

Abstract
Genetic ataxias are associated with mutations in hundreds of genes with high phenotypic overlap complicating the clinical diagnosis. Whole-exome sequencing (WES) has increased the overall diagnostic rate considerably. However, the upper limit of this method remains ill-defined, hindering efforts to address the remaining diagnostic gap. To further assess the role of rare coding variation in ataxic disorders, we reanalyzed our previously published exome cohort of 76 predominantly adult and sporadic-onset patients, expanded the total number of cases to 260, and introduced analyses for copy number variation and repeat expansion in a representative subset. For new cases (n = 184), our resulting clinically relevant detection rate remained stable at 47% with 24% classified as pathogenic. Reanalysis of the previously sequenced 76 patients modestly improved the pathogenic rate by 7%. For the combined cohort (n = 260), the total observed clinical detection rate was 52% with 25% classified as pathogenic. Published studies of similar neurological phenotypes report comparable rates. This consistency across multiple cohorts suggests that, despite continued technical and analytical advancements, an approximately 50% diagnostic rate marks a relative ceiling for current WES-based methods and a more comprehensive genome-wide assessment is needed to identify the missing causative genetic etiologies for cerebellar ataxia and related neurodegenerative diseases.
Keywords: ataxia, cerebellar ataxia, cerebellum, diagnostic testing, exome, gait disorders, genetics, genomics, neurogenetics, spastic paraparesis, spastic paraplegia, spinocerebellar ataxia
1 |. INTRODUCTION
Hereditary spinocerebellar ataxia (dominant SCA and recessive SCAR) and spastic paraplegia (HSP) are neurodegenerative disorders affecting the cerebellum, its pathways, and the corticospinal tracts that can result from mutations in one of the hundreds of genes (Online Mendelian Inheritance in Man [OMIM], https://www.omim.org/; OMIM, 2019). Both disorders manifest high degrees of phenotypic heterogeneity even with common specific causal mutations, necessitating genomic testing strategies to identify the relatively rare causal mutations that are pervasive in both disorders and heterogeneous in presentation (Anheim, Tranchant, & Koenig, 2012; Benini, Ben Amor, & Shevell, 2012; Brusse, Maat-Kievit, & van Swieten, 2007; Fogel & Perlman, 2006, 2007, 2011; Fogel, Satya-Murti, & Cohen, 2016; Klockgether, 2010; Manto & Marmolino, 2009). Both disorders include seemingly sporadic confirmed genetic causes at relatively high rates (~25%) in otherwise undiagnosed cases, commonly due to recessive inheritance, de novo mutations, or anticipation (Fogel et al., 2014; Nibbeling et al., 2017; Ohba et al., 2013; Pyle et al., 2015; Sawyer et al., 2013). In the last 5 years, whole-exome sequencing (WES) has come to the forefront of testing cases with suspected genetic causes of spinocerebellar ataxia or spastic paraplegia once the more common repeat expansion mutations have been ruled out (Fogel et al., 2014). WES offers cost-effective broad-coverage testing of almost all known coding variants due to single-nucleotide changes, small insertions/deletions, or proximal splice site variants (Fogel et al., 2016; Rexach, Lee, Martinez-Agosto, Nemeth, & Fogel, 2019). It also offers opportunities for the simultaneous analysis of comparator DNA (i.e., from parents or siblings) to facilitate identification of de novo or compound heterozygous mutations and rule out rare familial benign polymorphisms based on segregation with disease status. Our initial studies identified pathogenic/likely pathogenic variants in 21% (16/76) of properly selected ataxia/spasticity patients with an additional 40% (30/76) with variants of uncertain significance (VUS) requiring clinical follow-up (Fogel et al., 2014; Richards et al., 2015). Other more recent studies examining similar cohorts have increased this overall detection rate in hereditary ataxias to approximately 40–50% (Farwell et al., 2015; Nibbeling et al., 2017; Sawyer et al., 2013; Sun et al., 2018). Diagnostic rates are frequently higher in patients with positive family histories, when the probability for identifying a monogenic disorder is highest, and when multiple family members are available for testing, where interpretation of variants is improved by segregation relationships (Farwell et al., 2015; Fogel et al., 2014; Sawyer et al., 2013). Here we present a more extensive follow-up study with a larger cohort consisting primarily of adult-onset sporadic ataxia and spastic paraplegia cases with suspicion for a possible genetic etiology. Our study was intended to maximize diagnostic potential using genomic technologies including an assessment of copy number variation (CNV) and repeat expansion in a representative subset of the cohort. Additionally, we reanalyzed our previously reported cases to assess the effect of interval advances in variant annotation and gene discovery. Finally, we compare our results with other published studies to assess the overall diagnostic capability of current next-generation sequencing and WES pipelines and discuss approaches to further improvement of patient diagnosis.
2 |. METHODS
2.1 |. Patient enrollment and clinical assessment
This study comprises 184 index patients with a phenotypic range of either pure cerebellar ataxia, spasticity, or complex neurologic disorders linked to either condition. All patients had an extensive clinical evaluation to rule out the acquired causes of ataxia (Fogel et al., 2014). To qualify for this study, patients were required to have negative test results for the most common repeat expansion disorders (SCA1, SCA2, SCA3, SCA6, SCA7, and Friedreich ataxia) causing hereditary cerebellar ataxia (Fogel & Perlman, 2006, 2011; Fogel, Vickrey, Walton-Wetzel, Lieber, & Browner, 2013; Shakkottai & Fogel, 2013). Genetic counseling was provided for all patients both before and following the completion of the study. All patients enrolled in this study provided written informed consent. All methods in this study were approved by the Institutional Review Board of the University of California at Los Angeles.
2.2 |. Exome sequencing and data analysis
DNA samples were collected from the index patient and their family members based on family history and individual availability for exome sequencing. Exome capture was performed with commercially-available kits and sequencing was performed on the Illumina HiSeq platform with paired-end reads (Table S1). WES data analysis was conducted based on the Broad Institute’s Genome Analysis Toolkit (GATK3) version 3 best practices guidelines (DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013). Sequencing reads were mapped to the human genome (hs37d5) using the Burrows–Wheeler Aligner (Li & Durbin, 2009) and postprocessed with SAMtools (Li et al., 2009). Picard Tools (https://broadinstitute.github.io/picard/) was used to compute sequence alignment metrics and mark duplicate reads. The Qualimap tool was used to evaluate sequence alignment quality (Garcia-Alcalde et al., 2012; Okonechnikov, Conesa, & Garcia-Alcalde, 2015). The mean coverage of the protein-coding RefSeq genes was 94.3× with standard deviation 22.3× (range 50.3× to 204.5×). GATK was used for indel realignment, base quality score recalibration, joint genotyping, variant quality score recalibration, variant evaluation, and variant selection. Variant evaluation and selection were based off NCBI Reference Sequence Database RefSeq (https://www.ncbi.nlm.nih.gov/refseq; O’Leary et al., 2016) exon intervals. Variants were annotated with either the SNP & Variation Suite v8 or VarSeq v1 (both from Golden Helix, Inc., Bozeman, MT, www.goldenhelix.com). The Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) and the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/) public databases were used to filter for variants with a minor allele frequency ≤2% (Lek et al., 2016). Variants that were found to be disproportionately common among internal controls (e.g., batch effects) were excluded from the analysis. Phenotypic keywords provided by clinicians were used to generate gene lists from the OMIM (https://www.omim.org/; OMIM, 2019) and Human Gene Mutation Database Professional Version (HGMD; https://www.qiagenbioinformatics.com/products/human-gene-mutation-database/; Stenson et al., 2014) databases. Variants found within exons and splice regions of genes from these gene lists were first assessed for their clinical significance as previously described (Fogel et al., 2014; Richards et al., 2015). Variants were designated as pathogenic or likely pathogenic based on information from clinical databases such as HGMD, ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), or reports from published studies. Novel variants have been submitted to ClinVar as clinically appropriate. Comparator WES data were incorporated into the analysis when available (Table S2). If available, variants within linkage peaks (see below) were also prioritized for initial analysis. Subsequently, variants outside of the gene lists and linkage peaks were then analyzed. Variant classification and interpretation were based on the American College of Medical Genetics and Genomics guidelines (Richards et al., 2015). Sanger sequencing was used to confirm variant segregation with disease status if family members were available. Identified variants that had <Q500 were confirmed by Sanger sequencing (Strom et al., 2014). Statistical analysis of comparative diagnostic efficacy was performed using data from multiple previously published studies (Table S3).
2.3 |. Array genotyping and pedigree verification
Data from Illumina Infinium Human Exome v1–2 array and Human CytoSNP-12v1–0_D BeadChips (both Illumina, San Diego, CA) were generated for linkage analysis for selected index patients and their family members. The quality assessment included confirmation of sample identity and purity using the Error Rate In Sequencing (ERIS) pipeline. An “e-GenoTyping” approach was used to screen all sequence reads for exact matches to probe sequences defined by the variant and position of interest. Samples that passed quality control metrics of ERIS single nucleotide polymorphism (SNP) array concordance (>90%) and ERIS average contamination rate (<5%) were carried forward into quality filtering, pedigree validation, and linkage analysis. Insertions/deletions and nonautosomal polymorphisms and variants without an rs identifier were all removed. SNPs that had high missingness or mapped to identical locations (duplicates) were also removed. The prePRIMUS QC pipeline was used to estimate pairwise kinship and PRIMUS (Staples et al., 2014; Staples et al., 2016) was used to reconstruct and validate pedigrees. In the absence of array data, pedigree verification was approximated off WES variant calls by calculating the unadjusted Ajk statistic (Yang et al., 2010) with the relatedness algorithm from VCFtools (Danecek et al., 2011). Patient-reported sex was verified by exome data coverage of the sex chromosomes. Two sample swaps were confirmed and the corresponding pedigrees were updated for analysis.
2.4 |. Linkage analysis
Multifamily parametric linkage analysis was conducted with ALLEGRO (Gudbjartsson, Jonasson, Frigge, & Kong, 2000) using either a fully penetrant dominant model with no phenocopies (f0, f1, f2 = 0,1,1) or a recessive model (f0, f1, f2 = 0,0,1) corresponding to patterns of affection in the pedigree to identify overlapping haplotypes shared identically by descent (IBD). Identified candidate regions where IBD sharing was consistent with the model of inheritance for the disease were then used to prioritize variants for subsequent WES analyses. Because of power, the logarithm of the odds scores generated from these analyses did not meet genome-wide significance thresholds and thus was not considered.
2.5 |. CNV analysis
CNV analysis was conducted off WES data with read depth approaches using both Copy Number Inference From Exome Reads (CoNIFER; Krumm et al., 2012; O’Roak et al., 2012) and HMZDelFinder (Gambin et al., 2016) as well as exome hidden Markov Model (XHMM; Fromer & Purcell, 2014; Fromer et al., 2012; Poultney et al., 2013). A total of 53 families from the expanded cohort that had WES on one the following captured kits: Nextera Rapid Capture Exome, NimbleGen Seqcap EZ GSC VCRome, or Agilent SureSelect Human All Exon V4 Capture (Table S1) were selected for CNV analysis. In addition, 15 families from the original cohort (Fogel et al., 2014) that had WES on either Nextera Rapid Capture Exome or NimbleGen Seqcap EZ GSC VCRome capture kits were also included for CNV analysis. Cases were grouped by their capture kit platform for analysis. CNV calls in low complexity regions were removed. Intersecting CNV calls between CoNIFER and XHMM as well as CNV calls from HMZDelFinder were analyzed. All CNV calls were compared to the following CNV databases: ExAC CNV (Ruderfer et al., 2016), Database of Genomic Variant (DGV; MacDonald, Ziman, Yuen, Feuk, & Scherer, 2014) and DECIPHER (Firth et al., 2009). CNV calls that were present in multiple studies in DGV were classified as likely benign. CNV calls that were found in ExAC CNV were evaluated based on population and frequency information. If appropriate, biallelic inheritance was considered. CNV calls that were found in DECIPHER were evaluated based on reported phenotypes, overlapping gene annotation, and pathogenicity/contribution.
2.6 |. Short tandem repeat (STR) expansion detection analysis
STR expansion screening was conducted with STRetch (Dashnow et al., 2018) on the same families as the CNV analysis described above. BAM files were grouped by corresponding capture kit platforms and processed through the STRetch_exome_bam_pipeline.groovy pipeline. The EXOME_TARGET parameter was configured to the corresponding capture kit target coordinates for each run. A bed file with all STRs defined in the human genome was provided for the input_regions parameter. STRetch calls that had locus coverage lower than 3 and p_adj value >.05 were filtered. Only calls within genic regions were analyzed. Calls in known STR disease-causing genes were further evaluated for pathogenicity based on segregation of disease status (if applicable) and phenotype.
3 |. RESULTS
Overall, 49% of the cohort was female (90/184) with an average age of 50 years (standard deviation 20 years, range 2–88 years) and primarily of European (73%) descent (Tables S4–7). The majority of the cases were sporadic (124/184, 67%) and adult-onset (127/184, 69%; Table 1). Pathogenic or likely pathogenic variants were identified in 24% (44/184, Tables 1 and 2; Table S5) and VUS were identified in 23% (43/184, Tables 1 and 3; Table S6). Because VUS require clinical follow-up (e.g., additional confirmatory diagnostic testing, if available, or subsequent bioinformatic revaluation) we combined these categories to obtain an overall rate of 47% for identification of clinically relevant variation (87/184, Tables 1–3; Tables S5–7). There was no notable difference in overall observation of clinically relevant variants between familial (43%, 26/60) or sporadic cases (49%, 61/124) or in early-onset (58%, 33/57) versus adult-onset (43%, 54/127) cases (Tables 1–3; Tables S5 and S6). Generally, as expected, more pathogenic/likely pathogenic variants were found in familial (30%, 18/60) and early-onset cases (35%, 20/57) relative to sporadic and adult-onset cases (21%, 26/124 and 19%, 24/127, respectively; Tables 1–3; Tables S5 and S6). Pathogenic/likely pathogenic mutations were predominantly identified in genes associated with recessive disorders with compound heterozygous variants being the most common (41%), followed by heterozygous variants in genes associated with dominant disorders (25%) and homozygous recessive variants (23%, Table 1). De novo variation made up 7% of the pathogenic/likely pathogenic variants (Table 1). The most frequently encountered genes identified with pathogenic/likely pathogenic variants were SPG7 (20%, 9/44), CACNA1G (Ngo et al., 2018), and SYNE1 (7%, 3/44 each), and ITPR1, KCNA2, and SPG11 (5%, 2/44 each, Table 2) similar to the original cohort (Fogel et al., 2014).
TABLE 1.
Distribution of variant types detected by WES for expanded ataxia cohort
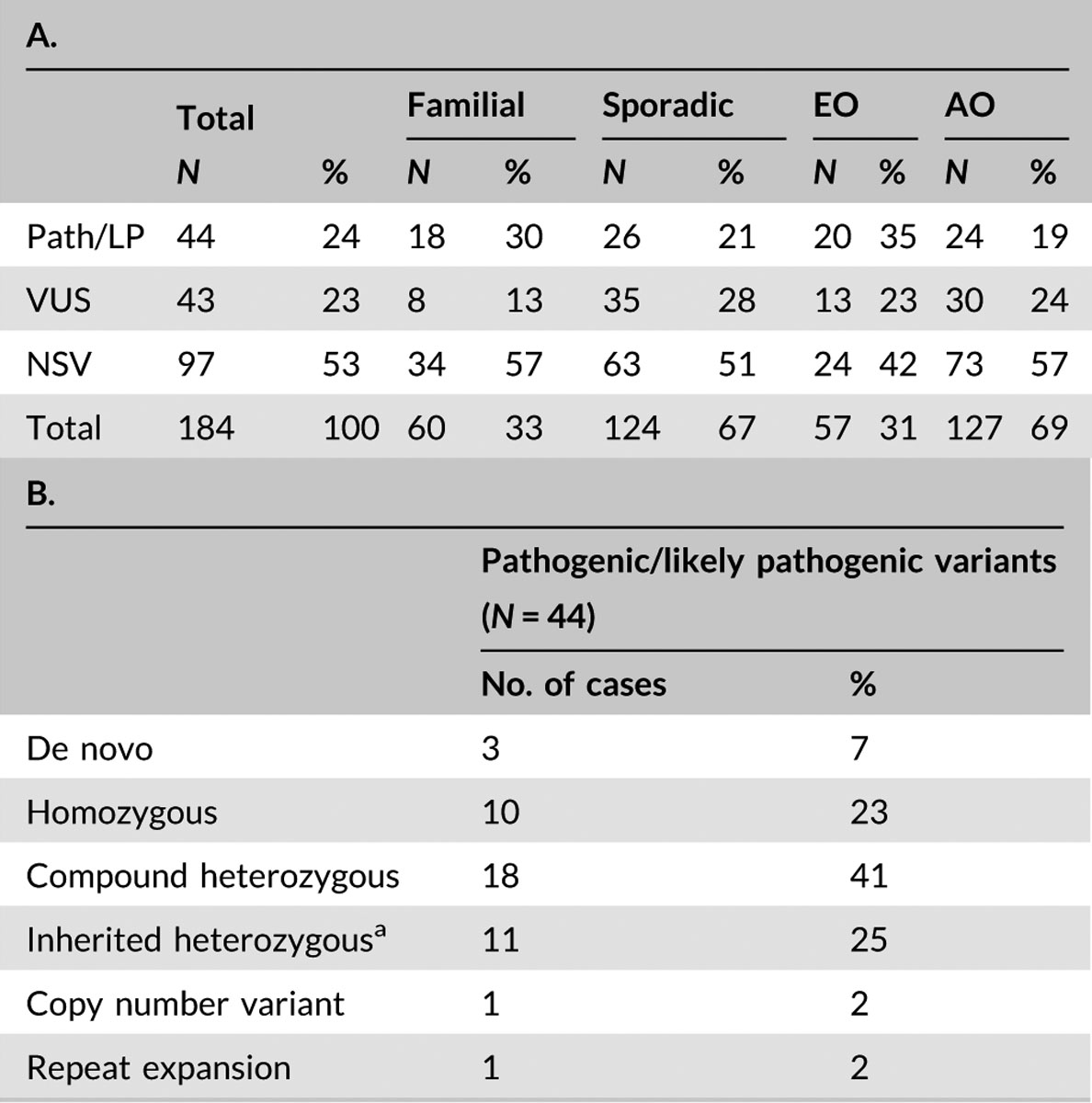 |
Abbreviations: AO, adult-onset; EO, early-onset (≤age 20 years); LP, likely pathogenic; N, number of index patients; NSV, no significant variants identified; Path, pathogenic; WES, Whole-exome sequencing.
Variants were considered inherited heterozygous unless parents or other family members were available for testing for de novo confirmation or variant was previously reported as de novo through another clinical test.
TABLE 2.
Patients with pathogenic/likely pathogenic variants identified
| Patient no./sex/age/onset | Family history | Primary symptom | Gene | Inheritance | cDNA | Protein | OMIM disease phenotype |
|---|---|---|---|---|---|---|---|
| ATX77/F/39/EO | Affected sister | Ataxia | SH3TC2 | AR | c.1546A>T c.1586G>A |
p.Lys516Ter p.Arg529His |
601596 |
| ATX78/F/59/AO | Multiple affected | Ataxia | ITPR1 | AD | c.(4494513_4735417)del | p.? | 606658 |
| ATX79/F/46/EO | Affected brother and sister | Ataxia | COQ8A | AR | c.74delC c.1555delA |
p.His26Thrfs*11 p.Thr519Profs*24 |
612016 |
| ATX80/F/50/EO | 2 Affected sisters | Ataxia | SPG11 | AR | c.6091C>T c.6526T>C |
p.Arg2031Ter p.Phe2176Leu |
604360 |
| ATX81/F/42/AO | Sporadic | Ataxia | ERCC4 | AR | c.2395C>T c.2117T>C c.1765C>G |
p.Arg799Trp p.Ile706Thr p.Arg589Gly |
278760 |
| ATX82/M/17/EO | Affected brother | Ataxia | AARS2 | AR | c.706delC c.984C>G |
p.Gln236Serfs*3 p.Ile328Met |
615889 |
| ATX83/F/41/EO | Affected sister | Ataxia | EXOSC3 | AR | c.395A>C c.572G>A |
p.Asp132Ala p.Gly191Asp |
614678 |
| ATX84/M/58/AO | Affected brother | Ataxia | ANO10 | AR | c.289delA homozygous |
p.Met97Ter | 613728 |
| ATX85/M/46/EO | Affected brother | Ataxia | KIF1C | AR | c.927_928insAAGGA homozygous |
p.Asn312Argfs*12 | 611302 |
| ATX86/M/8/EO | Sporadic | Ataxia | GOSR2 | AR | c.430G>T homozygous |
p.Gly144Trp | 614018 |
| ATX87/M/27/EO | Affected sister | Ataxia | DOCK6 | AR | c.2294G>A homozygous |
p.Arg765His | 614219 |
| ATX88/M/41/AOa | Multiple affected | Ataxia | CACNA1G | AD | c.5144G>A | p.Arg1715His | 616795 |
| ATX89/M/10/EO | Sporadic | Ataxia | KCNA2 | AD | c.890G>A | p.Arg297Gln | 616366 |
| ATX90/M/60/AO | Sporadic | Ataxia | SPG7 | AR | c.(1_?)del c.1529C>T |
p.? p.Ala510Val |
607259 |
| ATX91/M/28/AOa | Sporadic | Ataxia | CACNA1G | AD | c.5144G>A | p.Arg1715His | 616795 |
| ATX92/M/74/AOa | Multiple affected | Ataxia | CACNA1G | AD | c.5144G>A | p.Arg1715His | 616795 |
| ATX93/F/63/AO | Affected brother | Ataxia | SPG7 | AR | c.1454_1462delGGCGGGAGA c.1529C>T |
p.Arg485_Glu487del p.Ala510Val |
607259 |
| ATX94/M/77/AO | Affected sister | Ataxia | SPG7 | AR | c.1454_1462delGGCGGGAGA c.1529C>T |
p.Arg485_Glu487del p.Ala510Val |
607259 |
| ATX95/F/67/AO | Sporadic | Ataxia | SYNE1 | AR | c.16927C>T c.20236C>T |
p.Arg5643Trp p.Arg6746Trp |
610743 |
| ATX96/M/61/AO | Sporadic | Ataxia | SYNE1 | AR | c.2380_2381delinsAA c.23102G>A |
p.Ala794Lys p.Arg7701Gln |
610743 |
| ATX97/F/65/AO | Sporadic | Ataxia | SPG7 | AR | c.1529C>T homozygous |
p.Ala510Val | 607259 |
| ATX98/F/22/EO | Sporadic | Ataxia | WFS1 | AR | c.1082C>T homozygous |
p.Thr361Ile | 222300 |
| ATX99/M/61/EO | Sporadic | Ataxia | CACNA1A | AD | c.1997C>T | p.Thr666Met | 108500 |
| ATX100/M/57/AO | Sporadic | Ataxia | SPG7 | AR | c.1529C>T homozygous |
p.Ala510Val | 607259 |
| ATX101/F/43/EO | Sporadic | Ataxia | SYNE1 | AR | c.22958G>A c.13210A>T |
p.Trp7653Ter p.Lys4404Ter |
610743 |
| ATX102/M/70/AO | Sporadic | Ataxia | TMEM240 | AD | c.509C>T | p.Pro170Leu | 607454 |
| ATX103/F/71/AO | Sporadic | Ataxia | ALDH18A1 | AD | c.755G>A | p.Arg252Gln | 601162 |
| ATX104/F/48/AO | Multiple affected | Spasticity | SPAST | AD | c.1103T>C | p.Phe368Ser | 182601 |
| ATX105/F/35/EO | Sporadic | Spasticity | CYP7B1 | AR | c.392dupA c.825T>A c.889A>G |
p.Asn131Lysfs*3 p.Tyr275Ter p.Thr297Ala |
270800 |
| ATX106/M/51/AO | Affected mother (not examined) | Ataxia | SPG7 | AR | c.1454_1462delGGCGGGAGA c.1529C>T |
p.Arg485_Glu487del p.Ala510Val |
607259 |
| ATX107/F/27/EO | Sporadic | Spasticity | ATL1 | AD | c.742G>A | p.Glu248Lys | 182600 |
| ATX108/F/58/AO | Affected brother | Ataxia | SPG7 | AR | c.233T>A c.291_294delTACT |
p.Leu78Ter p.Thr98Serfs*8 |
607259 |
| ATX109/M/12/EO | Sporadic | Ataxia | ITPR1 | AD | c.7471G>A | p.Gly2491Arg | 206700 |
| ATX110/M/48/AO | Sporadic | Ataxia | SPG7 | AR | c.1529C>T c.1552+1G>T |
p.Ala510Val p.? |
607259 |
| ATX116/M/52/AO | Sporadic | Ataxia | ATXNOS8/ATXN8 | AD | Repeat expansion | CTG/CAG expansion | 608768 |
| ATX222/F/13/EO | Sporadic | Ataxia | SCN8A | AD | c.4850G>A | p.Arg1617Gln | 614306 |
| ATX223/M/30/EO | Sporadic | Spasticity | SPG11 | AR | c.6266dupT c.1013G>A |
p.Cys2090Valfs*14 p.Trp338Ter |
604360 |
| ATX224/M/38/AO | Sporadic | Ataxia | ABHD12 | AR | c.527G>A homozygous |
p.Gly176Glu | 612674 |
| ATX233/F/39/AOb | Multiple affected | Ataxia, spasticity | ELOVL4 | AD | c.512T>C | p.Ile171Thr | 133190 |
| ATX234/F/44/EO | Sporadic | Ataxia | SACS | AR | c.3484G>T c.4880_4881dupGT |
p.Glu1162Ter p.Gln1628Valfs*5 |
270550 |
| ATX237/F/69/AO | Sporadic | Spasticity | GBE1 | AR | c.986A>C homozygous |
p.Tyr329Ser | 263570 |
| ATX238/M/26/EO | Sporadic | Ataxia | SLC2A1 | AD | c.458G>A | p.Arg153His | 606777 612126 |
| ATX242/M/14/EO | Affected mother | Ataxia | KCNA2 | AD | c.890G>A | p.Arg297Gln | 616366 |
| ATX250/M/48/AO | Sporadic | Ataxia | SPG7 | AR | c.1529C>T homozygous |
p.Ala510Val | 607259 |
Note: Age reported in years.
Abbreviations: AD, autosomal dominant; AO, adult-onset; AR, autosomal recessive; cDNA, complementary DNA; EO, early-onset (≤ age 20 years); F, female; M, male; OMIM, Online Mendelian Inheritance in Man.
Family originally reported in Ngo et al. (2018).
Family originally reported in Xiao et al. (2019).
TABLE 3.
Patients with variants of uncertain significance identified
| Patient no./sex/age/onset | Family history | Primary symptom | Gene | Inheritance | cDNA | Protein | OMIM disease phenotype |
|---|---|---|---|---|---|---|---|
| ATX113/M/30/AO | Sporadic | Ataxia | FOXRED1 | AR | c.7C>T | p.Arg3Trp | 618241 |
| ATX115/F/ND/AO | Sporadic | Ataxia | CLCN2 | AR | c.1153C>T | p.Gln385Ter | 615651 |
| ATX117/F/48/AO | Sporadic | Ataxia | FUS | AD | c.253C>T | p.Gln85Ter | 608030 |
| ATX118/F/66/AO | Sporadic | Ataxia | ELOVL5 | AD | c.412C>T | p.Arg138Cys | 615957 |
| ATX119/F/49/AO | Sporadic | Ataxia |
SACS SPART |
AR AR |
c.2329T>C c.1939G>A |
p.Ser777Pro p.Val647Met |
270550 275900 |
| ATX120/F/53/EO | Affected sister | Ataxia | SPG11 | AR | c.2561C>T | p.Ala854Val | 604360 |
| ATX121/M/57/AO | Sporadic | Ataxia | PEX6 | AR | c.853C>G homozygous |
p.Pro285Ala | 614863 |
| ATX122/M/35/EO | Sporadic | Ataxia | PSAP | AR | c.1085A>T | p.Glu362Val | 249900 |
| ATX123/F/48/AO | Multiple affected | Ataxia | COQ2 | AD/AR | c.1178T>C | p.Val393Alaa | 146500 |
| ATX124/F/65/AO | Sporadic | Ataxia | KCNC3 | AD | c.691G>A | p.Gly231Ser | 605259 |
| ATX125/M/61/AO | Sporadic | Ataxia | PAX6 | AD | c.179A>G | p.Gln60Arg | 106210 |
| ATX128/M/50/AO | Sporadic | Ataxia | SYNE1 | AR | c.4843G>A c.17614T>C c.23102G>A |
p.Ala1615Thr p.Ser5872Pro p.Arg7701Gln |
610743 |
| ATX129/F/71/AO | Affected mother | Ataxia | EIF4G1 | AD | c.3470G>T | p.Arg1157Leu | 614251 |
| ATX130/M/42/AO | Sporadic | Ataxia | SYNE1 | AR | c.13696G>A c.14050C>T c.23102G>A |
p.Asp4566Asn p.Leu4684Phe p.Arg7701Gln |
610743 |
| ATX131/F/23/EO | Affected sister | Ataxia | ADAR | AR | c.577C>G | p.Pro193Ala | 615010 |
| ATX132/M/53/AO | Sporadic | Spasticity | SYNE1 | AR | c.19321G>T | p.Ala6441Ser | 610743 |
| ATX133/M/12/EO | Multiple affected | Ataxia | MYH14 | AD | c.1864G>A | p.Val622Ile | 614369 |
| ATX135/M/53/AO | Sporadic | Ataxia | USP9X | XLR | c.2515A>G | p.Ser839Gly | 300919 |
| ATX136/F/80/AO | Sporadic | Ataxia | CLCN1 | AD/AR | c.689G>A | p.Gly230Glu | 160800 255700 |
| ATX137/F/8/EO | Sporadic | Ataxia | SYNE1 | AR | c.2198A>G c.11440A>G |
p.Glu733Gly p.Thr3814Ala |
610743 |
| ATX138/M/72/AO | Sporadic | Spasticity | LAMA1 | AR | c.3724G>A c.5302G>A |
p.Ala1242Thr p.Val1768Met |
615960 |
| ATX139/M/56/AO | Sporadic | Ataxia | PNPLA6 | AR | c.2922_2923insAA | p.Thr975Lysfs*10 | 612020 |
| ATX140/M/60/AO | Sporadic | Spasticity |
SPTLC2 ZFYVE26 |
AD AR |
c.994G>T c.5518C>T |
p.Ala332Ser p.Arg1840Trp |
613640 270700 |
| ATX141/M/63/AO | Sporadic | Ataxia | HEXA | AR | c.739C>T | p.Arg247Trp | 272800 |
| ATX142/M /18/EO | Sporadic | Ataxia | BICD2 | AD | c.684T>A | p.Asp228Glu | 615290 |
| ATX143/M/56/AO | Sporadic | Spasticity | PNPLA6 | AR | c.2783G>C | p.Cys928Ser | 612020 |
| ATX144/M/56/AO | Sporadic | Ataxia | TDP1 | AR | c.346A>G | p.Ile116Val | 607250 |
| ATX145/M/54/AO | Sporadic | Ataxia | SYT14 | AR | c.573T>G | p.Asp191Glu | 614229 |
| ATX146/M/24/EO | Sporadic | Ataxia | TUBB4A | AD | c.439G>A | p.Gly147Arg | 612438 |
| ATX147/M/56/AO | Sporadic | Ataxia |
ZFYVE26 CACNA1G |
AR AD |
c.5584C>T c.6170G>C |
p.Arg1862Cys p.Gly2057Ala |
270700 616795 |
| ATX148/M/74/AO | Sporadic | Spasticity | GBA2 | AR | c.1196G>C | p.Arg399Pro | 614409 |
| ATX150/F/64/EO | Sporadic | Ataxia |
GRID2 SETX |
AR AR |
c.845C>T c.806C>T |
p.Thr282Met p.Ser269Leu |
616204 606002 |
| ATX151/M/62/AO | Affected father (not examined) | Ataxia | SACS | AR | c.8245A>G | p.Ile2749Val | 270550 |
| ATX226/M/55/AO | Multiple affected | Spasticity | EIF2B3 | AR | c.464G>A | p.Arg155His | 603896 |
| ATX227/M/20/EO | Sporadic | Ataxia | SPG7 | AR | c.488G>A | p.Gly163Asp | 607259 |
| ATX228/F/8/EO | Sporadic | Ataxia | TPP1 | AR | c.1496C>G | p.Pro499Arg | 609270 |
| ATX229/M/16/EO | Sporadic | Ataxia | SPTBN2 | AR | c.6577G>A | p.Ala2193Thr | 615386 |
| ATX230/F/16/EO | Sporadic | Ataxia | HTRA2 | AR | c. 346dupG c.1085T>C |
p.Ala116Glyfs*22 p.Ile362Thr |
617248 |
| ATX235/M/62/AO | Sporadic | Ataxia |
SYNE1 SACS |
AR AR |
c.12142G>A c.22432C>T c.9305T>A |
p.Glu4048Lys p.Arg7478Cys p.Leu3102Ter |
610743 270550 |
| ATX239/F/66/AO | Affected father (not examined) | Spasticity | SAMD9L | AD | c.3892C>G | p.Arg1298Gly | 159550 |
| ATX251/M/77/AO | Sporadic | Ataxia, Spasticity | ATP13A2 | AR | c.3057delC | p.Tyr1020Thrfs*3 | 617225 |
| ATX252/M/18/EO | Sporadic | Ataxia |
MFSD8 CTSF |
AR AR |
c.929G>A c.1046–2A>C |
p.Gly310Asp p.? |
610951 615362 |
| ATX260/F/56/AO | Sporadic | Ataxia | GRM1 | AD | c.2155A>C | p.Thr719Pro | 617691 |
Note: Age reported in years.
Abbreviations: AD, autosomal dominant; AO, adult-onset; AR, autosomal recessive; cDNA, complementary DNA; EO, early-onset (≤age 20 years); F, female; M, male; ND, not disclosed; OMIM, Online Mendelian Inheritance in Man; XLR, X-linked recessive.
Previously reported as a risk variant for multiple system atrophy (Zhao et al., 2016).
Following exome analysis, CNV and repeat expansion analysis was performed on a representative subset of the overall cohort (26%, 68/260) consisting of 53 undiagnosed families from the expanded cohort (29%, 53/184) and 15 undiagnosed families from the initial cohort (20%, 15/76) where WES data were available from multiple members to confirm disease segregation. A pathogenic CNV deletion was identified in two families (2.9%, Tables 1, 2, and 4; Table S5). In one family with six members affected by progressive adult-onset ataxia across two generations, a CNV deletion (exons 3–33) in the ITPR1 gene was identified. Similar deletions have previously been reported to cause SCA15/16 (van de Leemput et al., 2007). The CNV deletion was detected by CoNIFER and XHMM in both the patient and affected son’s WES data. Actual breakpoints were clinically confirmed through NGS and quantitative PCR. In the other family, a pathogenic deletion in the FARS2 gene was identified during the reanalysis of the original cohort, described in detail below. A pathogenic repeat expansion was also identified in one family (1.5%) by STRetch (Tables 1 and 2; Table S5). This expansion is associated with Spinocerebellar ataxia type 8 (Mundwiler & Shakkottai, 2018; Paulson, 2018) and was subsequently confirmed by clinical testing in the proband.
TABLE 4.
Reclassification of previously analyzed cases
| Variant summary | ||||||||
|---|---|---|---|---|---|---|---|---|
| Patient index | Pub. status | Reclassified status | Reason | OMIM disease phenotype | Gene | Genomic | cDNA | Protein |
| ATX12 | VUS | N | Variants too common in population | 270700 | ZFYVE26 | 14:68236320C>T 14:68265135G>A |
c.5612G>A c.1844C>T |
p.Cys1871Tyr p.Ser615Phe |
| N | Path | Known pathogenic variant, consistent phenotype | 617046 | FARS2 | 6:5369210C>A Conifer: 6:5109590–5613554del XHMM: 6:5404729–5431977del |
c.407C>A c.(613_46)del |
p.Pro136His p.? |
|
| ATX15a | N | Path | Known pathogenic variant, consistent phenotype, segregates with disease in family | 545000 | MT-TK | m.8344A>G | - | - |
| ATX19 | N | VUS | Rare variant, consistent phenotype | 617931 | PUM1 | 1:31532131C>T | c.283G>A | p.Gly95Arg |
| ATX23a | N | Path | Rare variants, consistent phenotype, segregates with disease in family | 616907 | CAPN1 | 11:64953668_64953669delAG 11:64974125_64974129delAGAGA |
c.618_619delAG c.1545_1549delAGAGA |
p.Gly208Glnfs*7 p.Lys517Cysfs*8 |
| ATX27 | VUS | N | Variant too common in population | 600224 | SPTBN2 | 11:66472288G>A | c.2459C>T | p.Thr820Met |
| ATX31 | N | VUS | Rare variants, consistent phenotype | 610743 | SYNE1 | 6:152470724C>T 6:152737976C>T |
c.24317G>A c.5617G>A |
p.Ser8106Asn p.Ala1873Thr |
| ATX36 | VUS | N | Variant does not segregate with disease in family | 222300 | WFS1 | 4:6302816C>G | c.1294C>G | p.Leu432Val |
| ATX38 | VUS | Pathb | Known pathogenic variant, consistent phenotypeb | 604360 | SPG11 | 15:44876420delCT | c.5456_5457delAG | p.Glu1819Alafs*10 |
| ATX39 | VUS | LP | Known pathogenic variant, consistent phenotype | 614436 | LRSAM1 | 9:130265074T>C | c.2068T>C | p.Cys690Arg |
| ATX40 | N | VUS | Rare variant, consistent phenotype | 615889 | AARS2 | 6:44269870C>T | c.2525G>A | p.Arg842Gln |
| ATX42 | N | VUS | Rare variant, consistent phenotype | 607259 | SPG7 | 16:89598384G>A | c.1060G>A | p.Gly354Arg |
| ATX45 | N | VUS | Rare variant, consistent phenotype | 615889 | AARS2 | chr6:44269119_44269120delGA | c.2680_2681delTC | p.Val895Alafs*10 |
| ATX50 | N | VUS | Rare variant, consistent phenotype | 213600 | SLC20A2 | 8:42297115C>A | c.787G>T | p.Val263Phe |
| ATX52 | VUS N |
N VUS |
Variant is too common in
population Rare variant, consistent phenotype |
612020 616439 | PNPLA6 TBK1 | 19:7607741C>T 12:64879243C>T |
c.1340C>T c.1198C>T |
p.Pro447Leu p.Pro400Ser |
| ATX64 | VUS | N | Variant is too common in population | 614251 | EIF4G1 | 3:184039843A>T | c.1492A>T | p.Ile498Phe |
| ATX66 | VUS | N | Variants too common in population | 263570 | GBE1 | 3:81640290A>C 3:81810551G>T |
c.1134T>G c.118C>A |
p.Ser378Arg p.Pro40Thr |
| ATX68 | VUS | N | Variants do not segregate with disease in family | 614895 271245 |
PRX TWNK |
19:40900066G>T 10:102749087C>T |
c.4193C>A c.1120C>T |
p.Ala1398Asp p.Arg374Trp |
| ATX74 | VUS | N | Variant is too common in population, does not segregate with disease in family | 613908 | TGM6 | 20:2375121T>G | c.31T>G | p.Trp11Gly |
Note: A Total of 76 cases from Fogel et al. (2014) were reanalyzed using current methods (see text). Population frequency determined by use of the ExAC (http://exac.broadinstitute.org/) and gnomAD (https://gnomad.broadinstitute.org/) databases.
Abbreviations: cDNA, complementary DNA; LP, likely pathogenic; N, nondiagnostic; OMIM, Online Mendelian Inheritance in Man; Path, pathogenic; VUS, variant of uncertain clinical significance.
These patients were originally reported with sporadic disorders but subsequently reclassified as familial when additional family members were clinically evaluated.
This recessive disorder was diagnosed clinically due to its distinctive phenotype, a second pathogenic variant is presumed to be noncoding.
Reanalysis of variants from the original cohort at 5 years resulted in an unchanged diagnostic interpretation in 58 of 76 cases (76%), and reclassification in the remaining 18 cases (24%, Table 4). In six subjects, variants previously classified as VUS were reclassified as benign based on either being too common in the human genome using updated ExAC/gnomAD frequency data or because they did not segregate with the disease when additional family members were tested. Conversely, six nondiagnostic cases were reclassified as having a reportable VUS based on interval evidence supporting disease. Two nondiagnostic cases and two with a previously reported VUS were reclassified as now having pathogenic/likely pathogenic variants based on interval updates. One patient had an originally reported VUS in the PNPLA6 gene reclassified as benign and a previously unreported VUS reported in the TBK1 gene due to a more consistent phenotype. Last, one patient had originally reported VUS in the ZFYVE26 gene reclassified as benign and a previously unreported pathogenic variant reported in the FARS2 gene (Sahai et al., 2018) due to a more consistent phenotype along with the identification of the second pathogenic variant through our CNV analysis (Table 4). As described above, the CNV analysis of 15 families from this original cohort (20%, 15/76) identified one (7%, 1/15) pathogenic/likely pathogenic variant. Together, these reclassifications resulted in a modest increase in pathogenic/likely pathogenic variant calls (28%, 21/76 vs. 21%, 16/76) and overall clinically relevant variants (63%, 48/76 vs. 61%, 46/76, Table 4). Combining these data with our expanded cohort, our collective observation of pathogenic/likely pathogenic variation is 25% (65/260) and clinically relevant variants were seen in 52% (135/260, Tables 1–4; Tables S5 and S6). For the combined cohort the most commonly identified genes with pathogenic/likely pathogenic variants were SPG7 (17%, 11/65), SYNE1 (9%, 6/65), CACNA1G, ITPR1, and SPG11 (5%, 3/65 each), and GBE1, KCNA2, SPAST, and WFS1 (3%, 2/65 each, Tables 2 and 4; Table S5; Fogel et al., 2014).
An important diagnostic question concerns how effective exome detection truly is for all known genetic ataxias. To estimate this, we examined the diagnostic rates for families with multiple affected members (either parents or siblings) where a Mendelian genetic etiology can most strongly be presumed. Next we combined our data with the clinically relevant exome diagnostic rates from five recent independent WES studies (Montaut et al., 2018; Nibbeling et al., 2017; Ohba et al., 2013; Pyle et al., 2015; Sawyer et al., 2013) of similar undiagnosed families with ataxia obtaining a mean diagnostic rate of 42% from 139 total families (58/139, Table 1 and Table S3). To assess the efficiency of exome sequencing as a diagnostic test we assumed all these families had a Mendelian genetic cause detectable by exome sequencing, thus setting the maximum diagnostic rate at 100% (adjusted to 96% for average exome coverage). Using a one proportion test, the observed diagnostic rate of 42% differs significantly from the maximum expected rate of 96% (z score = 32.5; p < .0001; 95% confidence interval = 34–51%). These data are sufficiently powered at this level of significance to show a significant discrepancy between the observed diagnostic rate and an expected rate as low as 51%. Therefore, even in a population with the strongest likelihood of having a Mendelian genetic cause identifiable by WES, half of the patients remain undiagnosed. Incorporating our data with that from a total of six additional studies that examined ataxia patients using NGS panel testing (Coutelier et al., 2018; Farwell et al., 2015; Marelli et al., 2016; Nemeth et al., 2013; Sun et al., 2018; van de Warrenburg et al., 2016) did not improve the maximum observed diagnostic confidence interval above 51% (Table 1 and Table S3). Incorporating sporadic cases from the above studies further weakens the maximum potential diagnostic rate (Table S3).
4 |. DISCUSSION
In this report, we performed exome sequencing on 184 patients with undiagnosed familial and sporadic ataxia and/or spastic paraplegia with suspicion for a genetic cause based on either familial inheritance patterns or negative screening for alternative acquired causes of ataxia (Fogel et al., 2014). CNV and repeat expansion analysis were also performed in a representative subset of the cohort, representing approximately one-third of undiagnosed cases. We identified pathogenic/likely pathogenic variants in 24% (44/184), and VUS in 23% (43/184) of cases for an overall clinically relevant detection rate of 47% (87/184). This included identification of a pathogenic CNV in one family and a pathogenic repeat expansion in another. Of note, 11 cases classified as nondiagnostic in this study were ultimately clinically diagnosed with multiple system atrophy, cerebellar type, and another case was identified with a VUS associated with increased risk for this condition (Gilman et al., 2008; Zhao et al., 2016). We also reclassified variants from our previously reported 76 cases (Fogel et al., 2014) based on current annotation, which has been shown to improve diagnosis over time (Alfares et al., 2018; Ewans et al., 2018; Fogel, 2018b; Fogel, Lee, Strom, Deignan, & Nelson, 2016; Fogel et al., 2016; Nambot et al., 2018; Rexach et al., 2019; Wright et al., 2018). This resulted in four cases previously classified as nondiagnostic or having a reportable VUS being reclassified with a pathogenic/likely pathogenic genetic variant. Six previous nondiagnostic cases were reclassified as having a reportable VUS, and six cases with variants previously designated as VUS or likely pathogenic reclassified as benign. Two cases had a previous VUS reclassified as benign but also had either a new VUS or pathogenic variant reported in a different gene. Despite these adjustments, our overall rate of detecting clinically relevant variants remained similar to our previous study (63%, 48/76 vs. 61%, 46/76) although pathogenic/likely pathogenic numbers improved (28%, 21/76 vs. 21%, 16/76). Combining these datasets, the overall detection of clinically relevant variants was 52% (135/260) with 25% (65/260) classified as pathogenic or likely pathogenic.
Collectively, numerous studies evaluating WES and NGS-based ataxia gene panels have achieved diagnostic rates of 32% across all patients (278/873) and 47% for familial cases (43/92), which improves slightly if focused on WES in familial cases (53%, 30/57, Table S3; Coutelier et al., 2018; Farwell et al., 2015; Keogh et al., 2015; Lee et al., 2014; Marelli et al., 2016; Montaut et al., 2018; Nemeth et al., 2013; Nibbeling et al., 2017; Ohba et al., 2013; Pyle et al., 2015; Sawyer et al., 2013; Sun et al., 2018; van de Warrenburg et al., 2016). By combining our current data with these studies we evaluated the capability of current exome sequencing and analysis pipelines to achieve a diagnosis of the remaining unsolved ataxia and spasticity cases. Even when combining the data from these multiple independent studies (Table S3) and assessing diagnostic rates in familial ataxia, that most likely to have an identifiable Mendelian cause, the maximum overall rate of detecting clinically significant findings consistently hovers at approximately 50%, well below even a conservative estimate of the expected rate.
While WES efficiently identifies certain types of disease-causing variants including small indels, single base changes disrupting protein function, and canonical splice acceptor/donor variants, the fact that approximately 50% of cases still remain unresolved highlights gaps in sensitivity of detecting of other classes of mutations (Rexach et al., 2019). In particular, many structural variants and repeat expansions present in the coding sequence are not detected well or reliably genome-wide by WES. For example, exome sequencing can be limited in the comprehensive detection of mutations within the mitochondrial genome and is incapable of detecting the majority of repeat expansions and noncoding genomic variation which can contribute to the modulation of gene expression (Rexach et al., 2019). With recent advances in analyses methods, WES can be used to detect CNV affecting multiple exons, as illustrated here, but continues to be limited for detecting small CNV (i.e., involving single-exons), which may be better identified with whole-genome sequencing (WGS) methods (Rexach et al., 2019). We assessed 26% (68/260) of our cohort for CNV and repeat expansions using the depth of read coverage data from WES, which resulted in the diagnosis of only three additional cases (4%). This suggests that the addition of detection methods for CNV and known repeat expansions to current diagnostic pipelines may not contribute much overall to the missing heredity. However, repeat expansions and single exon heterozygous deletions are challenging to reliably call from exome data so this may also indicate that current methods need further refinement. Extension to WGS using short read (to observe CNV) and long read (to observe CNV and repeat expansion) technologies in the evaluation of ataxia promises to improve detection of these genetic causes (Rexach et al., 2019). Noncoding variation, such as point mutations in promoter regions, splice sites, or other RNA-processing regulatory regions, all potentially detectable by WGS, must also account for some disease-causing mutations as well, but their categorization has remained difficult (Rexach et al., 2019).
The results of this study further emphasize the need for continued investigation into methods to complement and extend the diagnostic value of current next-generation sequencing datasets. It is certainly possible, and likely probable, that additional undiscovered genes responsible for these phenotypes exist and are causative in a percentage of our population. Even with the use of linkage analysis and the addition of multiple family members when available, such genes are challenging to detect if they cause extremely rare or private disorders. The development of collaborative resources to merge data and analysis from large cohorts of patients with ataxic phenotypes may aid in the discovery of such genes (Fogel, 2018a), as will a focus on mutation types not typically detected by exome sequencing. For example, novel disease-causing repeat expansion disorders continue to be described and subsequently identified in undiagnosed patients (Cortese et al., 2019; Ishikawa et al., 2011; Kobayashi et al., 2011; Rafehi et al., 2019; Seixas et al., 2017; Valera et al., 2017). Furthermore, variants whose effect is determined in combination with additional genes (digenic, polygenic), epigenetic, or environmental factors, or causal mutations in the noncoding genome that affect gene regulation would be difficult to detect by current DNA-only methods. The application of “multi-omic” strategies is increasingly being applied to facilitate variant interpretation by assessing their effects on the transcriptome, including alteration in messenger RNA splicing (Cummings et al., 2017; Elsaid et al., 2017; Kremer et al., 2017). Coupling next-generation sequencing methods with transcriptome analysis has already shown diagnostic utility in other rare diseases (Lee et al., 2019). In addition to WGS, new sequencing platforms and analysis strategies are in various stages of development that could facilitate the identification of rare or novel repeat expansions (Gymrek, Golan, Rosset, & Erlich, 2012; Rafehi et al., 2019), or smaller or more complex CNV or CNV mediated by difficult to map repeat elements (SINEs, LINEs, etc.; Turner et al., 2016). Additionally, pathway or network-based analytic methods have been utilized to identify rare or polygenic candidate disease genes based on their convergence upon common disease-associated biological pathways (Nibbeling et al., 2017). Finally, epidemiological and computational medicine approaches, which are increasingly enabled through large-scale precision health initiatives and availability of electronic medical records, have the potential to identify gene-environment interactions that have long eluded detection (Rexach et al., 2019).
Much of the above discussion still represents areas of advancing research investigation but, given the dynamic nature of the field, can be rapidly translated to clinical practice. For the clinician evaluating patients with ataxic and related disorders suspected to have a genetic etiology, standard of care would still include the use of exome sequencing (Fogel, 2018b; Fogel et al., 2014; Rexach et al., 2019) or a comprehensive next-generation sequencing panel targeting currently known ataxia genes (Sun et al., 2018) if exome sequencing is unavailable. However, once performed and if nondiagnostic, for the remaining undiagnosed familial cases with apparent monogenic inheritance, a key focus should be on repeating bioinformatic analysis at regular intervals, as well as the implementation of more comprehensive genomic tools and more complete methods to identify mutation types currently not observed in WES as they become clinically available.
5 |. CONCLUSION
Exome sequencing performed in a predominantly adult- and sporadic-onset cohort of 260 patients with cerebellar ataxia and/or spastic paraplegia observed clinically relevant genetic variation in 52% and pathogenic or likely pathogenic variants in 25% of cases, emphasizing the importance of coding variation to these disorders. However, improved annotation methods and the inclusion of CNV and repeat expansion analysis in a representative subset of this cohort did not dramatically improve overall diagnostic rates from prior studies, even among familial cases with the highest evidence for monogenic disorders. The discrepancy between observed and expected diagnostic rates in familial cases from this and other published studies supports a current diagnostic ceiling for exome sequencing of approximately 50%, suggesting that a critical limitation to genetic diagnosis in these patients rests on the high likelihood that missing pathogenic mutations lie outside the exome and must be identified by other methods and more comprehensive genome-wide strategies.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the University of California Los Angeles (UCLA) Clinical Neurogenomics Research Center for assistance with biobanking and exome sequencing, the UCLA Clinical Genomics Center for performing exome sequencing, and members of the UCLA Genomic Data Board for contributions to the bioinformatic analysis and interpretation of individual cases. BLF had full access to all the data in this study and takes responsibility for the integrity of the data and the accuracy of the data analysis. None of the funding agencies or sponsors had any role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication. This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk or via email from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by the Wellcome Trust. This work was supported by the National Institute for Neurological Disorders and Stroke (R01NS082094 to BLF) and the NIH/National Center for Advancing Translational Science UCLA Clinical and Translational Science Institute (CTSI) Grant Number UL1TR000124. This work was supported in part by grant UM1HG006542 (JRL) from the National Human Genome Research Institute (NHGRI)/National Heart, Lung, and Blood Institute to the Baylor Hopkins Center for Mendelian Genomics, R01NS058529, R35NS105078 (JRL), and R25NS065723 (JER) from the National Institute of Neurological Disorders and Stroke, and U54HG003273 (RAG) from NHGRI. JEP was supported by NHGRI K08HG008986. BLF acknowledges support through donations to the University of California by the Rochester Ataxia Foundation.
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Numbers: R01NS058529, R01NS082094, R25NS065723, R35NS105078; National Human Genome Research Institute, Grant/Award Numbers: K08HG008986, U54HG003273, UM1HG006542; Rochester Ataxia Foundation; NIH/National Center for Advancing Translational Science, Grant/Award Number: UL1TR000124
Footnotes
CONFLICT OF INTERESTS
JRL serves on the scientific advisory board for Baylor Genetics, has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. All other authors declare that there is no conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are not publicly available due to privacy restrictions but may be made available in whole or in part from the corresponding author upon reasonable request.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- Alfares A, Aloraini T, Subaie LA, Alissa A, Qudsi AA, Alahmad A, … Alfadhel M (2018). Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med. 20, 1328–1333. 10.1038/gim.2018.41 [DOI] [PubMed] [Google Scholar]
- Anheim M, Tranchant C, & Koenig M (2012). The autosomal recessive cerebellar ataxias. New England Journal of Medicine, 366(7), 636–646. 10.1056/NEJMra1006610 [DOI] [PubMed] [Google Scholar]
- Benini R, Ben Amor IM, & Shevell MI (2012). Clinical clues to differentiating inherited and noninherited etiologies of childhood ataxias. The Journal of Pediatrics, 160(1), 152–157. 10.1016/j.jpeds.2011.06.029 [DOI] [PubMed] [Google Scholar]
- Brusse E, Maat-Kievit JA, & van Swieten JC (2007). Diagnosis and management of early- and late-onset cerebellar ataxia. Clinical Genetics, 71(1), 12–24. 10.1111/j.1399-0004.2006.00722.x [DOI] [PubMed] [Google Scholar]
- Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yan YW, … Houlden H (2019). Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nature Genetics, 51(4), 649–658. 10.1038/s41588-019-0372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, … Ataxia N (2018). Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurology, 75(5), 591–599. 10.1001/jamaneurol.2017.5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, … MacArthur DG (2017). Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Science Translational Medicine, 9(386), eaal5209. 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, … Genomes Project Analysis G (2011). The variant call format and VCFtools. Bioinformatics, 27(15), 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dashnow H, Lek M, Phipson B, Halman A, Sadedin S, Lonsdale A, … Oshlack A (2018). STRetch: Detecting and discovering pathogenic short tandem repeat expansions. Genome Biology, 19(1), 121 10.1186/s13059-018-1505-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, … Daly MJ (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics, 43(5), 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaid MF, Chalhoub N, Ben-Omran T, Kumar P, Kamel H, Ibrahim K, … Aleem AA (2017). Mutation in noncoding RNA RNU12 causes early onset cerebellar ataxia. Annals of Neurology, 81(1), 68–78. 10.1002/ana.24826 [DOI] [PubMed] [Google Scholar]
- Ewans LJ, Schofield D, Shrestha R, Zhu Y, Gayevskiy V, Ying K, … Roscioli T (2018). Whole-exome sequencing reanalysis at 12 months boosts diagnosis and is cost-effective when applied early in Mendelian disorders. Genetics in Medicine, 20, 1564–1574. 10.1038/gim.2018.39 [DOI] [PubMed] [Google Scholar]
- Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, … Tang S (2015). Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: Results from 500 unselected families with undiagnosed genetic conditions. Genetics in Medicine, 17(7), 578–586. 10.1038/gim.2014.154 [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, … Carter NP (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. The American Journal of Human Genetics, 84(4), 524–533. 10.1016/j.ajhg.2009.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL (2018a). Collaborative science unites researchers and a novel spastic ataxia gene. Annals of Neurology, 83(6), 1072–1074. 10.1002/ana.25262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL (2018b). Genetic and genomic testing for neurologic disease in clinical practice. Handbook of Clinical Neurology, 147, 11–22. 10.1016/B978-0-444-63233-3.00002-6 [DOI] [PubMed] [Google Scholar]
- Fogel BL, Lee H, Deignan JL, Strom SP, Kantarci S, Wang X, … Nelson SF (2014). Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurology, 71(10), 1237–1246. 10.1001/jamaneurol.2014.1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, Lee H, Strom SP, Deignan JL, & Nelson SF (2016). Clinical exome sequencing in neurogenetic and neuropsychiatric disorders. Annals of the New York Academy of Sciences, 1366(1), 49–60. 10.1111/nyas.12850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, & Perlman S (2006). An approach to the patient with late-onset cerebellar ataxia. Nature Clinical Practice Neurology, 2(11), 629–635. 10.1038/ncpneuro0319 [DOI] [PubMed] [Google Scholar]
- Fogel BL, & Perlman S (2007). Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. The Lancet Neurology, 6(3), 245–257. 10.1016/S1474-4422(07)70054-6 [DOI] [PubMed] [Google Scholar]
- Fogel BL, & Perlman S (2011). Cerebellar disorders: Balancing the approach to cerebellar ataxia In Gálvez-Jiménez N, & Tuite PJ (), Uncommon Causes of Movement Disorders (1st ed, 198–216). Cambridge, NY: Cambridge University Press. [Google Scholar]
- Fogel BL, Satya-Murti S, & Cohen BH (2016). Clinical exome sequencing in neurologic disease. Neurology Clinical Practice, 6(2), 164–176. 10.1212/CPJ.0000000000000239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel BL, Vickrey BG, Walton-Wetzel J, Lieber E, & Browner CH (2013). Utilization of genetic testing before subspecialist referral for cerebellar ataxia. Genetic Testing and Molecular Biomarkers, 17(8), 588–594. 10.1089/gtmb.2013.0005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, … Purcell SM (2012). Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. The American Journal of Human Genetics, 91(4), 597–607. 10.1016/j.ajhg.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, & Purcell SM (2014). Using XHMM software to detect copy number variation in whole-exome sequencing data. Current Protocols in Human Genetics, 81, 7.23.1–7.23.21. 10.1002/0471142905.hg0723s81 23 21-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambin T, Akdemir ZC, Yuan B, Gu S, Chiang T, Carvalho CMB, … Lupski JR (2016). Homozygous and hemizygous CNV detection from exome sequencing data in a Mendelian disease cohort. Nucleic Acids Research, 45(4), gkw1237. 10.1093/nar/gkw1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, Tarazona S, … Conesa A (2012). Qualimap: Evaluating next-generation sequencing alignment data. Bioinformatics, 28(20), 2678–2679. 10.1093/bioinformatics/bts503 [DOI] [PubMed] [Google Scholar]
- Gilman S, Wenning GK, Low PA, Brooks DJ, Mathias CJ, Trojanowski JQ, … Vidailhet M (2008). Second consensus statement on the diagnosis of multiple system atrophy. Neurology, 71(9), 670–676. 10.1212/01.wnl.0000324625.00404.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudbjartsson DF, Jonasson K, Frigge ML, & Kong A (2000). Allegro, a new computer program for multipoint linkage analysis. Nature Genetics, 25(1), 12–13. 10.1038/75514 [DOI] [PubMed] [Google Scholar]
- Gymrek M, Golan D, Rosset S, & Erlich Y (2012). lobSTR: A short tandem repeat profiler for personal genomes. Genome Research, 22(6), 1154–1162. 10.1101/gr.135780.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa K, Durr A, Klopstock T, Muller S, De Toffol B, Vidailhet M, … Mizusawa H (2011). Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology, 77(20), 1853–1855. 10.1212/WNL.0b013e3182377e3a [DOI] [PubMed] [Google Scholar]
- Keogh MJ, Steele H, Douroudis K, Pyle A, Duff J, Hussain R, … Chinnery PF (2015). Frequency of rare recessive mutations in unexplained late onset cerebellar ataxia. Journal of Neurology, 262(8), 1822–1827. 10.1007/s00415-015-7772-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klockgether T (2010). Sporadic ataxia with adult onset: Classification and diagnostic criteria. The Lancet Neurology, 9(1), 94–104. 10.1016/S1474-4422(09)70305-9 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, … Koizumi A (2011). Expansion of Intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. The American Journal of Human Genetics, 89(1), 121–130. 10.1016/j.ajhg.2011.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, … Prokisch H (2017). Genetic diagnosis of Mendelian disorders via RNA sequencing. Nature Communications, 8, 15824 10.1038/ncomms15824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Sudmant PH, Ko A, O’Roak BJ, Malig M, Coe BP, … Eichler EE (2012). Copy number variation detection and genotyping from exome sequence data. Genome Research, 22(8), 1525–1532. 10.1101/gr.138115.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, … Nelson SF (2014). Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA, 312(18), 1880–1887. 10.1001/jama.2014.14604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Huang AY, Wang LK, Yoon AJ, Renteria G, Eskin A, … Nelson SF (2019). Diagnostic utility of transcriptome sequencing for rare Mendelian diseases. Genetics in Medicine, 10.1038/s41436-019-0672-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, … Exome Aggregation C (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, … Genome Project Data Processing, S. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25(16), 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, & Scherer SW (2014). The database of genomic variants: A curated collection of structural variation in the human genome. Nucleic Acids Research, 42(Database issue), D986–992. 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manto M, & Marmolino D (2009). Cerebellar ataxias. Current Opinion in Neurology, 22(4), 419–429. 10.1097/WCO.0b013e32832b9897 [DOI] [PubMed] [Google Scholar]
- Marelli C, Guissart C, Hubsch C, Renaud M, Villemin JP, Larrieu L, … Koenig M (2016). Mini-exome coupled to read-depth based copy number variation analysis in patients with inherited ataxias. Human Mutation, 37(12), 1340–1353. 10.1002/humu.23063 [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, … DePristo MA (2010). The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaut S, Tranchant C, Drouot N, Rudolf G, Guissart C, Tarabeux J, … Movement Disorders C (2018). Assessment of a targeted gene panel for identification of genes associated with movement disorders. JAMA Neurology, 75(10), 1234–1245. 10.1001/jamaneurol.2018.1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundwiler A, & Shakkottai VG (2018). Autosomal-dominant cerebellar ataxias. Handbook of Clinical Neurology, 147, 173–185. 10.1016/B978-0-444-63233-3.00012-9 [DOI] [PubMed] [Google Scholar]
- Nambot S, Thevenon J, Kuentz P, Duffourd Y, Tisserant E, Bruel AL, … Orphanomix Physicians, G. (2018). Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: Substantial interest of prospective annual reanalysis. Genetics in Medicine, 20(6), 645–654. 10.1038/gim.2017.162 [DOI] [PubMed] [Google Scholar]
- Nemeth AH, Kwasniewska AC, Lise S, Parolin Schnekenberg R, Becker EB, Bera KD, … Ragoussis J (2013). Next generation sequencing for molecular diagnosis of neurological disorders using ataxias as a model. Brain, 136(Pt 10), 3106–3118. 10.1093/brain/awt236awt236. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo K, Aker M, Petty LE, Chen J, Cavalcanti F, Nelson AB, … Fogel BL (2018). Expanding the global prevalence of spinocerebellar ataxia type 42. Neurology Genetics, 4(3), e232 10.1212/NXG.0000000000000232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibbeling EAR, Duarri A, Verschuuren-Bemelmans CC, Fokkens MR, Karjalainen JM, Smeets C, … Verbeek DS (2017). Exome sequencing and network analysis identifies shared mechanisms underlying spinocerebellar ataxia. Brain, 140(11), 2860–2878. 10.1093/brain/awx251 [DOI] [PubMed] [Google Scholar]
- O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, … Pruitt KD (2016). Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Research, 44(D1), D733–745. 10.1093/nar/gkv1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, … Eichler EE (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485(7397), 246–250. 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N, … Saitsu H (2013). Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics, 14(3–4), 225–232. 10.1007/s10048-013-0375-8 [DOI] [PubMed] [Google Scholar]
- Okonechnikov K, Conesa A, & Garcia-Alcalde F (2015). Qualimap 2: Advanced multisample quality control for high-throughput sequencing data. Bioinformatics, 32(2), btv566. 10.1093/bioinformatics/btv566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omim. ([accessed May 2019]). Online Mendelian Inheritance in Man, OMIM, from McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University; (Baltimore, MD: ) World Wide Web URL: http://omim.org/ [Google Scholar]
- Paulson H (2018). Repeat expansion diseases. Handbook of Clinical Neurology, 147, 105–123. 10.1016/B978-0-444-63233-3.00009-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poultney CS, Goldberg AP, Drapeau E, Kou Y, Harony-Nicolas H, Kajiwara Y, … Buxbaum JD (2013). Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. The American Journal of Human Genetics, 93(4), 607–619. 10.1016/j.ajhg.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle A, Smertenko T, Bargiela D, Griffin H, Duff J, Appleton M, … Chinnery PF (2015). Exome sequencing in undiagnosed inherited and sporadic ataxias. Brain, 138(Pt 2), 276–283. 10.1093/brain/awu348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NLM, Pope K, Smith KR, … Lockhart PJ (2019). Bioinformatics-based identification of expanded repeats: A nonreference intronic pentamer expansion in RFC1 causes CANVAS. The American Journal of Human Genetics, 105(1), 151–165. 10.1016/j.ajhg.2019.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach J, Lee H, Martinez-Agosto JA, Nemeth AH, & Fogel BL (2019). Clinical application of next-generation sequencing to the practice of neurology. The Lancet Neurology, 18(5), 492–503. 10.1016/S1474-4422(19)30033-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, … Committee ALQA (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, … Purcell SM (2016). Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nature Genetics, 48(10), 1107–1111. 10.1038/ng.3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai SK, Steiner RE, Au MG, Graham JM, Salamon N, Ibba M, & Pierson TM (2018). FARS2 mutations presenting with pure spastic paraplegia and lesions of the dentate nuclei. Annals of Clinical and Translational Neurology, 5(9), 1128–1133. 10.1002/acn3.598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Schwartzentruber J, Beaulieu CL, Dyment D, Smith A, Chardon JW, … Boycott KM (2013). Exome sequencing as a diagnostic tool for pediatric-onset ataxia. Human Mutation, 35, 45–49. 10.1002/humu.22451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seixas AI, Loureiro JR, Costa C, Ordonez-Ugalde A, Marcelino H, Oliveira CL, … Silveira I (2017). A pentanucleotide ATTTC repeat insertion in the noncoding region of DAB1, mapping to SCA37, causes spinocerebellar ataxia. The American Journal of Human Genetics, 101(1), 87–103. 10.1016/j.ajhg.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, & Fogel BL (2013). Clinical neurogenetics. Neurologic Clinics, 31(4), 987–1007. 10.1016/j.ncl.2013.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples J, Ekunwe L, Lange E, Wilson JG, Nickerson DA, & Below JE (2016). PRIMUS: Improving pedigree reconstruction using mitochondrial and Y haplotypes. Bioinformatics, 32(4), 596–598. 10.1093/bioinformatics/btv618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples J, Qiao D, Cho MH, Silverman EK, University of Washington Center for Mendelian, G., Nickerson DA, & Below JE (2014). PRIMUS: Rapid reconstruction of pedigrees from genome-wide estimates of identity by descent. The American Journal of Human Genetics, 95(5), 553–564. 10.1016/j.ajhg.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, & Cooper DN (2014). The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human Genetics, 133(1), 1–9. 10.1007/s00439-013-1358-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom SP, Lee H, Das K, Vilain E, Nelson SF, Grody WW, & Deignan JL (2014). Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genetics in Medicine, 16(7), 510–515. 10.1038/gim.2013.183gim2013183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, … Das S (2018). Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genetics in Medicine, 21, 195–206. 10.1038/s41436-018-0007-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner TN, Hormozdiari F, Duyzend MH, McClymont SA, Hook PW, Iossifov I, … Eichler EE (2016). Genome Sequencing of Autism-Affected Families Reveals Disruption of Putative Noncoding Regulatory DNA. The American Journal of Human Genetics, 98(1), 58–74. 10.1016/j.ajhg.2015.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera JM, Diaz T, Petty LE, Quintans B, Yanez Z, Boerwinkle E, … Fogel BL (2017). Prevalence of spinocerebellar ataxia 36 in a US population. Neurology Genetics, 3(4), e174 10.1212/NXG.0000000000000174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, … DePristo MA (2013). From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Current Protocols in Bioinformatics, 43, 11–33. 10.1002/0471250953.bi1110s43.1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, … Singleton AB (2007). Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLOS Genetics, 3(6), e108 10.1371/journal.pgen.0030108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Warrenburg BP, Schouten MI, de Bot ST, Vermeer S, Meijer R, Pennings M, … Kamsteeg EJ (2016). Clinical exome sequencing for cerebellar ataxia and spastic paraplegia uncovers novel gene-disease associations and unanticipated rare disorders. European Journal of Human Genetics, 24(10), 1460–1466. 10.1038/ejhg.2016.42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, … Firth HV (2018). Making new genetic diagnoses with old data: Iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genetics in Medicine, 20, 1216–1223. 10.1038/gim.2017.246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Binkley EM, Rexach J, Knight-Johnson A, Khemani P, Fogel BL, … Gomez CM (2019). A family with spinocerebellar ataxia and retinitis pigmentosa attributed to an ELOVL4 mutation. Neurology Genetics, 5(5), e357 10.1212/NXG.0000000000000357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, … Visscher PM (2010). Common SNPs explain a large proportion of the heritability for human height. Nature Genetics, 42(7), 565–569. 10.1038/ng.608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Yang X, Tian S, An R, Zheng J, & Xu Y (2016). Association of the COQ2 V393A variant with risk of multiple system atrophy in East Asians: A case-control study and meta-analysis of the literature. Neurological Sciences, 37(3), 423–430. 10.1007/s10072-015-2414-8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.