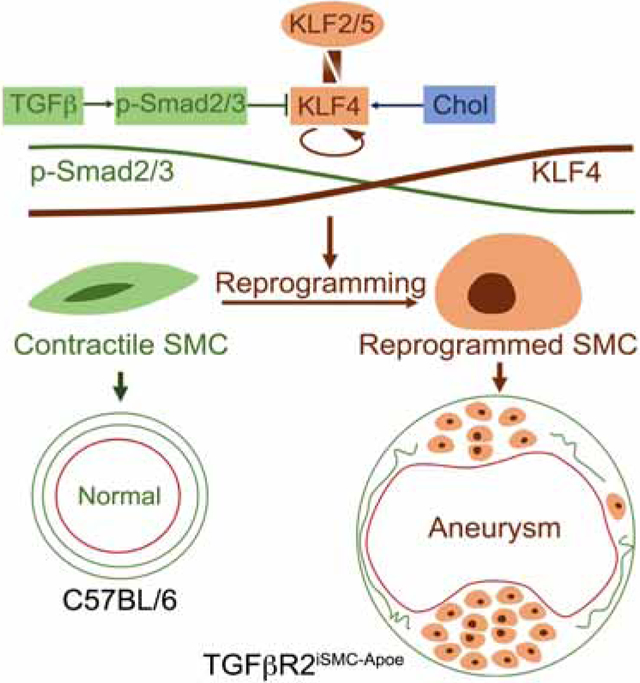
eTOC
Abrogation of TGFβ signaling in smooth muscle cells (SMCs) in combination with hypercholesterolemia results in formation of aortic aneurysms due to reprograming of a subset of normal SMC into mesenchymal-like stem cells that give rise to adipocytes, chondrocytes and osteoblasts, as well as into macrophage-like cells.
Introduction
Aortic aneurysms present a challenging set of problems. Despite their frequency and lethality, their molecular pathogenesis is poorly understood, and the only available therapies consist of surgical or endovascular interventions for advanced disease (Hiratzka et al., 2010). Aneurysms are observed both in young patients (e.g., Marfan and Loeys-Dietz syndromes) and in association with atherosclerosis and hypertension in older patients, emphasizing both genetic and environmental pathogenic factors. While there are differences in the presentation and pathoanatomy between aneurysms occurring in the ascending aorta and arch versus abdominal aortic aneurysms, common features involve the loss of elastin fibers, medial degeneration, and low grade aortic wall inflammation (Guo et al., 2006; Isselbacher, 2005). Whilst aneurysms associated with Marfan’s and Loeys-Dietz syndromes have been ascribed to activated SMC TGFβ signaling (Lindsay and Dietz, 2014), virtually nothing is known about aneurysm development in older patients with atherosclerosis, underscoring a need for a better understanding of basic aneurysm biology (Mallat et al., 2017). The present study was designed to define the molecular pathway leading to aneurysm formation in older atherosclerotic patients.
Using a combination of techniques, we show that a loss of smooth muscle TGFβ signaling input, when combined with hyperlipidemia, results in a transdifferentiation of small population of media SMCs to a mesenchymal stem cell (MSC)- like state and subsequent clonal differentiation into mesenchymal lineage cell types. The appearance and expansion of this cell population leads to the loss of elastin fibers, intramural calcifications combined with cartilage and bone formation, massive lipid uptake and extensive inflammation. These features closely mimic the structure of human aortic aneurysms. The key driver of this process appears to be a large (~100-fold) increase in KLF4 expression in combination with other KLF family members. A SMC-specific KLF4 knockout largely prevents aneurysm development in this model.
Results
To explore the pathogenesis of aortic aneurysms in patients with atherosclerosis, we examined SMC TGFβ signaling and expression of relevant genes in the media of normal human aortas and compared it to non-Marfan atherosclerotic ascending aorta aneurysms (Fig 1A and Table S1A Human subject characteristic) Histological analysis revealed expected changes, including elastin fibers breakdown, lipid accumulation and mineralization (Fig 1B). Bulk RNA-seq analysis was used to compare six ascending aorta media samples obtained from normal organ donors with an equal number of samples from patients with ascending aorta aneurysms, same patient cohort used for histologic evaluation (Table S1A Human subject characteristic). Compared to normal aorta media, aneurysm samples showed a clear reduction in TGFβ signaling and increased expression of inflammation-related genes (Fig 1C). Immunocytochemistry confirmed these findings, demonstrating a marked reduction in SMC p-Smad2 and TIMP3 levels, and increased expression of VCAM1 and MMP2 (Fig 1D and Mendeley Data Figure 1, http://dx.doi.org/10.17632/4mfgjpvvjm.1).
Figure 1: Inhibition of smooth muscle cell TGFβ signaling in hypercholesterolemic patients with aortic aneurysms.
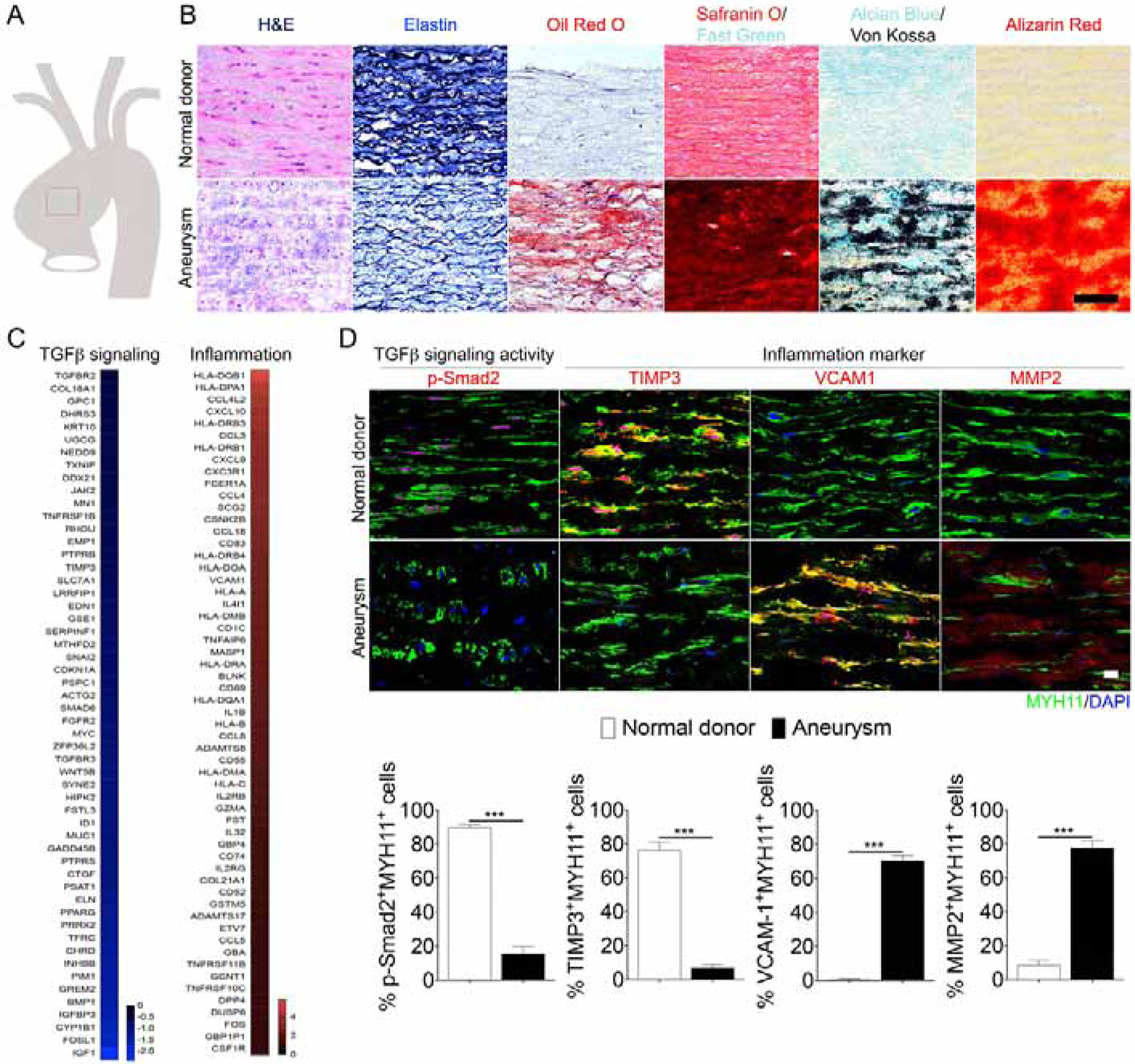
(A) Schematic drawings of defined human ascending aorta anatomical regions used for analysis. (B) Representative images of H&E, Elastin, Oil-Red-O, Safranin O, Alcian blue/Von Kossa, Alizarin Red-stained ascending aortas from normal donor and aortic aneurysm patients (Normal donor, N= 6; aneurysm patients, N=6). Scale bar: 50 μm. (C) Bulk RNA-seq analysis of ascending aorta media samples from normal organ donors and patients with ascending aorta aneurysms. (Normal donor, N= 6; aneurysm patients, N=6). Note strong inhibition of TGFβ-related genes and strong induction of inflammation-related genes. (D) (Upper panels) Histological analysis of human ascending aortas with TGFβ signaling (p-Smad2) and inflammation markers (TIMP3, VCAM1, MMP2) from normal donors (N=6) and aneurysm patients (N=6). Nuclei were stained with DAPI (blue). Scale bar: 16 μm. (Lower panels) Quantification of the number of ascending aortic media smooth muscle cells expressing MYH11 (green) and p-Smad2 (red) and inflammation markers (red) (***p<0.001; unpaired two-tailed Student’s t test). See also Figure S7 and Table S1A.
These data strongly implicate inhibition of SMC TGFβ signaling in atherosclerotic aortic aneurysm development. To test this hypothesis, we engineered transgenic mice on an Apoe−/− background carrying floxed Tgfbr2 and SMC lineage tracing mT/mG alleles under control of a myosin heavy chain (Myh11) promoter (Apoe−/−;Myh11CreERT2;mT/mGf/f;Tgfbr2f/f) hereafter called TGFβR2iSMC-Apoe (Fig S1A). Three different fate-mapped groups of mice were used as controls: Apoe−/− (Apoe−/−;Myh11CreERT2;mT/mGf/f), TGFβR2iSMC (Myh11CreERT2;mT/mGf/f;Tgfbr2f/f) and C57BL/6 background controls (Myh11CreERT2;mT/mGf/f). Tamoxifen administration at 6 weeks of age led to a complete deletion of Tgfbr2 in SMC (Fig S1B,C). The loss of Tgfbr2 significantly reduced expression of p-Smad2 in aortic SMC in vivo (Fig S1D) and prevented TGFβ1-dependent induction of Smad2/3 phosphorylation in aortic SMC in vitro (Fig S1E).
Following tamoxifen treatment, TGFβR2iSMC-Apoe and control mice were fed a high cholesterol high fat diet (HCHFD) for 4 months leading to expected changes in plasma total cholesterol and triglycerides levels (Fig S1F). When sacrificed after 4 months on HCHFD, all TGFβR2iSMC-Apoe mice exhibited severe aneurysmal enlargement and marked elongation of the entire aorta (Fig 2A–C). There was extensive neutral lipid accumulation, as shown by the Oil-Red-O (ORO) staining, throughout the aorta that was much more pronounced than in Apoe−/− or TGFβR2iSMC mice (Fig 2A–C). Morphometric analysis confirmed these observations: TGFβR2iSMC-Apoe mice showed a larger increase in aorta surface area, length and mean width compared to the other groups of mice (Fig S1G–L). In vivo micro-CT examination verified the presence of ascending and abdominal aortic aneurysm in all TGFβR2iSMC-Apoe mice with extensive areas of intraluminal calcifications (Fig 2D). In some mice, the aneurysms were particularly extensive, tortuous and highly reminiscent of advanced human aortic aneurysms (Fig 2De,f). Abdominal aorta aneurysms were found only in TGFβR2iSMC-Apoe mice (Fig 2C,D). In vivo ultrasound showed that while TGFβR2iSMC mice had slightly but not significant dilated ascending aortas compared to C57BL/6 controls (1.84±0.095 vs. 1.673±0.012 mm p=0.15), this was significantly less than aorta size in TGFβR2iSMC-Apoe mice (2.1±0.076 mm; p<0.01; Fig 2E). Time course analysis of disease development showed a significant increase in atherosclerosis burden, elongation, dilatation and unfolding of the aorta two and four months after the start of HCHFD in TGFβR2iSMC-Apoe compared to Apoe−/− or TGFβR2iSMC mice (Fig 2F,G, Fig S1G–I).
Figure 2: Smooth muscle cell Tgfbr2 knockout in Apoe−/− background accelerates aneurysm formation and atherosclerosis plaque development.
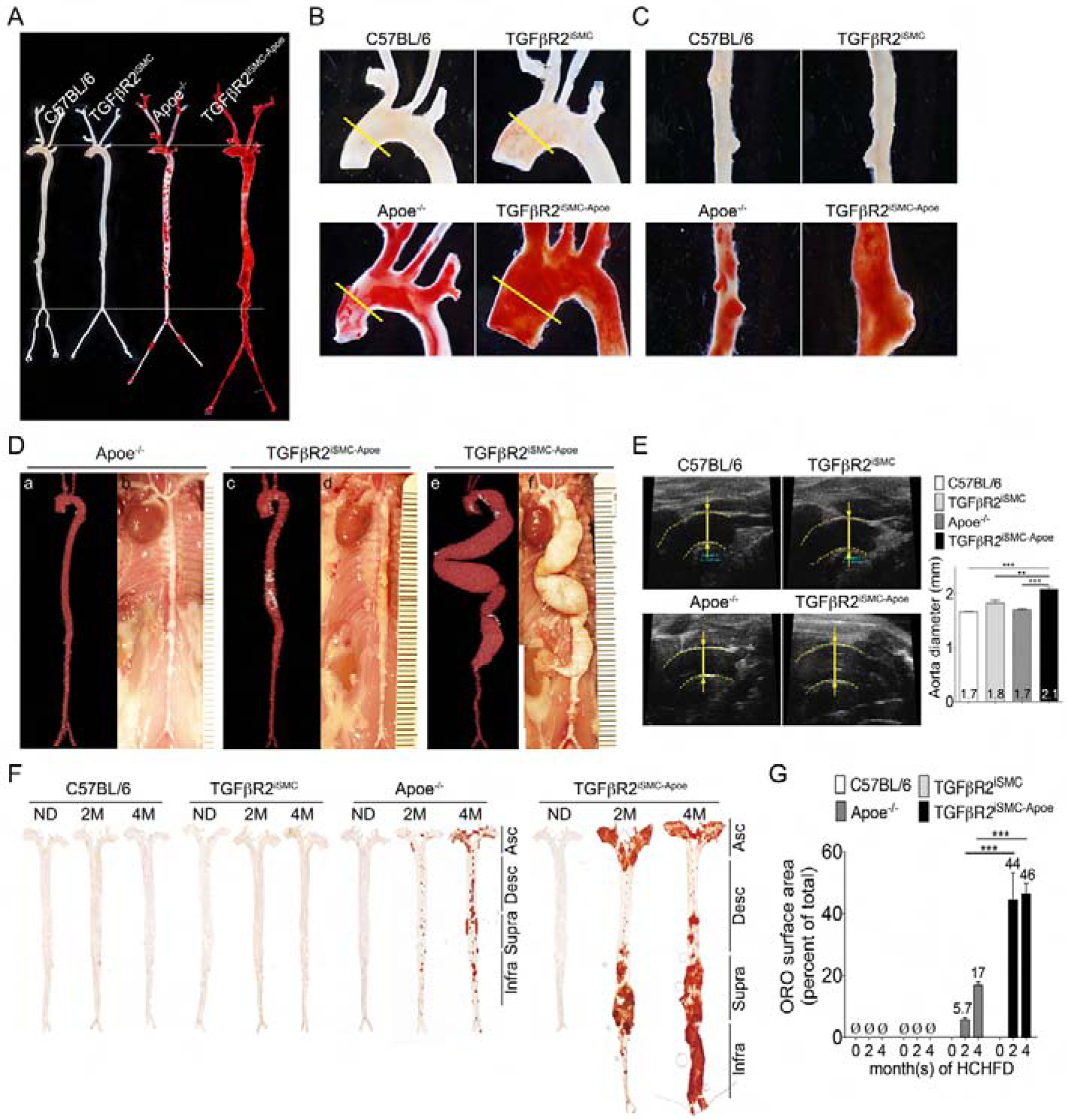
(A-C) Representative photomicrographs of Oil-Red-O stained whole aorta (A), aortic arch (B) or abdominal aorta (C) of C57BL/6, TGFβR2iSMC, Apoe−/−, and TGFβR2iSMC-Apoe mice after 4 months of high cholesterol high fat diet (HCHFD). N=4 mice/group. (D) Representative micro-CT images and gross view of aortas of Apoe−/− and TGFβR2iSMC-Apoe mice after 4 months of HCHFD. TGFβR2iSMC-Apoe mice exhibit 100% penetrance of the aneurysm phenotype. (a-b) typical Apoe−/−, (c-d) 90% of TGFβR2iSMC-Apoe, (e-f) 10% of TGFβR2iSMC-Apoe mice aorta (N=3 for Apoe−/− mice; N=4 for TGFβR2iSMC-Apoe mice). Calcium deposits are traced with white color. (E) Representative ultrasound images and ascending aorta diameters (yellow lines) of C57BL/6, TGFβR2iSMC, Apoe−/−, and TGFβR2iSMC-Apoe mice after 4 months of HCHFD. Data are shown as mean ± SEM (***p<0.001; unpaired two-tailed Student’s t test). N=3 mice/group. (F) Microphotographs of en face aortas from C57BL/6, TGFβR2iSMC, Apoe−/−, and TGFβR2iSMC-Apoe mice after 0, 2, 4 months of HCHFD stained with Oil-Red-O. (G) Lesion area quantification: % lesion area refers to Oil-Red-O stained as a % of the total aortic surface. All data shown as mean ± SEM (Ø: not detected; ***p<0.001; unpaired two-tailed Student’s t test) (for each time point N=3 for C57BL/6 mice; N=3 for TGFβR2iSMC mice; N=11 for Apoe−/− mice; and N=11 for TGFβR2iSMC-Apoe mice). See also Figure S1, Figure S2, Figure S3, Table S1B, and Table S1C.
The development of aneurysms in TGFβR2iSMC-Apoe mice was paralleled by a marked increase in lipid accumulation along the entire aortic tree that was particularly prominent in aneurysmal regions. In contrast, ORO-positive areas were absent in TGFβR2iSMC mice (Fig 2A–C;F,G). As expected, Apoe−/− mice demonstrated some ORO positive areas, though much less prominent than in TGFβR2iSMC-Apoe mice (Fig 2F,G). Kinetics of lipid deposition paralleled the elongation, dilatation and unfolding of the aorta observed in TGFβR2iSMC-Apoe mice.
To study this surprising increase in lipid accumulation in TGFβR2iSMC-Apoe mice, human aortic smooth muscle (HASMCs) were subjected to shRNA-mediated knockdown of TGFβR2 expression and cultured in the presence of cholesterol. This led to a marked increase in SMC acetylated LDL uptake and cholesterol ester content (Fig S1M–O). The in vivo observation of extensive accumulation of neutral lipids and foamy cells (perilipin A positive cells) in aortas isolated from TGFβR2iSMC-Apoe mice fed HCHFD (Fig S1P) is consistent with the in vitro data described above. We also observed a significant accumulation of ApoB, suggesting that TGFβR2 might also influence the extracellular matrix composition in the aorta facilitating pro-atherogenic lipoprotein retention (Fig S1P). In agreement with this, when mice lacking SMC TGFβR2 were injected with a fluorescently labelled LDL (DiI-LDL), there was a significant accumulation of LDL in the aorta (Fig S1Q).
Given a strong phenotype involving the ascending aorta and important differences between various aorta regions, the rest of the analysis deals with the ascending thoracic aorta. To test functional significance of these changes, we conducted ex vivo biomechanical analyses of ascending aortas one month after initiation of HCHFD. Hypercholesterolemia significantly impaired KCl-induced contraction of smooth muscle in the ascending aorta of TGFβR2iSMC and TGFβR2iSMC-Apoe mice as shown by smaller reductions in diameter (Fig S2A). However, only aortas of TGFβR2iSMC-Apoe mice on HCHFD exhibited impaired responses to phenylephrine (Fig S2B and Table S1B AAA active). To assess changes in aortic stiffness, passive mechanical metrics were evaluated at a pressure of 100 mmHg (Table S1C AAA passive). Ascending aortas of TGFβR2iSMC-Apoe mice on HCHFD demonstrated an increase in the circumferential material stiffness that was less marked in TGFβR2iSMC mice (Fig S2C). Increased circumferential stiffness in the ascending aorta indicates a propensity towards or presence of aneurysmal dilatation and is in the agreement with the appearance of frank aneurysms after 2 months of HCHFD. The increase in the unloaded outer diameter of the aortas in TGFβR2iSMC-Apoe mice on HCHFD (Fig S2D) indicates growth of the aorta, in agreement with morphologic observations. The increased vessel stiffness in TGFβR2iSMC-Apoe mice also caused systemic blood pressure (SBP) and pulse pressure to increase but not the pulmonary artery blood pressure (RVBP) (Fig S2E–F).
To confirm that these findings were not limited to the Apoe−/− phenotype, we tested the consequence of the loss of SMC Tgfbr2 in a different model of hypercholesterolemia. To this end, we crossed TGFβR2iSMC onto Ldlr−/− line generating TGFβR2iSMC-Ldlr mice. The mice were then started on HCHFD diet and morphological examinations were conducted 4 months later. These revealed the presence of TAA and AAA similarly to TGFβR2iSMC-Apoe mice with a 100% penetrance among all mice analyzed (Fig S3A–D). ORO staining showed marked lipid accumulation (Fig S3B,C) that led to a slight decrease in total serum cholesterol levels similar to TGFβR2iSMC-Apoe mice (Fig S3E). There were also increased aortic diameter and systolic and pulse pressures in TGFβR2iSMC-Ldlr compared to Ldlr−/− mice (Fig S3F–I).
Histological examination of ascending aneurysmal aortic sections from TGFβR2iSMC-Apoe and TGFβR2iSMC-Ldlr mice at the end of their HCHFD confirmed profound dilatation and increased media thickness (Fig 3A–B, Fig S3J,K). The medial layer exhibited prominent lipid uptake as shown by ORO staining (Fig 3A,B; Fig S3J,K). These medial lesions also associated with intramural calcification and areas of cartilage and bone formation (Safranin O, Alcian Blue, Von Kossa and Alizarin staining; Fig 3A,B; S3J,K). These hallmarks, typical of atherosclerotic lesions, developed specifically in the SMC medial layer, and were detected as early as 1 month after the initiation of the HCHFD in TGFβR2iSMC-Apoe mice (Mendeley data Figure 1, http://dx.doi.org/10.17632/f8s8gjyyw6.1).
Figure 3: Aortic smooth muscle cell reprogramming into osteoblast-, chondrocyte-, adipocyte-, and macrophage-like lineage cells in TGFβR2iSMC-Apoe mice.
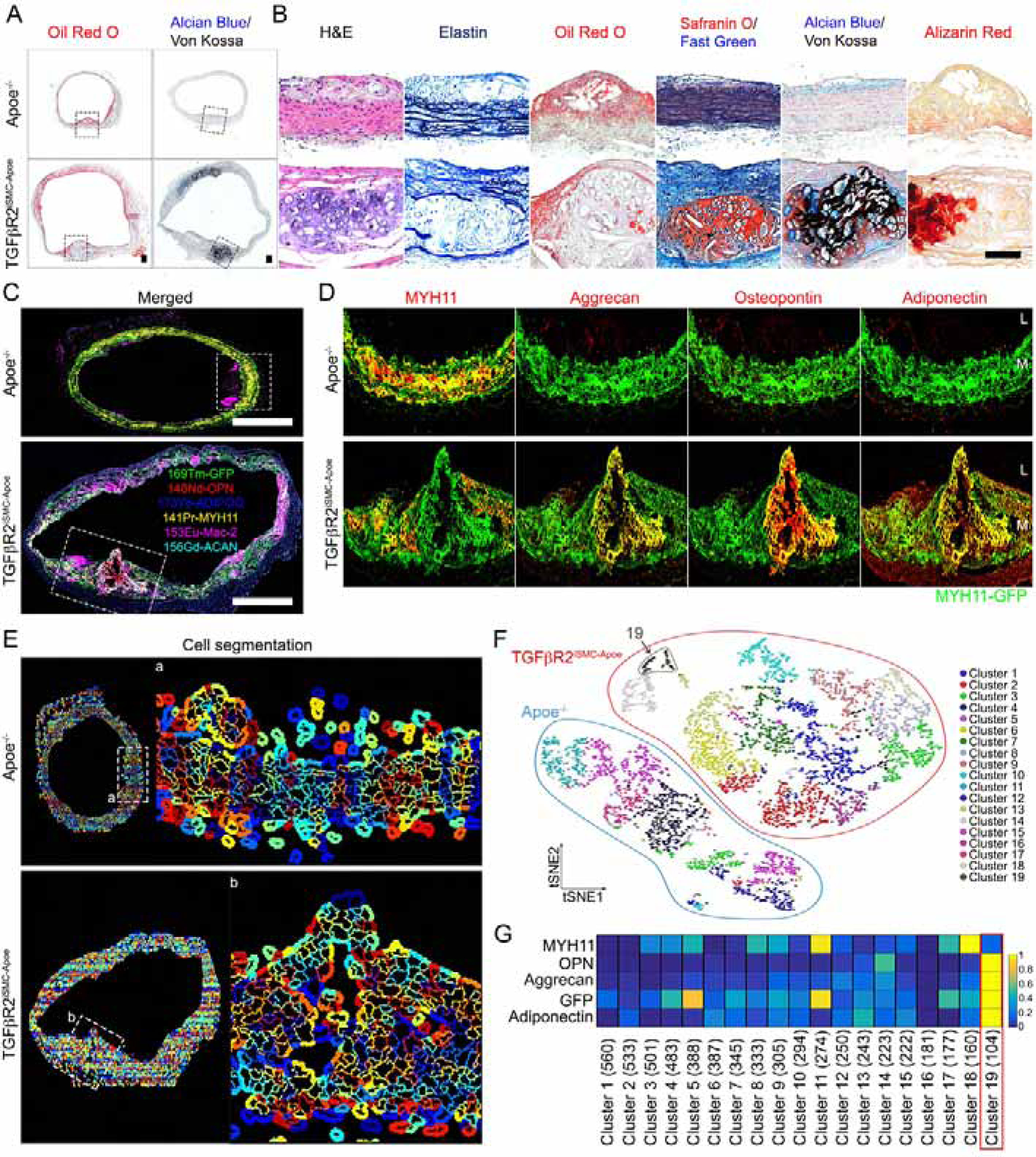
(A) Histologic analysis of mouse ascending aortas dissected from 24-week-old Apoe−/− and TGFβR2iSMC-Apoe mice after 4 months of high cholesterol high fat diet (HCHFD). Representative low-magnification images of Oil-Red-O (left) and Alcian Blue (stain mucopolysaccharides sialomucins for cartilage)/Von Kossa (stain calcium for bone) (right) stained mouse ascending aortas. N=6 mice/group. Scale bar: 200 μm. (B) Representative images of H&E, Elastin, Oil-Red-O, Safranin O (cartilage)/Fast Green, Alcian Blue (stain mucopolysaccharides sialomucins for cartilage)/Von Kossa (stain calcium for bone), and Alizarin Red (calcium)-stained mouse ascending aortas from Apoe−/− and TGFβR2iSMC-Apoe mice (N=6 mice/group). Scale bar: 50 μm. (C) Representative IMC images stained for GFP (green), MYH11 (yellow), Aggrecan (cyan), Osteopontin (red), Adiponectin (blue), and Mac-2 (magenta) from Apoe−/− and TGFβR2iSMC-Apoe ascending aorta are shown overlaid (N=6 mice/group). Scale bar: 500 μm. (D) A high-magnification of (C) showing separate channels of GFP (green), MYH11 (red), Aggrecan (red), Osteopontin (red), and Adiponectin (red). (E) Segmentation of individual cell membrane using CellProlifer. (F) The segmentation mask in (E) was used as the basis for analysis of raw data from this IMC experiment. Based on raw data from each of the four input channels, each individual cell was clustered by expression by histoCAT and manually assigned a phenotype. PhenoGraph clustering of all cell phenotype visualized as a distinct color on the tSNE plot. (G) Heatmap visualizing the marker intensity for each Phenograph. See also Figure S3 and Table S2A.
Given the similarities between the two models, we concentrated on analysis of mouse strains on the Apoe−/− background. To determine the origin of cell types responsible for these changes, we employed imaging mass spectroscopy (IMC, Fig 3C,D). These data are consistent with a sizeable proportion of cells expressing chondrocyte (aggrecan), osteoblast (osteopontin), and adipocyte (adiponectin) markers being of SMC origin as demonstrated by the presence of fate-mapping GFP signal (Fig 3C,D). To determine if there was a cell population expressing all of the mesenchymal markers, we used CellProfiler (McQuin et al., 2018; Stoter et al., 2019) and histoCAT software (Schapiro et al., 2017), a toolbox capable of multiparameter IMC data analysis (Fig 3E). t-Distributed Stochastic Neighbor Embedding (t-SNE) plot revealed genotype-specific segregation of SMC-derived cell clusters based on expression of SMC GFP cell fate signal, contractile marker Myh11 and mesenchymal lineage markers (Fig 3F). We observed a single cluster (cluster 19, 104 cells, 1.7% of the total cell population) in TGFβR2iSMC-Apoe that was made up of cells with a marked reduction or a complete absence of Myh11 expressing all the mesenchymal markers (Fig 3F,G). No such cells were seen in Apoe−/− aortic cross-sections. To confirm these results, we used conventional immunocytochemistry (ICH) with osteoblast-, chondrocyte-, adipocyte- and macrophage-specific antibodies in our mT/mG double-fluorescent reporter animals (Fig S4A). Staining with Myh11 antibody demonstrated continued, if somewhat reduced, expression in Apoe−/− mice and a severe reduction in TGFβR2iSMC-Apoe bone nodule area (Fig S4B) and TGFβR2iSMC-Ldlr mice (Mendeley Data Figure 1A, http://dx.doi.org/10.17632/x3r8fcnkpc.1). ICH confirmed and quantified these findings: GFP positive medial cells expressed high levels of the osteoblast (Runx2, Osteopontin), chondrocyte markers (SOX9, Lubricin), and adipocyte (Adiponectin, PPARγ), in TGFβR2iSMC-Apoe mice but not in Apoe−/− mice (Fig S4C–E and Mendeley Data Figure 1B–D, http://dx.doi.org/10.17632/x3r8fcnkpc.1).
To explain the appearance of multiple SMC-derived mesenchymal cell lineages, we looked for the presence of SMC-derived mesenchymal stem cell (MSC)-like cells that could function as precursors of these multiple lineages. IMC analysis of ascending aorta cross-sections obtained from TGFβR2iSMC-Apoe and Apoe−/− mice one month after initiation of HCHFD was done using multiple MSC markers antibodies (CD105, CD73, CD90, CD44 and Sca-1). There were large areas of overlap of MSC markers signals with the SMC-GFP fate-mapping signal, suggesting the presence of SMC-derived MSC-like cells in TGFβR2iSMC-Apoe mice (Mendeley Data Figure 2A, http://dx.doi.org/10.17632/f8s8gjyyw6.1). A CellProfiler/histoCAT analysis that combined staining with MSC and mesenchymal lineage marker antibodies, separated cells into 31 distinct clusters (Mendeley Data Figure 2B–C, http://dx.doi.org/10.17632/f8s8gjyyw6.1). One cluster (cluster 29) was made of SMC-derived cells expressing high level of both MSC and mesenchymal lineage markers and low level Myh11 (Mendeley Data Figure 2D, http://dx.doi.org/10.17632/f8s8gjyyw6.1). The location of cells in this cluster on the aortic cross-section is shown in Mendeley Data Figure 2E, http://dx.doi.org/10.17632/f8s8gjyyw6.1.
Cytometry by time-of-flight (CyTOF) analysis using viSNE, a visualization tool for high-dimensional single-cell data based on t-SNE algorithm (Amir el et al., 2013), was used to further verify these observations. After one month of HCHFD, a large pool of cells with variable level of GFP expression (yellow-to-red) is clearly visible in both Apoe−/− and TGFβR2iSMC-Apoe mice. A pool of GFP−/RFP+ cells (presumably macrophages and adventitial MSCs) is also detectable in both groups of mice (blue; arrow) (Mendeley Data Figure 2F, http://dx.doi.org/10.17632/f8s8gjyyw6.1). Expression of all MSC markers was much more abundant in GFP+ cells of TGFβR2iSMC-Apoe than Apoe−/− mice (Mendeley Data Figure 2G left, http://dx.doi.org/10.17632/f8s8gjyyw6.1). As expected, some of the GFP−/RFP+ cells also expressed these antigens, but their frequency was the same in both mouse groups (Mendeley Data Figure 2G right, http://dx.doi.org/10.17632/f8s8gjyyw6.1).
A similar analysis after 4 months of HCHFD demonstrated a marked increase in MSC marker expression in cell in the aortic media (Fig 4A). As at one month, a single cluster of cells prominently expressed both mesenchymal lineage and MSC markers (Fig 4B–E). CyTOF analysis showed a large expansion of the GFP−/RFP+ (blue) cell populations that reflected an increase in the size of neointima and adventitia (Mendeley Data Figure 2H, http://dx.doi.org/10.17632/f8s8gjyyw6.1). There were no differences in GFP−/RFP+ cell populations between the two strains (Mendeley Data Figure 2I right, http://dx.doi.org/10.17632/f8s8gjyyw6.1). A higher proportion of SMC-derived GFP+ cells from both TGFβR2iSMC-Apoe and Apoe−/−mice expressed MSC markers at 4 months than at 1 month. As before, all markers were expressed at much higher levels in SMC-derived cells in TGFβR2iSMC-Apoe compared to Apoe−/− mice (Mendeley Data Figure 2I left, http://dx.doi.org/10.17632/f8s8gjyyw6.1), consistent with IMC analysis. Z-stack confocal images confirmed the GFP+ smooth muscle cells expressing stem cells markers (CD105, CD73, CD90, Sca-1, CD44) in TGFβR2iSMC-Apoe compared to Apoe−/− mice at both 1 month and 4 months of HCHFD (Fig S5 and Mendeley Data Figure 3, http://dx.doi.org/10.17632/f8s8gjyyw6.1).
Figure 4: Aortic smooth muscle cells reprogramming into mesenchymal stem cells in TGFβR2iSMC-Apoe mice after 4 months of high cholesterol high fat diet.
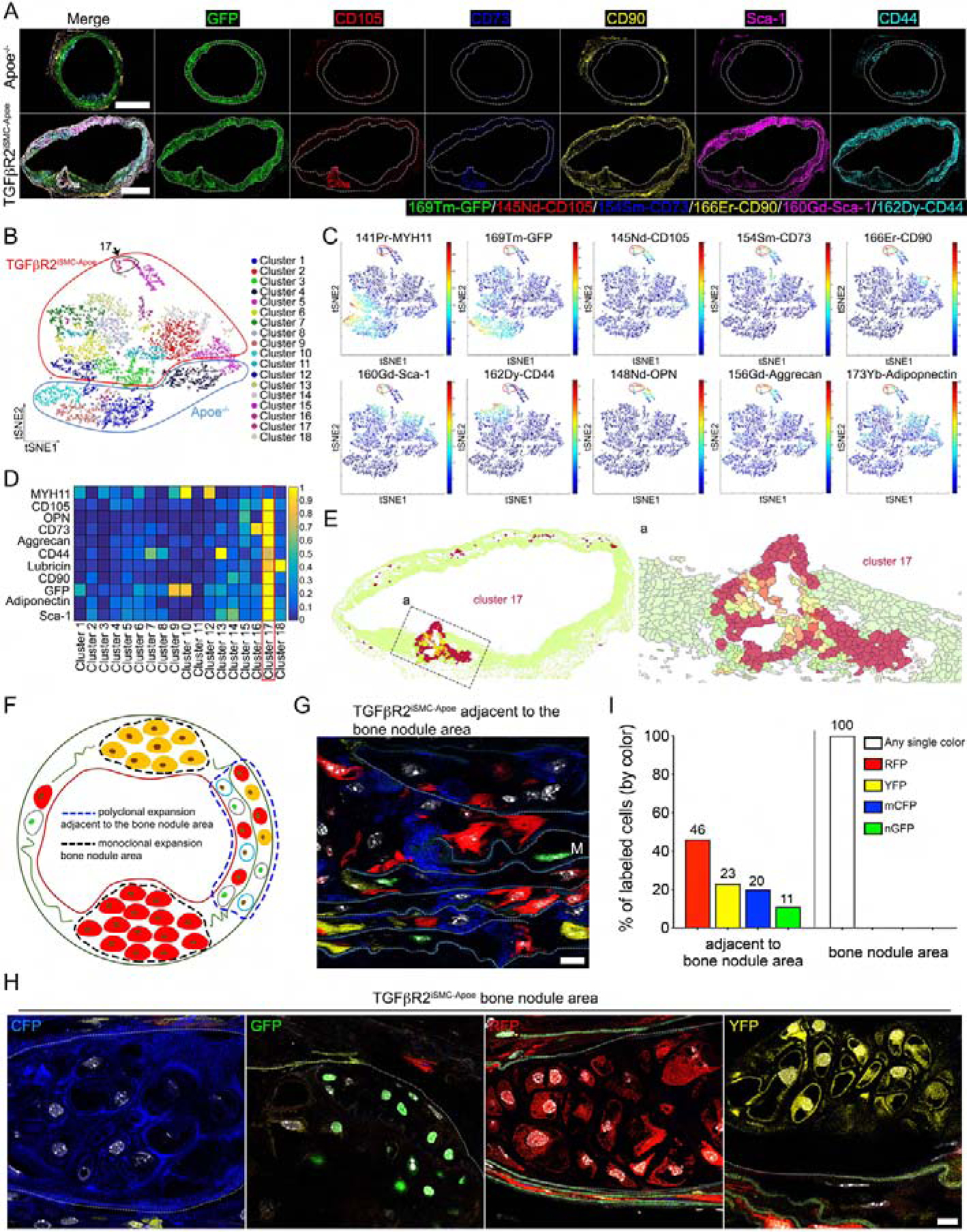
(A) Representative IMC images stained for GFP (green), CD105 (red), CD73 (blue), CD90 (yellow), Sca-1 (magenta), and CD44 (cyan) in the overlaid format and individual channel images (N=6 mice/group). Scale bar: 500 μm. (B) PhenoGraph clustering of all cell phenotype visualized as a distinct color on the tSNE plot. (C) tSNE plot of high-dimension single cell data. (D) Heatmap visualizing the marker intensity for each Phenograph. (E) (left) Cluster 17 location in the tissue section using HistoCAT spatial clustering analysis. (right) A high-magnification of (Ea) left. (F) A cartoon showing confetti color distribution in bone nodule area and adjacent to the bone nodule area. (G) TGFβR2iSMC-Apoe mice VSMCs adjacent to the bone nodule area are mixed labeled with red fluorescent protein (RFP), yellow fluorescent protein (YFP), nuclear (n) green fluorescent protein (GFP), or membrane associated (m) cyan fluorescent protein (CFP). Scale bar: 10 μm. (H) TGFβR2iSMC-Apoe mice VSMCs in the bone nodule media areas are labeled with single color fluorescent protein after 4 months of high cholesterol high fat diet. Scale bar: 10 μm. (I) Bar chart showing the proportions of each of the Confetti colors in TGFβR2iSMC-Apoe mice VSMCs in media ascending aorta adjacent to the bone nodule area and in bone nodule area. N=10 for TGFβR2iSMC-Apoe mice. See also Figure S4, Figure S5, and Table S2B.
To check if there was a clonal component to the origin of SMC-derived mesenchymal cell types, we replaced mTmG reporter strain TGFβR2iSMC-Apoe mice with Confetti, generating Myh11CreERT2;Tgfbr2f/f;Apoe−/−;Confettif/f and Myh11CreERT2;Apoe−/−:Confettif/f (control) lines (Mendeley Data Figure 4A, http://dx.doi.org/10.17632/f8s8gjyyw6.1). At 6 weeks of age Myh11 Cre was activated with tamoxifen and 2 weeks later the mice were placed on HCHFD (Mendeley Data Figure 4B, http://dx.doi.org/10.17632/f8s8gjyyw6.1). At sacrifice 4 months later, there was an expected frequency of confetti colors among medial aortic SMCs (Fig 4G&I and Mendeley Data Figure 4C, http://dx.doi.org/10.17632/f8s8gjyyw6.1). Remarkably, analysis of medial areas of calcification/bone formation sites similar to those shown in Fig 3A–C demonstrated monoclonal expansion in 9/10 samples with one sample showing the presence of two clones (Fig 4F&H&I, Mendeley Data Figure 4D, http://dx.doi.org/10.17632/f8s8gjyyw6.1).
To obtain molecular insights into the observed SMC to MSC-like-derived cell type transition, we performed a time series of single cell RNA-seq analysis (scRNA-seq) on GFP+ cells from the ascending aortas of TGFβR2iSMC-Apoe and Apoe−/− mice (Mendeley Data Figure 1A, http://dx.doi.org/10.17632/hrgxtyds44.1). The cells were obtained from mice prior to the start of HCHFD (T0), and one (T1), two (T2) and four (T3) months later. A novel deep learning approach (Mendeley Data Figure 1B–D, http://dx.doi.org/10.17632/hrgxtyds44.1, and Methods S1) generated cell clusters at each time point that almost completely segregated by genotype (Fig 5A). Prior to the initiation of HCHFD, clusters derived from TGFβR2iSMC-Apoe exhibited somewhat higher expression of genes associated with inflammation and inflammatory cells such as Vcam1, Ifitm3, Lgals3 and Ly96. Yet 1 month later (T1) TGFβR2iSMC-Apoe cluster T1.14 (Fig 5B) stood apart from the rest. Gene expression in this cluster showed a prominent decline in both contractile and synthetic SMC genes (Acta2, Myh11, Col1), and a significant increase in expression of MSC markers (Ly6a, Cd34, Cd44). Somewhat similar, albeit less pronounced, features we observed in Apoe−/− mice-derived clusters T1.8, T1.9, T1.11, and T1.13. These differences became even more pronounced 2 and 4 months later (Fig 5B).
Figure 5: Time series analysis of single cell RNA-sequencing data of smooth muscle cells isolated from mouse ascending aorta.
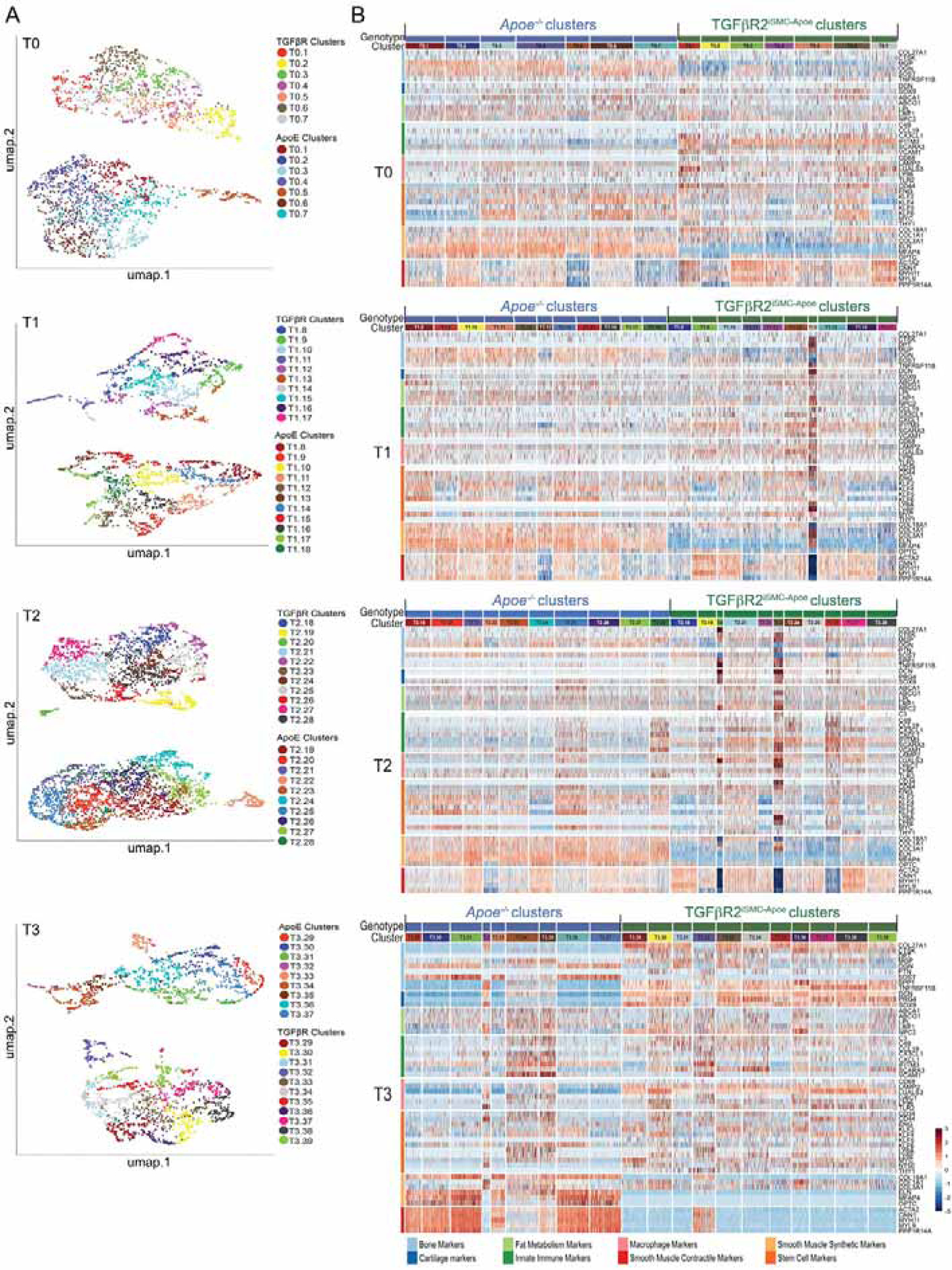
(A) UMAP representation of the scRNA-seq data showing all cell clusters across genotypes: Apoe−/− and TGFβRiSMC-Apoe. Labeled by genotype and specified by the time point: T0 (after 0 month of high cholesterol high fat diet), T1 (after 1 month of high cholesterol high fat diet), T2 (after 2 months of high cholesterol high fat diet), T3 (after 4 months of high cholesterol high fat diet). (B) Heatmap representation of scRNA gene expression for specific cell type markers. Cells are arranged by cell clusters and genotype. Genes are grouped and colored by biomarker class. The colors identifying the biomarker classes are on the left side of the heatmap and the color legend is located at the bottom of the heatmap. See also Methods S1.
To gain better insight into the time course of these changes we generated cell differentiation trajectory networks that show progression of gene expression changes in each cluster from T0 to T3 (Fig 6A). Importantly, all T3 nodes in TGFβR2iSMC-Apoe mice were derived from two T2 nodes (T2.20 and T2.23) that were, in turn, derived from a single T0 node (T0.6). In contrast, multiple T2 nodes give rise to T3 nodes in Apoe−/− mice. Analysis of temporal changes in gene expression in TGFβR2iSMC-Apoe mice nodes showed a gradual decrease in contractile SMC marker expression (Fig 6B) and appearance of mesenchymal lineage and MSC marker genes (Runx2, Sox9 and Pparγ shown as representative markers, Fig 6C–E). Thus, cluster T0.6 that shows normal Myh11 expression gives rise to cluster T1.14 that has reduced Myh11 expression that in turn gives rise to T2.20 and T2.23 with very low Myh11 expression and all of T3 cluster where Myh11 is virtually lost. At the same time, as Myh11 (as other contractile genes) expression decreases, there is a gradual increase in various mesenchymal lineage and MSC marker genes expression including such as Runx2, Sox9, and Pparγ.
Figure 6. Putative cell differentiation trajectory networks in Apoe−/− and TGFβR2iSMC-Apoe mice.
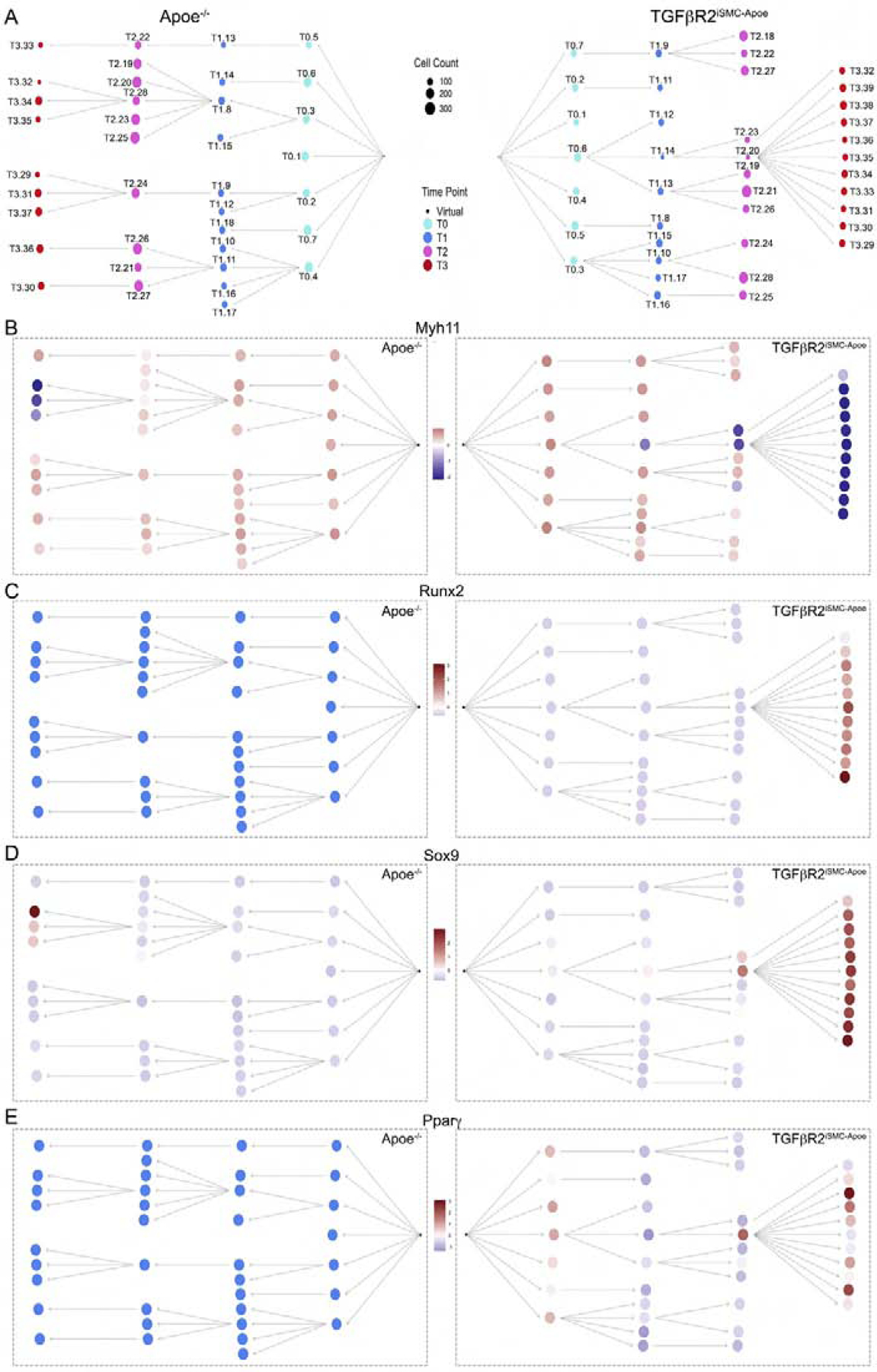
(A) Putative cell differentiation trajectory networks based on scRNA gene expression and segregated by genotype. Nodes represent cell clusters. Nodes are colored by time point and their size correspond to cell count. (B) Putative cell differentiation trajectory networks based on scRNA gene expression for Myh11, Runx2, Sox9, and Pparγ. Cell differentiation trajectories graphs designated by genotype, nodes represent cell clusters. Color represents the mean intensity of a winsorized Z-score (at −3 and 3) for each gene across each cell cluster for both genotypes. For each genotype, a virtual node was assumed under the hypothesis that at T0 the cells are already differentiated. The virtual nodes are represented as a black dot, which serves as a root for each branching network. See also Methods S1.
To gain further insight into this process and to associate these scRNA-seq findings with protein-expression-based analysis of expression, we looked for scRNA-seq based analogs to HistoCat-defined clusters with low level of Myh11 expression and high level MSC and MSC lineages marker expression (cluster 19 in Fig 3G and cluster 17 in Fig 4D from TGFβR2iSMC-Apoe mice), we examined the profile of all scRNA-seq clusters at the same T3 (4 months) time point with respect to the markers used in Fig 4D. One scRNA-seq cluster, TGFβR2iSMC-Apoe.T3_36, matched the defining expression profile found in HistoCat clusters 19 and 17 (Mendeley Data Figure 2A–B, http://dx.doi.org/10.17632/hrgxtyds44.1). We then studied all clusters across our scRNA-seq time analysis that manifested SMC-to-MSC transdifferentiation in the TGFβR2iSMC-Apoe genotype to identify a transition from cells with normal Myh11/low MSC expression to cells with low Myh11/high MSC marker expression. Clusters TGFβR2iSMC-Apoe.T0_6 and TGFβR2iSMC-Apoe.T1_14, which gave rise to all cells in the TGFβR2iSMC-Apoe genotype at T3, fit this profile (Mendeley Data Figure 2C–D, http://dx.doi.org/10.17632/hrgxtyds44.1). Interestingly, KLF4 expression is already somewhat increased in T0_6 (the only T0 cluster with increased KLF4 levels). These results are in agreement with IMC and Confetti fate mapping data demonstrating clonal origin of SMC-derived pathologic cell types.
Since the data presented above implicate the loss of SMC TGFβ signaling in SMC reprogramming, we next examined TGFβ-related transcriptional changes in these cells. To this end, we treated human aortic SMCs (HASMC) in vitro with TGFβ and mapped binding of SMAD2/3 (which reflects TGFβ activity) and Pol2-Ser2p (which reflects transcriptional activity) by ChIP-seq. SMAD2/3 bound to numerous promoter regions in the genome differentially regulating gene expression as shown by alterations in Pol2-Serp2 binding profile (Fig S6A and Mendeley Data Figure 3A–B, http://dx.doi.org/10.17632/hrgxtyds44.1) and analysis of bulk RNA sequencing (Fig S6B). Examination of the top 20 transcription factors identified by sequencing of SMAD2/3 binding regulatory regions, pointed to KLF4 gene as the most prominent gene suppressed by TGFβ (Fig S6C). Further analysis showed SMAD2/3 binding to multiple KLF4 gene regulatory elements that increased following TGFβ treatment, indicating that KLF4 is a direct target of SMAD2/3 (Fig S6D). At the same time, the amount of bound Pol2-Ser2p decreased, pointing to reduced transcriptional activity (Fig S6D). This was confirmed by bulk RNA-seq (Fig S6D). In contrast, SMAD2/3 did not bind to closely related KLF2 or KLF5 loci, indicating that these genes are not directly regulated by TGFβ (Mendeley Data Figure 3C, http://dx.doi.org/10.17632/hrgxtyds44.1).
To test the synergy between a decrease in SMC TGFβ signaling and hypercholesterolemia, we assessed the binding of SMAD2/3 and KLF4 to KLF4 regulatory regions in the presence or absence of cholesterol using ChIP-qPCR. As expected, TGFβR2 KD led to a profound reduction in SMAD2/3 binding to KLF4 regulatory elements. Interestingly, cholesterol treatment also had a significant (Fig S6E left). At the same time, while TGFβR2 KD or cholesterol exposure by themselves had little effect on KLF4 binding to KLF4 regulatory regions, a combination of these two factors dramatically increased KLF4 enrichment (Fig S6E right). qPCR analysis of gene expression confirmed a significant increase in SMC KLF4 (98.2 fold), KLF2 (11.7 fold) and KLF5 (2.3 fold) levels following TGFβR2 KD in the presence of cholesterol (Mendeley Data Figure 3D, http://dx.doi.org/10.17632/hrgxtyds44.1).
Immunocytochemical examination of KLF4 expression in mice confirmed a significant increase in expression in SMCs of TGFβR2iSMC-Apoe mice on HCHFD but not in TGFβR2iSMC or Apoe−/− mice (Fig S6F,G). This combination of TGFβR2 knockdown and cholesterol loading was also much more effective in inducing expression of several MSC- and mesenchymal lineage markers in HASMCs (Fig S6H), paralleling observations in TGFβR2iSMC-Apoe mice on HCHFD (Fig 2). To test the role played by KLF genes in SMC to MSC-like cell conversion, we carried out a rescue experiment in which HASMCs treated with cholesterol and subjected to TGFβR2 knockdown were subjected to shRNA-mediated knockdown of either KLF4 alone or in combination with KLF2 and KLF5. While KLF4 knockdown alone was partially effective, a simultaneous knockdown of all three KLFs fully suppressed the expression of MSC-derived lineages markers (Fig 6H–I, Mendeley Data Figure 3E, http://dx.doi.org/10.17632/hrgxtyds44.1). Since KLF4 is an immediate early gene (Godmann et al., 2010) and can be activated by the stress of SMC isolation from the aorta and subsequent manipulation, we used laser microdissection to verify the above observations (Mendeley Data Figure 2A, http://dx.doi.org/10.17632/x3r8fcnkpc.1). qPCR analysis of micro-dissected tissues confirmed the loss of Myh11 expression and a large increase of KLF4, 2 and 5 and MSC fate genes (Mendeley Data Figure 2B–D, http://dx.doi.org/10.17632/x3r8fcnkpc.1) and of MSC-derived cell fate markers (Mendeley Data Figure 2E–H, http://dx.doi.org/10.17632/x3r8fcnkpc.1) in TGFβR2iSMC-Apoe compared to Apoe−/− mice on HCHFD. Detection of BrdU and TUNEL staining after 4 months of HCHFD showed an increase in both cell proliferation and apoptosis in TGFβR2iSMC mice compared to Apoe−/− mice (Mendeley Data Figure 2I–K, http://dx.doi.org/10.17632/x3r8fcnkpc.1).
With these data in hand, we carried out in vivo KLF4 rescue of aneurysm development in TGFβR2iSMC-Apoe mice by crossing Klf4f/f mice on to the TGFβR2iSMC-Apoe strain. The Myh11Cre was activated, inducing excision of either Tgfbr2 alone or in combination with Klf4 at 6 weeks of age. The mice were then placed on HCHFD and the development of aneurysms was monitored over time. SMC-specific KLF4 knockout significantly reduced the development of both thoracic and abdominal aortic aneurysms and decreased overall atherosclerotic burden despite similar cholesterol levels (Fig 7A–E). In particular, there was a reduction in aortic length and diameter (Fig 7A–D), a decrease in wall calcifications and ossification, and reduced lipid deposition (Fig 7F,G) that was confirmed by immunocytochemical analysis of lineage markers (Fig 7H).
Figure 7. KLF4 is the key regulator controlling aortic smooth muscle cells reprogramming into mesenchymal stem cells in TGFβR2iSMC-Apoe mice.
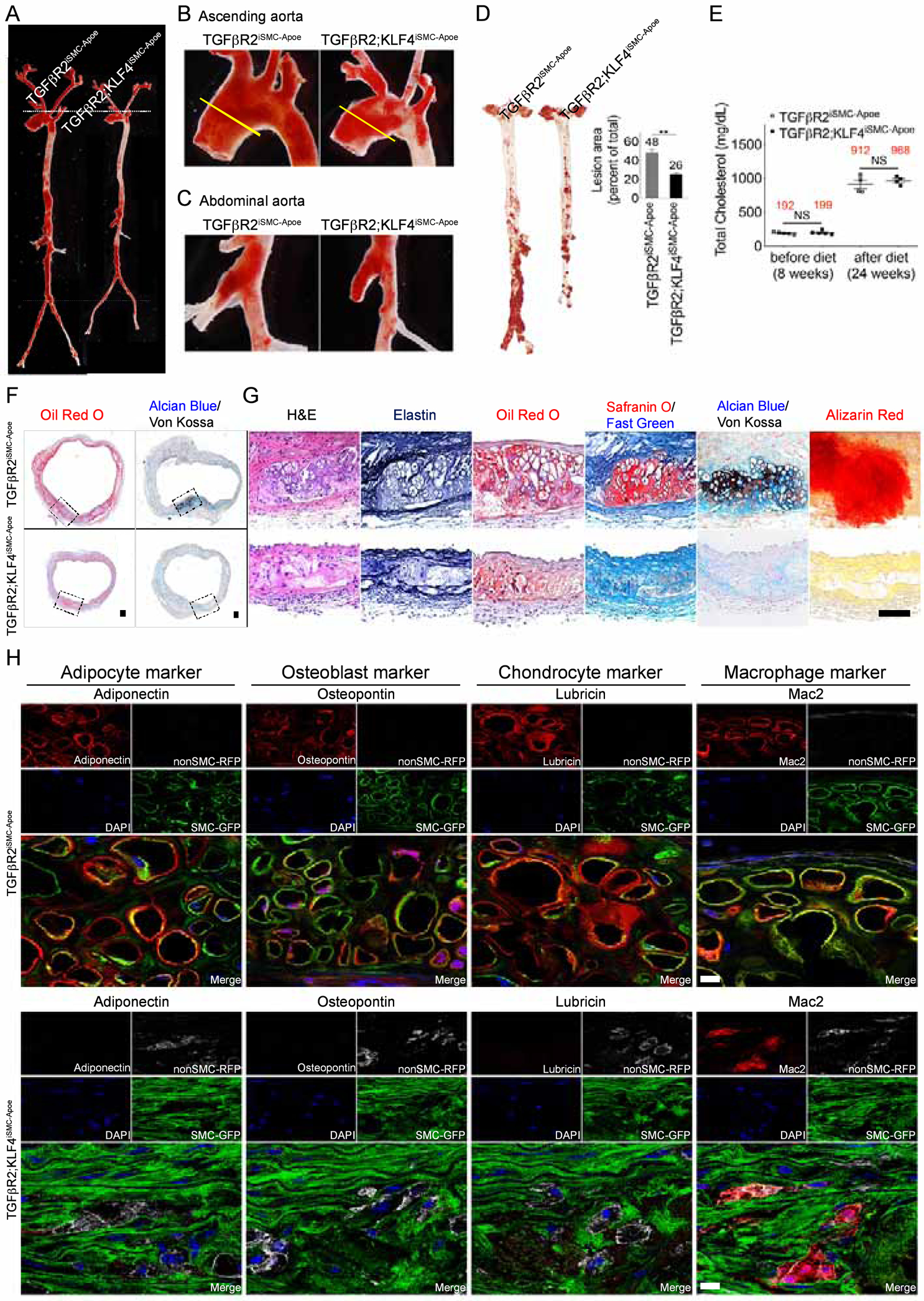
(A-C) Representative photomicrographs of Oil-Red-O stained whole aorta (A), aortic arch (B) or abdominal aorta (C) of TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice after 4 months of high cholesterol high fat diet (HCHFD). N=4 mice/group. (D) (left) Microphotographs of en face aortas from TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice after 4 months of HCHFD stained with Oil-Red-O. (right) Lesion area quantification: % lesion area refers to Oil-Red-O stained as a % of the total aortic surface. All data shown as mean ± SEM (**p<0.01; unpaired two-tailed Student’s t test). N=10 mice/group. (E) Total cholesterol levels from TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice after 4 months on a HCHFD (NS: not significant; unpaired two-tailed Student’s t test). N=5 for TGFβR2iSMC-Apoe mice; N=4 for TGFβR2;KLF4iSMC-Apoe mice. (F) Histologic analysis of mouse ascending aortas dissected from 24-week-old TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice after 4 months of high cholesterol high fat diet (HCHFD). Representative low-magnification images of Oil-Red-O (left) and Alcian Blue (stain mucopolysaccharides sialomucins for cartilage)/Von Kossa (stain calcium for bone) (right) stained mouse ascending aortas. N=6 mice/group. Scale bar: 200 μm. (G) Representative images of H&E, Elastin, Oil-Red-O, Safranin O (cartilage)/Fast Green, Alcian Blue (stain mucopolysaccharides sialomucins for cartilage)/Von Kossa (stain calcium for bone), and Alizarin Red (calcium)-stained mouse ascending aortas from TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice (N=6 mice/group). Scale bar: 50 μm. (H) Immunohistochemical staining of lineage-specific markers (Adipocyte, Osteoblast, Chondrocyte, Macrophage) in the ascending aortas of TGFβR2iSMC-Apoe and TGFβR2;KLF4iSMC-Apoe mice after 4 months of high cholesterol high fat diet using SP8 confocal microscope (N=6 mice/group). Scale bar: 10 μm. See also Figure S6.
Finally, we returned to human aneurysms to verify that these findings are indeed relevant to human disease. In agreement with findings in TGFβR2iSMC-Apoe mice, Z-stack confocal analysis of human ascending aorta aneurysms showed medial cells expressing various mesenchymal cell lineage markers (aggrecan, osteopontin, adiponectin and CD68) as well as MSC-like cell markers (CD105, CD73 and CD90) with focal loss of smooth muscle myosin heavy chain (SMMHC) encoded by MYH11 (Fig S7).
Discussion
The results of this study demonstrate that the loss of SMC TGFβ signaling in conjunction with hypercholesterolemia results in highly reproducible development of aortic aneurysms that closely resembles non-Marfan’s/Loeys-Dietz types of human disease- morphologically, biomechanically and molecularly. Aneurysm formation was driven by extensive reprogramming of contractile medial SMCs to MSC-like cells that was much more prevalent in TGFβR2iSMC-Apoe than Apoe−/− mice. A number of techniques, including serial scRNA-seq studies, CyTOF and IMC, demonstrated the presence of a distinct SMC-derived MSC-like cell population characterized by expression of antigens and transcription factors commonly associated with MSCs. The SMC-to-MSC fate change was accompanied by SMC reprogramming of a distinct subset of normal aortic SMCs into several mesenchymal-derived cell types including adipocytes, chondrocytes, osteoblasts, as well as macrophages that increased over time. Importantly, Confetti fate mapping demonstrated clonal origin of these cell transformations while serial scRNA-seq studies traced the development of MSC-like cells and MSC lineage cell populations to a single subset of SMC in the aorta prior to initiation of HCHFD. Interestingly, the predominant and the earliest conversion was to osteoblast/chondrocyte lineages in agreement with a recent observation of lower genetic barriers of MSC differentiation to osteoblast vs. adipocyte lineages (Rauch et al., 2019). Taken together, these data point to the existence of a “disease-prone” SMC population in the aorta that is poised to undergo SMC-to-MSC conversion and is responsible for aneurysm development in these settings.
In agreement with this high prevalence of SMC-to-MSC reprogramming, TGFβR2iSMC-Apoe mice exhibited extensive aneurysm development with all the key pathologic features, including ossification, lipid deposition and SMC-derived macrophage infiltration of the aorta. The result is a profound decrease in the population of contractile SMCs in the aorta, their replacement by non-muscle cell types, as well as intra-aortic ossification, cartilage formation, fat deposition and ongoing inflammation due to a large increase in macrophage population in the wall. These changes were reflected in mechanical properties of the aortic wall, including diminished contractile capacity, increased circumferential stiffness, increased systemic blood pressure, and increased pulse pressure. Overall, these events point to extensive SMC reprogramming as the main event driving aneurysm development and growth. Thus reprogrammed, SMCs may not be the only cell type contributing to aortic wall calcification as both adventitial (Kramann et al., 2016) and mesenchymal (Pillai et al., 2017) stromal cells have been previously identified as contributors to this process.
At the molecular level, SMC reprogramming was driven, at least in part, by a marked increase in expression of KLF4 and its family members KLF2 and KLF5. Critically, a combination of hypercholesterolemia and TGFβR2 disruption was required for this effect. Under normal conditions, p-Smad-2/3 directly binds to the KLF4 promoter and suppresses its expression. This suppression is lost following TGFβR2 disruption, leading to a small rise in KLF4 level. Hypercholesterolemia greatly augments decreased TGFβ signaling-driven induction of KLF4, resulting in a ~100-fold increase in KLF4 expression. In addition to KLF4, we also observed increases in KLF2 and, to a lesser extent, KLF5 levels. Unlike KLF4, these two KLF family members are not directly regulated by TGFβ signaling. Nevertheless, they appear functionally important given a known interaction between these three KLFs (Jiang et al., 2008). Indeed, a knockdown of all three KLFs was required to fully reverse HASMC reprogramming in vitro while a knockdown of KLF4 alone had only a partial effect and a known interaction between these three KLFs (Jiang et al., 2008)
In keeping with these observations, SMC KLF4 knockout in vivo lead to a significant but incomplete rescue of aneurysm development in TGFβR2iSMC-Apoe mice. These results are in keeping with the known role of KLF4 in induction of SMC transformation into osteogenic cells (Yoshida et al., 2012) and macrophages (Rong et al., 2003; Shankman et al., 2015) and with an observation that KLF4 deletion in SMC attenuates abdominal aortic aneurysm development following aortic wall injury (Salmon et al., 2013). However, the ability of a very high-level KLF4 expression to drive SMC reprogramming all the way to an MSC-like state is a new observation.
Importantly, molecular analysis of normal human aortas and ascending aortic aneurysms demonstrated the presence of all molecular features observed in TGFβR2iSMC-Apoe mice, including the loss of SMC p-Smad-2 indicating a decline in TGFβ signaling, and the presence of numerous MYH11+ cells expressing MSC markers in the aneurysmal, but not normal, aortic wall. Our findings point, therefore, to a combination of reduced TGFβ signaling and hypercholesterolemia as drivers of ascending aortic aneurysm development in patients with atherosclerosis.
Whereas clinical studies have linked hypercholesterolemia with abdominal aneurysms, the association with thoracic aortic aneurysms is much less certain (Hiratzka et al., 2010; Ladich et al., 2016) and no plausible mechanism has been proposed. In particular, the role of TGFβ signaling in aortopathies remains highly controversial. Whilst a significant body of literature considers activation of SMC TGFβ signaling the primary cause of TAA (Gallo et al., 2014; Lindsay and Dietz, 2014; Lindsay et al., 2012), other findings cast doubt on this assertion (Mallat et al., 2017). Indeed, disruption of TGFβR1 or TGFβR2 have been reported to lead to dilatation of the aorta and enhance the ability of angiotensin-2 to promote aneurysm development (Angelov et al., 2017; Hu et al., 2015; Li et al., 2014; Yang et al., 2016). However, the aortopathy observed in these studies bears little morphologic resemblance to human aneurysms, showing just the presence of modest laminin disruption and mild dilation but no other pathological features (Hu et al., 2015; Li et al., 2014). The implication is that a disrupted SMC TGFβ signaling is necessary but not sufficient to induce a full-blown aneurysm development. Yet aneurysms are minimal in Apoe−/− or Ldlr−/− mice in the absence of hypertension or mechanical wall damage (Ishibashi et al., 1994).
Our data demonstrate that hypercholesterolemia potentiates effects of disrupted SMC TGFβ signaling by promoting SMC reprogramming, leading to formation of aneurysms that fully match human pathologic and morphologic features even in the absence of hypertension or wall injury. Interestingly, a small “disease-prone” population of media SMCs is the source of these reprogrammed cells. This two-hit hypothesis provides an explanation for the relatively infrequent aneurysm occurrence in older patients with advanced atherosclerosis as well as the low frequency of both familial and spontaneous aneurysms in non-atherosclerotic settings. We speculate that any combination of events that result in very high-level expression of KLF4 in SMC will induce their reprogramming and lead to aneurysm development.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Simons (michael.simons@yale.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Generation of mice.
Myh11CreERT2;mT/mGf/f and Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice have been previously described (Li et al., 2014). Myh11CreERT2;mT/mGf/f and Myh11CreERT2;mT/mGf/f;Tgfbr2f/f strains had been back-crossed more than ten times to C57BL/6 background. Klf4f/f mice were obtained from Daniel Greif (Cardiovascular Research Center, Yale University). To generate Apoe−/−;Myh11CreERT2;mT/mGf/f and Apoe−/−; Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice, we mated Myh11CreERT2;mT/mGf/f and Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice with Apoe−/− mice (B6.129P2-Apoetm1Unc/J, Stock No: 002052). To generate Ldlr−/−;Myh11CreERT2;mT/mGf/f and Ldlr −/−; Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice, we mated Myh11CreERT2;mT/mGf/f and Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice with Ldlr mice (B6.129S7-Ldlrtm1Her/J, Stock No: 002207). To generate Apoe−/−;Myh11CreERT2;Confettif/f and Apoe−/−;Myh11CreERT2; Confettif/f;Tgfbr2f/f mice, we mated Myh11CreERT2; Apoe−/− and Myh11CreERT2; Apoe−/−;Tgfbr2f/f mice with Confettif/f mice (B6.129P2-Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J, Stock No: 017492). To generate Apoe−/−; Myh11CreERT2;mT/mGf/f;Tgfbr2f/f;Klf4f/f mice, we mated Apoe−/−; Myh11CreERT2;mT/mGf/f;Tgfbr2f/f mice with Klf4f/f mice. All mouse strains used in this study can be found in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Adiponectin | abcam | Cat#ab162551; RRID:AB_955649 |
| Mouse monoclonal anti-Adiponectin (clone 19F1) | abcam | Cat#ab22554; RRID:AB_447152 |
| Rabbit polyclonal anti-Aggrecan | NOVUS BIOLOGICALS | Cat#NB100-74350; RRID:AB_1048083 |
| Rabbit polyclonal anti-Apolipoprotein B | abcam | Cat#ab20737; RRID:AB_2056954 |
| Goat polyclonal anti-PECAM1 (clone M-20) | Santa Cruz | Cat#sc-1506; RRID:AB_2161037 |
| Mouse monoclonal anti-CD44 | NOVUS BIOLOGICALS | Cat#NBP1-47386, RRID:AB_10010339 |
| Mouse monoclonal anti-CD68 (clone KP1) | abcam | Cat#ab955, RRID:AB_307338 |
| Mouse monoclonal anti-CD73 (clone OTI1G2) | NOVUS BIOLOGICALS | Cat#NBP2-46259, RRID:AB_2813846 |
| Rat monoclonal anti-CD90 (clone IBL-6/23) | abcam | Cat#ab3105, RRID:AB_2287350 |
| Rat monoclonal anti-CD105 (clone MJ7/18) | BD Biosciences | Cat# 550546, RRID:AB_2277916 |
| Rabbit polyclonal anti-Collagen II | abcam | Cat# ab34712, RRID:AB_731688 |
| Rabbit polyclonal anti-GLI1 | NOVUS BIOLOGICALS | Cat# NB600-600, RRID:AB_2111758 |
| Rabbit polyclonal anti-HSP90AB1 (clone Ab-254) | Sigma-Aldrich | Cat# SAB4300541, RRID:AB_10629523 |
| Rabbit monoclonal anti-KLF4 | abcam | Cat# ab151733, RRID:AB_2721027 |
| Goat polyclonal anti-KLF4 | R and D Systems | Cat# AF3640, RRID:AB_2130224 |
| Rat monoclonal anti-MAC-2 (clone m3/38) | CEDARLANE | Cat# CL8942AP, RRID:AB_10060357 |
| Goat polyclonal anti-MAC2 | R and D Systems | Cat# AF1154, RRID:AB_663894 |
| Rabbit monoclonal anti-MAC-2 (clone D4I2R) | Cell Signaling Technology | Cat# 87985, RRID:AB_2800111 |
| Mouse monoclonal anti-Lubricin Antibody/Proteoglycan 4 (clone 9G3) | Millipore | Cat# MABT401, RRID:AB_2813847 |
| Rabbit polyclonal anti-MMP2 | abcam | Cat# ab37150, RRID:AB_881512 |
| Mouse monoclonal anti-MYH11 (clone 3F8) | abcam | Cat# ab682, RRID:AB_2298008 |
| Mouse monoclonal anti-MYH11 (clone 1G12) | abcam | Cat# ab683, RRID:AB_2235569 |
| Rabbit polyclonal anti-MYH11 | abcam | Cat# ab53219, RRID:AB_2147146 |
| Rabbit polyclonal anti-osteopontin | abcam | Cat# ab8448, RRID:AB_306566 |
| Rabbit polyclonal anti-Perilipin A | abcam | Cat# ab3526, RRID:AB_2167274 |
| Rat monoclonal anti-RNA pol II CTD phospho Ser2 (clone 3E10) | Active Motif | Cat# 61083, RRID:AB_2687450 |
| Rabbit polyclonal anti-Runx2 | NOVUS BIOLOGICALS | Cat# NBP1-77461, RRID:AB_11003000 |
| Rat monoclonal Anti-Ly-6A/E (Clone E13-161.7) | BD Biosciences | Cat# 553333, RRID:AB_394789 |
| Rabbit polyclonal anti-Smad2, phospho-specific (Ser465/467) | Sigma-Aldrich | Cat# AB3849, RRID:AB_177440 |
| Rabbit polyclonal anti-phospho-Smad2 (Ser465/467) | Cell Signaling Technology | Cat# 3101, RRID:AB_331673 |
| Rabbit monoclonal anti-Phospho-Smad2 (Ser465/467) (clone 138D4) | Cell Signaling Technology | Cat# 3108, RRID:AB_490941 |
| Rabbit polyclonal anti-Smad3, phospho (ser423 / ser425) | R and D Systems | Cat# AB3226, RRID:AB_562714 |
| Mouse monoclonal anti-Smad2/3 | BD Biosciences | Cat# 610843, RRID:AB_398162 |
| Rabbit monoclonal anti-Smad2/3 (clone D7G7) | Cell Signaling Technology | Cat# 8685, RRID:AB_10889933 |
| Goat polyclonal anti-Smad2/3 | R and D Systems | Cat# AF3797, RRID:AB_2270778 |
| Rabbit polyclonal anti-TGFβR1 | Santa Cruz Biotechnology | Cat# sc-398, RRID:AB_632493 |
| Rabbit polyclonal anti-TGFβR2 | Santa Cruz Biotechnology | Cat# sc-400, RRID:AB_632497 |
| Rabbit polyclonal anti-TIMP3 | abcam | Cat# ab39184, RRID:AB_2204971 |
| Mouse monoclonal anti-VCAM-1 (clone 6G9) | NOVUS BIOLOGICALS | Cat# NBP1-47491, RRID:AB_10010443 |
| Rat monoclonal anti-CD90 (clone 778053) | R and D Systems | Cat# MAB7335, RRID:AB_2687967 |
| Rabbit polyclonal anti-Osteopontin | abcam | Cat# ab8448, RRID:AB_306566 |
| Goat polyclonal anti-CD105 | R and D Systems | Cat# AF1320, RRID:AB_354735 |
| Goat polyclonal anti-GFP | R and D Systems | Cat# AF4240, RRID:AB_884445 |
| Rabbit monoclonal anti-PPAR gamma (clone c26H12) | Cell Signaling | Cat# 2435, RRID:AB_2166051 |
| Rabbit monoclonal anti-SOX9 (clone EPR14335-78) | abcam | Cat# ab185966, RRID:AB_2728660 |
| Rat monoclonal anti-BrdU [clone BU1/75 (ICR1)] | abcam | Cat# ab6326, RRID:AB_305426 |
| Rabbit polyclonal anti-GFP (Alexa Fluor 488) | Thermo Fisher Scientific | Cat# A-21311, RRID:AB_221477 |
| Chicken polyclonal anti-RFP | Rockland Immunochemicals | Cat# 600-901-379, RRID:AB_10704808 |
| Goat polyclonal anti-CD105-145Nd | This paper | N/A |
| Rat monoclonal anti-CD73-154Sm (clone TY/11.8) | Fluidigm | Cat# 3154019B, RRID:AB_2813854 |
| Monoclonal anti-Sca-1-160Gd (clone E13-161.7) | Harvard CyTOF Core | N/A |
| Monoclonal anti-CD44-162Dy (clone IM7) | Fluidigm | Cat#3162030B, RRID:AB_2814898 |
| Rat monoclonal anti-CD90-166Er | This paper | N/A |
| Monoclonal anti-GFP-169Tm (clone 5F12.4) | Fluidigm | Cat#3169009B, RRID:AB_2814899 |
| Rabbit polyclonal anti-Osteopontin-148Nd | This paper | N/A |
| Monoclonal anti-Mac2/Galectin-3-153Eu (clone M3/38) | Fluidigm | Cat# 3153026B, RRID:AB_2814900 |
| Rabbit polyclonal anti-Aggrecan-156Gd | This paper | N/A |
| Mouse monoclonal anti-Adiponectin-173Yb | This paper | N/A |
| Rabbit polyclonal anti-smooth muscle myosin heavy chain 11-141Pr | This paper | N/A |
| Goat polyclonal anti-CD105-145Nd | This paper | N/A |
| Mouse monoclonal anti-Lubricin-164Dy | This paper | N/A |
| Biological Samples | ||
| Healthy adult ascending aorta | This paper | |
| Aortic aneurysm patient tissue | This paper | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| eXIA™160 | Binitio Biomedical, Inc. | |
| Dil-Ac-LDL | Alfa Aesar | Cat# J65597 (BT-902) |
| Recombinant human TGFβ1 | BioLegend | Cat# 580702 |
| Tamoxifen | Sigma-Aldrich | Cat# T5648 |
| Oil red O | Sigma-Aldrich | Cat# O0625 |
| Corn oil | Sigma-Aldrich | Cat# C8267 |
| 5-Bromo-2’-deoxyuridine | Sigma-Aldrich | Cat# B9285 |
| Cell-ID™ Intercalator-Ir | Fluidigm | Cat# 201192A |
| Cell-ID™ Cisplatin | Fluidigm | Cat# 201064 |
| Critical Commercial Assays | ||
| KAPA HyperPrep Kit | Roche | Cat# 07962347001 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2, 4 rxns | 10x Genomics | Cat# PN-120267 |
| Chromium Single Cell A Chip Kit, 16 rxns | 10x Genomics | Cat# PN-1000009 |
| Chromium i7 Multiplex Kit, 96 rxns | 10x Genomics | Cat# PN-120262 |
| HCS LipidTOX™ Deep Red neutral lipid stain | Thermofisher Scientific | Cat# H34477 |
| In Situ Cell Death Detection Kit, TMR red | Roche | Cat# 12156792910 |
| Deposited Data | ||
| Human reference genome NCBI build 38, GRCh38 | Genome Reference Consortium | http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| Raw and analyzed data | This paper | GEO140947, GSE141031, GSE141032 |
| Processed data | This paper | GitHub |
| A study of SMC TGFb signaling in hypercholesterolemic patients with aortic aneurysms | This paper, Mendeley data | http://dx.doi.org/10.17632/x3r8fcnkpc.1 |
| A study of SMCs phenotype in TGFβR2iSMC-Apoe and TGFβR2iSMC-Ldlr mice | This paper, Mendeley data | http://dx.doi.org/10.17632/x3r8fcnkpc.1 |
| A study of SMCs phenotype in TGFβR2iSMC-Apoe mice using scRNAseq and ChIPseq | This paper, Mendeley data | http://dx.doi.org/10.17632/hrgxtyds44.1 |
| To study stem cell marker lineage marker gene expression and clonal expansion in TGFβR2iSMC-Apoe mice | This paper, Mendeley data | http://dx.doi.org/10.17632/f8s8gjyyw6.1 |
| Experimental Models: Cell Lines | ||
| Human: Aortic Smooth Muscle Cells (HASMC) | ThermoFisher SCIENTIFIC | C0075C |
| Human: 293T/17 [HEK 293T/17] | ATCC | ATCC Cat# CRL-11268, RRID:CVCL_1926 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Myh11CreERT2;mT/mGf/f;Tgfbr2f/f | Li et al., 2014 | N/A |
| Mouse: Klf4f/f | Sheikh et al., 2015 | N/A |
| Mouse: Apoe−/−;Myh11CreERT2;mT/mGf/f | This paper | N/A |
| Mouse: Apoe−/−;Myh11CreERT2;mT/mGf/f;Tgfbr2f/f | This paper | N/A |
| Mouse: Ldlr−/−;Myh11CreERT2;mT/mGf/f | This paper | N/A |
| Mouse: Ldlr −/−;Myh11CreERT2;mT/mGf/f;Tgfbr2f/f | This paper | N/A |
| Mouse: B6.129S7-Ldlrtm1Her/J | The Jackson Laboratory | JAX: 002207 |
| Mouse: Apoe−/−;Myh11CreERT2;Confettif/f | This paper | N/A |
| Mouse: Apoe−/−;Myh11CreERT2;Confettif/f;Tgfbr2f/f | This paper | N/A |
| Mouse: B6.129P2-Gt(ROSA)26Sortm1(CAG-Brainbow2.1)Cle/J | The Jackson Laboratory | JAX: 017492 |
| Mouse: Apoe−/−;Myh11CreERT2;mT/mGf/f;Tgfbr2f/f;Klf4f/f | This paper | N/A |
| Mouse: B6.129P2-Apoetm1Unc/J | The Jackson Laboratory | JAX: 002052 |
| Mouse: Myh11CreERT2;mT/mGf/f | Li et al., 2014 | N/A |
| Oligonucleotides | ||
| qRT-PCR primers | This paper | Table S3 |
| Recombinant DNA | ||
| VSVG | Addgene Plasmid | |
| Human KLF2: pLKO.1 human shRNA KLF2 | Sigma-Aldrich | |
| Human KLF4: pLKO.1 human shRNA KLF4 | Sigma-Aldrich | |
| Human KLF5: pLKO.1 human shRNA KLF5 | Sigma-Aldrich | |
| Human TGFβR1: pLKO.1 human shRNA TGFβR1 | Sigma-Aldrich | |
| Δ8.2 | Addgene Plasmid | |
| Software and Algorithms | ||
| FlowJo | Treestart | |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| VAE framework | Chen et al., 2019 | |
| Data and Code Availability | ||
| Methods S1 | This paper | Hyperparameter Search Model Parameters CellData tdTomato eGFP Sequences ClusterData GeneData |
| Table S1 | This paper | Table S1A: Human subject characteristics. Table S1B: Active biomechanical analysis. Table S1C: Passive biomechanical analysis. |
| Table S2 | This paper | Table S2A: IMC Cluster protein expression profile. Table S2B: IMC Cluster protein expression profile. |
| Table S3 | This paper | List of Primers used for qRT-PCR. |
Mouse genotyping.
Genotyping was performed by mouse ear DNA PCR analysis. Mouse ear DNA was isolated using the DNeasy Blood & Tissue kit (QIAGEN #69506). Genotyping was performed using the following PCR primers: MYH11-CreERT2 (5’-TGA CCC CAT CTC TTC ACT CC-3’, 5’-AAC TCC ACG ACC ACC TCA TC-3’, and 5’-AGT CCC TCA CAT CCT CAG GTT-3’), Tgfbr2fl/fl(5’-TAA ACA AGG TCC GGA GCC CA-3’, and 5’-ACT TCT GCA AGA GGT CCC CT-3’), Apoe (5’-GCC TAG CCG AGG GAG AGC CG-3’, 5’-TGT GAC TTG GGA GCT CTG CAG C-3’, and 5’-GCC GCC CCG ACT GCA TCT-3’), mT/mG (5’-CTC TGC TGC CTC CTG GCT TCT-3′, 5′-CGA GGC GGA TCA CAA GCA ATA-3′, and 5′-TCA ATG GGC GGG GGT CGT T-3′), Ldlr (5’-AATCCATCTTGTTCAATGGCCGATC-3’, 5’-CCATATGCATCCCCAGTCTT-3’, 5’GCGATGGATACACTCACTGC-3’), Confetti (5’-GAATTAATTCCGGTATAACTTCG-3′, 5’-AAAGTCGCTCTGAGTTGTTAT-3′, 5′-CCAGATGACTACCTATCCTC-3′), Klf4 (5’-CTGGGCCCCCACATTAATGAG-3’, 5’-TGACAGCCATGTCAGACTCGC-3’). PCR primers for Tgfbr2 exon2 mRNA Tgfbr2 (5’- GAC CAT CCA TCC ACT GAA ACA TTT TAA C -3’, 5’- GAC TTC ATG CGG CTT CTC ACA G-3’), Actb (5’- CCA GTT GGT AAC AAT GCC ATG T -3’, 5’- GGC TGT ATT CCC CTC CAT CG-3’. All animal procedures were performed under protocols approved by Yale University Institutional Animal Care and Use Committees.
Animal treatment.
Cre-Lox recombination was induced by tamoxifen (Sigma T5648) at 1 mg/day i.p. for 5 days versus vehicle (corn oil, Sigma C8267) alone. For high cholesterol high fat diet treatment, 8 week old mice were placed on a Western diet (40 kcal% Fat, 1.25% Cholesterol, 0% Cholic Acid) for 4, 8, or 16 weeks (Research Diets, product #D12108). A subgroup received 5-Bromo-2’-deoxyuridine (Sigma B9285) at 1 mg s.c. q.o.d. from 10 to 16 weeks of age.
METHOD DETAILS
Clonal analysis.
To determine whether Myh11-lineage SMC responded to loss of TGFβ signaling and hyperlipidemia by undergoing clonal expansion, we used Brainbow2.1 ‘Confetti’ lineage tracing to stochastically mark SMC with one of 4 hereditary fluorescent labels. 6 weeks old Apoe−/−;Myh11CreERT2;Confettif/f and Apoe−/−;Myh11CreERT2; Confettif/f;Tgfbr2f/f mice were treated with tamoxifen at 1 mg/day i.p. for 5 days. At 8 week old, mice were placed on a Western diet for 16 weeks. The ascending aorta were fixed, cryoprotected in a sucrose solution, embedded in OCT compound, and snap-frozen. The ascending aorta from both groups of mice were serially sectioned, washed, DAPI-stained, and mount for microscopy. Imaging was done using confocal laser scanning microscopy (SP8) with laser lines and detectors set for maximal sensitivity without spectral overlap for DAPI (405 laser, 417–508 nm), CFP (458 laser, 454–502 nm), GFP (488 laser, 498–506 nm), YFP (514 laser, 525–560 nm) and RFP (561 laser, 565–650 nm). Each marked cell was scored based on color (i.e., RFP, YFP, nGFP, mCFP).
In vivo micro-CT scanning:
In vivo micro-computed tomography angiography was under 1.5–2% isoflurane anesthesia using the U-SPECT-II/CT scanner (Milabs, Utrecht, the Netherlands) in a subset of randomly selected mice (3 control Apoe−/− and 4 TGFβR2iSMC-Apoe mice). Five minutes prior to image acquisition, mice were given an intrajugular bolus of Exia 160 (5 μL/g body weight, Binitio Biomedicals, Ontario, Canada). Micro-CT data were acquired with an x-ray source of 50 kVp tube voltage, 48 mA tube current, 11 × 11 detector binning model, 40 ms exposure per projection for contrast-enhanced CT acquisitions. A single frame of 480 projections for 4:58 min of continuous x-ray exposure was used without cardiac or respiratory gating. Volumetric micro-CT images were reconstructed using a Feldkamp algorithm with calibrated Hounsfield units. Image data were transferred to an image processing workstation (Advanced Workstation, version 4.4, GE Healthcare) for further reconstruction and quantitative analysis. The whole aortic tree was segmented and inner luminal diameter of the descending thoracic aorta was calculated.
In vivo LDL tissue uptake:
125 μl of DiI-LDL (300 μg) were injected into 12 weeks old mice (Apoe−/− and TGFβR2iSMC-Apoe) via jugular vein injection. After 18 hrs, mice were anesthetized and euthanized. Ascending aortas were perfused using 10 ml of PBS (Gibco 14190–144) followed by 10 ml of 4% paraformaldehyde (Polysciences, Inc. Cat #18814). Following Optimal Cutting Temperature medium (OCT) embedding, tissue sections of formalin-fixed ascending aorta were imaged using a Zeiss microscope.
Biomechanical Specimen Preparation:
Aortic tissue was harvested en bloc from all groups of mice. A 4-week time course of HCHFD provided a sufficient lipid challenge to elicit maladaptive cellular responses and matrix remodeling without inducing advanced plaque formation, which renders mechanical properties highly variable locally and complicates mechanical analysis (Hayenga et al., 2011). The ascending aorta was excised from the aortic root to the brachiocephalic branch. It was then gently cleaned of perivascular tissue by blunt dissection under a microscope, branches were ligated with individual threads of braided 9–0 nylon suture, and segments were mounted within a custom computer-controlled biaxial testing device (Gleason and Humphrey, 2005) in an oxygenated (95% O2 / 5% CO2) Krebs-Ringer solution at 37°C. This solution consisted of (in mM): 123 NaCl, 4.7 KCl, 1.2 MgCl2, 20 NaHCO3, 1.2 KH2PO4, 5.6 glucose, and 2.5 CaCl2. This device allows precise and highly reproducible control of the distending pressure and axial stretch (ratio of deformed to undeformed length) while measuring on-line the applied pressure, outer diameter, axial force, and applied axial stretch.
Biomechanical biaxial contractility assessment:
Upon verification of smooth muscle viability, the active in vivo axial stretch was estimated and defined as that value at which changes in axial force were minimal upon pressurization from 80 to 100 mmHg. Biaxial contractile responses were then measured in response to two agonists: 100 mM potassium chloride (KCl) and 1 μM phenylephrine (PE). All vessels were activated while held at the individual active in vivo axial stretch and a common pressure of 90 mmHg. Following completion of the active protocol, vessels were rendered passive by changing the bath to a Hank’s Buffered Saline Solution (HBSS) following previous studies (Bersi et al., 2017; Ferruzzi et al., 2011).
For the passive biaxial testing, vessels were subjected to seven different cyclic biaxial pressure-diameter and axial force-length protocols as described previously (Bersi et al., 2017). Briefly, unloaded geometry was recorded and the passive in vivo axial stretch, , of the segment estimated. Vessels were preconditioned at the estimated axial stretch, using cyclic pressurization from 80 to 120 mmHg, to minimize viscoelastic contributions to the mechanical behavior. The specimens were then subjected to three pressure-diameter tests, cyclically from 10 to 140 mmHg, at fixed axial stretches of , , and , as well as to four axial force-length tests, cyclically from 0 to 80 mN, at fixed luminal pressures of 10, 60, 100, and 140 mmHg.
To quantify the active properties, contractile response to KCl and PE was assessed as changes in outer diameter (in μm) and mean circumferential stress (in kPa) between relaxed (basal) and contracted (active) states. Basal conditions were defined when the sample was maintained in Krebs-Ringer solution at 37°C in the absence of agonists, and active conditions were defined as 15 minutes after introduction of agonists to the bath. Values of diameter and circumferential stress were normalized to the group-specific pre-constricted (basal) values determined when maintained in HBSS. Note that the circumferential stress, σϑ = Pa/h, where P is the applied (distending) pressure, a is the inner radius and h is the wall thickness, which can be computed from the measured quantities and by enforcing incompressibility.
Biomechanical quantification of passive mechanical properties:
Passive mechanical properties were quantified using a finite elasticity framework. Primary metrics of interest include circumferential material stiffness and elastically stored energy. Hence, we used a validated stored energy function W, namely,
where c, , and are material parameters determined during nonlinear regression analysis of the pressure-diameter and force-length data, Cis the right Green-Cauchy deformation tensor determined from diameter and length changes, Mi is a unit vector denoting the direction of the ith{i = 1,2,3,4} family of locally parallel collagen fibers in a reference configuration, and IC = trC and IVC = C: Mi⊗Mi are coordinate invariant measures of the deformation. A complete explanation of the mathematical framework can be found elsewhere (Baek et al., 2007). Values of Cauchy wall stress and associated biaxial material stiffness can be determined in terms of first and second derivatives of W, respectively, with respect to deformations.
Human specimen collection.
Investigators were on call with the surgical team and collected the ascending aorta. To minimize ex vivo artifacts, samples were removed within the operating room and immediately processed as frozen sections in OCT medium and, when of sufficient length, an additional segment was also fixed in formalin for later embedding, sectioning, and staining. Human aortas were obtained from the organ donors. Research protocols were approved by the Institutional Review Boards of Yale University and the New England Organ Bank. A waiver for consent was approved for surgical patients and written informed consent was obtained from a member of the family for deceased organ donors. Table S1A-Human subject characteristic-summarizes clinical characteristics of this patient group.
Lipid measurements.
Mice were fasted for 12–16 hrs overnight before blood samples were collected by retro-orbital venous plexus puncture, and plasma was separated by centrifugation. Total plasma cholesterol and triglycerides were enzymatically measured (Wako Pure Chemicals Tokyo, Japan) according to the manufacturer’s instructions.
Echocardiographic studies.
Mice body temperature was maintained with a heading pad. Mice were anesthetized with 2% isoflurane, maintained under anesthesia with 1% isoflurane, and examined. The mouse was placed chest up on an examination board interfaced with the Vevo 770. Warmed Aquasonic gel was applied over the thorax and a 30-MHz probe was positioned over the chest in a patasternal position. Long and short axis B-mode and M-mode images were recorded. All measurements were obtained from three to six consecutive cardiac cycles, and the averaged values were used for analysis. Upon completion of the procedure, the gel was wiped off and the animal was returned to its cage housed in a warm chamber (Chen et al., 2016b).
Hemodynamic Assessment.
The mice were anaesthetized by inhalation of isoflurane (1.5% vol/vol). Core body temperature was maintained at 37°C using a controlled heating pad. A 1.0-Fr Mikro-Tip pressure catheter (SPR-1000, Millar Instruments) was inserted into the left carotid artery to measure systemic arterial pressure (SBP). Afterwards the same catheter was inserted into the right jugular vein into the right ventricle for measurement of right ventricle pressure (RVBP). All hemodynamic measurements were performed with a PowerLab system using the LabChart 7.0 software (ADInstruments).
Analysis of plaque area and composition.
The plaque area and composition in C57BL/6, Apoe−/−, TGFβR2iSMC, TGFβR2iSMC-Apoe mice were analyzed after 4, 8, 12, or 16 weeks on a Western diet. Ldlr−/−, TGFβR2iSMC-Ldlr and TGFβR2;KLF4iSMC-Apoe mice were analyzed after 16 weeks on a Western diet. The ascending aorta, aortic arch, thoracic descending aorta, superior mesenteric artery, inferior mesenteric artery, and common iliac artery were fixed in 4% paraformaldehyde (Polysciences, Inc. Cat #18814), dehydrated overnight, and embedded in OCT. Serial cross sections (5 μm) were prepared for histological analysis. To quantify the surface area occupied by atherosclerosis, en face preparations of the aorta of C57BL/6, TGFβR2iSMC, Apoe−/−, TGFβR2iSMC-Apoe, Ldlr−/−, TGFβR2iSMC-Ldlr, and TGFβR2;KLF4iSMC-Apoe mice were stained with Oil Red O after 0, 8 or 16 weeks of Western diet. The surface lesion area was quantified with ImageJ software (NIH). The extent of lesion formation in each animal was expressed as percent lesion area per total area of the aorta. Aortic aneurysms were evaluated according to the Society for Vascular Surgery, International Society for Cardiovascular Surgery. The size of the aortic aneurysm was calculated from the circumference of the luminal surface (diameter = luminal circumference/π). Aneurysms were defined as an increase in vessel diameter of >50%.
Histomorphometry:
Aortas were fixed in 4% paraformaldehyde (PFA) overnight and embedded in paraffin. Serial 5 μm-thick transverse sections were stained in batches by Yale’s Research Histology Laboratory with hematoxylin and eosin (H&E), Elastin, Safranin O/Fast Green, Alcian Blue/Von Kossa, and Alizarin Red using standard techniques and an automated system. Digital H&E, Elastin, Safranin O/Fast Green, Alcian Blue/Von Kossa, and Alizarin Red-stained photographs of one section from each block were projected at final magnifications of X100 or X400. ImageJ software (NIH) was used for morphometric analyses. Morphometry of elastinstained transverse sections was performed using ImageJ software after outlining the internal and external elastic laminae. Lumen area was calculated within the internal elastic lamina (since it closely approximates the adjacent endothelial perimeter). Medial area was calculated between the internal and external elastic laminae.
Immunohistochemical staining.
Blocks were sectioned at 5 μm intervals using a Microm cryostat (for frozen blocks) or a Paraffin Microtome (for paraffin blocks). For frozen tissue sections, slides were fixed in acetone for 10 min at −20°C. For paraffin sections, slides were dewaxed in xylene, boiled for 20 min in citrate buffer (10 mM, pH 6.0) for antigen retrieval, and rehydrated. After washing three times with phosphate-buffered saline, tissue sections were incubated with primary antibodies diluted in blocking solution (10% BSA and horse serum in PBS) overnight at 4°C in a humidified chamber. For p-Smad2 staining, slides were denatured with 1.5 M HCl for 20 min prior to antibody labeling. Antibodies used in this study can be found in the Key Resources Table. Sections were washed three times with Tris-buffered saline, incubated with appropriate Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-conjugated secondary antibodies diluted 1:1000 in blocking solution for 1 hr at room temperature, washed again three times, and mounted on slides with ProLong Gold mounting reagent with DAPI (ThermoFisher Scientific P36935). All immunofluorescence micrographs were acquired using a Zeiss microscope or Leica SP8 confocal microscope. Images were captured using Volocity software (for Zeiss microscope) or Leica LAS-X (for Leica SP8 confocal microscope) and quantifications performed using ImageJ software (NIH).
Mass cytometry.
Antibodies used in this study can be found in the Key Resources Table. For CyTOF analysis, cells were washed, fixed and permeabilized using the Ebioscience Transcription Factor Fix/Perm Buffer for 45 min, washed in PBS/1% BSA/0.3% saponin, then labeled for intracellular markers. Cells were re-fixed in formalin, washed with Milli-Q water, and analyzed on a Helios CyTOF (Fluidigm) for mouse smooth muscle cells. Analyses were performed on cytometry data from 3–4 samples using the Barnes-Hut SNE implementation of t-SNE on Cytobank (http://www.cytobank.org) (Amir el et al., 2013; Rao et al., 2017). For Imaging Mass Cytometry (IMC), metal-labeled antibodies were commercially available or were obtained in carrier/protein-free buffer and then conjugated in-house using the MaxPar antibody conjugation kit according to the manufacturer’s protocol (Fluidigm). After we determined the percent yield by measurement of absorbance at 280 nm, the metal-labeled antibodies were diluted in Candor PBS Antibody Stabilization solution (Candor Bioscience) for long-term storage at 4°C. Tissue sections were blocked with 10% BSA and horse serum in PBS for 1 hr then incubated overnight at 4°C with antibodies. After a wash step with PBS, Iridium (191Ir and 193Ir) was added as a DNA1/DNA2 intercalator, and samples were incubated 2 hrs at room temperature. After being washed with PBS, samples were dried at room temperature before imaging mass cytometry (Fluidigm) (Giesen et al., 2014).
IMC image acquisition and image processing:
Samples were analyzed with Hyperion Imaging System (Fluidigm). The dried slide was loaded into the imaging module, where an optical previewing of the ROIs was recorded for laser ablation. For Figure 3C, areas of dimension 1158 × 1363 μm were acquired for Apoe−/− and 2352 × 11170 μm for TGFβR2iSMC-Apoe. Each ROI sample acquisition took 3.2 hours for Apoe−/− and 5.3 hours for TGFβR2iSMC-Apoe at an ablation frequency of 200Hz. For Mendeley Data Figure 2A http://dx.doi.org/10.17632/f8s8gjyyw6.1, areas of dimension 776 × 1090 μm were acquired for Apoe−/− and 1533 × 1354 μm for TGFβR2iSMC-Apoe. Each ROI sample acquisition took 1.73 hours for Apoe−/− and 4.2 hours for TGFβR2iSMC-Apoe at an ablation frequency of 200Hz. The resulting data files were stored in MCD binary format. The resolution of each IMC image shown is 1 μm for 1 pixel. Images were analyzed using MCD Viewer (Fluidigm, version 1.0). For all images shown, thresholding was performed in MCD Viewer with gamma set to 1, minimum color range set to zero, and maximum color range set to 100. Thresholding was performed to remove background signal, as assessed morphologically. For all comparative images, signal thresholds were set identically for each marker between samples to allow for unbiased comparison.
Image analysis software tools:
For each recorded ROI, 16-bit and 32-bit TIFF files were exported from MCD binary files using MCD™ Viewer 1.0 (Fluidigm). Cell-based morphological segmentation was performed with an image processing pipelines for the 16-bit images in and CellProfiler 3.1.8 (CP), a widely adopted software in the open source image analysis community which has been continually improved since its availability in 2005 to segment cell images using advanced algorithms. The segmentation pipeline was used to generate and save an unsigned 16-bit integer single cell mask TIFF image. Inputs of 16-bit TIFF images with their corresponding segmentation mask were uploaded in histoCAT (version 1.75), an open source software specifically developed for single-cell analysis of multiplexed cytometry data. Unbiased clustering was performed using the PhenoGraph algorithm and each cluster was assigned a phenotype based on its expression profile. Figure 3G Cluster 19 and Figure 4D Cluster 17 individual protein expression profiles are given in Table S2.
Mouse aorta digestion and single cell FACS sorting.
The ascending aorta of Apoe−/− and TGFβR2iSMC-Apoe mice were dissected from the mice and rinsed in cold PBS. The tissue was opened longitudinally and sliced into small fragments roughly 2 mm in length. The finely minced tissue was transferred to a digestion mix consisting of Hank’s balanced salt solution (Gibco) + 1 mg/ml collagenase type A (Sigma 10103578001) + 0.5 mg/ml elastase (Worthington LS006365) for 3 hrs at 37°C and pipetting every 30 min. DAPI (10 μg/ml; Sigma D9542) was used to detect dead cells. The cell suspension was passed through a 40 μm filter before sorting. A FACS machine (BD FACSAria) was used to sort GFP+RFP− live cells. Single cells were sorted into 0.4% BSA/PBS.
Chromatin immunoprecipitation (ChIP).
10×106 cells were first cross-linked with 1.5 mM EGS in PBS for 30 mins, followed by 1% paraformaldehyde for 10 minutes at room temperature. Glycine was added at a final concentration of 125 mM for 5 minutes at room temperature in order to quench the crosslinking reaction. The cross-linked material was washed once each with Buffer 1 (0.25% Triton, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH7.5) and Buffer 2 (200 mM NaCl, 10 mM EDTA, 0.5 mM EGTA, 10 mM Tris pH7.5) before resuspension in Shearing Buffer (0.1% SDS, 1 mM EDTA, 1 mM Tris-HCl pH7.6) with 1X Complete Protease Inhibitor Cocktail (Roche). Cell pellet was lysed in 130 μl SDS lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS, 50 mM PMSF) and sonicated using an S220 Focused-ultrasonicator (Covaris) to generate 200 to 600 bp fragments. Fragmented chromatin was centrifuged at 10,000 g for 5 minutes and the supernatants were diluted in ChIP dilution buffer (1% Triton, 150 mM NaCl, 20 mM Tris pH 8.0). Immunoprecipitation was performed by rotating samples at 4°C with magnetic beads (Dynabeads Protein A or G, Invitrogen) pre-bound with 5 μg of antibody. The beads were washed once each with low salt buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% Deoxycholate, 1% NP40, 1 mM EDTA), high salt buffer (50 mM Tris pH 8.0, 500 mM NaCl, 0.1% SDS, 0.5% Deoxycholate, 1% NP40, 1 mM EDTA), LiCl wash buffer (50 mM Tris pH 8.0, 250 mM LiCl, 0.5% Deoxycholate, 1% NP40, 1 mM EDTA), Morohashi RIPA buffer (50 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP40, 0.1% SDS), DOC/Triton Buffer (25 mM Tris pH7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton-X-100, 0.5% DOC), and twice with Tris-EDTA buffer (10 mM Tris pH 8.0, 1 mM EDTA). All washes took place on a rotator for 10 minutes at 4°C. Beads were treated twice with 100 μl elution buffer for 15 minutes on a shaker at room temperature, to obtain a total of 200 μl eluate. 8 μl of 5M NaCl was added to the eluate and the sample was reverse cross-linked overnight at 55°C. DNA was then purified using the QIAquick PCR Purification Kit (Qiagen). A list of all antibodies used in this study can be found in the Key Resources Table.
ChIP-qPCR.
ChIP DNA was analyzed by qPCR and data were normalized to input DNA. The locus-specific and non-specific binding primers used in qPCR are listed in Table S3.
ChIP-Seq analyses.
ChIP DNA samples was used in a 23.3 μl combined end repair and A-tailing reaction using a KAPA HyperPrep Kit (Roche 07962347001) for 30 minutes at 20°C followed by 30 minutes at 65°C. 10 μl of ligase buffer, 3.7 μl of Adapters and 3.3 μl ligase (KAPA Hyper Prep Kit) were added and incubated at 20°C for 4 hours. Double-stranded DNA fragments were purified from this reaction using KAPA Pure Beads (Roche 07983280001) and eluted in 22 μl 10 mM Tris pH8.0. Libraries were generated by PCR and size-selected on a 2% E-Gel EX agarose gel (Invitrogen) and fragments between 150 and 400 bp were extracted using a QIAEX II Gel Extraction Kit (Qiagen) performed at room temperature. Libraries were submitted to the Yale Center for Genome Analysis for quality control analyses and next-generation sequencing (Wang et al., 2018). Short reads from ChIP-Seq data sets were aligned to human reference genome GRCh38 (build 38) using BWA-MEM for PBS and TGFβ1 conditions. DeepTools (version 3.0.0) was then used to convert bam files to bigwig files using default options (Ramirez et al., 2016). The matrices were then computed and used for creating average profiles and signal visualization around promoter region of −5/+5kb and bin size of 25.
RNA-seq.
For population (bulk) RNA-seq, samples were lysed with RLT Plus buffer and RNA was extracted using the RNeasy Plus Mini Kit (Qiagen 74134). Full length RNA-seq libraries were prepared as previously described (Chen et al., 2019).
Droplet-based scRNA-seq library preparation and sequencing.
Sorted GFP+RFP− cells were processed for scRNA library preparation using the Chromium™ Single Cell Platform (10x Genomics) as per the manufacturer’s protocol. Briefly, single cell suspensions were partitioned into Gel Beads in Emulsion in the Chromium™ system (Chromium Single Cell 3’ Library & Gel Bead Kit v2, 4 rxns PN-120267, Chromium Single Cell A Chip Kit, 16 rxns PN-1000009, Chromium i7 Multiplex Kit, 96 rxns PN-120262) at Yale Center for Genome Analysis, followed by cell lysis and barcoded reverse transcription of RNA, cDNA amplification and shearing, and 5’ adaptor and sample index attachment. Final scRNA-seq libraries were sequenced on an Illumina NextSeq 500 (WuXiAppTec, China).
Bulk RNA-Seq data processing and differential analysis:
Next-generation whole-transcriptome sequencing was performed at the Broad Institute of MIT and Harvard. RNA-Seq reads from human samples (normal n=12 and aneurysm n=12) were aligned to a reference genome (human genome build 38 GRCh38) as previously described (Chen et al., 2019). The top 1000 differentially expressed genes (false discovery corrected p-value ≤ 0.01) from each comparison were used for further analysis.
Single cell RNA-Seq data processing:
To control for possible technical bias in our experimental design, data collection of transgenic mice at each timepoint was performed on the same day for both genotypes and by the same technician. This was followed by library preparation and sequencing using the same protocol and both genotypes for each timepoint were sequenced together as a single batch by the same technician. Single cell RNA sequencing (scRNA-Seq) was performed at the Shanghai Sequencing Lab of WuXiNextCODE using Illumina sequencers. We processed RNA-Seq short reads from each cell group using UMI-tools (version 0.5.3). Briefly, we first identified cell barcodes and UMI in the read sequence, which were then extracted from Read 1 and added to the Read 2 read name (Methods S1 CellData). We built a custom reference to include eGFP and tdTomato marker sequences (Methods S1 ddTomato eGFP Sequences) to the reference mouse genome build 38 (GRCm38) FASTA and annotation file (GENCODE mouse annotation, release M15). Splice aware aligner, STAR, was used to map extracted reads from Read 2 to the custom reference, followed by assigning reads to genes using the featureCounts tool from the subread package (version 1.6.0) (Liao et al., 2014). Quantification was then performed using UMI-tools count function to find the number of unique UMIs mapping to each gene. Cells with tdTomato expression were not detected. For each time point, count data were filtered by: (1) removing cells without a UMI count of the eGFP marker; (2) removing cells for which UMI count was less than 3,000 or total number of detected genes was less than 1,500; (3) removing genes that were not expressed in less than or equal to 5% of the remaining cells in at least one mouse genotype; and (4) filtering out cells in the Apoe−/− genotype with an ApoE UMI count greater than or equal to three, and cells in the TGFβR2iSMC-Apoe genotype with an ApoE and TGFβR2 UMI count greater than or equal to three (Mendeley Data Figure 4, http://dx.doi.org/10.17632/hrgxtyds44.1). In addition, we removed the ApoE and TGFβR2 UMI counts from the processed data matrix to prevent downstream analysis from segregating cells solely based on genotypes. Finalized UMI counts were normalized using the trimmed mean of M-values method (TMM) and used for downstream clustering and modeling. From the 2682 cells at the initiation of HCHFD (Apoe−/− n=1490, TGFβR2iSMC-Apoe n=1192), we included in 2542 cells (Apoe−/− n=1415, TGFβR2iSMC-Apoe n=1127) after filtering. From the 3049 cells one month after the initiation of HCHFD (Apoe−/− n=1599, TGFβR2iSMC-Apoe n=1450), we included 2790 cells (Apoe−/− n=1488, TGFβR2iSMC-Apoe n=1302) after filtering. From the 5491 cells two months after the initiation of HCHFD (Apoe−/− n=3065, TGFβR2iSMC-Apoe n=2426), we included 4912 cells (Apoe−/− n=2659, TGFβR2iSMC-Apoe n=2253) after filtering. From the 3280 cells four months after the initiation of HCHFD (Apoe−/− n=1528, TGFβR2iSMC-Apoe n=1752), we retained 2812 cells (Apoe−/− n=1235, TGFβR2iSMC-Apoe n=1577) after filtering. For Apoe−/−, from the filtered cells, a total of 2, 9, 27 and 21 cells did not have an eGFP UMI count at the initiation of HCHFD, one month, two months, and four months after initiation. In the case of TGFβR2iSMC-Apoe, from the filtered-out cells, a total of zero, 10, 3 and 35 cells did not have an eGFP UMI count at the initiation of HCHFD, one month, two months, and four months after initiation. For Apoe−/−, we retained a total of 9898, 9682, 9016 and 9481 genes, at the initiation of HCHFD, one month, two months, and four months after initiation. For TGFβR2iSMC-Apoe, we considered a total of 10077, 9706, 9553 and 9885 genes, at the initiation of HCHFD, and one month, two months, and four months after initiation.
Deep learning framework for single-cell RNA-seq analysis:
We carried out the unsupervised analysis and clustering of single cell RNA-seq data using a variational autoencoder (VAE) framework (Mendeley data Figure 1B http://dx.doi.org/10.17632/hrgxtyds44.1) as previously described (Chen et al., 2019). The hyper-parameters used in the analysis are presented in Methods S1 (Hyperparameter Search and Model Parameters). The code and version dependencies are available in https://github.com/wuxi-nextcode/CellStemCell_SmoothMuscle.
Data Assembly.
We applied our methodology to each time point combining the two genotypes. Genes not present in either sample were imputed as zero. The total number of genes used in the analysis were 10367, 10040, 9558 and 10742 at the initiation of HCHFD, and one month, two months, and four months after initiation, respectively.
Postulated differentiation networks of single cell RNA-Seq data:
We combined our results from the unsupervised analysis with Cell Differentiation Analysis Using Time-Series Single Cell RNA-seq Data (scdiff), which models differentiation networks from single cell time series RNA-seq data based on their expression similarity and potential protein-DNA interaction (Ding et al., 2018). Instead of using the clustering process implemented in scdiff, we imposed our results from our unsupervised analysis for each genotype independently. We then followed the scdiff workflow not allowing cells to relocate in future time points, under the assumption that at our initial time point the cells were differentiated. We used the parameters virtualAncestor=True, dsync=True, fChangeCut=1, together with the mouse TF-DNA interaction provided by the developers of scdiff. The code and version dependencies are available in https://github.com/wuxi-nextcode/CellStemCell_SmoothMuscle.
Data Assembly.
We ran analysis for each genotype taking the union of all the genes across the four time points. Genes not present in either sample were imputed as zero. In addition, we removed cells that were allocated in a cluster with genotype profile different than the cell genotype (Mendeley data Figure 1C–D http://dx.doi.org/10.17632/hrgxtyds44.1 and Methods S1 ClusterData and GeneData). We removed seven cells from Apoe−/− clusters (two cells from T2.22, and one cell from T0.4, T1.11, T2.24, T2.27 and T3.34), and three cells from TGFβR2iSMC-Apoe clusters (two cells from T0.1, and one cell from T1.17). For the Apoe−/− clusters we analyzed 6794 cells and 10338 genes, while for the TGFβR2iSMC-Apoe clusters we analyzed 6252 cells and 11221 genes.
Cells.
Primary mouse smooth muscle cells were isolated from mouse aorta as previously described. (Chen et al., 2016a) Aortas were predigested for 5 minutes at 37°C in HBSS (Gibco 14170–112) solution containing 1 mg/ml collagenase type A (Sigma 10103578001) to promote sharp removal of the adventitia under a dissecting microscope. The denuded vessels were transferred into 3 ml HBSS solution containing 2 mg/ml collagenase type A and 0.5 mg/ml elastase (Worthington LS006365) and incubated at 37°C for 30 minutes. Digestion was stopped with growth medium, the mixture was centrifuged, and the cells were resuspended in Claycomb medium (Sigma 51800C) supplemented with 10% fetal bovine serum (Life Technologies 16000–044), 10 units/ml Penicillin/ 10 μg/ml Strep (Gibco 15140–122), 2 mM L-glutamine (Gibco 25030–081), and cultured in 35 mm dishes. Primary mouse smooth muscle cells between passages 2 and 3 were used in all experiments.
Human 293T T17 cells (human embryonic kidney cells, ATCC CRL-11268) were maintained in Dulbecco’s modified Eagle’s medium (Gibco 11965–092) with 10% fetal bovine serum (Life Technologies 16000–044) and penicillin-streptomycin (15140–122, Gibco), and were grown at 37°C, 5% CO2. Human aortic smooth muscle cells (#C-007–5C), media (#M231–500), and supplements (SMGS: S-007–25; SMDS: S-008–5) were purchased from Thermofisher Scientific. The cells were grown at 37°C, 5% CO2 in Medium 231 supplemented with smooth muscle growth supplement (SMGS containing 4.9% FBS, 2 ng/ml FGF2, 0.5 ng/ml EGF, 5 ng/ml heparin, 2 μg/ml IGF-1, and 0.2 μg/ml BSA). For SMC differentiation, HASMC were incubated with Medium 231 containing smooth muscle differentiation supplement (SMDS containing 1% FBS and 30 μg/ml heparin) for different time points. Primary human aortic smooth muscle cells between passages 6 and 10 were used in all experiments.
Growth factor.
Recombinant human TGFβ1 (BioLegend 580702) solution in 30% Acetonitrile, 0.1% TFA (trifluoroacetic acid).
Generation of lentiviruses:
Human KLF2, KLF4, KLF5, and TGFβR2 shRNA lentiviral constructs were purchased from Sigma. For the production of shRNA lentivirus, 3.7 μg of Δ8.2, 0.2 μg of VSVG, and 2.1 μg of pLKO.1 carrying the control, KLF2, KLF4, KLF5, or TGFβR2 shRNA were co-transfected into 293T cells using X-tremeGENE 9 DNA Transfection Reagent (Sigma 6365787001). Forty-eight hr later the medium was harvested, cleared by 0.45 μm filter (PALL Life Sciences 4184), mixed with polybrene (5 μg/ml) (Sigma H9268), and applied to cells. After 6 hr incubation, the virus-containing medium was replaced by the fresh medium.
Beads uptake assay:
Primary human aortic smooth muscle cells were transduced with cholesterol, control, TGFβR2 shRNA, and KLF4 shRNA lentivirus. After 96 hrs of treatment, cells were change to fresh complete medium containing 1.5% vol/vol 0.84-μm polystyrene beads (Spherotech FP-0870–2) for 1.5 h. Cells were then stained with primary anti-LGALS3 antibody (1:50) overnight, secondary anti-goat Alexa568 antibody (1:1000) or secondary anti-Rabbit Alexa568 antibody (1:1000), finally counterstained with mounting solution with DAPI. Immunofluorescence images were acquired using a Leica SP8 confocal microscope and were captured using Leica LAS-X.
In vitro DiI-Ac-LDL labeling of smooth muscle cells:
For the Oil-Red-O staining, control and TGFβR2 knockdown HASMCs were seeded in 6-well culture dishes and were then incubated with Ac-LDL (10 μg/ml, 1.5 ml/well) for 72 h at 37°C. At the end of labeling, media were removed, cells were washed twice in probe-free medium, stained with Oil-Red-O for 30 min at room temperature, and pictures of the labeled cultures were taken. For Dil-Ac-LDL uptake assay, control and TGFβR2 knockdown HASMCs were seeded in 6-well culture dishes and were then incubated with DiI Ac-LDL (10 μg/ml, 1.5 ml/well) for different time points at 37°C. Then the labeled cells were washed once with PBS and then trypsinized to produce single cell suspensions. The trypsin was neutralized by washing the cells once in M231 containing 4.9% FBS. The Dil-Ac-LDL uptake was assessed using FACS (BD). Data analysis was performed using FlowJo analysis software (Treestart).
Cellular cholesterol esters measurement:
Control and TGFβR2 knockdown HASMCs were seeded in 6-well culture dishes and were then incubated with or without acetylated LDL (Ac-LDL; 10 μg/ml). After 72 hrs, intracellular total cholesterol, cholesterol esters, and free cholesterol content were measured using the Amplex Red Cholesterol Assay Kit (Molecular Probes; Invitrogen), according to the manufacturer’s instructions.
Analysis of multi-lineage differentiation:
For HASMC lineage differentiation experiments, after expansion in growth medium [Medium 231 supplemented with smooth muscle growth supplement (4.9% FBS, 2 ng/ml FGF2, 0.5 ng/ml EGF, 5 ng/ml heparin, 2 μg/ml IGF-1, and 0.2 μg/ml BSA)], primary human aortic smooth muscle cells were transduced with cholesterol, control, TGFβR2 shRNA, and KLF4 shRNA lentivirus for 96 hrs then were grown in StemPro osteogenic (Thermo Fisher Scientific A10072–01), chondrogenic (Thermo Fisher Scientific A10071–01), or adipogenic culture medium (Thermo Fisher Scientific A10070–01). Alizarin red (Electron Microscopy Sciences 26206–01) staining to detect matrix calcification was performed for 3 min on cultures grown in osteogenic medium for 21 days. Alkaline phosphatase staining to detect osteoblast was performed with the alkaline phosphatase kit and protocol (Promega S3771) on cultures grown in osteogenic medium for 21 days. Alcian blue (Electron Microscopy Sciences 26026–13) staining to detect chondrocyte proteoglycans was performed for 30 min on cultures in chondrogenic medium for 21 days. Oil red O (Sigma O0625) staining to detect adipocyte was performed for 30 min on cultures grown in adipogenic medium for 10 days. Images of 3 random fields were captured using a EVOS FL Cell Imaging System (ThermoFisher Scientific).
Laser microdissection (LMD), RNA isolation, and qRT-PCR:
For LMD, OCT-embedded frozen blocks were cut at 20 μm using the LMD6500 (Leica Microsystems) and collected into the cap of a 0.2 ml PCR microtube (11090615028007, Leica). The tissues were mounted on RNase-free POL Membrane Steel frame Slides (11505191, Leica). The presence of the laser-microdissected tissue sample in the cap of the microtube was confirmed by LMD microscopy. The laser-microdissected tissue samples were spun down to the bottom of microtubes by centrifugation at 15,000 xg for 1min. The precipitate was dissolved in 20 μl of lysis buffer from the PicoPure RNA Isolation Kit (Applied Biosystems/Life Technologies, cat. no. KIT0204). Isolation of RNA from LMD-harvested tissues was performed using the PicoPure RNA Isolation Kit according to manufacturer’s instructions. About 150 ng of DNAase-treated RNA was used as template for cDNA synthesis. Preamplification was performed on the cDNA samples prepared above. A mix was prepared containing 25 μL 2× TaqMan PreAmp Master Mix (4384266; Applied Biosystems) plus 12.5 μL 0.2× Pooled TaqMan Gene Expression Assays (0.05× each primer), and 12.5 μL cDNA sample. Following a brief vortex and centrifugation, the plate was transferred to a thermal cycler and subjected to the following thermal protocol: 95°C, 10 min; 10 cycles of (95°C, 15s; 60°C, 4 min); 99°C, 10 min. A mix was then prepared consisting of 1 μL TaqMan Gene Expression Assay, 5 μL preamplified cDNA products, 10 μL TaqMan Gene Expression Master Mix, plus 4 μL H2O, and 20 μL of this mix was dispensed to each well of a 96-well assay plate (MLL9601, Bio-Rad). PCR was performed using the thermal protocol (2 min of UNG incubation step at 50°C, 10 min of enzyme activation step at 95°C, followed by 40 cycles of PCR at 95°C for 15 s, 60°C for 60 s). Primers used in this study are listed in Table S3.
RNA isolation and qRT-PCR.
Cells were suspended in TRIzol Reagent (Invitrogen #15596018) and total RNA (QIAGEN #74134) were isolated according to the manufacturer’s instructions. Reverse transcriptions were performed by using iScript cDNA synthesis kit (Bio-Rad 170–8891). qRT-PCR was performed using Bio-Rad CFX94 (Bio-Rad) by mixing equal amount of cDNAs, iQ SYBR Green Supermix (Bio-Rad 170–8882) and gene specific primers (SABiosciences). Primers used in this study are listed in Table S3. All reactions were done in a 20 μl reaction volume in duplicate. Individual mRNA expression was normalized in relation to expression of endogenous β-actin. PCR amplification consisted of 10 min of an initial denaturation step at 95°C, followed by 46 cycles of PCR at 95°C for 15 s, 60°C for 30 s (for mRNA cDNA).
Western Blot Analysis.
Cells were lysed with HNTG lysis buffer (20 mM HEPES, pH 7.4/ 150 mM NaCl/ 10 % glycerol/ 1 % Triton-X 100/ 1.5 mM MgCl2/ 1.0 mM EGTA) containing complete mini EDTA-free protease inhibitors (Roche #11836170001) and phosphatase inhibitors (Roche #04906837001). 20 μg of total protein from each sample was resolved on Criterion TGX Precast Gels (Bio-Rad #567–1084) with Tris/Glycin/SDS Running Buffer (Bio-Rad #161–0772), transferred to nitrocellulose membranes (Bio-Rad #162–0094) and then probed with various antibodies. Chemiluminescence measurements were performed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific Prod #34080). Antibodies used in this study can be found in the Key Resources Table.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed with GraphPad Prism version 8 (GraphPad Software). Parameters such as sample size, the number of replicates, the number of independent experiments, measures of center, dispersion, and precision (mean ± SEM), and statistical significance are reported in Figures and Figure Legends. Results were considered statistically significant when P < 0.05 (*), P < 0.01 (**), P < 0.001 (***).
DATA AND CODE AVAILABILITY
High-throughput sequencing data associated with this study (scRNA-seq, ChIP-seq and bulk RNA-seq) have been deposited Gene Expression Omnibus (GEO140947, GSE141031, GSE141032). A detailed description of scRNAseq data analysis parameter can be found in Methods S1. All data associated with this study is publicly available from Mendeley http://dx.doi.org/10.17632/4mfgjpvvjm.1, http://dx.doi.org/10.17632/x3r8fcnkpc.1, http://dx.doi.org/10.17632/hrgxtyds44.1, http://dx.doi.org/10.17632/f8s8gjyyw6.1.
Supplementary Material
Highlights:
TGFβR2 ablation combined with hypercholesterolemia reprograms smooth muscle cells
Reprogrammed SMCs undergo clonal differentiation into varied mesenchymal lineages
Loss of normal aortic SMCs and increased non-SMC mass induces aortic aneurysms
ACKNOWLEDGMENTS
This work was supported, in part, by National Nature Science Foundation of China Grant 81600365 (GL), a Joint Biology Consortium Microgrant provided under NIH grant P30AR070253 (PYC), NIH grants NHLBI HL084312 (SB and EAF), NIH NCI R01CA212376-03S1 (ZT), R01 HL139796 and HL086418 (JH and GT). AI 089992 (RM), R35HL135820 (CF), HL135582 (MS and MAS), and S10 OD020142. We are grateful to R. Webber and L. Coon for maintaining the mice used in this study. We thank Ms. Shelly Ren (Yale CyTOF Facility), Eric Swanson (Fluidigm), and Andrew Draghi (Fluidigm).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Drs. J. Malagon-Lopez, S. Gujja and TW Chittenden are employees of WuxiNextCode, Inc.
SUPPLEMENTAL INFORMATION
Supplemental Tables
Table S1: Human and mouse aorta analysis
Table S2: IMC Cluster protein expression profile.
Table S3 (related to Quantitative Reverse Transcription PCR” in the STAR Methods): List of Primers used for qRT-PCR.
Supplemental methods
Methods S1 (related to Figure 5 and Figure 6): scRNAseq analysis parameters.
- Hyperparameter search
- Model Parameters
- CellData
- tdTomato eGFP Sequences
- ClusterData
- GeneData
References
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nature biotechnology 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelov SN, Hu JH, Wei H, Airhart N, Shi M, and Dichek DA (2017). TGF-beta (Transforming Growth Factor-beta) Signaling Protects the Thoracic and Abdominal Aorta From Angiotensin II-Induced Pathology by Distinct Mechanisms. Arterioscler Thromb Vasc Biol 37, 2102–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S, Gleason RL, Rjagopal KR, and Humphrey JD (2007). Theory of small on large: Potential utility in computations of fluid–solid interactions in arteries. Comput Methods Appl Mech Eng 196, 3070–3078. [Google Scholar]
- Bersi MR, Khosravi R, Wujciak AJ, Harrison DG, and Humphrey JD (2017). Differential cell-matrix mechanoadaptations and inflammation drive regional propensities to aortic fibrosis, aneurysm or dissection in hypertension. Journal of the Royal Society, Interface / the Royal Society 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P-Y, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, Gujja S, Cilfone NA, Kauffman KJ, Sun L, et al. (2019). Endothelial TGF-beta signaling drives vascular inflammation and atherosclerosis Nature Metabolism (in press). [DOI] [PMC free article] [PubMed]
- Chen PY, Qin L, Li G, Tellides G, and Simons M (2016a). Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFbeta)-dependent smooth muscle cell phenotype modulation. Scientific reports 6, 33407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PY, Qin L, Li G, Tellides G, and Simons M (2016b). Smooth muscle FGF/TGFbeta cross talk regulates atherosclerosis progression. EMBO molecular medicine. [DOI] [PMC free article] [PubMed]
- Ding J, Aronow BJ, Kaminski N, Kitzmiller J, Whitsett JA, and Bar-Joseph Z (2018). Reconstructing differentiation networks and their regulation from time series single-cell expression data. Genome Res. [DOI] [PMC free article] [PubMed]
- Ferruzzi J, Collins MJ, Yeh AT, and Humphrey JD (2011). Mechanical assessment of elastin integrity in fibrillin-1-deficient carotid arteries: implications for Marfan syndrome. Cardiovascular research 92, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, van Erp C, Gerber EE, Parker SJ, Sauls K, et al. (2014). Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. The Journal of clinical investigation 124, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schuffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nature methods 11, 417–422. [DOI] [PubMed] [Google Scholar]
- Gleason RL, and Humphrey JD (2005). Effects of a sustained extension on arterial growth and remodeling: a theoretical study. Journal of biomechanics 38, 1255–1261. [DOI] [PubMed] [Google Scholar]
- Godmann M, Kosan C, and Behr R (2010). Kruppel-like factor 4 is widely expressed in the mouse male and female reproductive tract and responds as an immediate early gene to activation of the protein kinase A in TM4 Sertoli cells. Reproduction 139, 771–782. [DOI] [PubMed] [Google Scholar]
- Guo DC, Papke CL, He R, and Milewicz DM (2006). Pathogenesis of thoracic and abdominal aortic aneurysms. Annals of the New York Academy of Sciences 1085, 339–352. [DOI] [PubMed] [Google Scholar]
- Hayenga HN, Trache A, Trzeciakowski J, and Humphrey JD (2011). Regional atherosclerotic plaque properties in ApoE−/− mice quantified by atomic force, immunofluorescence, and light microscopy. Journal of vascular research 48, 495–504. [DOI] [PubMed] [Google Scholar]
- Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr., Eagle KA, Hermann LK, Isselbacher EM, Kazerooni EA, et al. (2010). 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 121, e266–369. [DOI] [PubMed] [Google Scholar]
- Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, et al. (2015). Postnatal Deletion of the Type II Transforming Growth Factor-beta Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler Thromb Vasc Biol 35, 2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi S, Goldstein JL, Brown MS, Herz J, and Burns DK (1994). Massive xanthomatosis and atherosclerosis in cholesterol-fed low density lipoprotein receptor-negative mice. The Journal of clinical investigation 93, 1885–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isselbacher EM (2005). Thoracic and abdominal aortic aneurysms. Circulation 111, 816–828. [DOI] [PubMed] [Google Scholar]
- Jiang J, Chan YS, Loh YH, Cai J, Tong GQ, Lim CA, Robson P, Zhong S, and Ng HH (2008). A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 10, 353–360. [DOI] [PubMed] [Google Scholar]
- Kramann R, Goettsch C, Wongboonsin J, Iwata H, Schneider RK, Kuppe C, Kaesler N, Chang-Panesso M, Machado FG, Gratwohl S, et al. (2016). Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell stem cell 19, 628–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladich E, Yahagi K, Romero ME, and Virmani R (2016). Vascular diseases: aortitis, aortic aneurysms, and vascular calcification. Cardiovascular pathology : the official journal of the Society for Cardiovascular Pathology 25, 432–441. [DOI] [PubMed] [Google Scholar]
- Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, et al. (2014). Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. The Journal of clinical investigation 124, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lindsay ME, and Dietz HC (2014). The genetic basis of aortic aneurysm. Cold Spring Harbor perspectives in medicine 4, a015909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, et al. (2012). Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature genetics 44, 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat Z, Ait-Oufella H, and Tedgui A (2017). The Pathogenic Transforming Growth Factor-beta Overdrive Hypothesis in Aortic Aneurysms and Dissections: A Mirage? Circulation research 120, 1718–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai ICL, Li S, Romay M, Lam L, Lu Y, Huang J, Dillard N, Zemanova M, Rubbi L, Wang Y, et al. (2017). Cardiac Fibroblasts Adopt Osteogenic Fates and Can Be Targeted to Attenuate Pathological Heart Calcification. Cell stem cell 20, 218–232 e215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, et al. (2017). Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Haakonsson AK, Madsen JGS, Larsen M, Forss I, Madsen MR, Van Hauwaert EL, Wiwie C, Jespersen NZ, Tencerova M, et al. (2019). Osteogenesis depends on commissioning of a network of stem cell transcription factors that act as repressors of adipogenesis. Nature genetics 51, 716–727. [DOI] [PubMed] [Google Scholar]
- Rong JX, Shapiro M, Trogan E, and Fisher EA (2003). Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A 100, 13531–13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR Jr., Owens GK, and Ailawadi G (2013). KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation 128, S163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapiro D, Jackson HW, Raghuraman S, Fischer JR, Zanotelli VRT, Schulz D, Giesen C, Catena R, Varga Z, and Bodenmiller B (2017). histoCAT: analysis of cell phenotypes and interactions in multiplex image cytometry data. Nature methods 14, 873–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, et al. (2015). KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med 21, 628–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoter M, Janosch A, Barsacchi R, and Bickle M (2019). CellProfiler and KNIME: Open-Source Tools for High-Content Screening. Methods Mol Biol 1953, 43–60. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gearhart MD, Lee YW, Kumar I, Ramazanov B, Zhang Y, Hernandez C, Lu AY, Neuenkirchen N, Deng J, et al. (2018). A Non-canonical BCOR-PRC1.1 Complex Represses Differentiation Programs in Human ESCs. Cell stem cell 22, 235–251 e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Schmit BM, Fu C, DeSart K, Oh SP, Berceli SA, and Jiang Z (2016). Smooth muscle cell-specific Tgfbr1 deficiency promotes aortic aneurysm formation by stimulating multiple signaling events. Scientific reports 6, 35444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Yamashita M, and Hayashi M (2012). Kruppel-like factor 4 contributes to high phosphate-induced phenotypic switching of vascular smooth muscle cells into osteogenic cells. The Journal of biological chemistry 287, 25706–25714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data associated with this study (scRNA-seq, ChIP-seq and bulk RNA-seq) have been deposited Gene Expression Omnibus (GEO140947, GSE141031, GSE141032). A detailed description of scRNAseq data analysis parameter can be found in Methods S1. All data associated with this study is publicly available from Mendeley http://dx.doi.org/10.17632/4mfgjpvvjm.1, http://dx.doi.org/10.17632/x3r8fcnkpc.1, http://dx.doi.org/10.17632/hrgxtyds44.1, http://dx.doi.org/10.17632/f8s8gjyyw6.1.