

Abstract
Purpose
To identify the genetic cause of patients with primary ciliary dyskinesia (PCD) and male infertility from two unrelated Han Chinese families.
Methods
We conducted whole-exome sequencing of three individuals with PCD and male infertility from two unrelated Chinese families, and performed a targeted look-up for DNAAF6 variants in our previously reported cohort of 442 individuals (219 with isolated oligoasthenospermia and 223 fertile controls). Ultrastructural and immunostaining analyses of patients’ spermatozoa were performed. The pathogenicity of the variants was validated using patient’s spermatozoa and HEK293T cells. Intracytoplasmic sperm injection (ICSI) treatment was conducted in two patients.
Results
We identified one novel hemizygous frameshift variant (NM_173494, c.319_329del: p.R107fs) of DNAAF6 gene (previously named PIH1D3) in family 1 and one novel hemizygous missense variant (c.290G>T: p.G97V) in family 2. No hemizygous deleterious variants in DNAAF6 were detected in the control cohort of 442 individuals. Ultrastructural and immunostaining analyses of patients’ spermatozoa showed the absence of outer and inner dynein arms in sperm flagella. Both variants were proven to lead to DNAAF6 protein degradation in HEK293T cells. Both patients carrying DNAAF6 variants underwent one ICSI cycle and delivered one healthy child each.
Conclusion
We identified novel DNAAF6 variants causing male infertility and PCD in Han Chinese patients. This finding extended the spectrum of variants in DNAAF6 and revealed new light on the impact of DNAAF6 variants in sperm flagella.
Electronic supplementary material
The online version of this article (10.1007/s10815-020-01735-4) contains supplementary material, which is available to authorized users.
Keywords: Primary ciliary dyskinesia, Asthenozoospermia, DNAAF6 variant, Dynein arms, Sperm flagella
Introduction
Malfunctioning cilia are associated with approximately one hundred human genetic disorders, with an accumulated incidence of 1:1000 [1]. As the hallmark disease of motile cilia, primary ciliary dyskinesia (PCD; MIM: 244400) [2, 3] is a genetically heterogeneous recessive disorder that affects an estimated 1:15,000 live births with typical symptoms characterized by neonatal respiratory distress, chronic sinusitis, bronchiectasis, and situs inversus [4, 5]. To date, approximately 43 disease-causing genes accounting for approximately 70% of PCD cases have been implicated in humans [6]. The variants in five axoneme-related genes (DNAH5, DNAH11, DNAI1, CCDC39, and CCDC40) predominate in PCD [7–11] and only three X-linked PCD genes have been reported, namely OFD1, RPGR, and DNAAF6 [12–15]. Generally, males with PCD are infertile due to immotile spermatozoa [16]. However, since the male infertility is usually diagnosed after puberty, sperm parameters are not systematically investigated in previously identified PCD cases, leaving not only the records of male infertility incomplete but also the role of PCD-related genes in flagellar function unclear [17, 18].
DNAAF6 (MIM:300933, previously named PIH1D3) is located on chromosome Xq22.3 and contains 8 exons, encoding a protein containing 214 amino acids and a PIH1 domain [15]. DNAAF6 was identified as a dynein axonemal assembly factor (DNAAF) proposed to cooperate with heat shock protein (HSP) chaperones and other DNAAFs to promote subunit folding and cytoplasmic preassembly of dynein motors [19]. Dnaaf6 defective mice only presented with male infertility and Dnaaf6 defective spermatozoa were immotile and fragile while motile cilia remained unaffected [20]. Currently, three studies have reported that the loss-of-function variants in DNAAF6 cause the classic PCD phenotypes with ciliary immotility in humans due to the lack of dynein arms (DAs) and the male patients present infertility. These studies only focus on the impact of DNAAF6 variants in immotile cilia and respiratory tract phenotypes in patients [13, 15, 21]. However, no systematic studies yet exist on the ultrastructural defects of spermatozoa and asthenozoospermia phenotypes in human patients with DNAAF6 variants. Moreover, with the development of assisted reproductive technology (ART), asthenozoospermia patients can achieve pregnancy through intracytoplasmic sperm injection (ICSI) [22]. The impact of DNAAF6 variants in spermatozoa and the outcome of the ICSI treatment in DNAAF6-mutated male patients remain to be assessed.
In this study, we conducted a genetic analysis of three patients with PCD and male infertility from two unrelated Han Chinese families using whole-exome sequencing (WES), detecting two novel variants in DNAAF6 in three patients. The spermatozoa from PCD patients with DNAAF6 variants presented abnormal flagella with loss of DAs. We also validated the effects of these variants in patients’ spermatozoa and HEK293T cells. Our results indicated that the DNAAF6 variants might be responsible for PCD and male infertility.
Materials and methods
Patients and subjects
Three patients from two unrelated Han Chinese families were recruited to identify the causal genetic factor of infertility at the Reproductive and Genetic Hospital of CITIC-Xiangya (Changsha, Hunan, China). Family 1 included normal parents (first cousins married to each other, F1: I-1 and F1: I-2), their sons affected by PCD (F1: II-1, proband, and his brother F1: II-3), and their daughter devoid of known abnormalities (F1: II-2). Family 2 included normal parents (F2: I-1 and F2: I-2) and their son affected by PCD (F2: II-2).
A random cohort of 442 individuals (Han Chinese origin, described previously [23]), including 219 isolated oligoasthenospermia and 223 controls with normal fertility (had at least one offspring by natural fertilization), were used for variant screening. Abnormalities in somatic karyotypes and azoospermia factor (AZF) microdeletion on the Y chromosome were not detected. Other causes of infertility such as reproductive malformation, drug-use, and exposure to gonadotoxic factors were also excluded. Semen evaluations were performed according to World Health Organization (WHO, 2010) guidelines [24].
The present study was approved by the Institutional Ethics Committees of Central South University and the Reproductive Genetic Hospital of CITIC-Xiangya, China. All methods were performed in accordance with approved guidelines. Written informed consent was obtained from all individuals who participated in the study prior to commencement.
Whole-exome sequencing and bioinformatic analysis
Blood samples from participating individuals, including those of 3 individuals with PCD and male infertility were collected and genomic DNA was extracted using a DNA extraction kit (Qiagen, Hilden, Germany). This genomic DNA was subsequently used for whole-exome sequencing (WES) analysis. Library construction, WES, and data analysis were carried out by the Beijing Genomics Institute (BGI, Shenzhen, China) as described previously [25]. Variant identification was performed using the Genome Analysis Toolkit (GATK). Sequencing variants including single-nucleotide variants (SNVs) and small insertions or deletions (INDELs) were annotated using ANNOVAR software [26].
The most promising candidate pathogenic variants in the two families were screened using the following criteria: (1) variants with a minor allele frequency below 5% as reported by the public databases (dbSNP, GO-ESP, 1000G, and ExAC); (2) variants predicted to be deleterious using multiple tools (PolyPhen-2 (genetics.bwh.harvard.edu/pph2), Mutation Taster (www.mutationtaster.org/), and SIFT (sift.jcvi.org/)); and (3) phenotype relevance using comprehensive expression data (high expression in ciliated tissues) and model organism data [27]. We performed a targeted look-up for DNAAF6 variants in WES data from the 219 subjects with oligoasthenospermia and 223 subjects with normal fertility, described in our previous study [23].
Sanger sequencing
Specific PCR primers were designed to target the region of the variants in the candidate gene DNAAF6 with sequences as follows: PIH1D3-F1: 5′-ACTGAGTGTGAAGAAGAACAGGAG-3′, PIH1D3-R1: 5′-CCTTCCGTGTGTCACTGTAGTTT-3′. Amplified PCR products were analyzed via 2.0% agarose gel electrophoresis to determine band size, followed by bi-directional sequencing on an ABI 3730 automated sequencer (Applied Biosystems/Thermo Fisher Scientific, Foster City, CA, USA).
Transmission electron microscopy
Semen samples of normal fertile control and patients (F1: II-1 and F2: II-2) were treated as described previously [23]. Samples were fixed with glutaraldehyde (Sigma-Aldrich, St. Louis, MO, USA) and osmium tetroxide, followed by OsO4 and sucrose, then dehydrated using graded ethanol. Subsequently, the samples were embedded in Epon 812, dodecenylsuccinic anhydride, methyl-nadic anhydride, and dimethylaminomethyl phenol. Ultrathin (80 nm) sections were contrasted with uranyl acetate and lead citrate. An AHT7700 Hitachi electron microscope (Hitachi, Tokyo, Japan) and a MegaView Iii digital camera (Olympus Soft Imaging Solutions GmbH, Münster, Germany) were used to capture images.
Histological and immunofluorescence analysis
For hematoxylin and eosin (H&E) staining, sperm were dehydrated with graded ethanol, stained with H&E. For immunofluorescence analysis, the slides were incubated with primary antibodies (PIH1D3, DNAH5, DNALI1, AKAP4, TOMM20, RSPH1, SPAG6, and anti-acetylated tubulin monoclonal antibody, respectively) for 2.5 h at 37 °C. Detailed information on all antibodies is provided in Table S1. The slides were then incubated with secondary antibodies (Alexa Fluor® 488 anti-mouse IgG (A-21121) and Alexa Fluor® 555 anti-rabbit IgG (A31572)) for 1.5 h at 37 °C. Finally, all slides were stained using 2-(4-amidinophenyl)-1H-indole-6-carboxamidine (DAPI) for 5 min. An Olympus IX51 fluorescence microscope (Olympus, Tokyo, Japan) and VideoTesT-FISH 2.0 software (VideoTesT Ltd., St. Petersburg, Russia) were utilized to photograph fluorescence signals.
Expression vector construction
The full-length coding sequence of the human DNAAF6 (NM_173494) cDNA clone was purchased from GeneChem Co., Ltd. (Shanghai, China). Wild-type DNAAF6 was cloned to CMV-MCS-FLAG-SV40-Neomycin with FLAG-tag at the C-terminal end. Site-directed mutagenesis was performed to introduce both DNAAF6 variants (c.319_329del and c.290G>T) into the wild-type vector using a Mut Express II Fast Mutagenesis Kit V2 (Vazyme Biotech Co., Ltd., Nanjing, China) according to the manufacturer’s instructions. Wild-type and mutant clones were confirmed by Sanger sequencing.
Cell culture and transfection
Human embryonic kidney cells (HEK293T) obtained from China Center for Type Culture Collection (CCTCC) (Wuhan, China) were cultivated at 37 °C with 5% CO2 in a 6-mL petri dish supplemented with 10% fetal bovine serum (Gibco™/Thermo Fisher Scientific, Gaithersburg, MD, USA). When cells had grown to ~ 80% confluence, DNAAF6 wild-type and mutant plasmids were transiently transfected using Lipofectamine® 3000 reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Western blotting
Cells were collected after transfection for 48 h with DNAAF6 wild-type and mutant plasmids and homogenized with RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitor cocktail (Thermo Fisher Scientific). Lysates were centrifuged at 13,000×g for 15 min at 4 °C and supernatants were collected and mixed with 5× sodium dodecyl sulfate (SDS) loading buffer and heated at 100 °C for 10 min. The proteins were separated via 15% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) then transferred onto a polyvinylidene difluoride membrane and incubated overnight at 4 °C with anti-FLAG antibody (AB0008, Abways, Shanghai, China) and anti-GAPDH antibody (ab8245, Abcam, Cambridge. UK). Then, the membranes were incubated with secondary antibodies (goat anti-mouse IgG or goat anti-rabbit IgG, MultiSciences Biotech Co., Ltd., Hangzhou, China). Finally, the blots were revealed using the Pierce ECL Western Blotting Kit (Pierce Biotechnology, Rockford, IL, USA).
Intracytoplasmic sperm injection procedures
The wives of 2 individuals with DNAAF6 variants (F1: II-1 and F2: II-2) accepted intracytoplasmic sperm injection (ICSI) treatment. They underwent controlled ovarian hyperstimulation, oocyte retrieval, ICSI, and pregnancy confirmation as described previously [28, 29]. Viable spermatozoa for ICSI, selected from completely immotile spermatozoa, were stimulated using SperMagic® medium as our described previously [28, 29]. Pregnancy was confirmed via 2 hCG tests conducted 14 days following embryo transfer; clinical pregnancy was defined as, at minimum, a visible sac with a fetal heart beat detected via ultrasound screening after 28 days.
Results
Clinical findings
The first proband (F1: II-1) from a consanguineous family (Family 1) was 32 years old (Fig. 1a). Routine semen analysis of this subject revealed severe asthenoteratozoospermia with 100% immotile spermatozoa and comparatively low sperm count (5.2–11.2 × 106/mL) (Table 1). The bilateral testicular size, as measured by B ultrasonography, was normal. F1: II-1 exhibited classic PCD-like symptoms from early life based on his previous medical tests, including a positive history of chronic wet cough, nasal obstruction, pneumonia, and dextrocardia. However, F1: II-1 refused to a ciliary biopsy and nasal nitric oxide (nNO) test. The proband’s brother (F1: II-3) was also affected by primary infertility and exhibited classic PCD-like symptoms similar to the proband. F2: II-2 from a non-consanguineous family (Family 2) was 44 years old (Fig. 1a). Routine semen analysis of the subject revealed severe asthenoteratozoospermia with 91.4% immotile spermatozoa and a comparatively low sperm count (3.5–8.7 × 106/mL) (Table 1). F2: II-2 also exhibited classic PCD-like symptoms, including a positive history of chronic wet cough, nasal obstruction, pneumonia, and bronchiectasia, but he refused to a ciliary biopsy and nasal nitric oxide (nNO) test. Routine physical examination of these patients yielded normal results, including height, weight, hair distribution, mental state, and external genital organs.
Fig. 1.

Two DNAAF6 variants were identified in three patients with PCD and male infertility phenotypes. a Pedigree analysis of the three patients from two Han Chinese families. Black squares indicate male patients; black arrow, proband (P); circles with black spot, female heterozygous carriers; parallel lines, infertility. b Sanger sequencing results of the two variants from the two families. Red arrow indicates missense variant site; black line, frameshift variant site. NC, normal control. c Conservative analysis of the missense variant (c.290G>T: p.G97V). Red arrow, mutant amino acid site. d Positions of all reported variants in the DNAAF6 protein structure model. Green box, PIH domain; black, variants identified from other studies; red, variants identified in this study
Table 1.
Semen routine examination and intracytoplasmic sperm injection outcomes of the PCD couples
| Family | *Age | †Semen volume (mL) | †Sperm count (× 106/mL) | †Progressive motility (%) | Number of MII oocytes | Number of fertilized oocytes | Sperm retrieval technique | Number of transferred embryos | Characteristics of transferred embryos | Outcome of pregnancy |
|---|---|---|---|---|---|---|---|---|---|---|
| F1: II-1 |
M 32 F 28 |
2.5–3.8 | 5.2–11.2 | 0 | 13 | 9 | Ejaculation | 2 | 8-cell grade | Single birth |
| F2: II-2 |
M 44 F 35 |
1.5–2.8 | 3.5–8.7 | 8.6 | 15 | 13 | Ejaculation | 2 | 8-cell grade | Single birth |
*Age of the couple. M, male; F, female
†Semen parameters were evaluated according to the World Health Organization (WHO, 2010) guidelines
WES-based identification of DNAAF6 variants
WES was performed to identify the genetic source of PCD and male infertility in these patients. The results for both families were analyzed and filtered to exclude irrelevant variants according to the criteria described in Methods. Briefly, candidate variants were considered a priority when they met the following criteria: frequency below 5% in public databases, predicted to be deleterious, homozygous or compound heterozygous, and relevance to infertility or PCD phenotypes. After filtering, only two novel hemizygous variants (NM_173494, c.319_329del: p.R107fs for family 1 and c.290G>T: p.G97V for family 2) in DNAAF6, a known pathogenic gene for PCD [13, 15], fulfilled these criteria (Table 2). Neither hemizygous deleterious variants in DNAAF6 were assessed in our previously reported cohort of 442 individuals, including 219 with isolated oligoasthenospermia and 223 normal controls.
Table 2.
Variants identified by whole-exome sequencing in combination with cilia-related gene-filtering for the patients
| Patient | Gene | Position | RefSeq ID | AA alteration | Function | MinDepth | dbSNP ID | †1000 G | †GO-ESP | †ExAC | *Mutation Taster | *SIFT | *PolyPhen-2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
F1: II-1 F1: II-3 |
DNAAF6 | ChrX: 106462185 | NM_173494 | c.319_329del:p.R107fs | frameshift | 69 | - | - | - | - | D | - | - |
| F2: II-2 | DNAAF6 | ChrX: 106462157 | NM_173494 | c.G290T:p.G97V | missense | 49 | - | - | - | 0.00001201 | D | D | D |
*Mutation assessment by Mutation Taster, SIFT, and PolyPhen-2. D, disease causing; -, not available
†Frequency of corresponding mutations in 1000 Genomes, GO-ESP, and total ExAC Browser
Sanger sequencing and pedigree analysis were performed for the patients to confirm the DNAAF6 variants. Patient F1: II-1 and his brother F1: II-3 had inherited the frameshift variant (c.319_329del: p.R107fs) from their mother with heterozygous condition; their father and sister with normal phenotype were wild type. Co-segregation analysis supported a X-linked recessive genetic model and genotype–phenotype correlation (Fig. 1b). The missense variant (c.290G>T: p.G97V) of which position is highly conserved in different species was identified in patient F2: II-2 (Fig. 1c), but pedigree validation was failed to be performed due to unavailability of the family members (Fig. 1d). These results indicated that the two variants in DNAAF6 were associated with PCD and male infertility.
Patients with DNAAF6 variants lack dynein arms
We performed H&E staining to observe the morphological features of the mutated spermatozoa from F1: II-1 and F2: II-2. Most spermatozoa exhibited abnormal tail morphologies, including short, coiled, and curled tails (Fig. 2a). Transmission electron microscopy (TEM) analysis showed lack of inner dynein arms (IDAs) and outer dynein arms (ODAs) in the mutated spermatozoa from F1: II-1 and F2: II-2, while exhibiting normal “9 + 2” microtubule pairs (Fig. 2b). Immunostaining analyses of spermatozoa (F1: II-1) using the ODA marker DNAH5 and the IDA marker DNALI1 further confirmed the absence of ODA and IDA (Fig. 2c, d). We then assessed flagellum structure by immunofluorescence staining with AKAP4, TOMM20, RSPH1, and SPAG6—markers of the fibrous sheath, mitochondrial sheath, radial spokes, and central pair complex, respectively. The immunostaining of mutant spermatozoa (F1: II-1) with AKAP4, TOMM20, RSPH1, and SPAG6 yielded results similar to the normal controls (Fig. 3a–d). Our results showed that DNAAF6 variants only caused the absence of dynein arms (DAs) in flagella; other flagellum components including fibrous sheath, mitochondrial sheath, and radial spokes were not directly affected by variants in DNAAF6.
Fig. 2.

Abnormal morphology and ultrastructure of mutant spermatozoa. a H&E staining of spermatozoa from both patients (F1: II-1 and F2: II-2) and normal control (NC) indicated coiling tails and curly tails in DNAAF6 mutant spermatozoa. Scale bars, 10 μm. b TEM analysis indicated loss of IDAs and ODAs and normal microtubule pairs (9 + 2) in the spermatozoa from both patients. Scale bars, 200 nm; CPC, central pair complex; MT, peripheral microtubule doublet; ODA, outer dynein arms; IDA, inner dynein arms. c, d Immunofluorescence analysis showed significant decline with anti-DNAH5 and anti-DNALI1 antibodies (red) in the mutant spermatozoa from F1: II-1 compared with the normal control. The spermatozoa were stained with anti-α-tubulin antibody (green) as loading controls and 4′, 6-diamidine-2′-phenylindole dihydrochloride (DAPI, blue) as a nuclear marker. Scale bars, 5 μm; DIC, differential interference contrast
Fig. 3.

No abnormalities were found in other flagellum components. Immunostaining analyses with a anti-AKAP4, b anti-TOMM20, c anti-RSPH1, and d anti-SPAG6 antibodies (red) showed no significant abnormalities in fibrous sheath, mitochondrial sheath, radial spokes, and central pair complex of spermatozoa from F1: II-1 compared with the normal control (NC). Anti-α-tubulin antibodies (green) were used as loading controls and DAPI (blue) as a nuclear marker. Scale bars, 5 μm; DIC, differential interference contrast
Significantly decreased DNAAF6 protein in patient spermatozoa and in HEK293T cells
To assess the functional effects of the identified DNAAF6 variants in spermatozoa, we analyzed DNAAF6 expression by immunofluorescence analysis on spermatozoa from controls and patient F1: II-1. DNAAF6 was found to be localized on the midpiece of flagella in normal spermatozoa, but absent in DNAAF6-mutated spermatozoa (Fig. 4a). To further validate the effects caused by DNAAF6 variants in vitro, we constructed two mutant expression plasmids and transfected wild-type and mutant vectors into HEK293T cells. Western blot showed both c.290G>T (p.G97V) and c.319_329del (p.R107fs) variants caused DNAAF6 protein degradation, compared with that with wild-type proteins (Fig. 4b). These results suggested the missense variant (c.290G>T: p.G97V) and the frameshift variant (c.319_329del: p.R107fs) both caused extremely unstable proteins in spermatozoa and HEK293T cells.
Fig. 4.
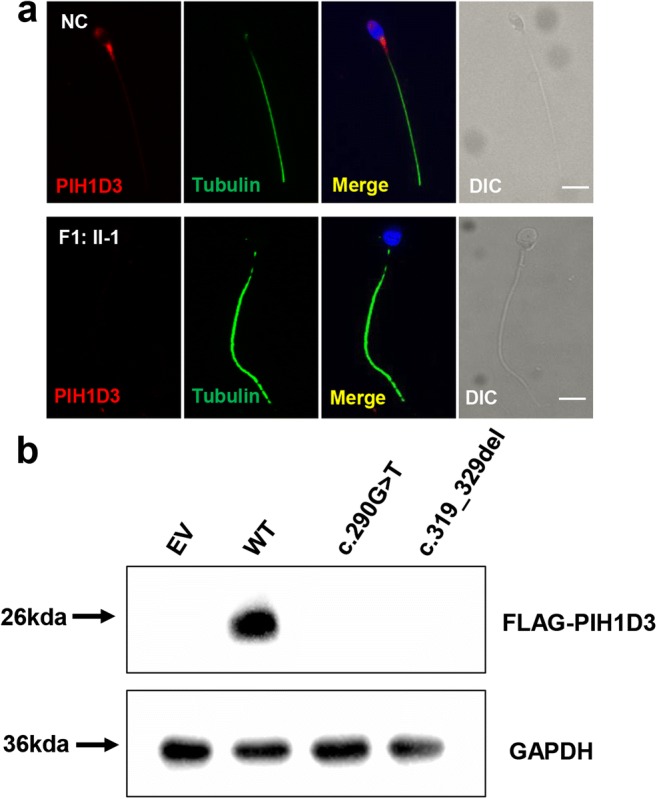
Expression levels of mutant DNAAF6 proteins in vitro and in spermatozoa. a DNAAF6 immunostaining in human spermatozoa from normal controls (NC) and F1: II-1 with anti-DNAAF6 (red) and anti-α-tubulin (green) antibodies. DNAAF6 protein located in midpiece of normal sperm flagella, but absent in mutant spermatozoa. Scale bars, 5 μm; DIC, differential interference contrast. b The effects of the variants on DNAAF6 protein levels by western blot in HEK293T cells transfected with empty vector (EV), wild-type (WT), and mutant DNAAF6 vectors. GAPDH was used as the loading control
ICSI outcome of both patients
Furthermore, ICSI was conducted for two couples using ejaculated spermatozoa with comparatively normal morphology. Both couples underwent one ICSI treatment cycle each; thirteen metaphase II oocytes were injected and nine oocytes were fertilized (2PN/injected oocytes = 69%) in family 1 (F1: II-1) and fifteen metaphase II oocytes were injected and thirteen oocytes were fertilized (2PN/injected oocytes = 87%) in family 2 (F2: II-2) (Table 1). Two embryos were subsequently transferred into the partner’s uterus and one embryo resulted in clinical pregnancy in both families. Both families ultimately delivered one healthy child each.
Discussion
In this study, two novel hemizygote variants (c.290G>T: p.G97V and c.319_329del: p.R107fs) in DNAAF6 were identified in three patients with PCD and male infertility from two unrelated Han Chinese families by WES and Sanger sequencing. TEM and immunostaining analyses of patients’ spermatozoa showed the absence of ODA and IDA in sperm flagella. Both variants impaired the stability of the DNAAF6 protein in HEK293T cells as well as in patient spermatozoa. These results revealed that both variants in DNAAF6 were associated with the PCD phenotypes and asthenozoospermia. Our study is also the first to report Chinese PCD patients with DNAAF6 variants.
DNAAF6 is a dynein axonemal assembly factor with only one PIH domain that participates in a co-chaperone complex with KTU/DNAAF2 and DYX1C1/DNAAF4 to regulate the dynein arm preassembly in cytoplasm by direct interaction with HSP90 and DNAI2 [13, 19]. To date, three groups have reported that variants in DNAAF6 caused PCD with outer and inner dynein arm defects. They identified a total of twelve variants in ten patients, including four nonsense variants, three frameshift variants, one splicing variant, one missense variant, and three gross deletions [13, 15, 21]. In this study, the frameshift variant (c.319_329del: p.R107fs) was predicted to produce truncated protein. The missense variant (c.290G>T: p.G97V) from F2: II-2 was located in a highly conserved region in the PIH domain [15]. This domain is a key non-catalytic subunit of the R2TP-HSP90 chaperoning complex [19]. Both variants were validated to impact the function of DNAAF6 protein in cells. These results suggested that the missense variant was also a main pathogenic variant type and that the region near the PIH domain may be the pathogenic hot spot combining all known variants.
We observed DNAAF6 staining only in the midpiece flagellum of human sperm, which is similar to the location of CCDC103, another cytoplasmatic dynein assembly factor [30]. In mice, Dnaaf6 was detected only in the cytoplasm of late pachytene spermatocytes to round spermatids [20]. We speculated that DNAAF6 also plays a role during flagellar development and was retained to the midpiece of flagella in chromatin condensation and the elimination of excess cytoplasm, while also participating in dynein arm preassembly in the cytoplasm of round sperm cells. The molecular pathways of DNAAF6 and other DNAAFs in sperm cells require further investigation.
For PCD patients, basal brushing biopsies were performed as a classical measurement to PCD diagnosis in previous reports [31–33]. However, it is known to cause trauma to patients. Our results showed that variants in DNAAF6 affected both IDAs and ODAs in the sperm flagella rather than only ODAs as previously described, the ultrastructural defects in DNAAF6-mutant spermatozoa showed no difference with those in DNAAF6-mutant cilia from other reports [13]. This result suggested that male PCD patients could be diagnosed by sperm flagella ultrastructural examination combined with known PCD pathogenic genes related to defects of both cilia and sperm flagella. It is essential to identify the effects of PCD pathogenic genes on spermatozoa, as such an assessment offers not only a potential non-invasive diagnostic method to PCD patients but also the possibility of treating male infertility.
From a clinical point of view, ICSI is currently the only way for couples to conceive when the male partner has been diagnosed with PCD and male infertility, due to the lack of empirical medicinal therapy to improve semen parameters. Specifically, the estimated fertilization and pregnancy rates among PCD patients were 55–65% and 35–45% when relying on ejaculated or testicular sources, respectively [34], while the overall live birth rate was only 39%. In this study, patients carrying DNAAF6 variants experience successful ICSI outcomes, indicating that ICSI may be a good choice for patients with DNAAF6 variants who want to have children. However, ICSI cycles are administered prior to genetic analysis on both patients; going forward, protocols should include variant screening of DNAAF6 in female partners and their offspring as well.
Overall, we validated two novel pathogenic variants in DNAAF6 from two unrelated Han Chinese families with PCD and male infertility. Patients who carried DNAAF6 variants have good ICSI outcomes. Our findings extend the mutational spectrums of DNAAF6 and have important implications for genetic counseling of patients with PCD and male infertility.
Electronic supplementary material
(DOC 43 kb)
Acknowledgments
The authors would like to thank all the families and individuals who participated in this study. We are grateful to the excellent technical support provided by Junpu Wang, as well as support from the clinical and nursing staff at the Reproductive and Genetic Hospital of CITIC-Xiangya.
Author contributions
Juan Du and Yue-Qiu Tan designed the study. Lanlan Meng, Dongyan Li, and Weili Wang performed the variant analysis. Ying Wang and Chaofeng Tu carried out the evaluation of the pathogenicity of variations and spermatozoa functional analyses. Hongchuan Nie, Huan Zhang, Guangxiu Lu, and Ge Lin worked on the clinical study. Ying Wang, Chaofeng Tu, Yue-Qiu Tan, and Juan Du wrote the paper. All authors read and approved the final manuscript.
Funding information
This work was supported by the National Key Research & Developmental Program of China (2018YFC1004900 to YQ.T), the National Natural Science Foundation of China (81771645 and 81971447 to YQ.T), the science and technology major project of the Ministry of Science and Technology of Hunan Province (2017SK1030 to YQ.T), the China Postdoctoral Science Foundation Funded Project (2019M662786 to CF.T), and the Graduate Research and Innovation Projects of Central South University (2019zzts998 to Y.W).
Data availability
The data and material that support the findings of this study are available from the corresponding author upon reasonable request.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ying Wang and Chaofeng Tu contributed equally to this work.
Contributor Information
Yue-Qiu Tan, Email: tanyueqiu@csu.edu.cn.
Juan Du, Email: tandujuan@csu.edu.cn.
References
- 1.Davis EE, Katsanis N. The ciliopathies: a transitional model into systems biology of human genetic disease. Curr Opin Genet Dev. 2012;22(3):290–303. doi: 10.1016/j.gde.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heydeck W, Fievet L, Davis EE, Katsanis N. The complexity of the cilium: spatiotemporal diversity of an ancient organelle. Curr Opin Cell Biol. 2018;55:139–149. doi: 10.1016/j.ceb.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stern BM, Sharma G. Ciliary dysfunction (Kartagener syndrome, primary ciliary dyskinesia) Treasure Island (FL): StatPearls; 2018. [Google Scholar]
- 4.Lucas JS, Burgess A, Mitchison HM, Moya E, Williamson M, Hogg C, et al. Diagnosis and management of primary ciliary dyskinesia. Arch Dis Child. 2014;99(9):850–856. doi: 10.1136/archdischild-2013-304831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mirra V, Werner C, Santamaria F. Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front Pediatr. 2017;5:135. doi: 10.3389/fped.2017.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fassad MR, Shoemark A, le Borgne P, Koll F, Patel M, Dixon M, Hayward J, Richardson C, Frost E, Jenkins L, Cullup T, Chung EMK, Lemullois M, Aubusson-Fleury A, Hogg C, Mitchell DR, Tassin AM, Mitchison HM. C11orf70 mutations disrupting the intraflagellar transport-dependent assembly of multiple axonemal dyneins cause primary ciliary dyskinesia. Am J Hum Genet. 2018;102(5):956–972. doi: 10.1016/j.ajhg.2018.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Djakow J, Svobodova T, Hrach K, Uhlik J, Cinek O, Pohunek P. Effectiveness of sequencing selected exons of DNAH5 and DNAI1 in diagnosis of primary ciliary dyskinesia. Pediatr Pulmonol. 2012;47(9):864–875. doi: 10.1002/ppul.22520. [DOI] [PubMed] [Google Scholar]
- 8.Antony D, Becker-Heck A, Zariwala MA, Schmidts M, Onoufriadis A, Forouhan M, Wilson R, Taylor-Cox T, Dewar A, Jackson C, Goggin P, Loges NT, Olbrich H, Jaspers M, Jorissen M, Leigh MW, Wolf WE, Daniels ML, Noone PG, Ferkol TW, Sagel SD, Rosenfeld M, Rutman A, Dixit A, O’Callaghan C, Lucas JS, Hogg C, Scambler PJ, Emes RD, Uk10k. Chung EM, Shoemark A, Knowles MR, Omran H, Mitchison HM. Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms. Hum Mutat. 2013;34(3):462–472. doi: 10.1002/humu.22261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dougherty GW, Loges NT, Klinkenbusch JA, Olbrich H, Pennekamp P, Menchen T, Raidt J, Wallmeier J, Werner C, Westermann C, Ruckert C, Mirra V, Hjeij R, Memari Y, Durbin R, Kolb-Kokocinski A, Praveen K, Kashef MA, Kashef S, Eghtedari F, Häffner K, Valmari P, Baktai G, Aviram M, Bentur L, Amirav I, Davis EE, Katsanis N, Brueckner M, Shaposhnykov A, Pigino G, Dworniczak B, Omran H. DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes. Am J Respir Cell Mol Biol. 2016;55(2):213–224. doi: 10.1165/rcmb.2015-0353OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zietkiewicz E, Nitka B, Voelkel K, Skrzypczak U, Bukowy Z, Rutkiewicz E, et al. Population specificity of the DNAI1 gene mutation spectrum in primary ciliary dyskinesia (PCD) Respir Res. 2010;11:174. doi: 10.1186/1465-9921-11-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olbrich H, Haffner K, Kispert A, Volkel A, Volz A, Sasmaz G, et al. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat Genet. 2002;30(2):143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 12.Ferrante MI, Zullo A, Barra A, Bimonte S, Messaddeq N, Studer M, et al. Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification. Nat Genet. 2006;38(1):112–117. doi: 10.1038/ng1684. [DOI] [PubMed] [Google Scholar]
- 13.Olcese C, Patel MP, Shoemark A, Kiviluoto S, Legendre M, Williams HJ, et al. X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3. Nat Commun. 2017;8:14279. doi: 10.1038/ncomms14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bukowy-Bieryllo Z, Zietkiewicz E, Loges NT, Wittmer M, Geremek M, Olbrich H, et al. RPGR mutations might cause reduced orientation of respiratory cilia. Pediatr Pulmonol. 2013;48(4):352–363. doi: 10.1002/ppul.22632. [DOI] [PubMed] [Google Scholar]
- 15.Paff T, Loges NT, Aprea I, Wu K, Bakey Z, Haarman EG, Daniels JMA, Sistermans EA, Bogunovic N, Dougherty GW, Höben IM, Große-Onnebrink J, Matter A, Olbrich H, Werner C, Pals G, Schmidts M, Omran H, Micha D. Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects. Am J Hum Genet. 2017;100(1):160–168. doi: 10.1016/j.ajhg.2016.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krausz C, Riera-Escamilla A. Genetics of male infertility. Nat Rev Urol. 2018;15(6):369–384. doi: 10.1038/s41585-018-0003-3. [DOI] [PubMed] [Google Scholar]
- 17.Ray PF, Toure A, Metzler-Guillemain C, Mitchell MJ, Arnoult C, Coutton C. Genetic abnormalities leading to qualitative defects of sperm morphology or function. Clin Genet. 2017;91(2):217–232. doi: 10.1111/cge.12905. [DOI] [PubMed] [Google Scholar]
- 18.El Khouri E, Thomas L, Jeanson L, Bequignon E, Vallette B, Duquesnoy P, et al. Mutations in DNAJB13, encoding an HSP40 family member, cause primary ciliary dyskinesia and male infertility. Am J Hum Genet. 2016;99(2):489–500. doi: 10.1016/j.ajhg.2016.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mali GR, Yeyati PL, Mizuno S, Dodd DO, Tennant PA, Keighren MA, et al. ZMYND10 functions in a chaperone relay during axonemal dynein assembly. Elife. 2018;7:e34389. doi: 10.7554/eLife.34389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenglan D, Kyosuke S, Yanick B, Ryo N, Yasuko A, Akemi F, et al. Pih1d3 is required for cytoplasmic preassembly of axonemal dynein in mouse sperm. J Cell Biol. 2014;204(2):203–213. doi: 10.1083/jcb.201304076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis SD, Rosenfeld M, Lee HS, Ferkol TW, Sagel SD, Dell SD, et al. Primary ciliary dyskinesia: longitudinal study of lung disease by ultrastructure defect and genotype. Am J Respir Crit Care Med. 2019;199(2):190–198. doi: 10.1164/rccm.201803-0548OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawasaki A, Okamoto H, Wada A, Ainoya Y, Kita N, Maeyama T, Edamoto N, Nishiyama H, Tsukamoto S, Joraku A, Waku N, Yoshikawa H. A case of primary ciliary dyskinesia treated with ICSI using testicular spermatozoa: case report and a review of the literature. Reproductive Medicine & Biology. 2015;14(4):195–200. doi: 10.1007/s12522-015-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Tu C, Nie H, Meng L, Li Y, Yuan S, et al. Biallelic mutations in CFAP65 lead to severe asthenoteratospermia due to acrosome hypoplasia and flagellum malformations. J Med Genet. 2019;56(11):750–757. doi: 10.1136/jmedgenet-2019-106031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sanchez-Alvarez J, Cano-Corres R, Fuentes-Arderiu X. A complement for the WHO laboratory manual for the examination and processing of human semen (first edition, 2010) EJIFCC. 2012;23(3):103–106. [PMC free article] [PubMed] [Google Scholar]
- 25.Tan YQ, Tu C, Meng L, Yuan S, Sjaarda C, Luo A, du J, Li W, Gong F, Zhong C, Deng HX, Lu G, Liang P, Lin G. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med. 2019;21(5):1209–1217. doi: 10.1038/gim.2017.130. [DOI] [PubMed] [Google Scholar]
- 26.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Mouse Genome Database G The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015;43(Database issue):D726–D736. doi: 10.1093/nar/gku967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong F, Li X, Zhang S, Ma H, Cai S, Li J, et al. A modified ultra-long pituitary downregulation protocol improved endometrial receptivity and clinical outcome for infertile patients with polycystic ovarian syndrome. Exp Ther Med. 2015;10(5):1865–1870. doi: 10.3892/etm.2015.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu YF, Zhou QW, Zhang SP, Lu CF, Gong F, Shi Y, Lu GX, Lin G. The clinical and neonatal outcomes after stimulation of immotile spermatozoa using SperMagic medium. Andrologia. 2018;50(7):e13056. doi: 10.1111/and.13056. [DOI] [PubMed] [Google Scholar]
- 30.Pereira R, Oliveira ME, Santos R, Oliveira E, Barbosa T, Santos T, et al. Characterization of CCDC103 expression profiles: further insights in primary ciliary dyskinesia and in human reproduction. J Assist Reprod Genet. 2019;36(8):1683–1700. doi: 10.1007/s10815-019-01509-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Höben IM, Hjeij R, Olbrich H, Dougherty GW, Nöthe-Menchen T, Aprea I, Frank D, Pennekamp P, Dworniczak B, Wallmeier J, Raidt J, Nielsen KG, Philipsen MC, Santamaria F, Venditto L, Amirav I, Mussaffi H, Prenzel F, Wu K, Bakey Z, Schmidts M, Loges NT, Omran H. Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body asymmetry due to defects of outer and inner dynein arms. Am J Hum Genet. 2018;102(5):973–984. doi: 10.1016/j.ajhg.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonnefoy S, Watson CM, Kernohan KD, Lemos M, Hutchinson S, Poulter JA, Crinnion LA, Berry I, Simmonds J, Vasudevan P, O’Callaghan C, Hirst RA, Rutman A, Huang L, Hartley T, Grynspan D, Moya E, Li C, Carr IM, Bonthron DT, Leroux M, Care4Rare Canada Consortium. Boycott KM, Bastin P, Sheridan EG. Biallelic mutations in LRRC56, encoding a protein associated with intraflagellar transport, cause mucociliary clearance and laterality defects. Am J Hum Genet. 2018;103(5):727–739. doi: 10.1016/j.ajhg.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kott E, Duquesnoy P, Copin B, Legendre M, Dastot-Lemoal F, Montantin G, et al. Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia. Am J Hum Genet. 2012;91(5):958–964. doi: 10.1016/j.ajhg.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coutton C, Escoffier J, Martinez G, Arnoult C, Ray PF. Teratozoospermia: spotlight on the main genetic actors in the human. Hum Reprod Update. 2015;21(4):455–485. doi: 10.1093/humupd/dmv020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOC 43 kb)
Data Availability Statement
The data and material that support the findings of this study are available from the corresponding author upon reasonable request.