

Abstract
To design and discover a new compound can used as a COX with TNF-α and IL-6 inhibitors is highly challenge. A series of spiroindolone-bearing benzofuran moieties were resynthesized from the chalcone-based benzo[b]furan with substituted isatin, and amino acids. The requisite spiroindolone analogues were tested for their potential inhibitory activities against lipid metabolizing enzymes such as cyclooxygenase COX-1, COX-2, and the release of pro-inflammatory cytokines interleukin IL-6, and tumor necrosis factor TNF-α. Among the tested compounds, 5a, 5c, 5h, 5i, 5l, and 5p exhibited COX-1 inhibitor selectively with percent of inhibition 40.81–83.4% and IC50 values ranging from 20.42 µM to 38.24 µM. In addition, all the synthesized target compounds possessed lipopolysaccharide-induced TNF-α, and IL-6 expression with a varying degree of COX-1 inhibition. Compounds 5d, 5e, 5f, 5g, and 5k markedly inhibited TNF-α, and IL-6 release in WI-38 fibroblast cells. Molecular docking of the most effective and highly selective compounds were investigated and shown important binding mechanisms which could affect pro-inflammatory enzymes and cytokines via the inhibition of COX-1, COX-2, IL-6, and TNF-α.
Keywords: Spirooxindole, Lipid metabolic enzymes, Pro-inflammatory cytokines, COX-1, COX-2, IL-6, TNF-α
1. Introduction
Several factors affect the development of cancer including the combination of environmental, and host factors. Inflammation has been linked and increased the risk for development different type of tumors such as gastric mucosal lymphoma, colon cancer, gastric cancer, and prostate cancer. Many factors lead to chronic inflammation such as microbial infections (e.g., Helicobacter pylori infection), autoimmune diseases (e.g., inflammatory bowel disease), inflammatory conditions (e.g., prostatitis), chemical compounds (e.g., ROS), or physical injuries (e.g., exposure to UV) (Balkwill and Mantovani, 2001, Koehne and Dubois, 2004, Chan et al., 2017, Mantovani et al., 2008).
There are two cyclooxygenase COX enzymes, namely cyclooxygenase-1 COX-1, and cyclooxygenase-2 COX-2. The cytoprotection process in the gastrointestinal tract and normal renal function is regulated by the constitutive isozyme COX-1. COX-2 is a proactive, and short-lived enzyme; its expression occurs as a pro-inflammatory response. COX-2 is involved in prostaglandin biosynthesis in inflammatory cells of the central nervous system (Marnett et al., 1999, Herschman, 1996). The activation of such enzymes stimulates intracellular signals (i.e., NFκB (Takeda et al., 2003, Ben-Neriah and Karin, 2011), p38, or MAPKs (Dhanasekaran and Johnson, 2007), which modify pro-inflammatory cytokine expression e.g., interleukin 1 beta (IL1β) (Church et al., 2008), interleukin 6 (IL6) (Trikha et al., 2003), and tumor necrosis factor alpha (TNF-α) (Tracey et al., 1994). These signals combined with cell adhesion proteins and chemokines leads to the stimulation and activation of immune cells. Therefore, many researchers focus on developing novel and selective inhibitors of pro-inflammatory enzymes and cytokines.
Oxindole derivatives are known as anti-tumor agents due to their tyrosine kinase inhibitory property. Additionally, the oxindole scaffold exhibits promising structural features and a good bioactivity profile including antiviral, antimicrobial, and local anesthetic properties (Galliford and Scheidt, 2007, Marti and Carreira, 2003, Trost and Brennan, 2009, Ding et al., 2005, Padwa and Pearson, 2002, Wade, 1991). Therefore, these compounds may further the design of novel lead compounds with anti-inflammatory, and anticancer activities. Recently, one such compound, JP-8g, exhibited high efficacy against cancer, and inflammation (Sun et al., 2014, Sun et al., 2015). Among synthetic compounds, oxindole gained attention as a potential anticancer drug. In this regard, compounds SPX-F (Jiang et al., 2010), and 4d (Barakat et al., 2017, Barakat et al., 2019) have exhibited efficacy for MDM2–p53 protein–protein interaction inhibitor and p53 reactivation in cancer cells (Fig. 1).
Fig. 1.

Representative examples of spirooxindoles and benzo[b]furan scaffolds.
Benzo[b]furan is an interesting pharmacophore which exists in several synthetic and natural hits and has pharmaceutical applications (Dawood, 2013). Some publications report the benzo[b]furan moiety to inhibit activities of human immunodeficiency virus (HIV) and hepatitis C virus (HVC). In addition, it also has possessed as antioxidant, cytotoxic, anti-inflammatory (e.g. ailanthoidol), antitumor, antimicrobial, antiplasmodial and, antitubercular activities (Dawood, 2013) (Fig. 1).
A derivative of celecoxib based a benzo[b]furan moiety was reported to demonstrate selective activity against COX-2 (Hassan et al., 2014). In addition, new molecules containing rhodanine and benzofuran scaffolds were designed, synthesized, and reported to exhibit dual COX-2, and 5-LOX inhibitory potential. Recent patent survey reported as a review focused on the benzofuran inhibitors (El-Miligy et al., 2017).
In this study, a series of compounds having the oxindole, and benzo[b]furan scaffolds were resynthesized according to Altowyan et al. (2019). These highly functionalized complex molecules, with different pharmacophores, may have potential applications in diverse, and economically relevant areas.
2. Materials and methods
The target compounds were resynthesized in accordance with a previous article (Altowyan et al., 2019). Additionally, all enzymatic assays and the molecular docking studies details have been provided in the supplementary information.
3. Results and discussion
3.1. Preparation of 5a-r
A number of spirooxindole benzo[b]furan scaffolds were resynthesized using the 1,3-dipolar cycloaddition reaction method. As shown in Fig. 2, the synthetic pathway consist of the reaction of chalcone 2a-f with dicarbonyl compounds (isatin, 5-chloroisatin, and 5-bromoisatin 3a-c) and amino acids including (S)-thiazolidine-4-carboxylic acid or (2S,3aS,7aS)-octahydro-1H-indole-2-carboxylic acid. In this manner, a number of spirooxindole based benzo[b]furan scaffolds (5a-r) were obtained in high analytical purity with complete stereoselective reaction (Scheme 1). The synthetic methodology was carried out under mild alcoholic conditions, which is considered an eco-friendly approach. The general reaction is shown below, as variations of all three reactants are possible. The mechanism of cyclization is in accordance to previously established methods (Islam et al., 2019, Barakat et al., 2018, Lotfy et al., 2018, Lotfy et al., 2019).
Fig. 2.

(A) Hydrophobic interactions embed compound 5i inside COX-1 receptor (ID: 5WBE). (B) Hydrophobic interactions embed compound 5i inside COX-2 receptor (ID: 3LN1). (C) Compound 5i is overlaid with celecoxib inside COX-2 receptor (ID: 3LN1). (D) Compound 5a with (ID: 5WBE) docked outside the receptor through formation of two HBs with ARG 120.
Scheme 1.

The resynthesized compounds 5a-r.
3.2. In vitro biological activity evaluation
3.2.1. COX-1 and COX-2 inhibitors
The COX inhibitory activity of resynthesized target compounds were screened in vitro for the two isoforms of COX (Abdellatif et al., 2018). COX (COX-1, and COX-2) at 40 μg/mL and the results obtained were shown in Table 1. The IC50 values were determined, and the data are summarized in Table 1. The data in Table 1 suggested that compounds 5a, 5c, 5h, 5i, 5l, and 5p possessed powerful COX-1 inhibitory abilities ranging from 40.81−83.4%. On the other hand, the inhibition of COX-2 was weaker, and was shown in the range of 1.11–20.56%. Indomethacin, acetyl-keto boswellic acid (AKBA), acetyl-β-boswellic acid, acetyl-α-boswellic acid, and β-boswellic acid were used as positive controls for COX-1. Celecoxib was used as a positive control for COX-2. In the case of COX-1 inhibition, the IC50 values recorded for the most active compounds were in the range of 20.42–38 µM; these included 5a, 5c, 5h, 5i, 5l, and 5p. Compound 5a (IC50 = 20.42 µM) exhibited the strongest inhibition of COX-1 between these series. It was observed that the analogs were less selective against COX-2 than against COX-1.
Table 1.
Results of percentage of inhibition of COX-1, and COX-2, IC50, selectivity COX-1/CO-2; percentage of inhibition of IL-6 (pg/ml), and TNF-α of the synthesized spirooxindoles based on benzo[b]furan scaffold 5a-r.
| # | Compound | COX-1% | COX-2% | COX-1 |
COX-2 |
Selectivityb IC50 COX-1/IC50 COX-2 |
IL-6 (pg/ml)% | TNF-α (pg/ml)% |
|---|---|---|---|---|---|---|---|---|
| IC50 (μM ± SD)a | ||||||||
| 1 | 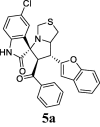 |
57.38 | 11.24 | 20.42 ± 0.55 | 88.09 ± 1.78 | 23% | 57.38 | 51.24 |
| 2 |  |
27.36 | 9.81 | NDc | ND | ND | 27.36 | 9.81 |
| 3 | 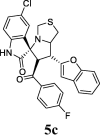 |
40.81 | 8.33 | 37 ± 1.54 | 76.17 ± 1.46 | 49% | 59.81 | 48.33 |
| 4 |  |
19.81 | 7.77 | ND | ND | ND | 76.81 | 67.77 |
| 5 | 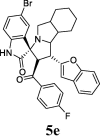 |
9.43 | 1.8 | ND | ND | ND | 82.43 | 71.8 |
| 6 | 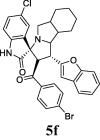 |
32.08 | 5.92 | ND | ND | ND | 63.08 | 55.92 |
| 7 | 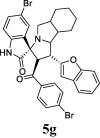 |
19.81 | 2.76 | ND | ND | ND | 81.81 | 78.76 |
| 8 | 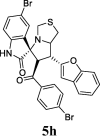 |
41.70 | 9.8 | 35.70 ± 0.51 | 84.66 ± 1.16 | 42% | 38.70 | 49.8 |
| 9 | 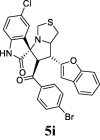 |
44.53 | 17.85 | 29.55 ± 0.19 | 71.25 ± 1.29 | 41% | 62.53 | 57.85 |
| 10 | 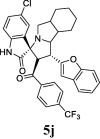 |
21.70 | 7.89 | ND | ND | ND | 77.70 | 67.89 |
| 11 | 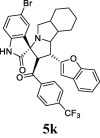 |
12.26 | 4.45 | ND | ND | ND | 87.26 | 74.45 |
| 12 | 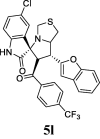 |
47.74 | 19.78 | 31.25 ± 0.24 | 58.75 ± 1.58 | 36% | 42.74 | 39.78 |
| 13 | 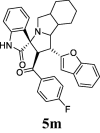 |
20.75 | 5.78 | ND | ND | ND | 79.54 | 75.78 |
| 14 | 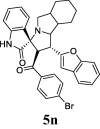 |
9.43 | 1.11 | ND | ND | ND | 82.12 | 77.11 |
| 15 | 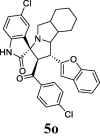 |
29.25 | 6.11 | ND | ND | ND | 76.81 | 66.11 |
| 16 | 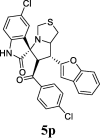 |
52.75 | 9.98 | 38.24 ± 0.96 | 85.70 ± 1.51 | 45% | 47.18 | 39.98 |
| 17 | 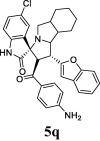 |
32.08 | 10.1 | ND | ND | ND | 67.43 | 60.1 |
| 18 | 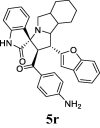 |
32.08 | 6.91 | ND | ND | ND | 64.81 | 62.91 |
| STD | Indomethacin | 87.31 | 18.76 | 0.24 ± 0.05 | 3.28 ± 0.09 | 7% | 7.7 | 8.1 |
| Acetyl-keto boswellic acid | 71.42 | 19.87 | 7.84 ± 0.13 | 82.52 ± 0.62 | 10% | 7.31 | 18.76 | |
| Acetyl-β-boswellic acid | 75.73 | 14.87 | 8.16 ± 0.10 | 72.52 ± 0.51 | 11% | 11.42 | 19.87 | |
| Acetyl-α-boswellic acid | 69.81 | 7.98 | 10.13 ± 0.17 | >100 | 10% | 15.73 | 14.87 | |
| β-Boswellic acid | 64.77 | 7.66 | 18.14 ± 0.42 | >100 | 18% | 19.81 | 27.98 | |
| Celecoxib | 8.7 | 92 | 29.19 ± 0.33 | 0.08 ± 0.44 | 365% | 24.77 | 25.16 | |
| Lipopolysaccharide (LPS) | – | – | – | – | – | 100 | 100 | |
COX-1, and COX-2 inhibitory activity is expressed as the mean ± SD of triplicate experiments. bSelectivity is defined as IC50 COX-1/IC50 COX-2. cND: not determined.
3.2.2. Inhibition of TNF-α and IL-6 release in LPS-stimulated cells
The inhibitory activity of the resynthesized compounds was examined against LPS-induced TNF-α, and IL-6 in WI-38 fibroblast cells. All the examined compounds inhibited LPS-induced TNF-α, and IL-6 expression to different extents. Compounds 5d, 5e, 5f, 5g, and 5k were the most effective among this compounds (Table 1).
3.3. Docking study
The studied compounds were subjected to molecular docking study to evaluate their activities as anti-inflammatory agents by analyzing their ability to inhibit COX-1 (PDB: ID 5wbe (Loll et al., 1996), and PDB: ID 1pgf (Wang et al., 2010), COX-2 (PDB: ID 3ln1) (He et al., 2005), and TNF-α (PDB: ID 2az5) (Wang et al., 2010). Moreover, COX-1/COX-2 selectivity was explained.
The docking mode of final compounds with COX-1(PDB: ID 5wbe) (Cingolani et al., 2017) showed that these compounds could be classified into two groups: Group (a) of compounds which interact with the protein inside the specific receptor of standards such as compound 5i (Fig. 2A–C), and the second group (b) of compounds where the standard ligand docked outside the receptor through formation of hydrogen bonding (HB) interaction such as compound 5a (Fig. 2D).
Compound 5i interacts with key residues of COX-1, and COX-2 receptors via hydrophobic interactions through the benzofuran, indole, and p-bromophenyl moieties, Fig. 2A, and B respectively. Compound 5i is overlaid with celecoxib inside the COX-2 receptor (ID: 3LN1). Biological studies revealed that both compounds (5i and 5p) are non-selective. This were clarified by their ability to adapt inside the receptors of both COX-1 and COX-2 using the same binding mode (hydrophobic interactions) (Fig. 2A–D). Compound 5a from the group (b) interacts with the receptor by formation of two HBs with Arg:120A via the oxygen of benzofuran moiety. This strong interaction helps to understand their selectivity.
The docking of the most active compounds with TNF-α (PDB: ID 2az5) was then studied. The standard ligand docked inside the receptor in a U-shape through the formation of hydrophobic interactions, Fig. 3A.
Fig. 3.

(A) Standard drug co-crystallized with (ID: 2az5) through the formation of hydrophobic-hydrophobic interactions. (B) Compound 5a with (ID: 2az5) docked outside the receptor through the formation of hydrophobic-hydrophobic interactions. (C) Compound 5a with (ID: 2az5) docked with receptor overlay with standard drugs through the formation of hydrophobic-hydrophobic interactions.
Compound 5a docked inside the receptor through the formation of hydrophobic-hydrophobic interactions. Moreover, it overlaid onto the standard drug via similar interactions with the receptor cleft, Fig. 3(B, C).
3.4. Structure activity relationship
The structure activity relationship (SAR) of the studied compounds, and their biological activities with COX-1, COX-2, and the release of IL-6, and TNF-α were evaluated. The data presented in Tables 1, and Fig. 4 demonstrate that:
-
(i)
Compounds 5a, 5c, 5h, 5i, 5l, and 5p which were synthesized based on the thiazole amino acid rather than fused cyclohexane-proline, lead to better inhibition of COX-1, COX-2, IL-6, and TNF-α. This could be due to the geometry of the chair form, and its role in positioning benzofuran inside the receptor.
-
(ii)
Compound 5a, with a chlorine atom on the oxindole moiety, and an un-substituted benzene on the benzoyl nucleus, showed the best, and selective results with 57.38% inhibition and IC50 = 20.42 ± 0.55 µM against COX-1 compared to the standard drug, celecoxib (8.7% inhibition and IC50 = 29.19 ± 0.33 µM).
-
(iii)
Compound 5c, with a chlorine atom on the oxindole moiety, and para fluoro-substituted benzene on the benzoyl nucleus, showed good inhibition with 40.81% and IC50 = 37 ± 1.54 µM against COX-1, on the other hands when the chlorine and fluorine atoms replaced with bromine there is not dramatically changes in the activity which has been shown slightly more inhibition with 41.70% and IC50 = 35.70 ± 0.51 µM but when chlorine combined with bromine in the hit 5i shown better activity (44.53% with IC50 = 29.55 ± 0.19 µM) than 5c and 5 h.
-
(iv)
Hits 5 l (para-CF3), and 5p (para-Cl) with the 5-chlorooxindole moiety, the percentage of inhibition increases and the IC50 decreases respectively.
-
(v)
Compound 5i, with a chlorine atom on the oxindole moiety and a bromo-substituted benzene on the benzoyl nucleus, showed the best results with 62.53% inhibition of IL-6 compared to the standard drug, celecoxib (7.7% inhibition).
-
(vi)
The benzofuran core structure plays a crucial rule in binding to the receptor via the formation of hydrogen bonds with the oxygen atom.
Fig. 4.

SAR of the studied compounds 5a-r.
4. Conclusions
The studied compounds 5a-r were resynthesized and evaluated in vitro against COX-1, and COX-2 enzymes. The in vitro assay results revealed that compounds 5a, 5c, 5h, 5i, 5l, and 5p fixed with a substituted (aryl or oxoindole) moiety were the most potent and selective for COX-1 inhibitor with IC50 values ranging between 20.42 and 38.24 µM. Additionally, compounds 5d, 5e, 5f, 5g, and 5k showed high potency against the release of pro-inflammatory cytokines such as IL-6, and TNF-α. The docking study reinforced the importance of the benzofuran moiety in the binding interaction especially through the formation of HB with receptors. This study illustrated that these could be useful lead compounds in the development of more potent and selective anti-inflammatory agents. Further in vivo studies will be considered for better understanding.
Declaration of conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This research was funded by deanship of scientific research at Princess Nourah Bint Abdulrahman University (Grant No#: 39-S-254).
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.sjbs.2020.02.010. The synthetic details of the studied compounds along with the biological activity assays and docking studies protocols are provided in the supplementary information.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Abdellatif K.R., Fadaly W.A., Elshaier Y.A. Non-acidic 1, 3, 4-trisubstituted-pyrazole derivatives as lonazolac analogs with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2018;77:568–578. doi: 10.1016/j.bioorg.2018.02.018. [DOI] [PubMed] [Google Scholar]
- Altowyan M.S., Barakat A. Spiroindolone analogues as potential hypoglycemic with dual inhibitory activity on α-amylase and α-glucosidase. Molecules. 2019;24:2342. doi: 10.3390/molecules24122342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F., Mantovani A. Inflammation and cancer: back to Virchow? The lancet. 2001;357(9255) doi: 10.1016/S0140-6736(00)04046-0. 5 39–545. [DOI] [PubMed] [Google Scholar]
- Barakat A., Islam M.S., Ghawas H.M. Design and synthesis of new substituted spirooxindoles as potential inhibitors of the MDM2− p53 interaction. Bioorg. Chem. 2019;86:598–608. doi: 10.1016/j.bioorg.2019.01.053. [DOI] [PubMed] [Google Scholar]
- Barakat, A., Islam, M.S., Al Majid, A.M., et.al. 2017. King Saud University, Substituted spirooxindoles. U.S. Patent 9,822,128.
- Barakat A., Islam M.S., Ghawas H.M. Substituted spirooxindole derivatives as potent anticancer agents through inhibition of phosphodiesterase 1. RSC Adv. 2018;8(26):14335–14346. doi: 10.1039/c8ra02358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Neriah Y., Karin M. Inflammation meets cancer, with NF-κB as the matchmaker. Nature Immunol. 2011;12(8):715. doi: 10.1038/ni.2060. [DOI] [PubMed] [Google Scholar]
- Chan A.T., Ogino S., Fuchs C.S. Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Eng. J. Med. 2017;356(21):2131–2142. doi: 10.1056/NEJMoa067208. [DOI] [PubMed] [Google Scholar]
- Cingolani Gino, Panella Andrea, Perrone Maria Grazia, Vitale Paola, Di Mauro Giuseppe, Fortuna Cosimo G., Armen Roger S., Ferorelli Savina, Smith William L., Scilimati Antonio. Structural basis for selective inhibition of Cyclooxygenase-1 (COX-1) by diarylisoxazoles mofezolac and 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (P6) European J. Med. Chem. 2017;138:661–668. doi: 10.1016/j.ejmech.2017.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church L.D., Cook G.P., McDermott M.F. Primer: inflammasomes and interleukin 1β in inflammatory disorders. Nat. Rev. Rheumatol. 2008;4(1):34. doi: 10.1038/ncprheum0681. [DOI] [PubMed] [Google Scholar]
- Dawood K.M. Benzofuran derivatives: a patent review. Exp. Opin. Ther. Patents. 2013;23(9):1133–1156. doi: 10.1517/13543776.2013.801455. [DOI] [PubMed] [Google Scholar]
- Dhanasekaran D.N., Johnson G.L. MAPKs: function, regulation, role in cancer and therapeutic targeting. Oncogene. 2007;26(22):3097. doi: 10.1038/sj.onc.1210395. [DOI] [PubMed] [Google Scholar]
- Ding K., Lu Y., Nikolovska-Coleska Z. Structure-based design of potent non-peptide MDM2 inhibitors. J. Amer. Chem. Soc. 2005;127(29):10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- El-Miligy M.M., Hazzaa A.A., El-Messmary H. New hybrid molecules combining benzothiophene or benzofuran with rhodanine as dual COX-1/2 and 5-LOX inhibitors: Synthesis, biological evaluation and docking study. Bioorg. Chem. 2017;72:102–115. doi: 10.1016/j.bioorg.2017.03.012. [DOI] [PubMed] [Google Scholar]
- Galliford C.V., Scheidt K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Ang. Chem. Int. Ed. 2007;46(46):8748–8758. doi: 10.1002/anie.200701342. [DOI] [PubMed] [Google Scholar]
- Hassan G.S., Abou-Seri S.M., Kamel G. Celecoxib analogs bearing benzofuran moiety as cyclooxygenase-2 inhibitors: design, synthesis and evaluation as potential anti-inflammatory agents. Euro. J. Med. Chem. 2014;76:482–493. doi: 10.1016/j.ejmech.2014.02.033. [DOI] [PubMed] [Google Scholar]
- Herschman H.R. Prostaglandin synthase 2. Biochim. Biophys. Acta (BBA)-Lipids and Lipid. Metabolism. 1996;1299(1):125–140. doi: 10.1016/0005-2760(95)00194-8. [DOI] [PubMed] [Google Scholar]
- He M.M., Smith A.S., Oslob J.D. Small-molecule inhibition of TNF-α. Science. 2005;310(5750):1022–1025. doi: 10.1126/science.1116304. [DOI] [PubMed] [Google Scholar]
- Islam M.S., Ghawas H.M., El-Senduny F.F. Synthesis of new thiazolo-pyrrolidine–(spirooxindole) tethered to 3-acylindole as anticancer agents. Bioorg. Chem. 2019;82:423–430. doi: 10.1016/j.bioorg.2018.10.036. [DOI] [PubMed] [Google Scholar]
- Jiang X., Cao Y., Wang Y. A unique approach to the concise synthesis of highly optically active spirooxazolines and the discovery of a more potent oxindole-type phytoalexin analogue. J. Amer. Chem. Soc. 2010;132(43):15328–15333. doi: 10.1021/ja106349m. [DOI] [PubMed] [Google Scholar]
- Koehne C.H., Dubois R.N. WB Saunders; 2004. COX-2 inhibition and colorectal cancer. In Seminars in oncology; pp. 12–21. [DOI] [PubMed] [Google Scholar]
- Lotfy G., Aziz Y.M., Said M.M. Synthesis of oxindole analogues, biological activity and in silico investigation. Chem. Select. 2019;4(35):10510–10516. [Google Scholar]
- Lotfy G., El Sayed H.S.H., Said M.M. Regio-and stereoselective synthesis of new spirooxindoles via 1, 3-dipolar cycloaddition reaction: anticancer and molecular docking studies. J. Photochem. Photobiol. B: Biol. 2018;180:98–108. doi: 10.1016/j.jphotobiol.2018.01.026. [DOI] [PubMed] [Google Scholar]
- Loll P.J., Picot D., Ekabo O. Synthesis and use of iodinated nonsteroidal antiinflammatory drug analogs as crystallographic probes of the prostaglandin H2 synthase cyclooxygenase active site. Biochemistry. 1996;35(23):7330–7340. doi: 10.1021/bi952776w. [DOI] [PubMed] [Google Scholar]
- Mantovani A., Allavena P., Sica A., Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- Marnett L.J., Rowlinson S.W., Goodwin D.C. Arachidonic acid oxygenation by COX-1 and COX-2 Mechanisms of catalysis and inhibition. J. Biol. Chem. 1999;274(33):22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- Marti C., Carreira E.M. Construction of Spiro [pyrrolidine-3, 3′-oxindoles]− Recent Applications to the Synthesis of Oxindole Alkaloids. Euro. J. Org. Chem. 2003;2003(12):2209–2219. [Google Scholar]
- Padwa, A., Pearson, W., 2002. Synthetic Applications of Dipolar Cycloaddition Chemistry Towards Heterocyclic and Natural Product Chemistry; Eds. WileyVCH; Weinheim.
- Sun Y., Liu J., Sun T. Anti-cancer small molecule JP-8g exhibits potent in vivo anti-inflammatory activity. Sci. Rep. 2014;4:4372. doi: 10.1038/srep04372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Liu J., Jiang X. One-step synthesis of chiral oxindole-type analogues with potent anti-inflammatory and analgesic activities. Sci. Rep. 2015;5:13699. doi: 10.1038/srep13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K., Kaisho T., Akira S. Toll-like receptors. Ann. Rev. Immunol. 2003;21(1):335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Tracey M.D., Cerami K.J., Ph D.A. Tumor necrosis factor: A pleiotropic cytokine and therapeutic target. Ann. Rev. Med. 1994;45(1):491–503. doi: 10.1146/annurev.med.45.1.491. [DOI] [PubMed] [Google Scholar]
- Trikha M., Corringham R., Klein B., Rossi J.F. Targeted anti-interleukin-6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin. Cancer Res. 2003;9(13):4653–4665. [PMC free article] [PubMed] [Google Scholar]
- Trost B.M., Brennan M.K. Asymmetric syntheses of oxindole and indole spirocyclic alkaloid natural products. Synthesis. 2009;2009(18):3003–3025. [Google Scholar]
- Wade, P. A. 1991. The Ganges In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Semmelhack, M. F., Eds.; Pergmon Press: Oxford, 4,1111.
- Wang J.L., Limburg D., Graneto M.J. The novel benzopyran class of selective cyclooxygenase-2 inhibitors. Part 2: the second clinical candidate having a shorter and favorable human half-life. Bioorg. Med. Chem. Lett. 2010;20(23):7159–7163. doi: 10.1016/j.bmcl.2010.07.054. [DOI] [PubMed] [Google Scholar]
Further Reading
- Lotfy G., Said M.M., El Sayed H.S.H. Synthesis of new spirooxindole-pyrrolothiazole derivatives: Anti-cancer activity and molecular docking. Bioorg. Med. Chem. 2017;25(4):1514–1523. doi: 10.1016/j.bmc.2017.01.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.