

Abstract
Life depends on a membrane's ability to precisely control the level of solutes in the aqueous compartments, inside and outside, bathing the membrane. The membrane determines what solutes enter and leave a cell. Transmembrane transport is controlled by complex interactions between membrane lipids, proteins, and carbohydrates. How the membrane accomplishes these tasks is the topic of this chapter.
Keywords: Biological membrane, Cells, Fick's laws, Membrane, Membrane transport, Semipermeability, Solutes
1. Introduction
Life depends on a membrane's ability to precisely control the level of solutes in the aqueous compartments, inside and outside, bathing the membrane. The membrane determines what solutes enter and leave a cell. Transmembrane transport is controlled by complex interactions between membrane lipids, proteins, and carbohydrates. How the membrane accomplishes these tasks is the topic of Chapter 19.
A biological membrane is semipermeable, meaning it is permeable to some molecules, most notably water, while being very impermeable to most solutes (various biochemicals and salts) found in the bathing solution. This very important concept of unequal transmembrane distribution and, hence, permeability between water and other solutes came out of the pioneering work of Charles Overton in the 1890s (see Chapter 2). How does a biological membrane accomplish semipermeability? The barrier to solute movement is largely provided by the membrane's hydrophobic core, a very thin (∼40 Å thick), oily layer. The inherent permeability of this core varies from membrane to membrane. Generally, the more tightly packed the lipids comprising the bilayer, the lower its permeability will be. Lipid bilayers are very impermeable to most solutes because of their tight packing. Fig. 19.1 depicts the membrane permeability of a variety of common solutes [1]. Note the data are presented as a log scale of solute permeability (P in cm/s) and ranges from Na+ = 10−12 cm/s to water = 0.2 × 10−2 cm/s, spanning almost 10 orders of magnitude!
Figure 19.1.
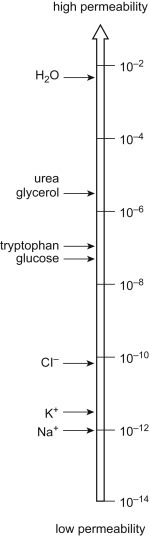
Log of the permeability (P in cm/s) across lipid bilayer membranes for common solutes ranging from Na+ (10−12 cm/s) to water (0.2 × 10−2 cm/s). This range spans almost 10 orders of magnitude [1].
Lipid bilayer permeability is not a constant but instead is affected by environmental factors. For example, LUVs (large unilamellar veicles) made from DPPC (16:0, 16:0 PC) have a sharp phase transition temperature, T m, of 41.3°C. At temperatures well below T m, the LUVs are in the tightly packed gel state and permeability is extremely low. At temperatures well above T m, the LUVs are in the loosely packed liquid disordered state (l d, also called the liquid crystalline state) and permeability is high. However, maximum permeability is not found in the l d state, but rather at the T m [2]. As the LUVs are heated from the gel state and approach the T m, domains of l d start to form in the gel state. Solutes can then pass more readily through the newly formed l d domains than the gel domains resulting in an increase in permeability. At T m there is a maximum amount of coexisting gel and l d state domains that exhibit extremely porous domain boundaries. It is through these boundaries that most permeability occurs. As the temperature is further increased, the LUVs pass into the l d state and the interface boundaries disappear, reducing permeability to that observed for the single-component l d state. Thus, maximum permeability is observed at the T m.
1.1. Fick's First Law
The tendency for solutes to move from a region of higher concentration to one of lower concentration was first defined in 1855 by the physiologist Adolf Fick (Fig. 19.2 ). His work is summarized in what is now the very well-known Fick's Laws of Diffusion [3]. The laws apply to both free solution and diffusion across membranes. Fick developed his laws by measuring concentrations and fluxes of salt diffusing between two reservoirs through connecting tubes of water.
Figure 19.2.

Adolf Fick, 1829–1901.
Fick's First Law describes diffusion as:
Where D = diffusion coefficient (bigger molecules have lower Ds); A = cross-sectional area over which diffusion occurs; dc/dx is the solute concentration gradient (diffusion occurs from a region of higher concentration to one of lower concentration).
The relationship between a solute's molecular weight and its diffusion coefficient is shown in Table 19.1 . Large solutes have low diffusion coefficients and therefore diffuse more slowly than small solutes. The diffusion rate for a particular solute under physiological conditions is a constant and cannot be increased. This defines the theoretical limit for an enzymatic reaction rate and also limits the size of a cell. If a solute starts at the center of a bacterial cell, it takes about 10−3 s to diffuse to the plasma membrane. For this reason, typical cells are microscopic (see Chapter 1). At about 3.3 pounds and the size of a cantaloupe, the largest cell on Earth today is the ostrich egg. However a fossilized dinosaur egg in the American Museum of Natural History in New York is about the size of basketball. Since an egg's only function is to store nutrients for a developing embryo, its size is many orders of magnitude larger than a normal cell.
Table 19.1.
Relationship Between a Solute's Molecular Weight and Its Diffusion Coefficient, D
| Compound | O2 | Acetyl choline | Sucrose | Serum albumin |
|---|---|---|---|---|
| D (cm2/s × 106) | 19.8 | 5.6 | 2.4 | 0.7 |
| Molecular weight | 32 | 182 | 342 | 69,000 |
1.2. Osmosis
Osmosis is a special type of diffusion, namely the diffusion of water across a semipermeable membrane. Water readily crosses a membrane down its potential gradient from high to low potential (Fig. 19.3 ) [4]. Osmotic pressure is the force required to prevent water movement across the semipermeable membrane. Net water movement continues until its potential reaches zero. An early application of the basic principles of osmosis came from the pioneering work on hemolysis of red blood cells by William Hewson in the 1770s (see Chapter 2). It has also been discussed that MLVs (multilamellar vesicles, liposomes) behave as almost perfect osmometers, swelling in hypotonic solutions and shrinking in hypertonic solutions (see Chapter 3) [5], [6]. Liposome swelling and shrinking can be easily followed by changes in absorbance due to light scattering using a simple spectrophotometer. Therefore, osmosis has been investigated for many years using common and inexpensive methodologies and a lot is known about the process.
Figure 19.3.
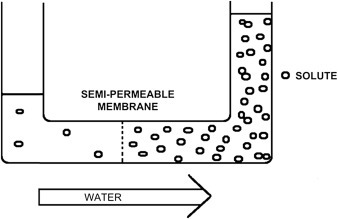
Osmosis and osmotic pressure. Water is placed in a U-shaped tube where each of the tube arms is separated by a semipermeable membrane with pores of a size that water can easily pass through but a solute cannot. Upon addition of the solute to the tube's right arm, water diffuses from left to right (high water potential to low). The column of water in the tube's right arm (the one containing the solute) rises until the extra weight of the column equals the osmotic pressure caused by the solute. A pump could then be used to counter the osmotic pressure whereupon the solution columns in the right and left arms of the tube are made the same. The pump pressure required to equalize the height of the two columns is the osmotic pressure [4]. Note a small amount of the solute leaks from right to left since no filter is perfect.
Membranes are rarely, if ever, perfectly semipermeable. Deviation from ideality is defined by a reflection coefficient (σ). For an ideal semipermeable membrane where a solute is totally impermeable, σ = 1. If a solute is totally permeable (its permeability is equal to water), σ = 0. Biological membranes are excellent semipermeable barriers with σ = 0.75 to 1.0.
2. Simple Passive Diffusion
Movement of solutes across membranes can be divided into two basic types: passive diffusion and active transport [7]. Passive diffusion requires no additional energy source other than what is found in the solute's electrochemical (concentration) gradient and results in the solute reaching equilibrium across the membrane. Passive diffusion can be either simple passive diffusion where the solute crosses the membrane anywhere by simply dissolving into and diffusing through the lipid bilayer, or facilitated passive diffusion where the solute crosses the membrane at specific locations where diffusion is assisted by solute-specific facilitators or carriers. Active transport requires additional energy, often in the form of ATP, and results in a nonequilibrium, net accumulation (uptake) of the solute on one side of the membrane. The basic types of membrane transport, simple passive diffusion, facilitated diffusion (by channels and carriers) and active transport are summarized in Fig. 19.4 [8]. There are countless different examples of each type of membrane transport process [7]. Only a few representative examples will be discussed here.
Figure 19.4.
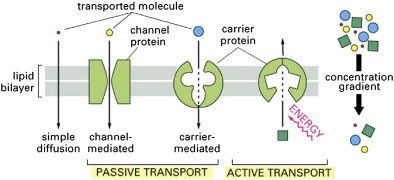
Basic types of membrane transport, simple passive diffusion, facilitated diffusion (by channels and carriers), and active transport [8].
Even simple passive diffusion requires energy to cross a bilayer membrane. In order to cross a membrane, the solute must first lose its waters of hydration, diffuse across the membrane, and then regain its waters on the opposite side. The limiting step involves the energy required to lose the waters of hydration. Table 19.2 shows the relationship between the waters of hydration (proportional to the number of —OH groups on a homologous series of solutes) and the activation energy for transmembrane diffusion. As the number of waters of hydration increases from glycol < glycerol < erythritol, the activation energy for diffusion also increases. The activation energy compares very well with the energy of hydration.
Table 19.2.
Relationship Between the Waters of Hydration (Number of —OH Groups on a Homologous Series of Solutes) and the Activation Energy for Transmembrane Diffusion
| Solute | Activation energy (kJ/mol) |
|---|---|
| Glycol (HO—CH2—CH2—OH) | 60 |
| Glycerol (HO—CH2—CH(OH)—CH2—OH) | 77 |
| Erythritol (HO—CH2—CH(OH)—CH(OH)—CH2—OH) | 87 |
However, water diffusion does not fit this model. Water permeability is just too high. Several possibilities have been suggested to account for the abnormally high membrane permeability of water:
-
1.
Water is very small and so it just dissolves in bilayers better than larger solutes.
-
2.
Due to its size, water can readily enter very small statistical pores (∼4.2 Å in diameter). Statistical pores result from the simultaneous lateral movement of adjacent membrane phospholipids in opposite directions. Statistical pores have only a fleeting existence and cannot be isolated or imaged.
-
3.
Passage down water chains.
-
4.
Water can be carried down kinks in acyl chains that result from acyl chain melting (see lipid melting in Chapter 9).
-
5.
Water may rapidly cross membranes through nonlamellar regions (eg, micelles, cubic or HII phase—see Chapter 10).
-
6.
High water permeability will occur at regions of packing defect (eg, surface of integral membrane proteins, boundary between membrane domains).
-
7.
Through pores or channels used to conduct ions.
-
8.
Through specific water channels known as aquaporins (see below, Chapter 19, Section 3.5).
The only molecules that can cross a membrane by simple passive diffusion are water, small noncharged solutes, and gasses. Charged or large solutes are virtually excluded from membranes and so require more than just simple passive diffusion to cross a membrane.
3. Facilitated Diffusion
Facilitated diffusion (also known as carrier-mediated diffusion) is, like simple passive diffusion, dependent on the inherent energy in a solute gradient. No additional energy is required to transport the solute and the final solute distribution reaches equilibrium across the membrane. Facilitated diffusion, unlike simple passive diffusion, requires a highly specific transmembrane integral protein or carrier to assist in the solute's membrane passage. Facilitators come in two basic types: carriers and gated channels. Facilitated diffusion exhibits Michaelis-Menton saturation kinetics (Fig. 19.5 , Part A, right), indicating the carrier has an enzyme-like active site. Like enzymes, facilitated diffusion carriers exhibit saturation kinetics and recognize their solute with exquisite precision, easily distinguishing chemically similar isomers like d-glucose from l-glucose. Fig. 19.5 (Part A) compares simple passive diffusion to facilitated diffusion. The figure is not to scale, however, as facilitated diffusion is orders of magnitude faster than simple passive diffusion.
Figure 19.5.
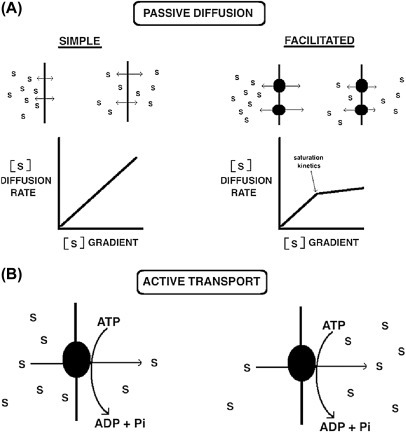
(A) Simple passive diffusion (top, left) and facilitated passive diffusion (top, right) both result in a final equilibrium distribution of a solute across the membrane. For a noncharged solute, the final distribution of the solute would find equal amounts of S on both sides of the membrane. Facilitated diffusion employs a specific transporter and exhibits Michaelis–Menten saturation kinetics. (A, center right) Active transport (bottom) utilizes energy, often in the form of ATP, to drive solute uptake against its gradient resulting in a net accumulation of the solute.
3.1. Glucose Transporter
A well-studied example of a facilitated diffusion carrier is the glucose transporter, or GLUT [9]. From the activation energies for transmembrane simple passive diffusion of glycol, glycerol and erythritol presented in Table 19.2, it can be estimated that the activation energy for glucose should be well over 100 kJ/mol, but instead it is only 16 kJ/mol. This large discrepancy is attributed to the presence of a glucose-facilitated diffusion carrier. Fig. 19.6 demonstrates the mode of action of one of these transporters, GLUT-1, from the erythrocyte [10]. GLUTs occur in nearly all cells and are particularly abundant in cells lining the small intestine. GLUTs are but one example in a superfamily of transport facilitators. GLUTs are integral membrane proteins whose membrane-spanning region is composed of 12 α-helices. GLUTs function through a typical membrane transport mechanism [10]. Glucose binds to the membrane outer surface site causing a conformational change associated with transport across the membrane. At the inner side of the membrane, glucose is released into the internal aqueous solution (Fig. 19.6).
Figure 19.6.
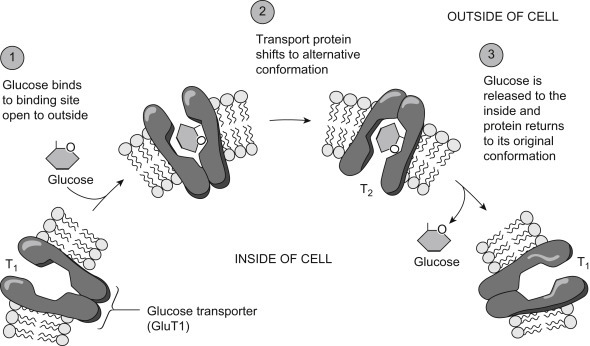
Glucose-facilitated diffusion transporter GLUT-1 [10].
3.2. Potassium Channels
In virtually all organisms there exists a wide variety of ion channels, the most widely distributed being potassium channels [11]. There are four basic classes of potassium channels, all of which provide essential membrane-associated functions including setting and shaping action potentials and hormone secretion:
-
1.
Calcium-activated potassium channel
-
2.
Inwardly rectifying potassium channel
-
3.
Tandem pore domain potassium channel
-
4.
Voltage-gated potassium channel
Potassium channels are composed of four protein subunits that can be the same (homotetramer) or closely related (heterotetramer). All potassium channel subunits have a distinctive pore-loop structure that sits at the top of the channel and is responsible for potassium selectivity [12]. This is often referred to as a selectivity or filter loop. The selectivity filter strips the waters of hydration from the potassium ion, allowing it into the channel. Farther down the structure is a 10-Å-diameter, transmembrane, water-filled central channel that conducts potassium across the membrane. Elucidating the three-dimensional structure of this important integral membrane protein by X-ray crystallography (Fig. 19.7 ) [12] was a seminal accomplishment in the field of membrane biophysics. For this work from 1998, Rod MacKinnon (Fig. 19.8 ) of Rockefeller University was awarded the 2003 Nobel Prize in Chemistry. Until the potassium channel work, just obtaining the structure of non–water-soluble proteins was next to impossible. MacKinnon's work elucidated not only the structure of the potassium channel but also its molecular mechanism. It has served as a blueprint for determining the structure of other membrane proteins and has greatly stimulated interest in the field.
Figure 19.7.
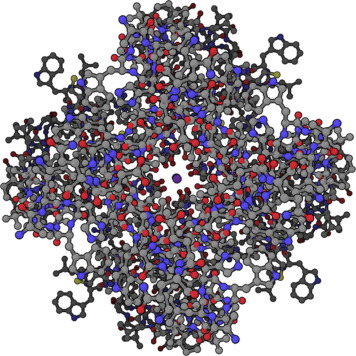
Three-dimensional structure of the potassium channel [12]. The channel itself is the clear opening in the center of the structure and a single K+ is shown in the center of the channel.
Figure 19.8.

Rod MacKinnon, 1956–.
3.3. Sodium Channel
In some ways, Na+ channels [13] parallel the action of K+ channels. They are both facilitated diffusion carriers that conduct the cation down the ion's electrochemical gradient. In excitable cells such as neurons, myocytes, and some glia, Na+ channels are responsible for the rising phase of action potentials (see Chapter 18). Therefore agents that block Na+ channels also block nerve conduction and so are deadly neurotoxins. There are two basic types of Na+ channels: voltage-gated and ligand-gated. The opening of a Na+ channel has a selectivity filter that attracts Na+. From there the Na+ ions flow into a constricted part of the channel that is about 3–5 Å wide. This is just large enough to allow the passage of a single Na+ with one attached water. Since the larger K+ cannot squeeze through, the channel is selective for Na+. Of particular interest are two extremely potent biological toxins, tetrodotoxin (TTX) and saxitoxin (STX) (Fig. 19.9 , [14]), that, in seafood, have killed and injured many humans. Both toxins shut down Na+ channels by binding from the extracellular side.
Figure 19.9.
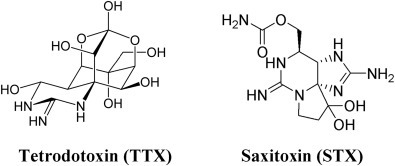
Structures of the extremely potent neurotoxins, tetrodotoxin (TTX) and saxitoxin (STX). Both neurotoxins function by blocking the Na+ channel.
TTX is encountered primarily in puffer fish but also in porcupine fish, ocean sunfish, and triggerfish. TTX (Fig. 19.9, left) is a potent neurotoxin that blocks Na+ channels while having no effect on K+ channels. Puffer fish is the second most poisonous vertebrate in the world, trailing only the Golden Poison Frog that is endemic to the rain forests on the Pacific Coast of Colombia. In some parts of the world puffer fish are considered to be a delicacy but must be prepared by chefs who really know their business, as a slight error can be fatal. Puffer poisoning usually results from consumption of incorrectly prepared puffer soup, and TTX has no known antidote!
Saxitoxin (STX, Fig. 19.9, right) is a Na+ channel–blocking neurotoxin produced by some marine dinoflagellates that can accumulate in shellfish during toxic algal blooms known as Red Tide. Saxitoxin is one of the most potent natural toxins, and it has been estimated that a single contaminated mussel has enough STX to kill 50 humans! STX's toxicity has not escaped the keen eye of the United States military, which has weaponized the toxin and given it the designation TZ.
3.4. Solute Equilibrium
The driving force for transmembrane solute movement by simple or passive diffusion is determined by the free energy change, ΔG.
Where ΔG is the free energy change; is the solute concentration on the right side of a membrane; [s o] is the solute concentration on the left side of a membrane; R is the gas constant; T is the temperature in K; Z is the charge of the solute; F is the Faraday; ΔΨ is the transmembrane electrical potential.
Solute movement will continue until ΔG = 0. If ΔG is negative, solute movement is left to right (it is favorable as drawn). If ΔG is positive, solute movement is right to left (it is unfavorable in the left-to-right direction) or energy must be added for the solute to go from left to right. The equation has two parts; a transmembrane chemical gradient and a transmembrane electrical gradient (ΔΨ). The net movement of a solute is therefore determined by a combination of the solute's chemical gradient and an electrical gradient inherent to the cell. If the solute has no charge, Z = 0 (as is the case for glucose) and the right hand part of the equation (ZFΔΨ) drops out. Therefore, the final equilibrium distribution of glucose across the membrane will have the internal glucose concentration equal to the external glucose concentration and is independent of ΔΨ, the electrical potential. At equilibrium for a noncharged solute, and ΔG can only be = zero if .
The situation for a charged solute like K+ is more complicated. The net ΔG is determined by both the chemical gradient and electrical gradient (ΔΨ). The ΔΨ results from the sum of all charged solutes on both sides of the membrane, not just K+. Therefore even if the K+ concentration is higher inside the cell than outside (the chemical gradient is unfavorable for K+ uptake), the ΔΨ may be in the correct direction (negative interior) and of sufficient magnitude to drive K+ uptake against its chemical gradient.
3.5. Aquaporins
Aquaporins are also known as water channels and are considered to be “the plumbing system for cells” [15], [16]. For decades it was assumed that water simply leaked through biological membranes by numerous processes described above (Chapter 19, Section 2). However, these methods of water permeability could not come close to explaining the rapid movement of water across some cells. Although it had been predicted that water pores must exist in very leaky cells, it was not until 1992 that Peter Agre (Fig. 19.10 ) at Johns Hopkins University identified a specific transmembrane water pore that was later called aquaporin-1. For this accomplishment Agre shared the 2003 Nobel Prize in Chemistry with Rod MacKinnon for his work on the potassium channel. Aquaporins are usually specific for water permeability and exclude the passage of other solutes. A type of aquaporin known as aqua-glyceroporins can also conduct some very small uncharged solutes such as glycerol, CO2, ammonia, and urea across the membrane. However, all aquaporins are impermeable to charged solutes. Water molecules traverse the aquaporin channel in single file (Fig. 19.11 ) [17].
Figure 19.10.

Peter Agre, 1949–.
Figure 19.11.
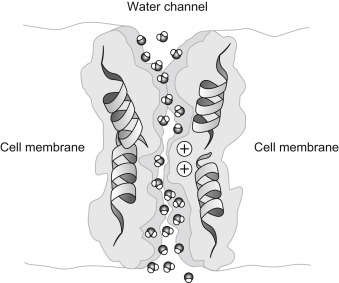
Aquaporin. Water molecules pass through the aquaporin channel in single file.
4. Active Transport
A characteristic of all living membranes is the formation and maintenance of transmembrane gradients of all solutes including salts, biochemicals, macromolecules, and even water. In living cells, large gradients of Na+ and K+ are particularly important. Typical cell concentrations are:
| Cell interior: | 400 mmol/L K+, 50 mmol/L Na+ |
| Cell exterior: | 20 mmol/L K+, 440 mmol/L Na+ |
Living cells will also have a ΔΨ from −30 to −200 mV (negative interior) resulting from the uneven distribution of all ionic solutes including Na+ and K+. The chemical and electrical gradients are maintained far from equilibrium by a multitude of active transport systems. Active transport requires a form of energy (often ATP) to drive the movement of solutes against their electrochemical gradient, resulting in a nonequilibrium distribution of the solute across the membrane. A number of nonexclusive and overlapping terms are commonly used to describe the different types of active transport. Some of these are depicted in Fig. 19.12 [18].
Figure 19.12.
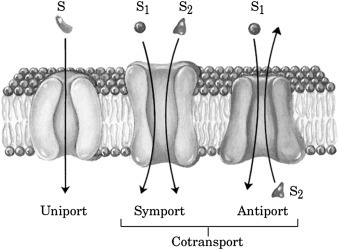
Basic types of active transport [18].
4.1. Primary Active Transport
Primary active transport is also called direct active transport or uniport. It involves using energy (usually ATP) to directly pump a solute across a membrane against its electrochemical gradient.
The most studied example of primary active transport is the plasma membrane Na+,K+-ATPase discussed below (Chapter 19, Section 4.2). Other familiar examples of primary active transport are the redox H+-gradient generating system of mitochondria (see Chapter 18), the light-driven H+-gradient generating system of photosynthetic thylakoid membranes, and the ATP-driven acid (H+) pump found in the epithelial lining of the stomach. There are four basic types of ATP-utilizing primary active transport systems (Table 19.3 ).
Table 19.3.
Four Types of ATP-Using Primary Active Transport Systems
| ATP-using primary active transport systems | Example |
|---|---|
| P-type | Na+,K+-ATPase Ca2+ pump H+ acid pump |
| F-type | Mitochondrial ATP synthase Chloroplast ATP synthase |
| V-type | Vacuolar ATPase |
| ABC (ATP binding cassette transporter) | Many |
4.2. Na+,K+-ATPase
Arguably the most important active transport protein is the plasma membrane-bound Na+,K+-ATPase. This single enzyme accounts for one-third of human energy expenditure and is often referred to as the “pacemaker for metabolism.” As a result the Na+,K+-ATPase has been extensively studied for more than 50 years. The enzyme was discovered in 1957 by Jens Skou (Fig. 19.13 ) who, 40 years later, was awarded the 1997 Nobel Prize in Chemistry.
Figure 19.13.

Jens Skou, 1918–.
As is often the case in biochemistry, a serendipitous discovery of a natural product from the jungles of Africa has been instrumental in unraveling the enzyme's mechanism of action. The compound is ouabain (Fig. 19.14 ), a cardiac glycoside first discovered in a poison added to the tip of Somali tribesmen's hunting arrows. In fact the name ouabain comes from the Somali word waabaayo that means “arrow poison.” The sources of ouabain are ripe seeds and bark of certain African plants and ouabain is potent enough to kill a hippopotamus with a single arrow. For decades after its discovery, ouabain was routinely used to treat atrial fibrillation and congestive heart failure in humans. More recently, ouabain has been replaced by digoxin, a structurally related, but more lipophilic cardiac glycoside.
Figure 19.14.
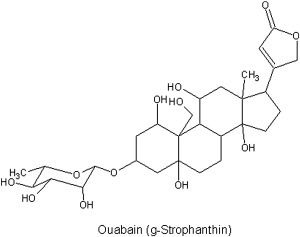
Structure of ouabain.
There are several important observations about Na+,K+-ATPase that had to be factored in before a mechanism of action could be proposed. These include:
-
1.
Na+,K+-ATPase is an example of active antiport and primary active transport.
-
2.
Na+,K+-ATPase is inhibited by ouabain, a cardiac glycoside.
-
3.
Ouabain binds to the outer surface of Na+,K+-ATPase and blocks K+ transport into the cell.
-
4.
Na+ binds better from the inside.
-
5.
K+ binds better from the outside.
-
6.
ATP phosphorylates an aspartic acid on the enzyme from the inside.
-
7.
Phosphorylation is related to Na+ transport.
-
8.
Dephosphorylation is related to K+ transport.
-
9.
Dephosphorylation is inhibited by ouabain.
-
10.
Three Na+ ions are pumped out of the cell as two K+ ions are pumped in, driven by hydrolysis of one ATP.
-
11.
Na+,K+-ATPase is electrogenic.
Mechanism of Na+,K+-ATPase [19] is based on toggling back and forth between two conformational states of the enzyme, ENZ-1 and ENZ-2 (Fig. 19.15 ). Three Na+s bind from the inside to Na+,K+-ATPase in one conformation (ENZ-1). This becomes phosphorylated by ATP causing a conformation change producing ENZ-2∼P. ENZ-2∼P does not bind Na+, but does bind two K+ ions. Therefore, three Na+ ions are released to the outside and two K+ ions are bound from the outside, generating ENZ-2∼P (2K+). Upon hydrolysis of ∼P, Na+,K+-ATPase (ENZ II) reverts back to the original ENZ-1 conformation that releases two K+ ions and binds three Na+ ions from the inside. Ouabain blocks the dephosphorylation step.
Figure 19.15.
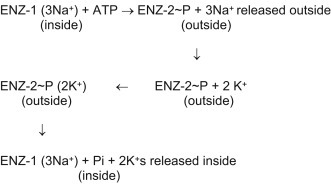
Mechanism of the Na+,K+-ATPase.
4.3. Secondary Active Transport
Secondary active transport (also known as cotransport) systems are composed of two separate functions. The energy-dependent movement of an ion (eg, H+, Na+, or K+) generates an electrochemical gradient of the ion across the membrane. This ion gradient is coupled to the movement of a solute in either the same direction (symport) or in the opposite direction (antiport, see Fig. 19.12, [18]). Movement of the pumped ion down its electrochemical gradient is by facilitated diffusion. The purpose of both types of co-transport is to use the energy in an electrochemical gradient to drive the movement of another solute against its gradient. An example of symport is the SGLT1 (sodium-glucose transport protein-1) in the intestinal epithelium [20]. SGLT1 uses the energy in a downhill transmembrane movement of Na+ to transport glucose across the apical membrane against an uphill glucose gradient so that the sugar can be transported into the bloodstream.
4.4. Bacterial Lactose Transport
The secondary active symport system for lactose uptake in Escherichia coli is shown in Fig. 19.16 [21]. Lactose uptake is driven through a channel by a H+ gradient generated by the bacterial electron transport system [22]. The free energy equation for transport described above can be rearranges for cases employing H+ gradients (see Chapter 18) to:
Where is the proton motive force; ΔΨ is the transmembrane electrical potential; R is the gas constant; T is the temperature in °K; n is the solute charge (+1 for protons); F is the Faraday; ΔpH is the transmembrane pH gradient.
Figure 19.16.
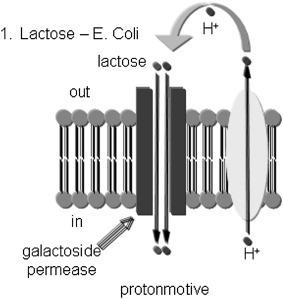
Lactose transport system in Escherichia coli[21]. Uptake of lactose is coupled to the movement of an H+ down its electrochemical gradient. This is an example of active transport, co-transport, and active.
It is the force on an H+ (called the proton motive force) that drives lactose uptake. Note that the ability to take up lactose is a combination of the electrical gradient and the pH gradient. Although lactose uptake is directly coupled to H+ transmembrane movement, it is possible to take up lactose even if the pH gradient is zero (ie, if the ΔΨ is sufficiently large).
4.5. Vectorial Metabolism, Group Translocation
Over 50 years ago, Peter Mitchell (see Chapter 18, Fig. 18.26) recognized the importance of what he termed “vectorial metabolism” [23], [24]. Water-soluble enzymes convert substrate to product without any directionality. Mitchell proposed that many enzymes are integral membrane proteins that have a specific transmembrane orientation. When these enzymes convert substrate to product they do so in one direction only. This enzymatic conversion is therefore unidirectional, or “vectorial.” Mitchell expanded this basic concept into his now famous “chemiosmotic hypothesis” for ATP synthesis in oxidative phosphorylation (Chapter 18) [25], [26]. For this revolutionary idea Mitchell was awarded the 1997 Nobel Prize in Chemistry.
Vectorial metabolism has been used to describe the mechanism for several membrane transport systems. For example, it has been reported in some cases the uptake of glucose into a cell may be faster if the external source of glucose is sucrose rather than free glucose. Through a vectorial transmembrane reaction, membrane-bound sucrase may convert external sucrose into internal glucose plus fructose more rapidly than the direct transport of free glucose through its transport system.
Mitchell defined one type of vectorial transport as group translocation, the best example being the PTS (phosphotransferase system) discovered by Saul Roseman in 1964.
PTS is a multicomponent active transport system that uses the energy of intracellular phosphoenol pyruvate (PEP) to take up extracellular sugars in bacteria. Transported sugars include glucose, mannose, fructose, and cellobiose. Components of the system include both plasma membrane and cytosolic enzymes. PEP is a high-energy phosphorylated compound (ΔG of hydrolysis is −61.9 kJ/mol) that drives the system. The high-energy phosphoryl group is transferred through an enzyme bucket brigade from PEP to glucose producing glucose-6-phosphate in several steps (PEP → EI → HPr →EIIA → EIIB → EIIC → glucose-6-phosphate). The sequence is depicted in more detail in Fig. 19.17 [27]. HPr stands for heat-stable protein that carries the high-energy ∼P from EI (enzyme-I) to EIIA. EIIA is specific for glucose and transfers ∼P to EIIB that sits next to the membrane where it takes glucose from the transmembrane EIIC and phosphorylates it producing glucose-6-phosphate. Although it is glucose that is being transported across the membrane, it never actually appears inside the cell as free glucose but rather as glucose-6-phosphate. Free glucose could leak back out of the cell via a glucose transporter, but glucose-6-phosphate is trapped inside the cell where it can rapidly be metabolized through glycolysis. Group translocation is defined by a transported solute appearing in a different form immediately after crossing the membrane.
Figure 19.17.
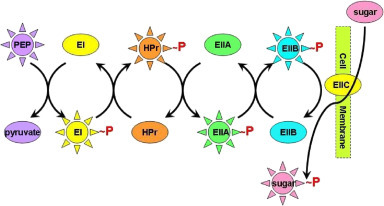
The bacterial PTS system for glucose transport [27].
5. Ionophores
The term ionophore means “ion bearer.” Ionophores are small, lipid-soluble molecules, usually of microbial origin, whose function is to conduct ions across membranes [28], [29]. They are facilitated diffusion carriers that transport ions down their electrochemical gradient. Ionophores can be divided into two basic classes: channel formers and mobile carriers (Fig. 19.18 ) [30]. Channel formers are long lasting, stationary structures that allow many ions at a time to rapidly flow across a membrane. Mobile carriers bind to an ion on one side of a membrane, dissolve in and cross the membrane bilayer and release the ion on the other side. They can only carry one ion at a time. Four representative ionophores will be discussed: the K+ ionophore valinomycin, the proton ionophore 2,4-dinitrophenol, synthetic crown ethers, and the channel-forming ionophore nystatin (Fig. 19.19 ).
Figure 19.18.
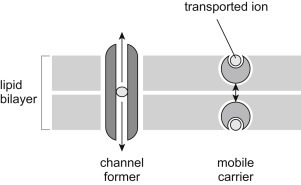
Two basic types of ionophores: channel formers (left) and mobile carriers (right) [30].
Figure 19.19.
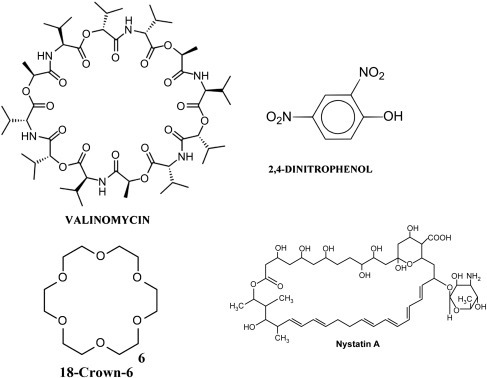
Representative examples of ionophores: the K+ ionophore valinomycin, the proton ionophore 2,4-dinitrophenol, the synthetic crown ether 18-crown-6, and the channel forming ionophore nystatin.
5.1. Valinomycin
Superficially valinomycin resembles a cyclic peptide (Fig. 19.19). However, upon closer examination the ionophore is actually a 12-unit (dodeca) depsipeptide where amino acid peptide bonds are alternated with amino alcohol ester bonds. Therefore the linkages that hold the molecule together alternate between nitrogen esters (peptide bonds) and oxygen esters. The units that comprise valinomycin are d- and l-valine (hence the name “valinomycin”), hydroxyvaleric acid and l-lactic acid. The circular structure is a macrocyclic molecule with the 12 carbonyl oxygens facing the inside of the ring where they chelate a single K+. The outside surface of valinomycin is coated with nine hydrophobic side chains of d- and l-valine and l-hydroxyvaleric acid. The polar interior of valinomycin precisely fits one K+. The binding constant for K+-valinomycin is 106 while Na+-valinomycin is only 10. This emphasizes the high selectivity valinomycin has for K+ over Na+. Valinomycin, therefore, has an oily surface that readily dissolves in a membrane lipid bilayer, carrying K+ across the membrane down its electrochemical gradient.
Valinomycin was first recognized as a potassium ionophore by Bernard Pressman in the early 1960's [31], [32]. He reported that valinomycin, a known antibiotic, stimulated K+ uptake and H+ efflux from mitochondria. Many studies showed that valinomycin dissipates essential transmembrane electrochemical gradients causing tremendous metabolic upheaval in many organisms including microorganisms. It is for this reason that valinomycin was recognized as an antibiotic long before it was identified as an ionophore. Currently several ionophores are added to animal feed as antibiotics and growth enhancing additives [33]. Recently valinomycin has been reported to be the most potent agent against SARS-CoV (severe acute respiratory-syndrome coronavirus), a severe form of pneumonia first identified in 2003 [34].
5.2. 2,4-Dinitrophenol
2,4-Dinitrophenol (DNP, Fig. 19.19) is considered to be the classic uncoupler of oxidative phosphorylation (see Chapter 18). It is a synthetic lipid-soluble proton ionophore that dissipates proton gradients across bioenergetic membranes (mitochondrial inner, thylakoid, bacterial plasma). An uncoupler is therefore an H+-facilitated diffusion carrier. Elucidating the role of DNP in uncoupling oxidative phosphorylation was an essential component in support of Peter Mitchell's chemiosmotic hypothesis [25]. Electron movement from NADH or FADH2 to O2 via the mitochondrial electron transport system generates a considerable amount of electrical energy that is partially captured as a transmembrane pH gradient (see Chapter 18). The movement of H+s back across the membrane, driven by the electrochemical gradient, is through a channel in the F1ATPase (an F-type primary active transport system discussed above, (Chapter 19, Section 4.1)) that is coupled to ATP synthesis. DNP short-circuits the H+ gradient before it can pass through the F1ATPase, thus uncoupling electron transport, the energy source for the H+ gradient, from ATP synthesis. Therefore, in the presence of DNP, electron transport continues, even at an accelerated rate, but ATP production is diminished. The energy that should have been converted to chemical energy in the form of ATP is then released as excess heat.
This combination of properties led to the medical application of DNP to treat obesity from 1933 to 1938 [35]. Upon addition of DNP:
-
•
The patient became weak due to low ATP levels.
-
•
Breathing increased due to increased electron transport to rescue ATP production.
-
•
Metabolic rate increased.
-
•
Body temperature increased due to inability to trap electrical energy as chemical energy in the form of ATP, releasing heat.
-
•
Body weight decreased due to increased respiration burning more stored fat.
DNP was indeed a successful weight loss drug. Two of the early proponents of DNP use as a diet drug, Cutting and Tainter at Stanford University, estimated that more than 100,000 people in the United States had tested the drug during its first year in use [35]. DNP, however, did have one disturbing side effect—death! Fatality was not caused by a lack of ATP, but rather by a dangerous increase in body temperature (hyperthermia). In humans, 20–50 mg/kg of DNP can be lethal. Although general use of DNP in the United States was discontinued in 1938, it is still employed in other countries and by bodybuilders to eliminate fat before competitions.
5.3. Crown Ethers
Crown ethers are a family of synthetic ionophores that are generally similar in function to the natural product valinomycin [36]. The first crown ether was synthesized by Charles Pederson (Fig. 19.20 ) while working at DuPont in 1967. For this work Pedersen was co-awarded the 1987 Nobel Prize in Chemistry. Crown ethers are cyclic compounds composed of several ether groups. The most common crown ethers are oligomers of ethylene oxide with repeating units of (—CH2CH2O—)n where n = 4 (tetramer), n = 5 (pentamer), or n = 6 (hexamer). Crown ethers are given structural names, X-crown-Y, where X is the total number of atoms in the ring and Y is the number of these atoms that are oxygen. Crown refers to the crown-like shape the molecule takes. Crown ether oxygens form complexes with specific cations that depend on the number of atoms in the ring. For example, 18-crown-6 (Fig. 19.19) has high affinity for K+, 15-crown-5 for Na+, and 12-crown-4 for Li+. Like valinomycin, the exterior of the ring is hydrophobic, allowing crown ethers to dissolve in the membrane lipid bilayer while carrying the sequestered cation down its electrochemical gradient. It is now possible to tailor make crown ethers of different sizes that can encase a variety of catalysts for phase transfer into the bilayer hydrophobic interior where they can be used to catalyze reactions inside the membrane.
Figure 19.20.

Charles Pedersen, 1904–1989.
5.4. Nystatin
Nystatin (Fig. 19.19) is a channel-forming ionophore that creates a hydrophobic pore across a membrane [37], [38]. Channel-forming ionophores allow for the rapid facilitated diffusion of various ions that depend on the dimensions of the pore. Nystatin, like other channel-forming ionophores (eg, amphotericin B and natamycin), is a commonly used antifungal agent. Finding medications that can selectively attack fungi in the presence of normal animal cells presents a difficult challenge since both cell types are eukaryotic. Bacteria, being prokaryotes, are sufficiently different to present a variety of anti-bacterial approaches not amenable to fungi. However, fungi do have an Achilles heel. Fungal plasma membranes have as their dominant sterol ergosterol, not the animal sterol cholesterol (see Chapter 5). Nystatin binds preferentially to ergosterol, thus targeting fungi in the presence of animal cells. When present at sufficient levels, nystatin complexes with ergosterol and forms transmembrane channels that lead to K+ leakage and death of the fungus. Nystatin is a polyene antifungal ionophore that is effective against many molds and yeast including Candida. A major use of nystatin is as a prophylaxis for AIDS patients who are at risk for fungal infections.
6. Gap Junctions
Gap junctions are a common structural feature of many animal plasma membranes [39], [40]. In plants similar structures are known as plasmodesmata. Gap junctions were introduced earlier in Chapter 11 (see Fig. 11.6). Gap junctions represent a primitive type of intercellular communication that allows transmembrane passage of small solutes like ions, sugars, amino acids, and nucleotides while preventing migration of organelles and large polymers like proteins and nucleic acids. Gap junctions connect the cytoplasms of two adjacent cells through nonselective channels. Connections through adjacent cells are at locations where the gap between cells is only 2–3 nm. This small gap is where the term “gap junction” originated. Gap junctions are normally clustered from a few to over a 1000 in select regions of a cell plasma membrane.
Early experiments involved injecting fluorescent dyes, initially fluorescein (molecular weight 300), into a cell and observing the dye movement into adjacent cells with a fluorescence microscope [41], [42]. Currently Lucifer Yellow has become the fluorescent dye of choice for gap junction studies, replacing fluorescein. At first, the dye only appeared in the initially labeled cell. With time, however, the dye was observed to spread to adjacent cells through what appeared to be points on the plasma membrane. These points were later recognized as gap junctions. By varying the size of the fluorescent dye, it was shown that there was an upper size limit for dye diffusion. Solutes had to have a molecular weight of less than ∼1200 to cross from one cell to another [41].
Although gap junctions were obviously channels that connected the cytoplasms of adjacent cells, it was years before their structure, shown in Fig. 19.21 , was determined [43], [44]. Each channel in a gap junction is made up of 12 proteins called connexins. Six hexagonally arranged connexins are associated with each of the adjacent cell plasma membranes that the gap junction spans. Each set of six connexins is called a connexon and forms half of the gap junction channel. Therefore, one gap junction channel is composed of 2 aligned connexons and 12 connexins. Each connexin has a diameter of about 7 nm and the hollow center formed between the 6 connexins (the channel) is about 3 nm in diameter. Gap junctions allow adjacent cells to be in constant electrical and chemical communication with one another. Of particular importance is the rapid transmission of small second messengers, such as inositol triphosphate (IP3) and Ca2+.
Figure 19.21.
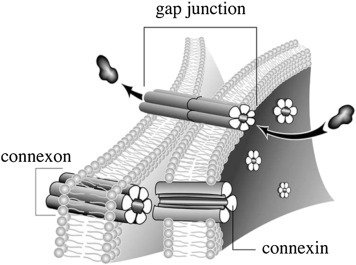
Gap junction [43]. Six connexins form a connexon and one connexon from each cell unite to form a gap junction.
It appears that all cells in the liver are interconnected through gap junctions. This presents a possible dilemma. If even a single cell is damaged, deleterious effects may be rapidly spread throughout the entire liver. Preventing this is one important function of Ca2+. Extracellular Ca2+ is ∼10−3 mol/L while intracellular levels are maintained at ∼10−6 mol/L. If a cell is damaged, Ca2+ rushes in, dramatically increasing intracellular Ca2+. Gap junction channels close if intracellular Ca2+ reaches 10−3 mol/L, thus preventing the spread of damage.
Gap junctions are particularly important in cardiac muscle as the electrical signals for contraction are passed efficiently through these channels [45]. As would be expected, malfunctions of gap junctions lead to a number of human disorders including demyelinating neurodegenerative diseases, skin disorders, cataracts, and even some types of deafness.
7. Other Ways to Cross the Membrane
There are several other ways that solutes, including large macromolecules, can cross membranes. These methods include receptor-mediated endocytosis (RME, discussed in Chapter 17), phagocytosis, pinocytosis, exocytosis, and membrane blebbing. These methods involve large sections of a membrane containing many lipids and proteins.
Two similar transport processes that have been known for a long time are pinocytosis and phagocytosis [46]. Both involve nonspecific uptake (endocytosis) of many things from water and ions through to large macromolecules and, for phagocytosis, even whole cells. Pinocytosis is Greek for “cell drinking” and involves the plasma membrane invaginating a volume of extracellular fluid and anything it contains including water, salts, biochemicals and even soluble macromolecules. Phagocytosis is Greek for “cell eating” and involves the plasma membrane invaginating large insoluble solids.
7.1. Pinocytosis
Pinocytosis is a form of endocytosis involving fluids containing many solutes. In humans, this process occurs in cells lining the small intestine and is used primarily for absorption of fat droplets. In endocytosis the cell plasma membrane extends and folds around desired extracellular material, forming a pouch that pinches off creating an internalized vesicle (Fig. 19.22 , [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45], [46], [47]). The invaginated pinocytosis vesicles are much smaller than those generated by phagocytosis. The vesicles eventually fuse with the lysosome whereupon the vesicle contents are digested. Pinocytosis involves a considerable investment of cellular energy in the form of ATP and so is many 1000 times less efficient than RME (see Chapter 17). Also, in sharp contrast to RME, pinocytosis is nonspecific for the substances it accumulates. Pinocytosis is not a recent discovery as it was first observed decades before the other transport systems discussed in Chapter 19. Its discovery is attributed to Warren Lewis in 1929.
Figure 19.22.
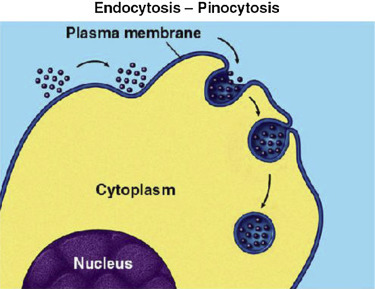
Pinocytosis, a type of endocytosis. An invagination of the plasma membrane encapsulates many water-soluble solutes ranging in size from salts to macromolecules.
7.2. Phagocytosis
Phagocytosis is a type of endocytosis that involves uptake of large solid particles, often >0.5 mm [47]. The particles are aggregates of macromolecules, parts of other cells, and even whole microorganisms and, in contrast to pinocytosis (shown in Fig. 19.22), phagocytosis has surface proteins that specifically recognize and bind to the solid particles. Fig. 19.23 [48] depicts events in phagacytosis. Phagocytosis is a routine process that ameba and ciliated protozoa use to obtain food. In humans, phagocytosis is restricted to specialized cells called phagocytes that include white blood cell neutrophils and macrophages. As with pinocytosis, phagocytosis generates intracellular vesicles called phagosomes that have sequestered solid particles they transport to the lysosome for digestion. Phagocytosis is a major mechanism used by the immune system to remove pathogens and cell debris. In fact, very early studies of the immune system led Elie Metchnikoff to discover phagocytosis in 1882. For this work Metchnikoff shared the 1908 Nobel Prize in Medicine with Paul Ehrlich.
Figure 19.23.
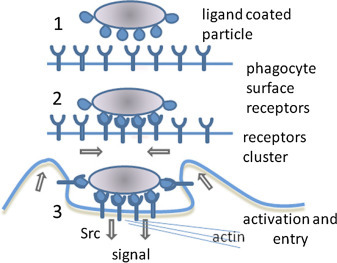
Phagocytosis, a type of endocytosis that involves uptake of large solid particles.
7.3. Exocytosis
Exocytosis is the process by which cells excrete waste and other large molecules from the cytoplasm to the cell exterior [49] and therefore is the opposite of endocytosis. Exocytosis generates vesicles referred to as secretory or transport vesicles (Chapter 17). In exocytosis, intracellular (secretory) vesicles fuse with the plasma membrane and release their aqueous sequestered contents to the outside at the same time that the vesicular membrane hydrophobic components (mostly lipids and proteins) are added to the plasma membrane (Fig. 19.24 , [50]). Steady state composition of the plasma membrane results from a balance between endocytosis and exocytosis. The resultant process of plasma membrane recycling is amazingly fast. For example, pancreatic secretory cells recycles an amount of membrane equal to the whole surface of the cell in ∼90 min. Even faster are macrophages that can recycle contents of their plasma membrane in only 30 min.
Figure 19.24.
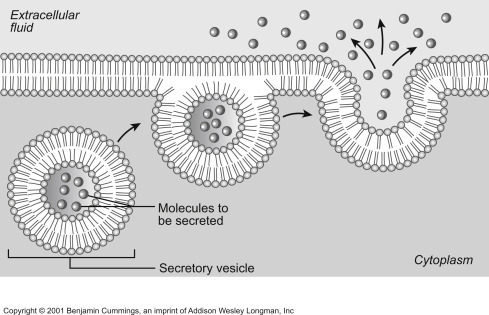
Exocytosis. Intracellular secretory vesicles fuse with the plasma membrane releasing their water-soluble contents to the outside and adding membrane material to the plasma membrane [50].
Before approaching the plasma membrane for fusion, exocytosis vesicles had a prior life that is considered in Chapter 17. The vesicles must first dock with the plasma membrane, a process that keeps the two membranes separated by <5–10 nm. During docking, complex molecular rearrangements occur to prepare the membranes for fusion. The process of vesicle fusion and release of aqueous compartment components is driven by SNARE proteins (see Chapters 10 and 17Chapter 10Chapter 17) [51], [52].
7.4. Blebbing
Blebbing of the plasma membrane is a morphological feature of cells undergoing late stage apoptosis (programmed cell death, see Chapter 24) [53]. A bleb is an irregular bulge in the plasma membrane of a cell caused by localized decoupling of the cytoskeleton from the plasma membrane. The bulge eventually blebs off from the parent plasma membrane taking part of the cytoplasm with it. It is clear in Fig. 19.25 [54] that the plasma membrane of an apoptotic cell is highly disintegrated and has lost the integrity required to maintain essential transmembrane gradients. Blebbing is also involved in some normal cell processes, including cell locomotion and cell division.
Figure 19.25.
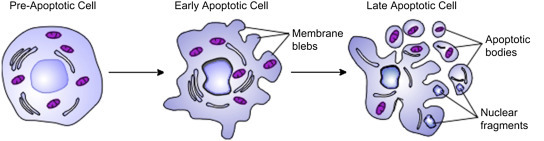
Membrane blebbing during apoptosis [54].
8. Summary
Carefully controlled solute movement into and out of cells is an essential feature of life. There are many ways solutes are transported across the thin (∼40 Å) membrane hydrophobic barrier. Transport is divided into passive diffusion and active transport. A biological membrane is semipermeable, being permeable to some molecules, most notably water (osmosis), while being very impermeable to most solutes that require some form of transporter. Passive diffusion (simple and facilitated) only requires the energy inherent in the solute's electrochemical gradient and results in its equilibrium across the membrane. In contrast, active transport requires additional energy (ie, ATP), and results in a nonequilibrium, net accumulation of the solute. Passive transport can involve simple diffusion or facilitated carriers including ionophores and channels. Active transport comes in many, often complex forms. Examples of active transport include primary active transport (uniport), secondary active transport (co-transport, antiport), and group translocation. Besides the multitude of transport systems, transport can be accomplished by gap junctions, receptor mediated endocytosis, phagocytosis, pinocytosis, exocytosis, and apoptotic membrane blebbing.
Chapter 20 will discuss bioactive lipids, highly specialized lipids that are functional at very low levels. Discussed bioactive lipids include ceramides, diacylglycerol, eicosanoids, steroid hormones, and phosphatidic acid.
References
- 1.Alberts B., Bray D., Lewis J., Raff M., Roberts K., Watson J.D. Molecular biology of the cell. 3rd ed. Garland Science; New York: 1994. Principles of membrane transport. [Google Scholar]
- 2.Blok M.C., van Deenen L.L.M., de Gier J. Effect of the gel to liquid crystalline phase transition on the osmotic behaviour of phosphatidylcholine liposomes. Biochim Biophys Acta. 1976;433:1–12. doi: 10.1016/0005-2736(76)90172-3. [DOI] [PubMed] [Google Scholar]
- 3.Miller F.P., Vandome A.F., McBrewster J. VDM Publishing House Ltd; 2010. Fick's laws of diffusion; p. 76. [Google Scholar]
- 4.Water systems: aqua technology for the 21st century. Introductory information on reverse osmosis.
- 5.Bangham A.D., de Gier J., Greville G.D. Osmotic properties and water permeability of phospholipids liquid crystals. Chem Phys Lipids. 1967;1:225–246. [Google Scholar]
- 6.De Gier J. Osmotic behaviour and permeability properties of liposomes (review) Chem Phys Lipids. 1993;64:187–196. doi: 10.1016/0009-3084(93)90065-b. [DOI] [PubMed] [Google Scholar]
- 7.Baldwin S.A. Oxford University Press; USA: 2000. Membrane transport: a practical approach; p. 320. [Google Scholar]
- 8.Alberts B., Bray D., Johnson A., Lewis J., Raff M., Roberts K. Essential cell biology. 2nd ed. Garland Publishing: Taylor Francis Group; 2004. Figure 12.4. Comparison of passive and active transport. [Google Scholar]
- 9.Huang S., Czech M.P. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 10.Biologia Medica . 2010. Transporte de Glucosa: GLUT y SGLT Seminarios de Biología Celular y Molecular – USMP Filial Norte. [Google Scholar]
- 11.Lippiat J.D. vol. 491. Humana Press; New York (NY): 2009. Potassium channels. methods and protocols; p. 302. (Series in methods in molecular biology). [Google Scholar]
- 12.Doyle D.A., Morais C.J., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 13.Goldin A.L. Resurgence of sodium channel research. Ann Rev Physiol. 2001;63:871–894. doi: 10.1146/annurev.physiol.63.1.871. [DOI] [PubMed] [Google Scholar]
- 14.Penzotti J.L., Fozzard H.A., Lipkind G.M., Dudley S.C. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys J. 1998;75(6):2647–2657. doi: 10.1016/S0006-3495(98)77710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noda Y., Sohara E., Ohta E., Sasaki S. Aquaporins in kidney pathophysiology. Nat Rev Nephrol. 2010;6:168–178. doi: 10.1038/nrneph.2009.231. [DOI] [PubMed] [Google Scholar]
- 16.Hill A.E., Shachar-Hill B., Shachar-Hill Y. What are aquaporins for? J Membr Biol. 2004;197:1–32. doi: 10.1007/s00232-003-0639-6. [DOI] [PubMed] [Google Scholar]
- 17.Kungl Vetenskapsakademien, The Royal Swedish Academy of Sciences . 2003. The Nobel prize in chemistry: Peter Agre, Roderick MacKinnon.Nobelprize.org The official web site of the Nobel Prize. [Google Scholar]
- 18.Nelson D.L., Cox M.M. Lehninger: principles of biochemistry. Worth Publishers; 2006. Chapter 10. Biological membranes and transport. Solute transport across membranes. Figure 10-21. [Google Scholar]
- 19.Lopina O.D. Na+, K+ ATPase: structure, mechanism, and regulation. Membr Cell Biol. 2000;13:721–744. [PubMed] [Google Scholar]
- 20.Wright E.M. Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- 21.Jakubowski H. Chapter 9 – signal transduction. A. Energy transduction: uses of ATP. 2002. http://employees.csbsju.edu/hjakubowski/classes/ch331/signaltrans/olsignalenergy.html [Google Scholar]
- 22.Martin S.A. Nutrient transport by ruminal bacteria: a review. J Anim Sci. 1994;72:3019–3031. doi: 10.2527/1994.72113019x. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell P., Moyle J. Group-translocation: a consequence of enzyme-catalysed group-transfer. Nature. 1958;182:372–373. doi: 10.1038/182372a0. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell P., Moyle J. Coupling of metabolism and transport by enzymic translocation of substrates through membranes. Proc R Phys Soc Edinb. 1959;28:19–27. [Google Scholar]
- 25.Mitchell P., Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–139. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- 26.Mitchell P. Proton current flow in mitochondrial systems. Nature. 1967;214:1327–1328. doi: 10.1038/2141327a0. [DOI] [PubMed] [Google Scholar]
- 27.Herzberg O., Canner D., Harel M., Prilusky J., Hodis E. Proteopedia, Weizmann Institute of Science in Israel; 2011. Enzyme I of the phosphoenolpyruvate: sugar phosphotransferase system. [Google Scholar]
- 28.Pressman B.C. Biological applications of ionophores. Annu Rev Biochem. 1976;45:501–530. doi: 10.1146/annurev.bi.45.070176.002441. [DOI] [PubMed] [Google Scholar]
- 29.Szabo G. Structural aspects of ionophore function. Fed Proc. 1981;40:2196–2201. [PubMed] [Google Scholar]
- 30.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Molecular biology of the cell. 4th ed. Garland Scientific; New York: 2002. Principles of membrane transport. Figure 11.5. [Google Scholar]
- 31.Moore C., Pressman B.C. Mechanism of action of valinomycin on mitochondria. Biochem Biophys Res Comm. 1964;15:562–567. [Google Scholar]
- 32.Pressman B.C. Induced active transport of ions in mitochondria. Proc Natl Acad Sci USA. 1965;53:1076–1083. doi: 10.1073/pnas.53.5.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Page S.W. Avcare Limited; Canberry (Australia): 2003. The role of enteric antibiotics in livestock production. [Google Scholar]
- 34.Cheng Y.Q. Deciphering the biosynthetic codes for the potent anti-SARS-CoV cyclodepsipeptide valinomycin in Streptomyces tsusimaensis ATCC 15141. Chembiochem. 2006;7:471–477. doi: 10.1002/cbic.200500425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tainter M.L., Stockton A.B., Cutting W.C. Use of dinitrophenol in obesity and related conditions: a progress report. J Am Med Assoc. 1933;101:1472–1475. [Google Scholar]
- 36.Huszthy P., Toth T. Synthesis and molecular recognition studies of crown ethers. Period Polytech. 2007;51:45–51. [Google Scholar]
- 37.Borgos S.E.F., Tsan P., Sletta H., Ellingsen T.E., Lancelin J.-M., Zotchev S.B. Probing the structure−function relationship of polyene macrolides: engineered biosynthesis of soluble nystatin analogues. J Med Chem. 2006;49:2431–2439. doi: 10.1021/jm050895w. [DOI] [PubMed] [Google Scholar]
- 38.Lopes S., Castanho M.A.R.B.J. Revealing the orientation of nystatin and amphotericin B in lipidic multilayers by UV-Vis linear dichroism. Phys Chem B. 2002;106:7278–7282. [Google Scholar]
- 39.Revel J.P., Karnovsky M.J. Hexagonal array of subunits in intracellular junctions of the mouse heart and liver. J Cell Biol. 1967;33:C7–C12. doi: 10.1083/jcb.33.3.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.In: Peracchia C, editor. Gap junctions: molecular basis of cell communication in health and disease. Benos D, series editor. Current topics in membranes and transport, series ed. New York: Elsevier Publishing; 1999. 648 pp.
- 41.Simpson I., Rose B., Loewenstein W.R. Size limit of molecules permeating the junctional membrane channels. Science. 1977;195:294–296. doi: 10.1126/science.831276. [DOI] [PubMed] [Google Scholar]
- 42.Imanaga I., Kameyama M., Irisawa H. Cell-to-cell diffusion of fluorescent dyes in paired ventricular cells. Am J Physiol Heart Circ Physiol. 1987;252:H223–H232. doi: 10.1152/ajpheart.1987.252.1.H223. [DOI] [PubMed] [Google Scholar]
- 43.Echevarria W., Nathanson M.H. Chapter: channels and transporters. Molecular pathogenesis of cholestasis. Madame Curie Bioscience Database. 2003. Gap junctions in the liver. [Google Scholar]
- 44.Cao F., Eckert R., Elfgang C., Nitsche J.M., Snyder S.A., Hulsen D.F. A quantitative analysis of connexin-specific permeability differences of gap junctions expressed in HeLa transfectants and Xenopus oocytes. J Cell Sci. 1998;111:31–43. doi: 10.1242/jcs.111.1.31. [DOI] [PubMed] [Google Scholar]
- 45.Jongsma H.J., Wilders R. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193–1197. doi: 10.1161/01.res.86.12.1193. [DOI] [PubMed] [Google Scholar]
- 46.Aderem A., Underhill D.M. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 47.Underhill D.M., Ozinsky A. Phagocytosis of microbes: complexity in action. Ann Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 48.Ernst J.D., Stendahl O., editors. Phagocytosis of bacteria and bacterial pathogenicity. Cambridge University Press; 2006. p. 6. [Google Scholar]
- 49.Li L., Chin L.-S. The molecular machinery of synaptic vesicle exocytosis. Cell Mol Life Sci. 2003;60:942–960. doi: 10.1007/s00018-003-2240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benjamin Cummings an imprint of Addison Wesley Longman. 2001. http://www.highlands.edu/academics/divisions/scipe/biology/faculty/harnden/2121/notes/cell.htm Cell and cell structure. IV. Membrane transport processes. [Google Scholar]
- 51.Sudhof T.C., Rothman J.E. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wightman R.M., Haynes C.L. Synaptic vesicles really do kiss and run. Nat Neurosci. 2004;7:321–322. doi: 10.1038/nn0404-321. [DOI] [PubMed] [Google Scholar]
- 53.Coleman M.L., Sahai E.A., Yeo M., Bosch M., Dewar A., Olson M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK1. Nat Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- 54.O'Day D. 2011. Human Development, Bio 380F. As modified from: Walker NI, Harmon BV, Gobe, GC, Kerr JF. Patterns of cell death. Methods Achiev Exp Pathol 1988;13:18–54. [PubMed]