

SUMMARY
Netherton syndrome (NS) is a monogenic skin disease resulting from loss of function of lymphoepithelial Kazal-type-related protease inhibitor (LEKTI-1). In this study we examine if bacteria residing on the skin are influenced by the loss of LEKTI-1 and if interaction between this human gene and resident bacteria contributes to skin disease. Shotgun sequencing of the skin microbiome demonstrates that lesional skin of NS subjects is dominated by Staphylococcus aureus (S. aureus) and Staphylococcus epidermidis (S. epidermidis). Isolates of either species from NS subjects are able to induce skin inflammation and barrier damage on mice. These microbes promote skin inflammation in the setting of LEKTI-1 deficiency due to excess proteolytic activity promoted by S. aureus phenol-soluble modulin α as well as increased bacterial proteases staphopain A and B from S. aureus or EcpA from S. epidermidis. These findings demonstrate the critical need for maintaining homeostasis of host and microbial proteases to prevent a human skin disease.
Graphical Abstract

In Brief
Williams et al. show how an abnormal skin microbiome promotes inflammation associated with Netherton syndrome, a monogenic disorder in the human protease inhibitor SPINK5. Subjects with Netherton syndrome have excess colonization by S. aureus or S. epidermidis that then produce and promote increased protease production in the epidermis.
INTRODUCTION
The identity and abundance of microbes on epithelial surfaces have been linked to important outcomes in human disorders, such as inflammatory bowel diseases, neurological disorders, obesity, cancer, diabetes, autoimmune disorders, and skin diseases (Gilbert et al., 2018; Helmink et al., 2019; Kho and Lal, 2018; Schmidt et al., 2018). Symptoms associated with microbial dysbiosis are generally thought to reflect the influence of specific members of the microbial community on health. For example, the presence of Staphylococcus aureus (S. aureus) on subjects with atopic dermatitis (AD) is strongly associated with disease (Alsterholm et al., 2017; Gong et al., 2006; Kong et al., 2012; Leyden et al., 1974; Paller et al., 2019). However, even in this example, S. aureus is not universally found on all AD subjects, and the role of the microbe in AD pathogenesis continues to be debated (Paller et al., 2019). It also remains unclear why S. aureus promotes inflammation in the absence of infection in AD but not in healthy individuals. Although much progress has been made in the past decade toward understanding the roles of the microbiome in human health, the inability to connect host genotypes to microbial functions has made it difficult to establish causality for microbes in human inflammatory diseases.
The contribution of the host to control the composition of the resident microbiome is poorly understood. In the case of AD, mutations in the skin barrier protein filaggrin (FLG) represent the most significant known genetic risk factor (Esparza-Gordillo et al., 2009; Morar et al., 2007). FLG mutations in AD have also been associated with an increase of S. aureus colonization in subjects (Clausen et al., 2017). However, not all subjects with FLG mutations have dysbiosis of the microbiome, and most AD subjects do not have FLG mutations. The complexity of a multifactorial disease such as AD makes it difficult to connect skin inflammation to microbial dysbiosis through a human genetic abnormality. Several murine models have been useful to show how host genetic modifications can influence microbial colonization or inflammation, but much work remains to establish this link in humans (Kobayashi et al., 2015; Nakatsuji et al., 2016). Overall, there is a paucity of examples of a canonical pathway connecting a human gene to microbial dysbiosis and subsequent disease symptoms.
In this study, we investigated subjects with the autosomal-recessive skin disease Netherton syndrome (NS) to understand the relationship between human genetic mutations and the role of the microbiome in skin disease. NS subjects have a single gene mutation in the serine protease inhibitor Kazal type 5 gene (SPINK5) that leads to a loss of function of the protein lymphoepithelial Kazal-type-related protease inhibitor (LEKTI-1) (Chavanas et al., 2000). Mechanistically, this mutation leads to an increase in epidermal serine protease activity that subsequently leads to skin barrier damage and inflammation. The mouse Spink5 knockout is lethal shortly after birth because of a severely impaired epidermal skin barrier (Bonnart et al., 2010; Briot et al., 2009; Descargues et al., 2005). In humans, NS subjects typically have a generalized skin inflammatory phenotype at birth that reflects the abnormal development of the epidermal barrier due to the lack of LEKTI-1 activity. However, patients with NS improve with age, and adults do not show a generalized skin phenotype. Adults with NS typically have limited and distinct skin locations with epidermal breakdown, and this clinical phenotype changes over time. On the basis of this clinical finding, we investigated if the function of the local skin microbiome could contribute to the localized and transient nature of skin disease in adult NS subjects.
Recently S. aureus was shown to damage normal human keratinocytes through the release of bacterial proteases and the production of phenol-soluble modulin alpha (PSMα) peptides that induce human endogenous serine protease activity on the skin surface (Liu et al., 2017; Nakagawa et al., 2017; Nakatsuji et al., 2016; Williams et al., 2017, 2019). These findings showed that bacterial products can negatively modulate the host skin barrier either by direct action of their own secreted proteases or by controlling host protease responses.
On the basis of the variable clinical phenotype of adult NS subjects and previous findings with S. aureus, we hypothesized that the mutation of the human protease inhibitor gene SPINK5 could enable the skin microbiome to further exacerbate the clinical phenotype of NS beyond the effects on epidermal differentiation observed in newborns. This hypothesis was further supported by the fact NS subjects can also frequently become infected by S. aureus (Chao et al., 2005; Renner et al., 2009; Zhvania et al., 2017). Unexpectedly, all ten NS subjects assessed in our study were dominated by S. aureus and/or Staphylococcus epidermidis (S. epidermidis). Furthermore, aside from the previously established capacity of S. aureus to alter proteolytic balance in the epidermis, we discovered that an S. epidermidis cysteine protease may also play an important role in damaging the skin and inducing inflammation. Overall, this study allows for a better understanding of how interactions between systems of microbial and human proteases activity are important to maintain homeostasis and contributes to disease in NS.
RESULTS
The Skin Microbiome of NS Is Distinct from Healthy Skin
Swabs were collected from the skin of ten subjects with NS whose clinical diagnosis was confirmed by the presence of causative mutations in SPINK5. Swabs were collected on different anatomical areas of the trunk and extremities according to the presence of a lesion. Several subjects were also screened at multiple time points. Areas sampled included the abdomen, thigh, and arm, which have similar microbial content under healthy conditions (Table S1). The number of collected samples varied from one subject to another depending of the number of visits the subject had during the time of the study. The microbiota from individual swab samples were studied by both deep shotgun sequencing and culturing of live individual bacterial isolates (Figures 1A and 1B; Figure S1). To permit adequate depth of sequencing, only samples with less than 98% of human genomic DNA contamination were fully sequenced (five healthy subjects and six NS subjects). Meta-analysis of the assembled contigs revealed major differences between the healthy and NS cohorts but a similarity between bacterial DNA sequences from lesional and non-lesional sites within the NS group (Figures 1C–1E; Figure S2). Of the 40 most abundant bacterial species and strains, Cutibacterium acnes predominated on healthy control subjects, except for one subject with Enhydrobacter aerosaccus, while both the lesional and non-lesional skin of NS subjects had increased staphylococcal species, including both S. epidermidis and S. aureus (Figure 1F; Figure S3). Overall the most abundant species on either healthy or NS skin samples was frequently greater than 30% of the entire community. These data show a large difference between the compositions of the overall bacterial community of NS subjects compared with controls. This difference in bacterial communities between healthy skin and NS exceeds prior reports of the difference in the microbiome between subjects with AD and controls (Byrd et al., 2018).
Figure 1. Netherton Skin Microbiome Differs from Healthy Skin.
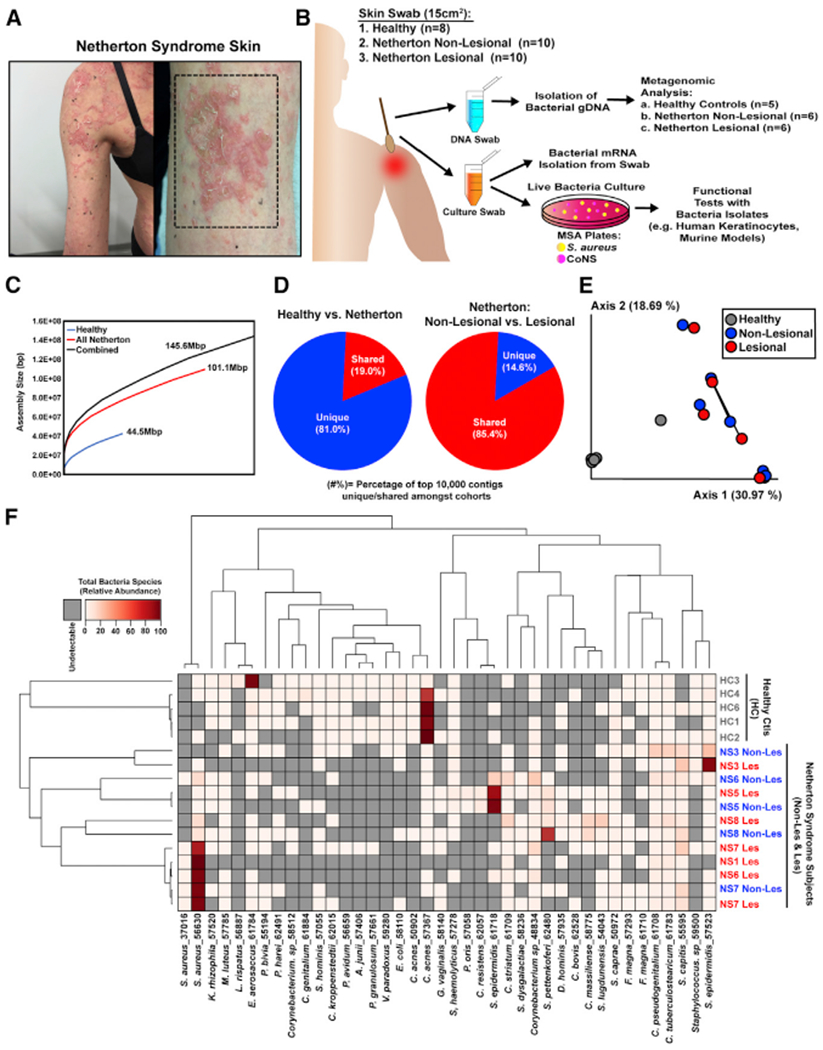
(A) Representative picture of Netherton syndrome skin with severe disease.
(B) Workflow of Netherton skin microbiome collection and analysis.
(C) QUAST plots to assess the size of contigs for all assemblies. Three different co-assemblies were performed: reads from all samples (healthy and infected) (black), reads from only the Netherton cohort (red), and reads from only the healthy samples (blue).
(D) Pie chart representing the percentage of the top 10,000 contigs unique (blue) or shared (red) between healthy subjects and Netherton syndrome patients (left chart) and between Netherton syndrome non-lesional skin and lesional skin (right chart).
(E)Principal-coordinates analysis (PCoA) plot of beta diversity among samples using the Bray-Curtis dissimilarity metric. Each dot represents an individual swab. Swabs fromlesional and non-lesional skin from the same subject are connected by a black line.
(F) Hierarchical clustering of samples showing the top 40 most prevalent species across all samples.
See also Figures S1–S3.
Analysis of the diversity of staphylococcal species on NS and healthy controls showed an increase in the relative abundance of S. aureus and S. epidermidis on NS subjects regardless of whether the swabs were collected from lesional or non-lesional sites (Figure 2A; Figure S1). However, analysis of the absolute abundance of total live staphylococci and S. aureus on skin by colony counting showed a significant increase on the lesional skin of NS subjects compared with non-lesional skin sites and skin from healthy controls (Figure 2B). Also, all of the NS subjects in this study were colonized by S. aureus at least at one time point from multiple collections (Figure 2C; Figure S4). Together, these findings unexpectedly showed that S. aureus and S. epidermidis dominate NS skin in terms of both absolute abundance of live colonies and relative species abundance as determined by shotgun metagenomic analysis.
Figure 2. Staphylococcus aureus Colonization Is Increased on Netherton Syndrome Skin.
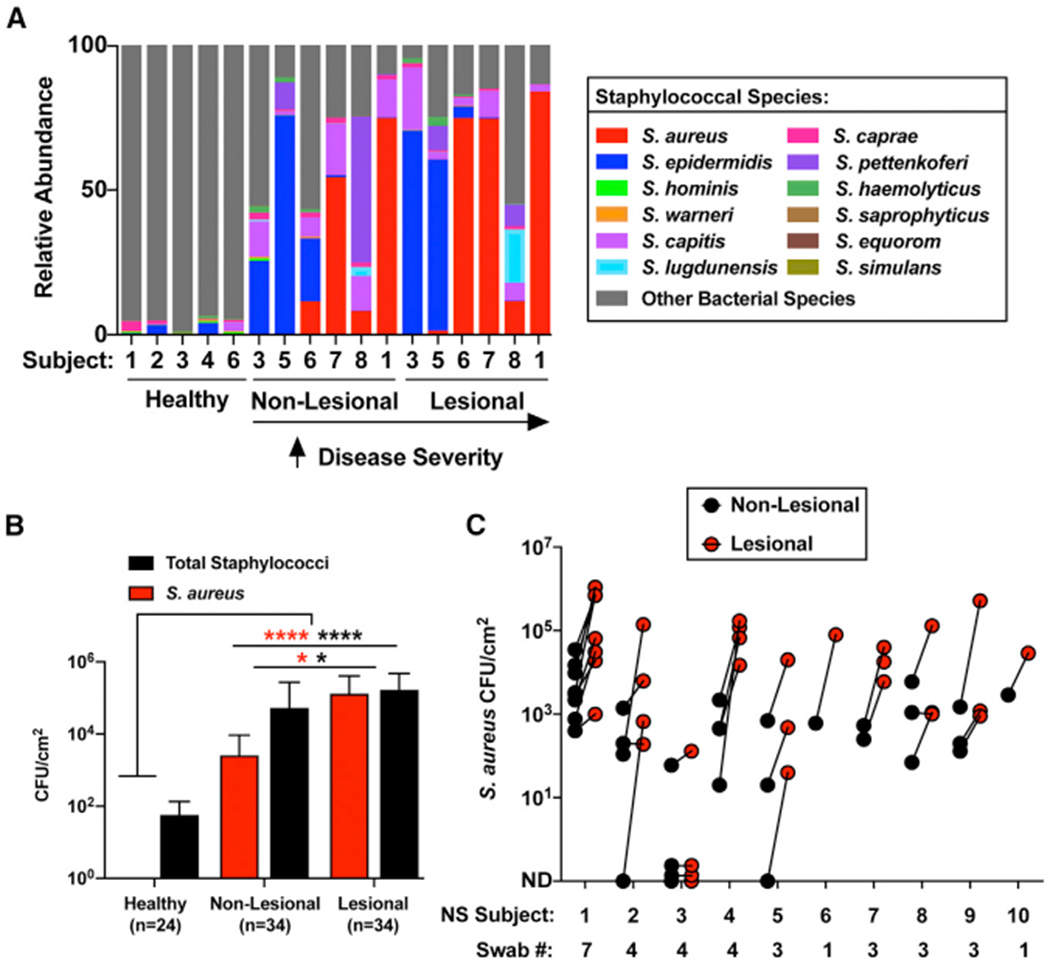
(A) Percentage relative abundance of staphylococcal species within the total bacterial population on healthy controls, NS non-lesional, and NS lesional skin. NS subjects are arranged according to disease severity.
(B) S. aureus (red) and total staphylococci (black) colony-forming units (CFUs) per square centimeter of skin from healthy controls and NS non-lesional and lesional skin (n, number of swabs assessed per condition). Results represent mean ± SEM, and the non-parametric unpaired Kruskal-Wallis test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(C) S. aureus CFUs per square centimeter of skin of NS non-lesional (black) and lesional (red) skin swabs at different visits (swab number) for each subject within the NS cohort. Each dot represents a swab sample. Different numbers of swabs were collected for the different subjects depending on the number of visits they had during the time of the study.
See also Figure S4.
S. aureus Phenol-Soluble Modulin α Promotes Epidermal Protease Activity that Is Amplified in NS
To identify the mechanism by which S. aureus could promote disease in NS, we examined the S. aureus virulence factor PSMα. This peptide has been shown to induce activity of several serine proteases of the kallikrein (KLK) family in human keratinocytes (Williams et al., 2017,2019). We hypothesized that induced expression of human proteases by PSMα might be amplified by the lack of the serine protease inhibitor LEKTI-1 in NS subjects. Our metagenomic analysis showed that DNA for the psmα operon was elevated in all samples in which S. aureus colonization was present (subjects 1, 6, 7, and 8), and a strong correlation between the presence of psmα and disease severity was observed (Figure 3A). psmα mRNA extracted from skin swabs also correlated with the abundance of S. aureus colonization on NS skin (Figures 3B and 3C). Analysis of individual S. aureus colonies further revealed that expression of the psmα operon varied between isolates and that this expression correlated with the capacity of an isolate to induce endogenous serine protease activity in primary human keratinocytes (Figure 3D).
Figure 3. Staphylococcus aureus PSMα Is Increased on Netherton syndrome Skin and Promotes Epidermal Protease Activity.
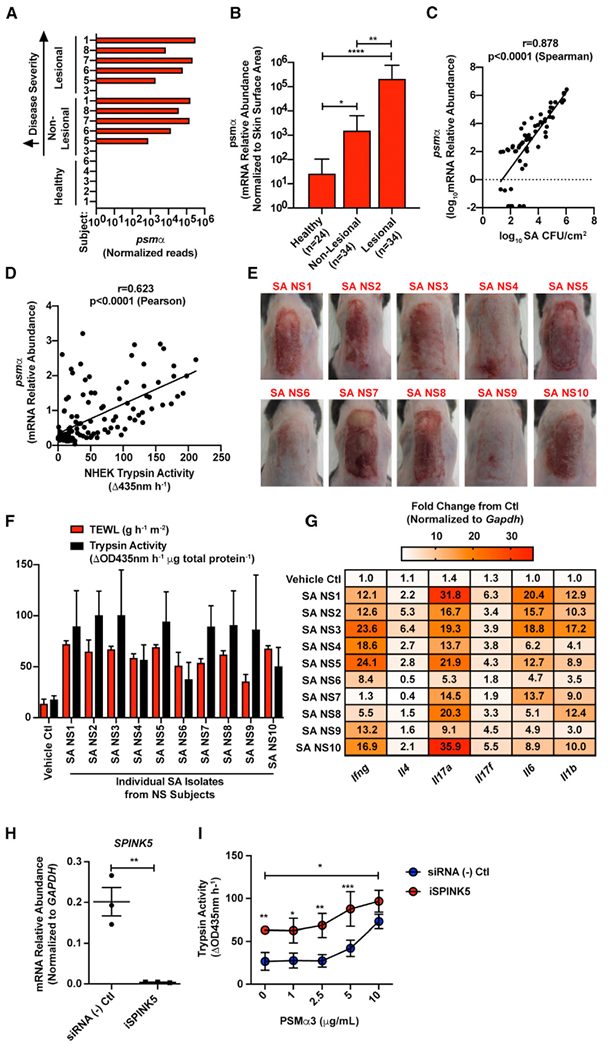
(A) Normalized counts of the gene psmα detected from metagenomic samples.
(B) Relative abundance of S. aureus psmα mRNA isolated from skin swabs of healthy and NS non-lesional and lesional skin (n, number of swabs assessed per condition). Results represent mean ± SEM, and a non-parametric unpaired Kruskal-Wallis test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(C) Spearman correlation between S. aureus (SA) CFU/cm2 and the relative abundance of S. aureus psmα mRNA isolated from skin swabs. Each dot represents an individual swab.
(D) Pearson correlation between the trypsin activity induced in neonatal human epidermal keratinocytes (NHEKs) after culture for 24 h with 5% supernatant of clinical S. aureus (SA) isolates from NS skin and the relative abundance of psmα mRNA level in the same SA isolates. Each dot represents an individual SA isolate.
(E–G) Epicutaneous application of 1e7 CFU/cm2 of S. aureus clinical isolates on murine back skin for 48 h (n = 3 per group). For each NS subject, one lesional S. aureus isolate with a high psmα expression was selected for mouse skin application.
(E) Visual representation of murine back skin after 48 h colonization with 1e7 CFU/cm2 SA isolates. (F and G) Analysis of (F) epidermal barrier damage (TEWL), trypsin activity, and (G) qPCR analysis of inflammatory cytokines stimulated in murine skin by clinical SA NS isolates 1–10. qPCR cytokine levels (Ifng, Il4, Il17a, Il17f, Il6, and Il1b) are normalized to the housekeeping gene Gapdh.
(H) Relative abundance of SPINK5 mRNA in NHEKs that were treated with scrambled control or SPINK5 siRNA (iSPINK5) (n = 3 per condition). Each dot represents an individual sample. Results represent mean ± SEM, and Student’s t test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(I) Trypsin activity from conditioned medium of NHEKs that were pretreated with scrambled control or SPINK5 siRNA (iSPINK5) and then cultured for 24 h with S. aureus synthetic PSMα3 peptide (0, 1, 2.5, 5, and 10 μg/mL) (n = 4 per condition). Results represent mean ± SEM, and two-way ANOVA was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
In (E)–(I), experiments are representative of two independent experiments. See also Figure S5.
To further establish if S. aureus isolates from NS skin could damage the skin and therefore participate in disease, we selected one lesional S. aureus isolate per subject to apply epicutaneously to murine back skin. As expected, similar to what was observed with S. aureus laboratory strains or isolates from AD skin (Byrd et al., 2018; Liu et al., 2017; Nakagawa et al., 2017; Nakatsuji et al., 2016; Williams et al., 2017, 2019), all the tested S. aureus isolates induced visual skin inflammation and barrier damage as assessed by increased transepidermal water loss (TEWL) and endogenous trypsin activity (Figures 3E and 3F). Furthermore, skin exposed to S. aureus from NS also demonstrated increased mRNA for cytokines linked to general inflammation, including Il1b, Il6, Il17a/f, and Ifng (Figure 3G). These findings demonstrate that S. aureus NS isolates have the capacity to promote skin barrier damage and inflammation in vivo. Next, to determine the relevance of the increased expression of S. aureus psmα in NS subjects, normal human keratinocytes were exposed to PSMα3 peptide in culture following inhibition of SPINK5 by small interfering RNA (siRNA). As expected, the NS keratinocyte model lacked SPINK5 mRNA expression, and this correlated with increased baseline trypsin activity from the cells. However, PSMα3 significantly increased keratinocyte trypsin activity in SPINK5-deficient cells (Figures 3H and 3I) above normal cells. These data demonstrate how PSMα peptides from S. aureus can exacerbate proteolytic activity in NS skin because of the unopposed induction of epidermal serine protease activity by this bacterial toxin.
S. aureus Cysteine Proteases Staphopain A and B Are Associated with Disease in NS
In addition to the keratinocyte protease activity induced by PSMα, staphylococci also secrete several proteases that could interact with LEKTI and participate in damaging the epidermis in NS (Nakatsuji et al., 2016; Williams et al., 2019). To determine the effect of an S. aureus protease on skin barrier damage and inflammation, we assessed individual protease gene deletions in a parental strain of S. aureus USA300 LAC. Individual deletions of the genes encoding the cysteine proteases staphopain A and B (scpA, sspB) did not significantly decrease skin damage induced by S. aureus after topical application on mouse skin, but a combined knockout of both cysteine proteases reduced inflammation and increased TEWL associated with loss of skin barrier integrity (Figures 4A and 4B). Similar amounts of bacteria were found on mouse skin after treatment, showing that this effect was not due to a difference in levels of colonization by the mutant strains of bacteria (Figure S5). The abundance of gDNA for these cysteine proteases was increased in NS subjects with elevated S. aureus abundance in the metagenomic dataset (Figure 4C). In parallel with these findings, both scpA and scpB showed significantly increased abundance of mRNA on lesional NS skin compared with non-lesional and healthy controls and correlated with the absolute abundance of S. aureus found on NS subjects (Figures 4D and 4E). These findings show that S. aureus cysteine proteases staphopain A and B can also contribute to skin barrier damage on subjects with NS.
Figure 4. Staphylococcus aureus Staphopain A (scpA) and B (sspB) Are Increased in Netherton Syndrome and Induce Epithelial Barrier Damage.
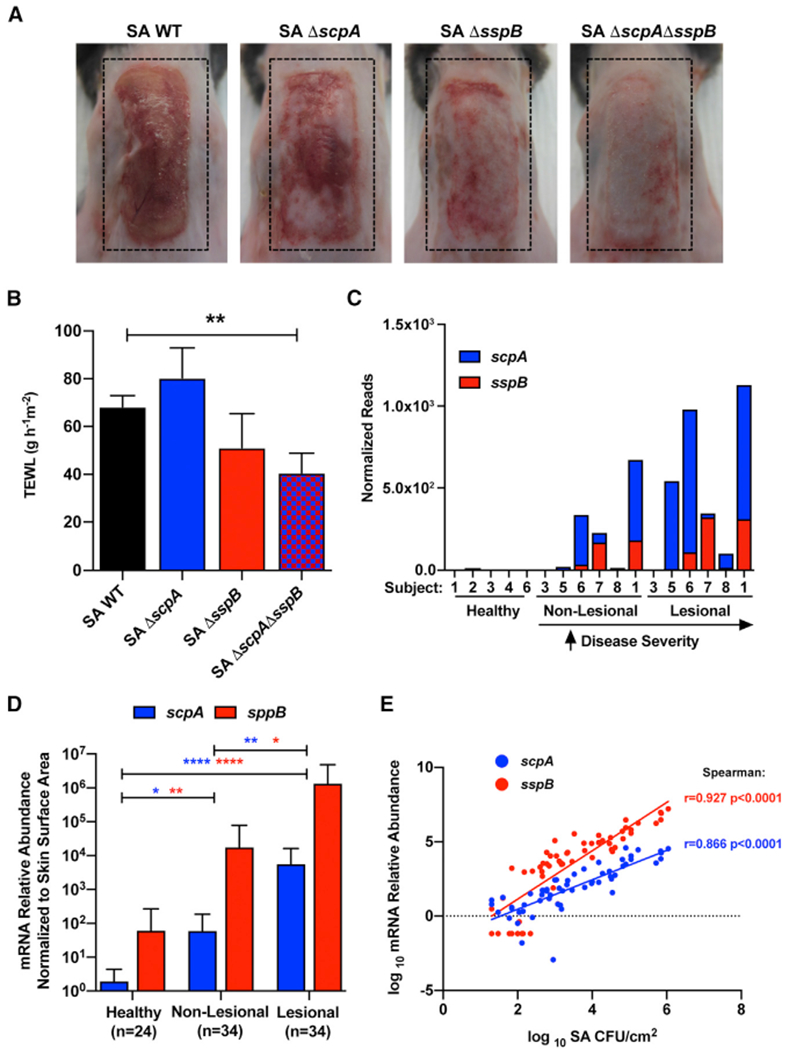
(A and B) (A) Representative picture and (B) TEWL measurement of female C57BL/6J murine back skin after epicutaneous application of 1e7 CFU/cm2 of S. aureus (SA) wild-type (WT), SA scpA knockout (−ΔscpA), SA sspB knockout (ΔsspB), or SA scpA/sspB double knockout (ΔscpAΔsspB) for 48 h (n = 5 per group). Results represent mean ± SEM, and one-way ANOVA was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(C) Number of reads from metagenomic data corresponding to S. aureus scpA (red) and sspB (blue) genes normalized per library size for each sample.
(D) Relative abundance of S. aureus scpA (red) and sspB (blue) mRNA isolated from swabs of healthy control and NS non-lesional and lesional skin normalized to skin area (n, number of individual skin swabs per condition). Results represent mean ± SEM, and a non-parametric unpaired Kruskal-Wallis test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(E) Spearman correlation between the relative abundance of either scpA (red) or sspB (blue) mRNA and S. aureus (SA) CFU/cm2 from all skin swabs. Each dot represents an individual swab. Results are represented as mean ± SEM.
In (A) and (B), data are representatives of at least two independent experiments. See also Figure S5.
S. epidermidis Cysteine Protease EcpA Is Associated with Disease in NS and Can Promote Skin Damage
Because a predominance of colonization by S. epidermidis was also seen in NS, we sought to determine how this species might contribute to disease. A potential role for S. epidermidis was particularly evident in NS subject 3 (NS3), who displayed low levels of S. aureus colonization but elevated S. epidermidis relative to controls (Figures 2A and 2C). S. epidermidis is known to secrete a single cysteine protease termed extracellular cysteine protease A (EcpA) that has similarity to S. aureus cysteine proteases staphopain A (ScpA) and staphopain B (SspB) (Dubin et al., 2001; Oleksy et al., 2004). Alignment of amino acid sequences of the active cysteine proteases showed homology between S. aureus staphopains A and B and S. epidermidis EcpA (Figure 5A; Figure S6). Using a single-gene deletion knockout for the EcpA gene (ecpA) in S. epidermidis, we established that S. epidermidis can also drive skin barrier damage and inflammation on murine back skin through the expression of this cysteine protease (Figures 5B and 5C). The difference of effect between the two bacteria was not due to a different level of colonization (Figure S5A). Analysis of the human skin swab samples showed increased S. epidermidis gDNA absolute abundance levels on both non-lesional and lesional skin that were comparable to increases in S. aureus compared with healthy control subjects (Figure 5D). Furthermore, mRNA abundance for ecpA found on NS skin swabs was significantly elevated on lesional skin, and the relative abundance correlated with the gDNA absolute abundance of S. epidermidis found on the skin (Figures 5E and 5F). These data revealed that subject NS3 had the highest abundance of both S. epidermidis and ecpA mRNA and displayed the least amount of live S. aureus (Figure 2C). The analysis of individual S. epidermidis isolates from subject NS3 then revealed that activity of EcpA varied between isolates (Figure 5G). To determinate if S. epidermidis isolates from NS skin could damage the skin, an S. epidermidis isolate selected for having EcpA activity was applied on mice. Similar to what was observed with S. aureus clinical isolates, this S. epidermidis isolate induced skin damage measured by increased TEWL and increased endogenous trypsin activity (Figures 5H and 5I). Moreover, S. epidermidis isolate NS3 2 also increased the mRNA levels of Il1b, Il6, and Il17a/f but not Ifng or Il4 (Figure 5J). These findings suggest that S. epidermidis can contribute to skin damage when present on NS skin through the expression of the cysteine protease EcpA.
Figure 5. Staphylococcus epidermidis Colonization Is Increased in Netherton Syndrome Skin and Can Induce Epithelial Barrier Damage through the Expression of the Cysteine Protease EcpA.
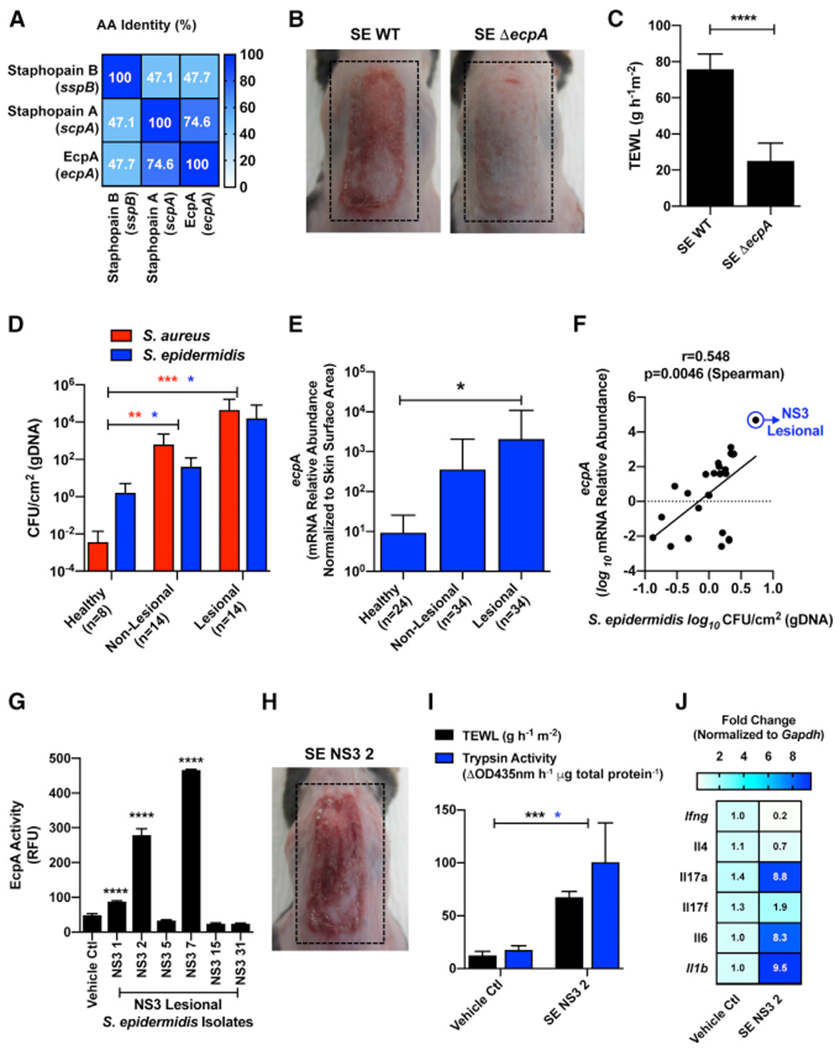
(A) Percentage amino acid sequence identity of the mature forms of the two S. aureus secreted cysteine proteases staphopain A (scpA) and staphopain B (sspB) and the S. epidermidis secreted cysteine protease EcpA (ecpA).
(B and C) (B) Representative pictures and (C) TEWL measurement of female C57BL/6J murine back skin after epicutaneous application of 1e7 CFU/cm2 of S. epidermidis (SE) wild-type (WT) or SE ecpA knockout (ΔecpA) strains for 48h (n = 5 per group). Results represent mean ± SEM, and Student’s t test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(D) Measurement of gDNA absolute abundance of S. epidermidis (blue bars) and S. aureus (red bars) CFU/cm2 on NS (non-lesional and lesional) versus healthy skin (n, number of individual skin swabs per condition). Results represent mean ± SEM, and a non-parametric unpaired Kruskal-Wallis test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(E) Relative abundance of S. epidermidis ecpA mRNA isolated from swabs of healthy control and NS non-lesional and lesional skin normalized to skin area (n, number of individual skin swabs per condition). Results represent mean ± SEM, and a non-parametric unpaired Kruskal-Wallis test was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(F) Spearman correlation between the relative abundance of S. epidermidis ecpA mRNA and S. epidermidis CFU/cm2 (gDNA) from skin swabs.
(G) Assessment of subject NS3 isolated S. epidermidis isolates from lesional skin swabs for specific cleavage of EcpA substrate (n = 3). Results represent mean ± SEM, and a one-way ANOVA was used to determine statistical significance: *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
(H) Representative picture of murine back skin after 48 h colonization with 1e7 CFU/cm2 of clinical S. epidermidis isolate NS3 2 (SE. NS3 2).
(I and J) Analysis of epidermal barrier damage (TEWL), trypsin activity, and qPCR analysis of inflammatory cytokines stimulated in murine skin by S. epidermidis isolate SE. NS3 2. qPCR cytokine levels (Ifng, Il4, Il17a, Il17f, Il6, and Il1b) are normalized to the housekeeping gene Gapdh.
In (B), (C), and (G)–(J), data are representatives of at least two independent experiments. See also Figures S5 and S6.
DISCUSSION
A fundamental understanding of how commensal microbial gene products interact with the human genome is generally lacking. In this study, we focused on subjects with an autosomal-recessive skin disease that results from loss-of-function mutations in a single gene, the serine protease inhibitor SPINK5. This syndrome was of interest because although it is a germline mutation, the severity and location of inflammation vary over time, thus suggesting the potential that this single-gene mutation enables a dynamic epidermal microbial community to influence the observed phenotype. Bacteria present on subjects with NS were similar to one another but differed greatly from bacteria present on the skin of healthy control subjects. Staphylococcal species, in particular either S. aureus or S. epidermidis, were predominant on NS subjects. Genomic and functional analysis identified specific gene products from these bacteria that altered the proteolytic activity of the epidermis and therefore were relevant to subjects with loss of SPINK5. This human disease demonstrates how products of the human and microbial genomes can interact and illustrates the functional consequences of loss of homeostasis driven by a defect in this system. The epidermal ecosystem is multidimensional, and our findings show how several bacterial genes from different species can contribute to a disease phenotype. Overall we provide an important model of how communication with commensal microbes influences human health.
This study presents one of the most deeply sequenced skin microbiome datasets to date, which has allowed metagenomic co-assembly. The QUAST analysis of the co-assemblies shows that the major shared genomes were highly covered, as does the psmα contig analysis. The strain resolved phylogenetic profiling was conducted using software explicitly designed for this analysis (Nayfach et al., 2016). Despite the findings of the metagenomic sequencing presented in this study, we do recognize several potential shortcomings. As with other skin microbiome studies, a lack of ability to remove the majority of human DNA contamination leads to difficulty in assessing bacteria reads with enough coverage for assembly. Sampling of the NS subject cohort was also not contained to a single common site, because of the sporadic nature of NS skin lesions. Skin swabs were collected based upon the availability of lesions at each visit. These swab sites were predominately on the abdomen and local extremities (e.g., shoulder, arm, thigh), which all have similar baseline healthy microbiomes and are thus relevant to compare. Age can also lead to varying skin microbiome populations. It has been observed that younger patients have significantly less Propionibacterium on the skin surface than older patients (Oh et al., 2012). Subject ages in this study varied drastically, ranging from 12 to 47 years, with the presence of younger subjects making it difficult to include age-matched healthy control subjects. Despite these challenges in sample collection with a rare skin disease, all sites sampled displayed a clear phenotype of increased staphylococci colonization that differed greatly from normal skin (age range 27–52 years) and often was predominated by S. aureus. This does not occur in the healthy population despite age or swab site, and the percentage of NS individuals who were positive for S. aureus (>90%) greatly exceeded how often AD subjects are colonized by S. aureus (>50%). Overall, these findings provide a highly informative deep shotgun sequencing metagenomic analysis of a skin disease and suggest that the gene mutation in NS results in a unique skin microbiome.
Several of the observations made in this study were unexpected. S. aureus was known to promote damage to keratinocytes and induce skin inflammation (Liu et al., 2017; Nakagawa et al., 2017; Nakatsuji et al., 2016; Williams et al., 2017, 2019) but was not known to play such an important role in NS. Although the presence of S. aureus on NS subject skin was already reported in the literature (Chao et al., 2005; Renner et al., 2009; Śmigiel et al., 2017), the present study showed that NS subjects appeared to be almost universally colonized by S. aureus. Furthermore, we demonstrated that S. aureus isolates from NS subjects have similar effect on mouse skin to what was previously observed with isolates from AD skin (Williams et al., 2019). PSMα from isolates of both diseases induced serine protease activity and promoted skin damage. Moreover, S. aureus staphopain A (scpA) and staphopain B (sspB), two cysteine proteases previously underappreciated for their virulence capacity, were shown to contribute to this response. Human keratinocytes showed increased protease activity when SPINK5 was silenced and treated with PSMα3, thus showing in human cells how the loss-of-function mutations in SPINK5 found in NS subjects will increase their susceptibility to proteolytic damage promoted by S. aureus. In agreement with our data, a case report showed a very significant improvement of symptoms and quality of life of a subject with NS after treatment with an anti-staphylococcal bacteriophage preparation (Zhvania et al., 2017). Altogether, these findings demonstrate how S. aureus present on NS skin is a major factor that exacerbates the pathogenesis of NS.
An important and unexpected observation from this study was also that S. aureus is not the only member of the complex skin microbiome that promotes inflammation in NS. Metagenomic analysis of NS subjects found an increase in the relative abundance of S. epidermidis, with one subject being preferentially dominated by S. epidermidis. We demonstrated that an S. epidermidis NS isolate was able to induce skin damage in mice. This finding was very surprising, as S. epidermidis was previously regarded as one of the key members of the normal skin microbiota and not recognized to have pathogenic effects when colonizing only the skin surface. In contrast, S. epidermidis has been hypothesized to have multiple beneficial effects for the host (Byrd et al., 2018; Nakatsuji and Gallo, 2019; Stacy and Belkaid, 2019). Various S. epidermidis strains have been shown to have the capability to tune skin immunity, to promote wound repair, to limit pathogen infections, and to protect against skin tumors (Lai et al., 2009, 2010; Linehan et al., 2018; Naik et al., 2015; Nakatsuji et al., 2017, 2018). Under some conditions, such as those that were apparently present on NS3, S. epidermidis may effectively outcompete S. aureus for dominance on the skin (Nakatsuji et al., 2017). Although S. epidermidis can also be an invasive pathogen (Dong et al., 2018; Le et al., 2018; Otto, 2009; Uçkay et al., 2009), this behavior is opportunistic and occurs typically in a setting of immunosuppression or implanted foreign devices.
The cysteine protease EcpA was shown to be a major factor used by S. epidermidis to damage the skin, as its deletion in a laboratory strain prevented this effect, and several clinical isolates expressing EcpA had a similar capacity to promote epidermal injury. It is unlikely that the capacity of S. epidermidis to induce skin inflammation is specific to NS isolates. As EcpA expression is under the control of the accessory gene regulatory (agr) quorum sensing system of S. epidermidis (Olson et al., 2014), we speculate that either the density of S. epidermidis on normal skin is too low to activate the agr quorum sensing system, or other commensal coagulase-negative staphylococci can inhibit its agr system when the relative abundance of S. epidermidis is low. This could explain why on healthy skin S. epidermidis does not produce sufficient EcpA to induce disease. Alternatively, LEKTI-1 has also been shown to have the capacity to inhibit cysteine proteases (Bennett et al., 2010). Thus, in NS subjects, the loss of a single gene expressing LEKTI-1 could lead to a skin microenvironment that promotes excess activity of EcpA. This may occur in association with S. aureus, or in some cases, such as subject NS3, skin lesions may be induced by S. epidermidis acting alone. The contribution of other bacterial proteases or toxins to this system cannot be excluded and are considered to be likely (Cheung et al., 2014; Qin et al., 2017; Queck et al., 2009).
NS has similarities to AD but also has important differences. For instance, NS subjects show large dysbiosis across both non-lesional and lesional skin, while AD subjects mostly display dysbiosis only on lesional sites (Byrd et al., 2017). AD subjects display increased S. epidermidis and S. aureus skin colonization during disease flares (Byrd et al., 2017), a response similar to what we observed here on NS subjects. Interestingly, all NS subjects in this study revealed high amounts of S. aureus, but many AD subjects are not culture positive for S. aureus. Interestingly, our findings show how “commensal” S. epidermidis strains may behave on the skin in a similar way to S. aureus to disrupt proteolytic balance and promote inflammation. Therefore, consideration should also be given to S. epidermidis as a pathogen in AD. Further research into the negative effects of S. epidermidis in AD and other disorders is needed.
This study of subjects with NS provides insight into host genetic mechanisms behind changes in the human skin microbiome. In the case of NS, loss of a single serine protease inhibitor is enough to enable dysbiosis, but it is unclear why normal appearing skin on NS subjects also has an abnormal microbiome. Other host factors may be involved in control of the microbial community on the skin. The presence of the gene products identified here from S. aureus and S. epidermidis appears to contribute to the pathogenesis of skin lesions in the adult subjects, but it does not answer why dysbiosis is established. We hypothesize that the proteolytic imbalance from loss of SPINK5 influences innate immune function and the physical barrier. For example, serine proteases promote activation and inactivation of the antimicrobial peptide LL-37 (Yamasaki et al., 2006). LL-37 is an important antimicrobial peptide in the skin against S. aureus (Larrick et al., 1995; Noore et al., 2013). Elevated serine protease activity promotes cleavage of LL-37 (Morizane et al., 2010). Once S. aureus colonization is established, its own secreted proteases can also degrade LL-37 (Sieprawska-Lupa et al., 2004; Sonesson et al., 2017). Thus, it is possible that elevated endogenous serine protease activity due to loss of SPINK5 can prevent host immunity from establishing a healthy homeostasis. As the epidermis matures, established changes to the microbial ecosystem can then further enable manifestations of disease in NS in a localized and transient pattern. Because LEKTI-1 deficiency is lethal in mice (Descargues et al., 2005), efforts are currently being made to develop a viable mouse model for NS to further investigate this question.
In conclusion, this work illustrates how the rare monogenetic skin disease NS results in the establishment of a distinct bacterial community on the skin of adult subjects that is dominated by S. aureus and S. epidermidis. Our data suggest that specific gene products from these microbes may promote the barrier disruption that is characteristic of this disorder. These observations reveal an unanticipated pathway for Staphylococci to contribute to skin inflammation in this disease. Importantly, this work also illustrates how a single genetic modification in a host extracellular protease inhibitor affects the complex ecosystem of the skin. These observations uncover potential areas for future therapeutic interventions in NS.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Richard Gallo (rgallo@ucsd.edu). All bacteria strains used in this study are available from the lead contact while the metagenomic dataset is deposited to NCBI and available according the Bioproject number PRJNA551026.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Human subjects
All experiments involving human subjects were sponsored by INSERM Institute (C12-56, ID-RCB Number: 2013-A00275-40) and were carried out according to protocols approved by the French Agence Nationale de Sécurité du Médicament (ANSM; Project #131066B42) and by the Institutional Review Board (Project #101-13). Skin swabs from 10 subjects with NS (non-lesional and lesional; ages ranging from 12-47 years old listed in Table S1) and 8 healthy subjects (ages ranging from 27-52 years old listed in Table S2) were used in this study (Table S1). Both NS and healthy groups included both male and female subjects. NS subjects were confirmed clinically according to at least 3 of the following parameters: (i) scaly erythroderma in the first year of life, (ii) ichthyosis lineariz circumflexa; (iii) hair shaft defect including trichorrhexis invaginata, and (iii) atopic manifestations (elevated total IgE serum levels). Second, NS subjects were confirmed due to analysis of subject mutations to the SPINK5 gene by direct sequencing of the coding region and adjacent intronic sequences of SPINK5 as reported previously (Bitoun et al., 2002). To compare the severity between individuals, a severity score (Figure S1) based on the following criteria: scaly erythroderma and ichthyosis lineariz circumflexa (absent = 0, minimum = 1, moderate = 2, severe = 3, very severe = 4) was used.
Bacteria preparation
All S. aureus and S. epidermidis strains were grown overnight (18h) to stationary phase in 3% tryptic soy broth (TSB) at 300RPM in a 37°C incubator unless stated otherwise. This growth time for all staphylococci indicated approximately an OD600nm reading of 10 and 1e9 CFU. For treatment of bacterial supernatant on human keratinocytes, overnight cultured bacteria were pelleted (15min, 4,000RPM, RT) followed by filter sterilization of the supernatant (0.22 μm). For mouse experiments with live bacteria colonization, bacterial CFU was approximated by OD600nm prior to application to mouse back skin followed by confirmation of the actual CFU the following day.
Mouse model of epicutaneous bacteria exposure
Age-matched 8-10 weeks old female C57BL/6J mice were used in all experiments (n = 3-5 per condition). Mice were co-housed with 3-5 mice per cage in all experiments. All animal experiments were approved by the UCSD (University of California, San Diego) Institutional Animal Care and Use Committee (Protocol#S09074). A previously described mouse model of epicutaneous bacterial exposure was used (Williams et al., 2019). Briefly, the dorsal skin of anesthetized mice (2% isoflurane) was shaved and depilated using Nair cream for 2min followed by immediate removal with sterile alcohol wipes. The skin barrier was allowed to recover from hair removal for 24h prior to application of bacteria. S. aureus and S. epidermidis (1e7 CFU) in 3%TSB was applied to murine skin for 48h at a 100 μL volume on a 2x1cm piece of sterile gauze. A bio-occlusive dressing (Tegaderm; 3M) along with a flexible fabric Band-Aid was applied on top of gauze to hold in place for duration of the treatment.
Normal human keratinocyte model
Normal neonatal human epidermal keratinocytes (NHEKs; Thermo Fisher Scientific) were cultured in Epilife complete medium containing 60 μM CaCl2 (Thermo Fisher Scientific) supplemented with 1x Epilife Defined Growth Supplement (EDGS; Thermo Fisher Scientific) and 1x antibiotic-antimycotic (PSA; 100U/mL penicillin, 100U/mL streptomycin, 250ng/mL amphotericin B; Thermo Fisher Scientific) at 37°C, 5%CO2. NHEKs were only used for experiments between passages 3-5. NHEKs were grown to approximately 80% confluency followed by differentiation in high calcium (2mM CaCl2) EpiLife complete medium for 48h. For bacterial supernatant treatments, differentiated NHEKs were treated with sterile-filtered bacterial supernatant at 5% by volume to Epilife medium for 24h.
SPINK5 gene silencing human keratinocyte model
NHEKs grown to 50% confluency were treated for 24h with 20nM SPINK5 silencer select siRNA or a siRNA scrambled (-) control (ThermoFisher Scientific) using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) and OptiMEM medium (GIBCO). After 24h, remaining siRNA was removed and NHEKs were supplemented with fresh Epilife complete medium for 48h until cells reached about a complete monolayer followed by addition high calcium Epilife complete medium 2mM CaCl2 for an additional 48h. Finally, S. aureus synthetic PSMα3 peptide (10 μg/mL) was added to differentiated NHEKs for an additional 24h prior to analysis.
METHOD DETAILS
Collection of skin microbiome from human subjects
For all subjects with Netherton syndrome, collection of surface bacteria was done from a pre-measured area (15cm2) of both lesional and non-lesional skin, on different anatomical areas according to lesion location and at different time points (Table S1). For the healthy subjects, surface bacteria were similarly collected on matching anatomical areas (Table S2). For collection of skin microbiome DNA, swab head was soaked in 1mL of molecular biology grade TE buffer (Invitrogen) containing 0.1% Triton X-100 and 0.05% Tween-20 and surface bacteria were collected by rubbing the pre-measured skin area with 50 strokes of constant pressure. Swab head was placed in a microcentrifuge tube and immediately stored at −80°C for further analysis. For collection of skin microbiome RNA and live bacteria, swab head was soaked in 3%TSB and 16.67% glycerol solution and surface bacteria were collected as described above. Swab head was placed in a microcentrifuge tube containing 1 mL of 3%TSB and 16.67% glycerol solution. Following 1 min of vortexing samples, the bacterial suspensions were stored at −80°C for further analysis. After skin microbiome collection, the skin of subjects was cleaned with an alcohol swab.
Microbiome metagenomic analysis
DNA extraction, library preparation, and shotgun sequencing
Microbiome DNA was extracted from skin swab using the PureLink Microbiome DNA Purification Kit according to manufacturer’s instructions (Thermo Fisher Scientific). After purification, samples were treated with NEBNext microbiome DNA enrichment kit according to manufacturer’s instructions (New England Biolabs) except that SPRI select beads (Beckman Coulter) were used for the final clean-up step and 40μL of Low EDTA TE buffer (Swift Biosciences) to elute the samples. The concentration of samples was monitored before and after enrichment using Qubit dsDNA HS assay kit (Invitrogen). 300bp fragments were generated by sonication. Next-generation sequencing (NGS) libraries were then prepared using Accel-NGS 2S Plus DNA Library Kit and 2S Indexed Adaptor Kit according to manufacturer’s instructions (Swift Biosciences) and using SPRI select beads (Beckman Coulter) for all clean-up steps. Before indexing, the quality and fragment-size of the libraries were checked using High Sensitivity D1000 ScreenTape according to manufacturer’s instructions (Agilent Technologies). Finally, HiSeq 2500 150bp paired-end sequencing (Illumina) was performed. 17 samples in total (12 NS lesional (n = 6) and non-lesional (n = 6) and 5 healthy controls) were selected for metagenomic sequencing based upon < 98% human DNA contamination. Cumulatively, the total number of reads across all samples was 2.6x108 reads (with an average of 1.5x107 reads per sample.
Quality control, host removal and metagenomic co-assembly
Raw metagenomic reads were trimmed for quality by removing low quality reads, followed by removal of reads that mapped to the human genome (assembly19) using KneadData (version 0.5.4) with default parameters. Post quality trimming, the percentage of host contamination among subjects ranged from < 10% in some samples to > 80% in a few others, accounting for an average of approximately 1x107 reads per sample. The total number of reads after discarding those that mapped back to the human reference was 4.1x107, with an average of 2.4x106 reads per sample. The host decontaminated reads from all samples (skin conditions including both NS syndrome (non-lesional and lesional) and healthy samples) were then co-assembled using metaSPAdes (version 3.11.1) on k-mer sizes of k = 21,33,55,77. Furthermore, reads across all samples were also combined and co-assembled on the basis of skin condition; i.e., healthy, lesional, and non-lesional to compare total size for the assembled cohorts and assess cumulative genome length and subsequent genome overlap between them (Table S3).
Taxonomic composition using species and strain level profiling
Species level taxonomic profiling was performed on the post-processed reads in order to examine the community composition using MIDAS (version 1.3.0) by leveraging approximately 30,000 bacterial reference genomes, clustered into 5,952 species groups. Relative taxonomic abundances were estimated for each of the bacterial species groups, or strains, across 30 universal single copy marker genes included in the marker_genes reference database by mapping reads to gene-specific, species-level mapping thresholds (94.5%–98% nucleotide identity). Hierarchical clustering was performed for the top 40 most prevalent species were visualized for the entire cohort using “dplyr” to filter the data frame and “heatmap3” with the “average” linkage method for generating the heatmap in R (version 3.4.1). MIDAS also allowed for pan-genomic profiling by mapping metagenomic reads to species of interest to quantify the abundance of these pangenomic genes, with at least one mapped read, across all samples. Results were visualized using “dplyr” and “reshape2” to manipulate the data and “ggplot2” for generating the plots in R.
Community richness and diversity based on ordination analyses
Microbial community richness and diversity metrics were examined using multidimensional scaling techniques like Principal Coordinates Analysis (PCoA) on the read counts matrix previously generated by taxonomic species level profiling for all samples. Beta diversity metrics like bray-curtis dissimilarity index and alpha diversity metrics like shannon, chao1 and observed_otus were calculated on reads from all species that had an abundance of at least 1.0 across all samples. These metrics were then explored and visualized in QIIME 2 (version 2019.1). Between and within beta diversity based on sample groupings by skin condition (healthy/non-lesional/lesional) and subject code was also explored.
Identification of staphylococcal virulence factors among subjects
Each contig from the all-inclusive co-assembly was screened for the presence and expression of specific virulence factors produced by S. aureus and S. epidermidis that are vital for pathogenicity, specifically the following six genes: (1) Phenol-soluble modulin α (PSMα1: fMGIIAGIIKVIKSLIEQFTGK (Williams et al., 2019) (2) Staphopain A (scpA [GenBank: CAD61962.1]) (3) Staphopain B (sspB [GenBank: AAG45844.1]) (4) Extracellular cysteine protease (ecpA [GenBank: AJ298299]. Assembly ORFs were called on all contigs using FragGeneScan (version 1.16), and read counts for each ORF across all samples were obtained by mapping reads to predicted ORFs using clc_ref_assemble_long in CLC Assembly Cell (CLC bio, version 3.22.55705) using the method as described in (Dupont et al., 2015). These ORFs were also searched against PhyloDB (version 1.076) using Blast (E-value threshold < 1e-03) to establish phylogenetic annotation. Additionally, the ORFs were also searched against the Pfam and TIGRfam custom database using HMMER (version 3.0) to establish functional annotation. For Pfam and TIGRFAM assignments, only matches with scores above the model trusted cut-off score were considered. Finally, all phylogenetic and functional annotation results were merged with the read counts for all ORFs across all samples.
Quantification of live staphylococci and collection of isolates from skin swabs
For quantification of live staphylococci and collection of clinical isolates, swabs were rapidly thawed, vortexed, serial diluted and plated onto mannitol salt agar (MSA) selection plates supplemented with 3% egg yolk. After overnight incubation at 37°C, the number of colony-forming unit (CFU) was determined. S. aureus was distinguished from coagulase-negative staphylococci (CoNS) according to mannitol metabolism and the egg yolk reaction as described previously (Iwase et al., 2010; Nakatsuji et al., 2017). For each swab, various CoNS and S. aureus isolates were picked, cultured overnight at 37°C in 3%TSB and stored at −80°C in 3%TSB and 16.67% glycerol solution for further analyses.
Quantification of staphylococcal genomic DNA (gDNA)
The absolute abundance of S. aureus and S. epidermidis gDNA in the microbial DNA elution prepared from skin swabs was determined by quantitative real-time PCR (qPCR) as previously described (Nakatsuji et al., 2013, 2016). Briefly, qPCR was performed with Power SYBR Green Master mix (Applied Biosystems) using S. epidermidis and S. aureus specific primers, targeting the S. epidermidis gseA gene and S. aureus femA gene respectively. To determine the relative CFU of S. aureus or S. epidermidis specific DNA, a standard curve was generated with gDNA extracted from known CFUs of S. aureus (ATCC113) or S. epidermidis (ATCC12228), respectively. The specificity of all primer pairs was confirmed by melting curve analysis and comparison with standard curves.
Transepidermal water loss measurement
To determine damage to the epidermal skin barrier, transepidermal water loss (TEWL) of murine skin treated for 48h with S. aureus or S. epidermidis was measured using a TEWAMETER TM300 (C & K).
RNA isolation and quantitative real-time PCR
All RNA was isolated using the Purelink RNA isolation kit according to manufacturer’s instructions (Thermo Fisher Scientific). For mouse tissue, about 0.5cm2 full thickness skin was bead beat in 750 μL of RNA lysis buffer (2x 30sec with 5min on ice after each, 2.0mm zirconia bead). Tissue was then centrifuged (10min, 13,000RPM, 4°C), followed by adding 350 μL of clear lysate to 70% EtOH and column based isolation of RNA. For isolation of human skin swab microbiome RNA, skins swabs were vortexed (30sec) followed by incubation of 250 μL with RNAprotect reagent (QIAGEN) for 10min prior to centrifugation (10min, 13,000RPM, RT), resuspension in 750 μL of RNA lysis buffer, and bead beating (2x 1min with 5min on ice after each) using lysing matrix B tubes. Samples were then centrifuged again and 350 μL of clear lysate was added to 70% EtOH as above. After RNA isolation, samples were quantified with a Nanodrop (ThermoFisher Scientific), and 500ng of bacterial or mouse RNA was reverse-transcribed using the iScript cDNA synthesis kit (Bio-Rad). qPCR reactions were ran on a CFX96 Real-Time Detection System (Bio-Rad). For mouse samples, gene-specific primers and TaqMan probes (Thermo Fisher Scientific) were used with GAPDH as a housekeeping gene. For bacterial RNA, SYBR Green qPCR Master Mix (Biotool) was used along with specific primers. The mRNA relative abundance of the genes sspB, scpA and ecpA were normalized to the overall skin area swabbed.
NHEK and murine skin trypsin activity analysis
NHEK conditioned medium was added at 50 μL to black 96 well black bottom plates (Corning) followed by addition of 150 μL of the peptide Boc-Val-Pro-Arg-AMC (trypsin-like substrate, BACHEM) at a final concentration of 200 μM in 1x digestion buffer (10 mM Tris-HCl pH7.8, Teknova) and incubated at 37°C for 24h. Relative fluorescent intensity (excitation: 354nm, emission: 435nm) was analyzed with a SpectraMAX Gemini EM fluorometer (Thermo Fisher Scientific). For murine skin trypsin activity analysis, 0.5cm2 full-thickness skin was bead beat (2.0mm zirconia beads, 2x 30sec with 5min on ice after each) in 1mL of 1M acetic acid followed by an overnight rotation at 4°C. Samples were centrifuged (10min, 13,000RPM, 4°C), added to a new microcentrifuge tube followed by protein concentration using a speedvac to remove all remaining acetic acid. Proteins were re-suspended in molecular grade water (500 μL) and rotated overnight at 4°C followed by another centrifugation. Clear protein lysates were added to a new tube, and BCA (Bio-rad) analysis used to determine protein concentration. Finally, 10 μg of total protein was added to a 96 well plate followed by analysis with the trypsin substrate as above.
Staphylococcus epidermidis protease EcpA activity analysis
For the measurement of S. epidermidis protease activity, bacteria were grown for 24h in 3%TSB at 37°C before preparation of filtered-sterilized supernatant. The supernatant was tested for ecpA activity using a specific FRET substrate with the sequence (5-FAM)-Lys-Leu-Leu-Asp-Ala-Ala-Pro-Lys-(QXL520)-OH (AnaSpec, Fremont, CA) (Olson et al., 2014). 25 μl of S. epidermidis supernatant was added to black 96 well black bottom plates (Corning) followed by addition of 25 μL of 1x digestion buffer (10 mM Tris-HCl pH7.8, Teknova) containing the ecpA FRET substrate (1nM final). Relative fluorescent intensity (excitation: 485nm, emission: 538nm) was measured with a SpectraMAX Gemini EM fluorometer (Thermo Fisher Scientific) at t = 0 and after incubation at 37°C for 24h.
Mature cysteine protease Staphopain A, Staphopain B and EcpA sequence alignment
The amino acid sequence of the mature forms of the two S. aureus cysteine proteases Staphopain A (UniProtKB/Swiss-Prot: P81297.2) and Staphopain B (UniProtKB/Swiss-Prot: P0C1S6.1) and the S. epidermidis cysteine protease EcpA (UniProtKB/Swiss-Prot: P0C0Q0.1) were aligned using Geneious R11.1.5 (https://www.geneious.com).
QUANTIFICATION AND STATISTICAL ANALYSIS
All figures utilized non-parametric unpaired Kruskal Wallis analysis, Student’s t tests, and One/Two-way ANOVAs for statistical analysis as indicated in the figure legends. All statistical analysis was performed using GraphPad Prism Version 8.0 (GraphPad, La Jolla, CA). All data is presented as mean ± standard error of the mean (SEM) and a P value ≤ 0.05 considered significant.
DATA AND CODE AVAILABILITY
The accession number for the Netherton syndrome high-throughput shotgun sequencing metagenomic datasets generated in the course of this project have been deposited at the National Center for Biotechnology Information Sequence Read Archive under BioProject ID: PRJNA551026. Any further details regarding these datasets will be made available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Staphylococcus aureus NS1 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS2 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS3 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS4 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS5 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS6 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS7 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS8 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS9 | Gallo (UCSD) | This Study |
| Staphylococcus aureus NS10 | Gallo (UCSD) | This Study |
| Staphylococcus aureus USA300 WT (AH1263) | Horswill (UC Denver) | Mootz et al., 2015 |
| Staphylococcus aureus USA300 ΔscpA (AH1825) | Horswill (UC Denver) | Mootz et al., 2015 |
| Staphylococcus aureus USA300 ΔsspB (AH2594) | Horswill (UC Denver) | Mootz et al., 2015 |
| Staphylococcus aureus USA300 ΔscpAΔsspB (AH2595) | Horswill (UC Denver) | Mootz et al., 2015 |
| Staphylococcus epidermidis NS3 1 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis NS3 2 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis NS3 5 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis NS3 7 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis NS3 15 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis NS3 31 | Gallo (UCSD) | This Study |
| Staphylococcus epidermidis 1457 (AH2490) | Horswill (UC Denver) | Olson et al., 2014 |
| Staphylococcus epidermidis 1457 ΔecpA (AH2924) | Horswill (UC Denver) | Olson et al., 2014 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1M Tris-HCl, pH 7.8 solution | Teknova | Cat# T1078 |
| 2-mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Antibiotic-Antimycotic (100X) | GIBCO | Cat# 15240062 |
| Bacto Agar | BD Biosciences | Cat# 214010 |
| Boc-Val-Pro-Arg-AMC hydrochloride salt | BACHEM | Cat# I-1120 |
| Calcium chloride dihydrate | Sigma-Aldrich | Cat# 22,350-6 |
| Defined Trypsin Inhibitor (DTI) | GIBCO | Cat# R-007-100 |
| DPBS | GIBCO | Cat# 14190-144 |
| EpiLife Defined Growth Supplement (EDGS) | GIBCO | Cat# S-012-5 |
| EpiLife complete medium, with 60 M calcium | GIBCO | Cat# MEPI500CA |
| Ethyl alcohol, Pure | Sigma-Aldrich | Cat# E7023 |
| Formalin | Azer Scientific | Cat# PFNBF-20 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778030 |
| Low EDTA TE buffer | Swift Biosciences | Cat# 90296 |
| Lysing Matrix B | MP Biomedical | Cat# 116911050-CF |
| Molecular biology grade TE buffer | Invitrogen | Cat# AM9849 |
| OptiMEM medium | GIBCO | Cat# 31985062 |
| PSMα3peptide:fMEFVAKLFKFFKDLLGKFLGNN | LifeTein | N/A |
| RNAlater Stabilization Solution | Invitrogen | Cat# AM7021 |
| RNAprotect Bacteria Reagent | QIAGEN | Cat# 76506 |
| SPRI Select beads | Beckman Coulter | Cat# B23317 |
| Tryptic soy Broth (TSB) | Sigma-Aldrich | Cat# T8907-1KG |
| Trypsin/EDTA solution | GIBCO | Cat# R-001-100 |
| UltraPure distilled water | Invitrogen | Cat# 10977-015 |
| Critical Commercial Assays | ||
| 2S Indexed Adaptor Kit | Swift Biosciences | Cat# 26596 |
| Accel-NGS 2S Plus DNA Library Kit | Swift Biosciences | Cat# 21096 |
| EnzCheck Protease assay kit | ThermoFisher Scientific | Cat# E6638 |
| iSCRIPT cDNA synthesis Kit | BIO-RAD | Cat# 178891 |
| High Sensitivity D1000 ScreenTape | Agilent Technologies | Cat# 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent Technologies | Cat# 5067-5585 |
| NEBNext Microbiome DNA Enrichment Kit | New England Biolabs | Cat# E2612 |
| Power SYBR Green Master Mix | Applied Biosystems | Cat# 4367659 |
| PureLink RNA Mini Kit | Invitrogen | Cat# 12183025 |
| PureLink Microbiome DNA Purification Kit | Invitrogen | Cat# A29790 |
| SYBR Green qPCR Master Mix (2X) | Biotool | Cat# B21204 |
| Qubit dsDNA HS assay kit | Invitrogen | Cat# Q32851 |
| Deposited Data | ||
| Metagenomic Sequencing Dataset | This Study | BioProject ID: PRJNA551026 |
| Experimental Models: Cell Lines | ||
| Human Epidermal Keratinocytes, neonatal (HEKn) | GIBCO | Cat# C0015C |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Strain: 000664 |
| Oligonucleotides | ||
| S. aureus femA Forward: AACTGTTGGCCACTATGAGT | Paule et al., 2004 | N/A |
| S. aureus femA Reverse: CCAGCATTACCTGTAATCTCG | Paule et al., 2004 | N/A |
| S.epidermidis gseA Forward: ATGAAAAAGAGATTTTTATCT | Ikeda et al., 2004 | N/A |
| S.epidermidis gseA Reverse: GTTTGGTGACACTCTTAAG | Ikeda et al., 2004 | N/A |
| S. epidermidis ecpA Forward: TGTGCTTAAAACGCCACGTA | Olson et al., 2014 | N/A |
| S. epidermidis ecpA Reverse: GTATAGCCGGCACACCAACT | Olson et al., 2014 | N/A |
| S. aureus psmα Forward: TAAGCTTAATCGAACAATTC | Sully et al., 2014 | N/A |
| S. aureus psmα Reverse: CCCCTTCAAATAAGATGTTCATATC | Sully et al., 2014 | N/A |
| S. aureus scpA Forward: CTATTGCAAACGCTGAGAGC | Mootz et al., 2015 | N/A |
| S. aureus scpA Reverse: ACGTACGTCAGTAGGAACACTCTT | Mootz et al., 2015 | N/A |
| S. aureus sspB Forward: CAGCAAATTGTTGTTGTGCTAG | Ma et al., 2012 | N/A |
| S. aureus sspB Reverse: AAGCCAAAGCCGATTCACACTC | Ma et al., 2012 | N/A |
| TaqMan Murine Gapdh primers | Mm99999915_g1 (Applied Biosystems) | N/A |
| TaqMan Murine Il6 primers | Mm00446190_m1 (Applied Biosystems) | N/A |
| TaqMan Murine Ifng primers | Mm01168134_m1 (Applied Biosystems) | N/A |
| TaqMan Murine Il4 primers | Mm00445259_m1 (Applied Biosystems) | N/A |
| TaqMan Murine Il17a primers | Mm00439618_m1 (Applied Biosystems) | N/A |
| TaqMan Murine Il17f primers | Mm00521423_m1 (Applied Biosystems) | N/A |
| TaqMan Murine Il1b primers | Mm00434228_m1 (Applied Biosystems) | N/A |
| TaqMan Human GAPDH primers | Hs02786624_g1 (Applied Biosystems) | N/A |
| Taqman Human SPINK5 primers | Hs00928570_m1 (Applied Biosystems) | N/A |
| Silencer Select SPINK5 siRNA | s21667 (ThermoFisher) | Cat#4392420 |
| Software and Algorithms | ||
| CLC Assembly Cell (CLC bio, version 3.22.55705) | QIAGEN | https://digitalinsights.qiagen.com/products-overview/analysis-and-visualization/qiagen-clc-assembly-cell/ |
| Clustal Omega (version 1.2.1) | Sievers et al., 2011 | http://www.clustal.org/omega/ |
| FragGeneScan (version 1.16) | Kim et al., 2015 | https://github.com/hallamlab/FragGeneScanPlus |
| Geneious R11.1.5 | Geneious | https://www.geneious.com |
| GraphPad Prism (version 5.01) | GraphPad Software | https://www.graphpad.com/ |
| HMMER (version 3.0) | Eddy, 1998 | http://hmmer.org/ |
| KneadData, (version 0.5.4) | https://bitbucket.org/biobakery/kneaddata/wiki/Home | |
| metaSPAdes (version 3.11.1) | Nurk et al., 2017 | http://cab.spbu.ru/software/meta-spades/ |
| MIDAS (version 1.3.0) | Nayfach et al., 2016 | https://github.com/snayfach/MIDAS |
| PhyloDB (version 1.076) | https://scripps.ucsd.edu/labs/aallen/data/ | |
| QIIME 2 (version 2019.1) | https://qiime2.org/ | |
| R (version 3.4.1) | R Core team, 2017 | https://cran.r-project.org/bin/windows/base/old/3.4.1/ |
Highlights.
Netherton subjects have increased S. aureus or S. epidermidis on their skin
S. aureus PSMα induces greater protease activity in keratinocytes deficient in SPINK5
S. epidermidis EcpA protease damages the epidermis
S. aureus and S. epidermidis proteases induce skin inflammation in mice
ACKNOWLEDGMENTS
R.L.G. was supported by the National Institutes of Health (NIH) grants R37AI052453, R01AR076082, R01AR074302, and R01AR069653 and by grant U19 AI117673 from the Atopic Dermatitis Research Network (ADRN).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.021.
DECLARATION OF INTERESTS
R.L.G. is a co-founder, scientific advisor, consultant, and has equity in MatriSys Biosciences and is a consultant, receives income, and has equity in Sente. All other authors declare no competing interests.
REFERENCES
- Alsterholm M, Strömbeck L, Ljung A, Karami N, Widjestam J, Gillstedt M, Åhren C, and Faergemann J (2017). Variation in Staphylococcus aureus colonization in relation to disease severity in adults with atopic dermatitis during a five-month follow-up. Acta Derm. Venereol 97, 802–807. [DOI] [PubMed] [Google Scholar]
- Bennett K, Callard R, Heywood W, Harper J, Jayakumar A, Clayman GL, Di WL, and Mills K (2010). New role for LEKTI in skin barrier formation: label-free quantitative proteomic identification of caspase 14 as a novel target for the protease inhibitor LEKTI. J. Proteome Res 9, 4289–4294. [DOI] [PubMed] [Google Scholar]
- Bitoun E, Chavanas S, Irvine AD, Lonie L, Bodemer C, Paradisi M, Hamel-Teillac D, Ansai S, Mitsuhashi Y, Taieb A, de Prost Y, Zambruno G, Harper JI, and Hovnanian A (2002). Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol 118, 352–361. [DOI] [PubMed] [Google Scholar]
- Bonnart C, Deraison C, Lacroix M, Uchida Y, Besson C, Robin A, Briot A, Gonthier M, Lamant L, Dubus P, et al. (2010). Elastase 2 is expressed in human and mouse epidermis and impairs skin barrier function in Netherton syndrome through filaggrin and lipid misprocessing. J. Clin. Invest 120, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, Dubus P, and Hovnanian A (2009). Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med 206, 1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng WI, Conlan S, Belkaid Y, Segre JA, and Kong HH; NISC Comparative Sequencing Program (2017). Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med 9, eaal4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Belkaid Y, and Segre JA (2018). The human skin microbiome. Nat. Rev. Microbiol 16, 143–155. [DOI] [PubMed] [Google Scholar]
- Chao SC, Richard G, and Lee JY (2005). Netherton syndrome: report of two Taiwanese siblings with staphylococcal scalded skin syndromeand mutation of SPINK5. Br. J. Dermatol 152, 159–165. [DOI] [PubMed] [Google Scholar]
- Chavanas S, Bodemer C, Rochat A, Hamel-Teillac D, Ali M, Irvine AD, Bonafé JL, Wilkinson J, Taïeb A, Barrandon Y, et al. (2000). Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat. Genet 25, 141–142. [DOI] [PubMed] [Google Scholar]
- Cheung GY, Joo HS, Chatterjee SS, and Otto M (2014). Phenol-soluble modulins—critical determinants of staphylococcal virulence. FEMS Microbiol. Rev 38, 698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen ML, Edslev SM, Andersen PS, Clemmensen K, Krogfelt KA, and Agner T (2017). Staphylococcus aureus colonization in atopic eczema and its association with filaggrin gene mutations. Br. J. Dermatol 177, 1394–1400. [DOI] [PubMed] [Google Scholar]
- Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, Elias P, Barrandon Y, Zambruno G, Sonnenberg A, and Hovnanian A (2005). Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat. Genet 37, 56–65. [DOI] [PubMed] [Google Scholar]
- Dong Y, Speer CP, and Glaser K (2018). Beyond sepsis: Staphylococcus epidermidis is an underestimated but significant contributor to neonatal morbidity. Virulence 9, 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin G, Chmiel D, Mak P, Rakwalska M, Rzychon M, and Dubin A (2001). Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol. Chem 382, 1575–1582. [DOI] [PubMed] [Google Scholar]
- Dupont CL, McCrow JP, Valas R, Moustafa A, Walworth N, Goodenough U, Roth R, Hogle SL, Bai J, Johnson ZI, et al. (2015). Genomes and gene expression across light and productivity gradients in eastern subtropical Pacific microbial communities. ISME J. 9, 1076–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (1998). Profile hidden Markov models. Bioinformatics 14, 755–763. [DOI] [PubMed] [Google Scholar]
- Esparza-Gordillo J, Weidinger S, Fölster-Holst R, Bauerfeind A, Ruschendorf F, Patone G, Rohde K, Marenholz I, Schulz F, Kerscher T, et al. (2009). A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat. Genet 41, 596–601. [DOI] [PubMed] [Google Scholar]
- Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, and Knight R (2018). Current understanding of the human microbiome. Nat. Med 24, 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong JQ, Lin L, Lin T, Hao F, Zeng FQ, Bi ZG, Yi D, and Zhao B (2006). Skin colonization by Staphylococcus aureus in patients with eczema and atopic dermatitis and relevant combined topical therapy: a double-blind multicentre randomized controlled trial. Br. J. Dermatol 155, 680–687. [DOI] [PubMed] [Google Scholar]
- Helmink BA, Khan MAW, Hermann A, Gopalakrishnan V, and Wargo JA (2019). The microbiome, cancer, and cancer therapy. Nat. Med 25, 377–388. [DOI] [PubMed] [Google Scholar]
- Ikeda Y, Ohara-Nemoto Y, Kimura S, Ishibashi K, and Kikuchi K (2004). PCR-based identification of Staphylococcus epidermidis targeting gseA encoding the glutamic-acid-specific protease. Can. J. Microbiol 50, 493–498. [DOI] [PubMed] [Google Scholar]
- Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, and Mizunoe Y (2010). Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 465, 346–349. [DOI] [PubMed] [Google Scholar]
- Kho ZY, and Lal SK (2018). The human gut microbiome—a potential controller of wellness and disease. Front. Microbiol 9, 1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Hahn AS, Wu S, Hanson NW, Konwar KM, and Hallam SJ (2015). FragGeneScan-Plus for Scalable High-Throughput Short-Read Open Reading Frame Prediction (IEEE; ). [Google Scholar]
- Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, Kong HH, Amagai M, and Nagao K (2015). Dysbiosis and Staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity 42, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, et al. ; NISC Comparative Sequence Program (2012). Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 22, 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR, Hooper LV, Schmidt RR, von Aulock S, et al. (2009). Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med 15, 1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, Di Nardo A, and Gallo RL (2010). Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J. Invest. Dermatol 130, 2211–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larrick JW, Hirata M, Zhong J, and Wright SC (1995). Anti-microbial activity of human CAP18 peptides. Immunotechnology 1, 65–72. [DOI] [PubMed] [Google Scholar]
- Le KY, Park MD, and Otto M (2018). Immune evasion mechanisms of Staphylococcus epidermidis biofilm infection. Front. Microbiol. 9, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyden JJ, Marples RR, and Kligman AM (1974). Staphylococcus aureus in the lesions of atopic dermatitis. Br. J. Dermatol 90, 525–530. [DOI] [PubMed] [Google Scholar]
- Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, Sen SK, Shaik J, Smelkinson M, Tamoutounour S, et al. (2018). Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 172, 784–796.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, Kao T, Lee SK, Cai SS, Miller RJ, et al. (2017). Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe 22, 653–666.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Xu Y, Yestrepsky BD, Sorenson RJ, Chen M, Larsen SD, and Sun H (2012). Novel inhibitors of Staphylococcus aureus virulence gene expression and biofilm formation. PLoS ONE 7, e47255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootz JM, Benson MA, Heim CE, Crosby HA, Kavanaugh JS, Dunman PM, Kielian T, Torres VJ, and Horswill AR (2015). Rot is a key regulator of Staphylococcus aureus biofilm formation. Mol. Microbiol 96,388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morar N, Cookson WO, Harper JI, and Moffatt MF (2007). Filaggrin mutations in children with severe atopic dermatitis. J. Invest. Dermatol 127, 1667–1672. [DOI] [PubMed] [Google Scholar]
- Morizane S, Yamasaki K, Kabigting FD, and Gallo RL (2010). Kallikrein expression and cathelicidin processing are independently controlled in keratinocytes by calcium, vitamin D(3), and retinoic acid. J. Invest. Dermatol 130, 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, Conlan S, Himmelfarb S, Byrd AL, Deming C, et al. (2015). Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 520, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, Saijo S, Inohara N, Otto M, Matsue H, et al. (2017). Staphylococcus aureus virulent PSMalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 22, 667–677.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, and Gallo RL (2019). The role of the skin microbiome in atopic dermatitis. Ann. Allergy Asthma Immunol 122, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, and Gallo RL (2013). The microbiome extends to subepidermal compartments of normal skin. Nat. Commun 4, 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, Hata TR, and Gallo RL (2016). Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J. Invest. Dermatol 136, 2192–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, Shafiq F, Kotol PF, Bouslimani A, Melnik AV, et al. (2017). Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci. Transl. Med 9, eaah4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsuji T, Chen TH, Butcher AM, Trzoss LL, Nam SJ, Shirakawa KT, Zhou W, Oh J, Otto M, Fenical W, et al. (2018). A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci. Adv 4, eaao4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, and Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noore J, Noore A, and Li B (2013). Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 57, 1283–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, and Pevzner PA (2017). meta-SPAdes: a new versatile metagenomic assembler. Genome Res. 27,824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J, Conlan S, Polley EC, Segre JA, and Kong HH (2012). Shifts in human skin and nares microbiota of healthy children and adults. Genome Med. 4, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksy A, Golonka E, Bańbuła A, Szmyd G, Moon J, Kubica M, Greenbaum D, Bogyo M, Foster TJ, Travis J, and Potempa J (2004). Growth phase-dependent production of a cell wall-associated elastinolytic cysteine proteinase by Staphylococcus epidermidis. Biol. Chem 385, 525–535. [DOI] [PubMed] [Google Scholar]
- Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Büttner H, Dunman PM, Rohde H, Cech NB, Fey PD, and Horswill AR (2014). Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J. Bacteriol 196, 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M (2009). Staphylococcus epidermidis—the ‘accidental’ pathogen. Nat. Rev. Microbiol 7, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, Luger T, and Irvine AD (2019). The microbiome in patients with atopic dermatitis. J. Allergy Clin. Immunol 143, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paule SM, Pasquariello AC, Hacek DM, Fisher AG, Thomson RB Jr., Kaul KL, and Peterson LR (2004). Direct detection of Staphylococcus aureus from adult and neonate nasal swab specimens using real-time polymerase chain reaction. J. Mol. Diagn 6, 191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Da F, Fisher EL, Tan DC, Nguyen TH, Fu CL, Tan VY, McCausland JW, Sturdevant DE, Joo HS, et al. (2017). Toxin mediates sepsis caused by methicillin-resistant Staphylococcus epidermidis. PLoS Pathog. 13, e1006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queck SY, Khan BA, Wang R, Bach TH, Kretschmer D, Chen L, Kreiswirth BN, Peschel A, Deleo FR, and Otto M (2009). Mobile genetic element-encoded cytolysin connects virulence to methicillin resistance in MRSA. PLoS Pathog. 5, e1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner ED, Hartl D, Rylaarsdam S, Young ML, Monaco-Shawver L, Kleiner G, Markert ML, Stiehm ER, Belohradsky BH, Upton MP, et al. (2009). Comèl-Netherton syndrome defined as primary immunodeficiency. J. Allergy Clin. Immunol 124, 536–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt TSB, Raes J, and Bork P (2018). The human gut microbiome: from association to modulation. Cell 172, 1198–1215. [DOI] [PubMed] [Google Scholar]
- Sieprawska-Lupa M, Mydel P, Krawczyk K, Wójcik K, Puklo M, Lupa B, Suder P, Silberring J, Reed M, Pohl J, et al. (2004). Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother 48, 4673–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śmigiel R, Królak-Olejnik B, Śniegórska D, Rozensztrauch A, Szafrańska A, Sasiadek MM, and Wertheim-Tysarowska K (2017). Is c.1431-12G>A a common European mutation of SPINK5? Report of a patient with Netherton Syndrome. Balkan J. Med. Genet 19, 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonesson A, Przybyszewska K, Eriksson S, Mörgelin M, Kjellström S, Davies J, Potempa J, and Schmidtchen A (2017). Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci. Rep 7, 8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacy A, and Belkaid Y (2019). Microbial guardians of skin health. Science 363,227–228. [DOI] [PubMed] [Google Scholar]
- Sully EK, Malachowa N, Elmore BO, Alexander SM, Femling JK, Gray BM, DeLeo FR, Otto M, Cheung AL, Edwards BS, et al. (2014). Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 10, e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uçkay I, Pittet D, Vaudaux P, Sax H, Lew D, and Waldvogel F (2009). Foreign body infections due to Staphylococcus epidermidis. Ann. Med 41, 109–119. [DOI] [PubMed] [Google Scholar]
- Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, and Gallo RL (2017). Staphylococcus aureus induces increased serine protease activity in keratinocytes. J. Invest. Dermatol 137, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, Sanford JA, O’Neill AM, Liggins MC, Nakatsuji T, et al. (2019). Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci. Transl. Med 11, eaat8329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki K, Schauber J, Coda A, Lin H, Dorschner RA, Schechter NM, Bonnart C, Descargues P, Hovnanian A, and Gallo RL (2006). Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 20, 2068–2080. [DOI] [PubMed] [Google Scholar]
- Zhvania P, Hoyle NS, Nadareishvili L, Nizharadze D, and Kutateladze M (2017). Phage therapy in a 16-year-old boy with Netherton syndrome. Front. Med. (Lausanne) 4, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the Netherton syndrome high-throughput shotgun sequencing metagenomic datasets generated in the course of this project have been deposited at the National Center for Biotechnology Information Sequence Read Archive under BioProject ID: PRJNA551026. Any further details regarding these datasets will be made available upon request.