

SUMMARY
Receptor-interacting protein kinase 1 (RIPK1) and 3 (RIPK3) are well known for their capacity to drive necroptosis via mixed-lineage kinase-like domain (MLKL). Recently, RIPK1/3 kinase activity has been shown to drive inflammation via activation of MAPK signaling. However, the regulatory mechanisms underlying this kinase-dependent cytokine production remain poorly understood. In the present study, we establish that the kinase activity of RIPK1/3 regulates cytokine translation in mouse and human macrophages. Furthermore, we show that this inflammatory response is downregulated by type I interferon (IFN) signaling, independent of type I IFN-promoted cell death. Specifically, low-level constitutive IFN signaling attenuates RIPK-driven activation of cap-dependent translation initiation pathway components AKT, mTORC1, 4E-BP and eIF4E, while promoting RIPK-dependent cell death. Altogether, these data characterize constitutive IFN signaling as a regulator of RIPK-dependent inflammation and establish cap-dependent translation as a crucial checkpoint in the regulation of cytokine production.
In Brief
Balancing inflammatory responses is critical for host survival. Muendlein et al. show that constitutive type I IFN signaling inhibits translation machinery activated downstream of the kinase activity of RIPK1/3, preventing the production of a subset of inflammatory cytokines. This work identifies cap-dependent translation as a checkpoint in regulation of RIPK1/3-kinase-dependent inflammation.
Graphical Abstract
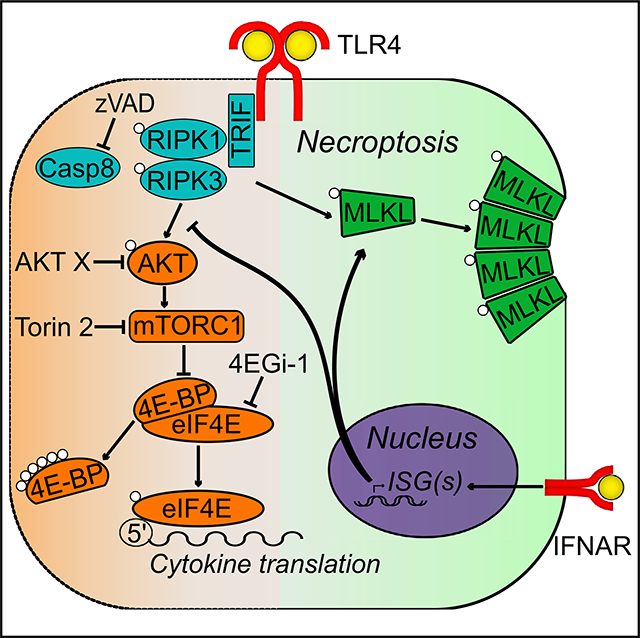
INTRODUCTION
Cell death and inflammation are two largely interconnected processes crucial for host defense against pathogen infection, as elimination of infected cells limits pathogen replication and promotes host survival. Often, receptor-interacting protein kinase 1 (RIPK1) and 3 (RIPK3) stand at the crux of these two processes because of their role in modulating inflammatory signaling and a form of cell death known as necroptosis. Studied comprehensibly downstream of TNF receptor 1 (TNF-R1), RIPK1 acts as a scaffold in a kinase-independent manner to drive NF-κB- and MAPK-mediated activation of inflammatory programs (Hsu et al., 1996; Ting et al., 1996; Kim et al., 2001; Micheau and Tschopp, 2003; Lee et al., 2004; Orozco and Oberst, 2017).
However, in cases in which caspase-8 (CASP8) is inhibited such as with pan-caspase inhibitor z-VAD-FMK (zVAD), TNF-R1 ligation results in RIPK1 and RIPK3 phosphorylation and kinase activation (Cho et al., 2009; He et al., 2009; Pasparakis and Vandenabeele, 2015; Silke et al., 2015). RIPK3 phosphorylates the pseudokinase mixed-lineage kinase-like domain (MLKL), inducing MLKL dimerization and oligomer formation. Translocation of MLKL oligomers to the plasma membrane results in cell death via pore formation and membrane disruption, accompanied by the release of cytosolic components (Sun et al., 2012; Murphy et al., 2013; Linkermann and Green, 2014; Wang et al., 2014).
In addition to downstream of death receptor signaling, necroptosis can be induced in various cell types by signaling through Toll-like receptors (TLRs) such as TLR3 and TLR4. In the case of TLR stimulation by double-stranded RNA (dsRNA) (TLR3) or bacterial lipopolysaccharide (LPS) (TLR4), the RHIM (RIP homotypic interacting motif) domain-containing endosomal adaptor TRIF (TIR domain-containing adaptor-inducing interferon [IFN]-β) is engaged to recruit RIPK1/3 to drive necroptosis. Because of the presence of a RHIM domain in TRIF, RIPK3 can also be directly recruited, leading to RIPK1-independent necroptosis (He et al., 2011; Kaiser et al., 2013; Schworer et al., 2014).
Similarly, the cytosolic DNA sensor and RHIM domain-containing protein ZBP1 (Z-DNA-binding protein 1) is capable of driving necroptosis and promoting host survival in response to viral infection, through interactions with RIPK3, independent of RIPK1 (Upton et al., 2012, 2017; Kuriakose et al., 2016; Kuriakose and Kanneganti, 2018). In addition to its role downstream of viral infection, a plethora of bacteria, including Salmonella enterica, Staphylococcus aureus, Staphylococcus pneumonia, Serratia marcescens, Listeria monocytogenes, and uropathogenic Escherichia coli, induce necroptosis (Robinson et al., 2012; González-Juarbe et al., 2015; Ahn and Prince, 2017). Thus, necroptosis is an important aspect of the host response to viral and bacterial assaults. Indeed, to thwart host responses, numerous viruses and bacteria have evolved mechanisms to inhibit necroptosis (Upton et al., 2008; Li et al., 2013; Pearson et al., 2017).
In addition to induction of necroptosis, pathogenic infection also drives the production of type I IFNs downstream of TRIF and cGAS/STING activation by pathogenic components (Vilcek, 2006; Sun et al., 2013; Wu et al., 2013). Type I IFNs are a family of proteins including IFNβ that bind the transmembrane IFNα receptor (IFNAR) (Thomas et al., 2011). Stimulation of IFNAR leads to activation of Janus kinase (JAK) and the transcription factors STAT1/2, which regulate a class of IFN-stimulated genes (ISGs) that serve many antiviral and antibacterial functions (Battistini, 2009; Schneider et al., 2014; Mostafavi et al., 2016). IFN signaling has been shown to regulate necroptosis and regulate the levels and activation of the key necroptotic effectors MLKL, RIPK3, and ZBP1(Fu et al., 1999; Thapa et al., 2013; McComb et al., 2014; Sarhan et al., 2018; Ingram et al., 2019). In the absence of activation or pathogen infection, sufficient levels of these effectors are sustained by so-called constitutive (or tonic) IFN signaling, the origins of which remain poorly understood (Vogel et al., 1979; Yoshida et al., 2005; Abt et al., 2012; Ahn et al., 2012). Therefore, the processes of necroptosis and IFN signaling represent inseparable arms of the host defense mechanism against infection.
To complicate matters further, in addition to driving necroptosis, the kinase activity of RIPK1/3 has been recently reported to promote inflammatory cytokine transcription in response to TNF/zVAD and LPS/zVAD stimulation (Wallach et al., 2011; Christofferson et al., 2012; Najjar et al., 2016; Shutinoski et al., 2016). In extension of these findings, in the present study, we show a role for the kinase activity of RIPK1/3 in promoting cap-dependent cytokine translation in response to LPS/zVAD. Additionally, we identify a role for constitutive IFN signaling in the inhibition of RIPK1/3 kinase-dependent cytokine translation. Specifically, low constitutive IFN signaling leads to increased activation of mammalian target of rapamycin complex 1 (mTORC1) pathway components, resulting in hyper-phosphorylation of eIF4E-binding proteins (4E-BP) and increased eukaryotic translation initiation factor 4E (eIF4E)-mediated cytokine translation.
RESULTS
RIPK Promotes Cytokine mRNA and Protein Production in LPS-Activated MOLF Macrophages
Inhibition of CASP8 with the pan-caspase inhibitor zVAD in LPS-activated macrophages (LPS/zVAD [LZ]) leads to the kinase activation of RIPK1/3 and the induction of necroptosis (He et al., 2011). We have recently reported that in addition to inducing cell death, LZ treatment also leads to the RIPK1/3-kinase-dependent transcriptional upregulation of cytokines, including IFNβ, downstream of TRIF in C57BL/6J (B6) BMDMs (bone marrow-derived macrophages) (Najjar et al., 2016; Saleh et al., 2017).
We have previously taken advantage of the genetic diversity of wild-derived MOLF/Ei (MOLF) mice for mapping and identification of genes conferring resistance to necroptosis in MOLF macrophages (Schworer et al., 2014; Sarhan et al., 2018). Here, to extend our line of inquiry to the genetic level, we compared LZ-driven inflammatory responses in BMDMs from MOLF and B6 mice using TNF-α and CXCL-1 as a readout. We observed that LZ treatment resulted in enhanced upregulation of cytokines at the mRNA level in B6 and MOLF BMDMs and peritoneal macrophages (Figures 1A and S1A), which was completely reversed with the addition of an inhibitor of RIPK1 kinase activity, Nec-1s (LZNs) (Figures 1A and S1A). BMDMs from mice lacking RIPK1 or RIPK3 kinase activity (RIP1/3 Ki) failed to upregulate cytokine mRNA in response to LZ, further confirming the requirement for RIPK kinase activity in the inflammatory phenotype (Figure 1A). Interestingly, LZ-induced upregulation of cytokine mRNA did not correlate with increased protein production in B6, as only MOLF BMDMs and peritoneal macrophages responded to LZ treatment with robust, Nec-1s-reversible production of cytokines at the protein level (Figures 1B and S1B). We have termed this MOLF-specific increase in cytokine and chemokine production upon LZ treatment “RIPK-kinase-dependent inflammation” or the “RIPK-kinase-dependent inflammatory phenotype.”
Figure 1. RIPK Promotes Cytokine mRNA and Protein Production in LPS-Activated MOLF Macrophages.

(A and B) TNF-α and CXCL-1 mRNA (A) and protein (B) levels in unstimulated, LPS-, LZ-, LZN-, Z-, and N-stimulated B6, MOLF, RIP1 Ki, RIP3 Ki, and Mlkl−/− BMDMs.
(C) Cell death as measured by propidium iodide incorporation over 5 h in B6, MOLF, RIP1 Ki, and Mlkl−/− BMDMs.
(D) RIP1, total, and phospho-MLKL levels in B6 and MOLF BMDMs stimulated as indicated with LZ ± 5 IU IFNβ priming overnight.
(E) IFNb, ISG15, and IRF7 mRNA levels as a readout for constitutive IFN signaling in B6 and MOLF, BMDMs, I.P. macrophages, and blood.
(F and G) Total STAT1 (F) and ISG15 and IRF7 mRNA (G) levels as a readout of reconstitution of constitutive IFN signaling in B6 and MOLF BMDMs stimulated as indicated with LZ ± 5 IU IFNβ priming overnight.
In all panels, BMDMs were stimulated with LPS, LPS/zVAD (LZ), LPS/zVAD/Nec-1 s (LZNs), zVAD (Z), or Nec-1 s (Ns) as indicated. ELISA and qPCR data are shown as ±SD from three independent experiments compared using two-way ANOVA: n.s. (p > 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001. Kinetic cell death and western blots are representative of three or more independent experiments. Kinetic data are presented as the mean ± SD of triplicate wells.
See also Figure S1.
MOLF BMDMs are resistant to LZ-induced necroptosis compared with B6 (Figure 1C), because of a hypomorphic allele of Sting (stimulator of IFN genes), which regulates MLKL, an important effector of necroptosis, as well as a number of other ISGs (Surpris et al., 2015; Sarhan et al., 2018). Although total MLKL levels were reduced in MOLF macrophages, MLKL phosphorylation and upstream RIPK1 phosphorylation were uninhibited, indicating that RIPK1 kinase activity was functional in MOLF macrophages (Figure 1D) (Sarhan et al., 2018). To determine if resistance to necroptosis in MOLF permitted increased cytokine production at the protein level, we investigated whether BMDMs deficient for MLKL were capable of producing cytokines in a RIPK-kinase-dependent manner. As expected, Mlkl−/− BMDMs were completely resistant to LZ-induced necroptosis (Figure 1C) but nevertheless recapitulated the inflammatory phenotype of B6 BMDMs (Figures 1A and 1B), suggesting that the ability of MOLF BMDMs to respond to LZ with robust cytokine production was not due to their relative resistance to necroptotic cell death.
Because of their hypomorphic allele of Sting, MOLF mice lack constitutive (tonic) type I IFN signaling, exemplified by low baseline levels of IFNb, ISG15, and IRF7 mRNAs detected in BMDMs, peritoneal macrophages, and blood (Figure 1E) and low levels of total STAT1 compared with B6 (Figure 1F). However, MOLF macrophages were responsive to IFN signaling, as STAT1 and ISG levels were rescued with low-dose overnight priming with recombinant IFNβ (Figures 1F and 1G). Therefore, we hypothesized that constitutive IFN signaling may play a role in regulating RIPK-kinase-dependent cytokine production, in addition to its role as a regulator of necroptosis.
Constitutive IFN Signaling Downregulates RIPK-Dependent Inflammation
Accordingly, we examined RIPK-dependent inflammation in two mouse models deficient for IFN signaling, Ifnar−/− and Ifnb−/− BMDMs. Both Ifnar−/− and Ifnb−/− exhibited increased RIPK-kinase-dependent cytokine and chemokine production at both the protein and mRNA level (Figures 2A and S2A). Additionally, inhibition of IFN signaling with IFNAR-blocking antibody or via the JAK1/2 inhibitors ruxolitinib or baricitinib was sufficient to induce RIPK-kinase-dependent inflammation in B6 BMDMs (Figure S2B). Importantly, the addition of low-dose exogenous IFNβ (5 IU/mL) completely abrogated RIPK-dependent cytokine protein production in Ifnb−/− but not in Ifnar−/− BMDMs, confirming the specificity of the inhibitory effect of IFN signaling on RIPK-kinase-dependent inflammation (Figure 2B). However, the addition of exogenous IFN had no effect on cytokine mRNA levels, hinting at regulation at the level of translation (Figure S2C). Similar to MOLF, Ifnar−/− and Ifnb−/− BMDMs exhibited resistance to LZ-induced necroptosis, which could be abrogated with the addition of low-dose exogenous IFNβ (Figure S2D). As we have shown previously (Sarhan et al., 2018), Ifnb−/− BMDMs displayed decreased total and phosphorylated MLKL but uninhibited RIPK1 phosphorylation (Figure S2E). Because IFNβ priming promotes necroptosis in Ifnb−/− BMDMs (Figures 2C and S2D), we separated the effects of IFN signaling on cell death and inflammation by generating Mlkl−/− Ifnb−/− double-knockout mice. BMDMs from these mice were resistant to necroptotic cell death, independent of IFNβ priming (Figure 2C). Nevertheless, we observed RIPK-dependent inflammation in these cells (Figure 2D). Similar to Ifnb−/−, this inflammation could be abrogated with low-dose IFNβ priming. Together these data show that constitutive IFN signaling is involved in the translational control of RIPK-kinase-dependent inflammation, independent of necroptosis.
Figure 2. Constitutive IFN Signaling Downregulates RIPK-Dependent Inflammation.

(A) TNF-α and CXCL-1 protein levels in unstimulated, LPS-, LZ-, LZN-, Z-, and N-stimulated B6, Ifnar−/−, and Ifnb−/− BMDMs.
(B) TNF-α and CXCL-1 protein levels after indicated stimulations ± 5 IU IFNβ overnight priming in B6, Ifnar−/−, and Ifnb−/− BMDMs.
(C) Cell death as measured by propidium iodide incorporation over 6 h in B6, Ifnb−/−, Mlkl−/− Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs stimulated with LZ ± 5 IU IFNβ priming overnight.
(D) TNF-α and CXCL-1 protein levels after indicated stimulations ± 5 IU IFNβ priming overnight in B6, Ifnb−/−, Mlkl−/− Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs.
In all panels, BMDMs were stimulated with LPS, LZ, LZNs, Z, or Ns as indicated. ELISA and qPCR data are shown as ±SD from three independent experiments compared using two-way ANOVA: n.s. (p > 0.05), *p < 0.05, and ****p < 0.0001. Kinetic cell death experiments are representative of three or more independent experiments and presented as the mean ± SD of triplicate wells.
See also Figures S2 and S3.
To further understand the mechanistic link between RIPK-dependent inflammation and constitutive IFN signaling, we crossed RIP1 Ki mice with Ifnb−/− mice to produce a progeny homozygous for Rip1K45A/K45A and Ifnb−/−. As expected, the BMDMs from these mice were resistant to LZ-induced necroptosis and failed to induce RIPK-dependent inflammation at the protein level (Figures 2C and 2D). Importantly, addition of exogenous IFNβ had no effect on the phenotype, suggesting that IFN signaling exerts its inhibitory effect on RIPK-dependent inflammation at a level below RIPK1. These data provide support for a model in which, in the absence of constitutive IFN signaling and CASP8 activity, the kinase activity of RIPK1 contributes to the transcriptional and translational activation of inflammatory cytokines in response to LPS. The inhibitory effect of the IFN signaling on RIPK-kinase-dependent inflammation is exerted at the level of translation and below RIPK1 activity.
To extend our findings to TNFR- and TLR3-mediated necroptosis, we stimulated B6 and Ifnb−/− BMDMs with TNF and zVAD (TZ) or polyinosine-polycytidylic acid (pI:C) and zVAD (PZ) respectively. Both TZ and PZ treatment induced RIPK-kinase-dependent TNF protein production in Ifnb−/−-deficient but not B6 BMDMs, indicating that the inflammatory phenotype was not specific to TLR4 (Figure S3A). Similarly to LZ treatment, TZ and PZ increased TNF mRNA production that was abrogated in the absence of RIPK1 kinase activity (Figure S3B).
IFN Signaling and Translation Initiation Pathways Regulate RIPK-Dependent Inflammation
To identify the global effect of IFN signaling on cytokine production downstream of RIPK1, we compared the upregulation of mRNA in response to either LPS or LZ in B6, Ifnb−/−, and RIP1 Ki BMDMs. The effect of the RIPK-kinase-dependent inflammation was calculated as log2(LZ/LPS). The RNA sequencing (RNA-seq) analysis was complemented with a protein cytokine array comparison between these strains, and cytokines were grouped as either upregulated or downregulated at the mRNA level after LZ compared with LPS treatment in B6 (Figure 3A). RNA-seq data confirmed transcriptional upregulation of a subset of cytokines including TNF and CXCL-1 in both B6 and Ifnb−/−, but not in RIP1 Ki BMDMs (Figure 3B). In contrast, only Ifnb−/− BMDMs were able to upregulate translation of the same subset of inflammatory cytokines, indicating an inhibitory effect of IFN signaling on the machinery regulating the translation of these cytokines (Figure 3C). Finally, there was no global defect on protein synthesis as evaluated via puromycin incorporation, which suggested that the inhibitory effect of IFN signaling specifically affects translation of the identified inflammatory cytokines that are dependent on RIPK kinase activity (Figure 3D). To further understand why Ifnb−/− but not B6 BMDMs produce RIPK-kinase-dependent inflammatory cytokine protein, we identified the pathways that were specifically upregulated in LZ-stimulated Ifnb−/− BMDMs (Figure 3E). Interestingly, the most activated pathways included eIF2, eIF4, and mTOR signaling pathways, which may point to a potential mechanism of regulation at the level of translation.
Figure 3. IFN Signaling and Translation Initiation Pathways Regulate RIPK-Dependent Inflammation.

(A) Schematic of sorting process used to compare RNA sequencing and cytokine array results from LPS- and LZ-stimulated B6, Ifnb−/−, and RIP1 Ki BMDMs
(B and C) RNA (log2 FPKM [fragments per kilobase of transcript per million mapped reads] LZ/LPS) (B) and protein (log2 signal intensity LZ/LPS) (C) for cytokines upregulated and downregulated in LZ compared with LPS treatment at the mRNA level for B6, Ifnb−/−, and RIP1 Ki BMDMs.
(D) Extent of global translation as measured by puromycin incorporation in unstimulated, LPS- and LZ-stimulated B6, Ifnb−/−, and RIP1 Ki BMDMs.
(E) Ingenuity Pathway Analysis of RNA sequencing data depicting top ten upregulated pathways for LZ-stimulated Ifnb−/− macrophages.
Statistical significance was determined using Student’s t test: n.s. (p > 0.05) and ****p < 0.0001.
IFN Signaling Antagonizes RIPK in the Activation of eIF4E and mTORC1-Mediated Translation
Protein kinase R (PKR) is an ISG that has been shown to phosphorylate the translation initiation factor eIF2α, leading to inhibition of translation in conditions of stress or infection (Dalet et al., 2015). We identified eIF2α signaling as a highly upregulated pathway in LZ-treated Ifnb−/− BMDMs (Figure 3E), and PKR interacts with RIPK1, leading to necroptosis (Thapa et al., 2013), thus suggesting that PKR might be involved in the inhibition of translation in constitutive IFN-sufficient B6 BMDMs. However, we were unable to detect any RIPK or IFN-dependent phosphorylation of eIF2α, and inhibition of PKR with imidazole-oxindole PKR inhibitor C16 did not result in an increase in RIPK-kinase-dependent inflammation in B6 BMDMs (Figures S4A and S4B).
The major rate-limiting step of protein synthesis is translation initiation, in which the eukaryotic initiation factor 4F (eIF4F) complex assembles at the 5′ end of the mRNA transcript, a process driven in part by cap-binding protein eIF4E (Gingras et al., 1999; Poulin and Sonenberg, 2013). To look for the role of eIF4E in RIPK-kinase-dependent inflammation, we compared the effect of inhibition of eIF4E on RIPK-kinase-dependent inflammation in Ifnb−/−, B6, and RIP1 Ki Ifnb−/− BMDMs using the eIF4E inhibitor 4EGi-1(Moerke et al., 2007). 4EGi-1 showed no effect on TNF and CXCL-1 production in LZ-treated B6 and RIP1 Ki Ifnb−/− BMDMs but abrogated the LZ-induced inflammatory phenotype of Ifnb−/− BMDMs (Figure 4A). Importantly, treatment with 4EGi-1 had no effect on RIPK-kinase-dependent mRNA levels (Figure S4C).
Figure 4. IFN Signaling Antagonizes RIPK in the Activation of eIF4E- and mTORC1-Mediated Translation.

(A) TNF-α and CXCL-1 protein levels after indicated stimulations ± treatment with eIF4E inhibitor 4EGi-1 in B6, Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs.
(B) p-eIF4E and total 4E-BP levels in B6, Ifnb−/−, and RIP1 Ki BMDMs stimulated as indicated ± 5 IU IFNβ priming overnight.
(C) TNF-α and CXCL-1 protein levels after indicated stimulations ± treatment with mTORC1 inhibitor Torin 2 in B6, Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs.
(D and E) Cell death as measured by propidium iodide incorporation over 6 h in B6, Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs stimulated with LZ ± treatment with 4EGi-1 (D) or Torin 2 (E).
(F) TNF-α and CXCL-1 protein levels after indicated stimulations ± overnight treatment with IFNAR-blocking antibody (αIFNAR) in Akt1−/− and wild-type (WT) littermate BMDMs.
In all panels, BMDMs were stimulated with LPS, LZ, LZNs, Z, Ns, or LZN as indicated. ELISA and qPCR data are shown as ±SD from three independent experiments compared using two-way ANOVA: n.s. (p > 0.05), *p < 0.05, and ****p < 0.0001. Western blot and kinetic cell death experiments are representative of three or more independent experiments. Kinetic data are presented as the mean ± SD of triplicate wells.
See also Figures S4 and S5.
To look for a specific eIF4E activation event that might be affected by IFN signaling, we investigated phosphorylation of eIF4E on Ser 209, which has been associated with an increased rate of translation (Poulin and Sonenberg, 2013). eIF4E phosphorylation was present at similar levels in LPS-, LZ-, and LZN-activated B6 BMDMs (Figure 4B). In contrast, IFN deficiency resulted in significantly enhanced LZ-specific phosphorylation, which was attenuated upon treatment with exogenous IFNβ, as well with the RIP1 kinase inhibitor Nec-1 (Figure 4B). In further support of the RIPK1-kinase-specific effect of eIF4E modifications, all enhanced eIF4E phosphorylation was attenuated in RIP1 Ki BMDMs (Figure 4B). This IFN-dependent eIF4E phosphorylation, which occurred only in the context of RIP1 kinase active macrophages, further specified the role of IFN signaling in RIPK-kinase-dependent translation.
Working upstream in the pathway, eIF4E activity is controlled in part by the eIF4E-binding proteins (4E-BPs), which inhibit eIF4E-mediated translation (Gingras et al., 1999). The binding capabilities of 4E-BPs are regulated by phosphorylation at six Ser/Thr residues, with hyper-phosphorylation resulting in weaker binding and greater eIF4E activation (Gingras et al., 2001; Moerke et al., 2007; Spriggs et al., 2010). Therefore, we investigated the levels of total 4E-BP1 in B6 and Ifnb−/− BMDMs. We observed an increase in the phosphorylation of 4E-BP1 (γ form) in Ifnb−/− but not B6 LZ-treated BMDMs, which was abrogated by the addition of 5 IU/mL IFNβ priming (Figure 4B). Furthermore, phosphorylation of 4E-BP1 in RIP1 Ki BMDMs was completely unchanged by LZ treatment or addition of IFNβ (Figure 4B). Therefore, this RIPK-kinase-dependent increase in phosphorylation in Ifnb−/− BMDMs but not in B6 BMDMs indicated an inhibition of the cap-dependent translation inhibitor 4E-BP1 and may explain the increased cytokine translation observed under these conditions. Conversely, the levels of the hypo-phosphorylated (α form) of 4E-BP1 that were present in LZ-treated B6 BMDMs were absent in Ifnb−/− BMDMs but were rescued with IFNβ priming (Figure 4B). These data suggest that constitutive IFN inhibits RIPK-kinase-driven translation of cytokines via abrogation of 4E-BP1 inactivation.
The eIF4E inhibitor 4E-BP1 is in turn regulated by mTORC1, which phosphorylates and inactivates 4E-BP1 (Brunn et al., 1997; Gingras et al., 1998). Given that mTOR signaling was one of the top upregulated pathways identified in our pathway analysis (Figure 3E), we examined whether mTORC1 facilitates RIPK-kinase-driven inflammation. Accordingly, we treated B6, Ifnb−/−, and RIP1 Ki Ifnb−/− BMDMs with the mTORC1 inhibitor Torin 2 (Figure 4C). Treatment with Torin 2 abrogated the LZ-induced cytokine response in Ifnb−/− at the protein level (Figure 4C). Similar to the eIF4E inhibition experiments described above, mTORC1 inhibition had no effect on cytokine mRNA levels, or on the cytokine protein production in B6 and RIP1 Ki Ifnb−/− BMDMs (Figures S4D and 4C). Furthermore, treatment with Torin 2 resulted in the presence of only the α form of 4E-BP1 in B6, Ifnb−/−, and RIP1 Ki BMDMs, as well as the elimination eIF4E hyper-phosphorylation in LZ-treated Ifnb−/− BMDMs (Figure S4E).
Treatment of Ifnb−/− but not RIP1 Ki Ifnb−/− BMDMs with 4EGi-1 and Torin 2 enhanced LZ-induced necroptosis to levels near B6, hinting toward a potential overlap in the pathways regulating RIPK-dependent pro-inflammatory cytokine signaling and cell death (Figures 4D and 4E).
The serine/threonine kinase AKT is an upstream positive regulator of mTORC1 and has also been shown to be involved in 4E-BP phosphorylation and interaction with RIPK1 (McNamara et al., 2013; Liu et al., 2014; Gingras et al., 1998). Accordingly, we tested if inhibition of AKT with pan AKT inhibitor AKT X affects RIPK-kinase-dependent cytokine protein production in Ifnb−/− BMDMs (Figure S5A). AKT inhibition eliminated RIPK-kinase-dependent cytokine production at the protein level in Ifnb−/−, while having no effect in B6, or RIP1 Ki Ifnb−/−, or on cytokine mRNA levels (Figures S5A and S5B). Similar to 4EGi-1 and Torin 2 treatment, treatment with AKT X enhanced LZ-induced necroptosis in Ifnb−/− but not RIP1 Ki Ifnb−/− BMDMs to levels near B6 (Figure S5C).
To extend our investigation, we compared the effect of blocking IFN signaling in B6 and AKT-deficient BMDMs. As anticipated, inhibition of IFN signaling with IFNAR-blocking antibodies resulted in a significant increase in TNF and CXCL-1 production in LZ-induced B6 BMDMs (Figure 4F). In contrast, inhibition of constitutive IFN signaling by IFNAR-blocking antibody pretreatment in Akt1−/− BMDMs failed to produce enhanced RIPK-kinase-dependent cytokine protein production but exhibited enhanced RIPK-kinase-dependent cytokine transcript levels, implicating AKT1 as a translational regulator of RIPK-kinase-dependent inflammation (Figures 4F and S5D). Interestingly, RIPK-kinase-dependent cytokine production was not attenuated in Akt3−/− BMDMs, indicating a role for AKT1 specifically in the translation of RIPK-kinase-dependent cytokine transcripts (Figure S5E).
On the basis of these results, we propose that constitutive IFN signaling inhibits the RIPK-activated AKT1/mTORC1 pathway, preventing 4E-BP phosphorylation and thus inhibiting RIPK-kinase-dependent cytokine production.
4E-BP Activity Blocks RIPK-Dependent Cytokine Translation
Our data so far show that phosphorylation of 4E-BP plays a pivotal role in RIPK-kinase-dependent cytokine translation, with hypo-phosphorylated 4E-BP serving as a brake against enhanced cytokine production in the presence of constitutive IFN. To mechanistically connect 4E-BP and IFN signaling, we used 4E-BP triple-knockout (TKO) mice deficient for all three isoforms of 4E-BP. Encouragingly, the TKO BMDMs exhibited enhanced RIPK-kinase-dependent cytokine production that, unlike in Ifnb−/− BMDMs, was unaffected by IFNβ priming (Figure 5A). This supports the mechanism that IFN signaling acts upstream of 4E-BP to prevent translation. In further confirmation, we treated 4E-BP TKO BMDMs with the eIF4E inhibitor 4EGi-1 or mTORC1 inhibitor Torin 2 (Figures 5B and 5C). eIF4E inhibition eliminated RIPK-kinase-dependent cytokine production in the 4E-BP TKO, as expected on the basis of the role of eIF4E downstream of 4E-BP (Figure 5B). Conversely, inhibition of mTORC1 had no effect on RIPK-kinase-dependent cytokine production in 4E-BP TKO BMDMs because of the absence of downstream target 4E-BP (Figure 5C). Unlike Ifnb−/−, 4E-BP TKO BMDMs were susceptible to LZ-induced necroptosis, indicating that 4E-BP and downstream cytokine production do not play a role in the regulation of cell death (Figure 5D). So far, we have shown that 4E-BP suppresses RIPK-kinase-dependent inflammation at the level of translation initiation. We have also shown that 4E-BP and mTORC1 are in the same pathway in the context of RIPK-kinase-dependent inflammation, with 4E-BP downstream of mTORC1. Finally, we confirmed the inhibitory role of constitutive IFN signaling with respect to this mTORC1/eIF4E-driven translation pathway.
Figure 5. 4E-BP Activity Blocks RIPK-Dependent Cytokine Translation.

(A) TNF-α and CXCL-1 protein levels after indicated stimulations ± 5 IU IFNβ priming overnight in B6, Ifnb−/−, 4E-BP TKO, and RIP1 Ki Ifnb−/− BMDMs.
(B and C) TNF-α and CXCL-1 protein levels after indicated stimulations ± treatment with 4EGi-1 (B) or Torin-2 (C) in B6, 4E-BP TKO, and RIP1 Ki Ifnb−/− BMDMs.
(D) Cell death as measured by propidium iodide incorporation over 6 h in B6, Ifnb−/−, 4E-BP TKO, and RIP1 Ki BMDMs stimulated with LZ.
In all panels, BMDMs were stimulated with LPS, LZ, LZNs, Z, or Ns as indicated. ELISA data are shown as ±SD from three independent experiments compared using two-way ANOVA: n.s. (p > 0.05) and ****p < 0.0001. Kinetic cell death experiments are representative of three or more independent experiments and presented as the mean ± SD of triplicate wells.
IFN Signaling Regulates RIPK-Dependent Cytokine Translation in Human PBMCs
In confirmation of the relevance of our findings to human biology, we activated human PBMC (peripheral blood mononuclear cell)-derived macrophages with LZ. In comparison with LPS alone, LZ induced cytotoxicity in PBMCs (Figure 6A), which was blocked by the addition of Nec-1s, thus implicating the death as RIPK-mediated necroptosis. In agreement with our findings in mouse BMDMs, the extent of cell death was dependent on IFN signaling, as human cells treated with IFNAR-blocking antibodies were protected from LZ-induced necroptosis (Figure 6A). When we measured the production of CXCL-1 in IFNAR-blocking antibody treated cells, we were intrigued to see substantial levels of CXCL-1 protein produced in a RIPK-kinase-dependent manner, which suggested that IFN signaling plays an inhibitory role similar to that observed in mouse BMDMs (Figure 6B). Furthermore, as in mouse cells, LZ-induced human cells produced elevated CXCL mRNA levels independent of IFN signaling (Figure 6C). Downstream of RIPK1, inhibition of eIF4E and mTORC1 attenuated CXCL-1 protein, but not mRNA production, thus supporting the role for these proteins in promoting RIPK-kinase-dependent inflammation, as we established in the mouse model (Figures 6D and 6E).
Figure 6. IFN Signaling Regulates RIPK-Dependent Cytokine Translation in Human PBMCs.

(A) Cell death as measured by propidium iodide incorporation over 8 h in human PBMC-derived macrophages stimulated as indicated ± overnight treatment with IFNAR-blocking antibody.
(B and C) CXCL-1 protein (B) and mRNA (C) levels in human PBMC-derived macrophages stimulated as indicated ± overnight treatment with IFNAR-blocking antibody
(D and E) CXCL-1 protein (D) and mRNA (E) levels in human PBMC-derived macrophages stimulated as indicated ± overnight treatment with IFNAR-blocking antibody and/or treatment with 4EGi-1 or Torin 2.
In all panels, human PBMC-derived macrophages were stimulated with LPS, LZ, or LZNs as indicated. ELISA and qPCR data are shown as ±SD from three independent experiments compared using two-way ANOVA: n.s. (p > 0.05), **p < 0.01, ***p < 0.001, and ****p < 0.0001. Kinetic cell death experiments are representative of three or more independent experiments and presented as the mean ± SD of triplicate wells.
DISCUSSION
It has been well described that independent of kinase functionality, RIPK1 is capable of acting as a scaffold to drive NF-κB- and MAPK-mediated inflammatory cytokine production downstream of TNF-R1 and TLR/TRIF (Hsu et al., 1996; Ting et al., 1996; Kim et al., 2001; Meylan et al., 2004). However, distinct from RIPK1 scaffold functions, our work shows that the kinase activity of RIPK1/3 contributes to the production of cytokines at the level of translation in response to LPS/zVAD, which is an extension of earlier reports characterizing the importance of the kinase activity of RIPK1/3 in the regulation of cytokine transcription (Najjar et al., 2016). Specifically, we show that RIPK1/3 kinase activity was required for the phosphorylation of 4E-BP and the initiation of cap-dependent translation of a subset of inflammatory cytokines downstream of AKT and mTORC1 activation. Altogether, our work contributes to our understanding of the diversity of effects of RIPK kinase activity in host defense.
This translational effect, driven by RIPK1/3 kinase activity, was inhibited in the context of constitutive (tonic) IFN signaling in B6 BMDMs. In contrast, in BMDMs lacking constitutive IFN, RIPK-kinase-dependent transcriptional upregulation of cytokine mRNAs was complemented with successful translation. In this regard, our study extends upon initial reports characterizing RIPK-kinase-dependent transcriptional cytokine upregulation in response to LPS in B6 (Najjar et al., 2016; Saleh et al., 2017) and explains that the inflammatory response dependent on the kinase activity of RIPK is limited to transcriptional activation in the presence of constitutive IFN signaling. More specifically, constitutive IFN inhibited RIPK1/3 kinase-dependent activation of AKT1 and mTORC1, preventing the phosphorylation 4E-BP and limiting the translation of cytokine mRNAs. However, when constitutive IFN signaling was abrogated, such as in Ifnar−/−, Ifnb−/−, or MOLF macrophages, RIPK1/3 kinase-dependent activation of AKT1 and mTORC1 resulted in phosphorylation of 4E-BP and dissociation of 4E-BP from eIF4E, leading to enhanced cytokine mRNA translation.
Furthermore, constitutive IFN signaling regulates the cells’ decision to undergo death or mount a potent inflammatory response. Lack of IFN signaling not only prevents necroptosis via low MLKL levels (Sarhan et al., 2018) but also promotes a robust inflammatory response dependent on the kinase activity of RIPK1/3 and via activation of the AKT1/mTORC1 signaling axis. This is in contrast with higher IFN signaling in B6 BMDMs, which undergo rapid MLKL-mediated necroptosis and experience diminished translation of cytokines. Although MLKL is one of the key mediators of necroptosis and is maintained by constitutive IFN signaling (Thapa et al., 2013; Sarhan et al., 2018), it is dispensable in the context of the inflammatory response mediated by the kinase activity of RIPK. Therefore, IFN signaling is acting on a yet unknown ISG to inhibit translation downstream of RIPK kinase activity, at the level of AKT1/mTORC1 pathway components.
It is well known that IFN signaling regulates the expression of a number of ISGs capable of inhibiting the translation of viral and in some cases host mRNA transcripts in response to various DNA and RNA viruses (Li et al., 2015). Viral RNA translation can be inhibited via PKR-driven phosphorylation of eIF2α, as well as IFN-mediated inhibition of cap-dependent translation machinery, and in some cases has been shown to be repressed by STING signaling (Chee and Roizman, 2004; Tesfay et al., 2008; Reynaud et al., 2015; Franz et al., 2018). Although a role for IFN signaling in the regulation of translation has been well documented, in this report we demonstrate a role for IFN in specifically regulating the translation of host cytokine transcripts that are dependent on the kinase activity of RIPK.
Finally, our results show that the regulation of cytokine translation by the kinase activity of RIPK and constitutive IFN signaling is conserved between mouse and human macrophages in response to LPS/zVAD treatment. This is of physiological relevance, as numerous bacteria are capable of inducing RIPK-driven necroptosis. Gram-negative bacteria, including Staphylococcus aureus and Serratia marcescens, rely on pore-forming toxins to drive macrophage and epithelial cell death that is abrogated by the inhibition of RIPK1 and RIPK3 kinase activity as well as MLKL in models of bacterial pneumonia (González-Juarbe et al., 2015, 2017; Kitur et al., 2015). Interestingly, in the context of Staphylococcus aureus pulmonary infection, RIPK3 deficiency was shown to reduce CXCL-1, TNF, IL1α, and IL1β levels detected in bronchoalveolar lavage and improve host survival (Kitur et al., 2015). In the case of cutaneous infection, RIPK3 deficiency reduced GM-CSF and IL1β production and increased bacterial clearance, while MLKL deficiency increased inflammatory signaling, and bacterial loads remained high (Kitur et al., 2016). Taken together, these results indicate a probable role for the kinase activity of RIPK in the regulation of inflammation in the context of bacterial infection and suggest that RIPK1/3 may drive inflammatory responses independently of cell death machinery. Therefore, our understanding of the mechanisms regulating the extent of the inflammatory response downstream of RIPK1/3 activation could be pertinent in the setting of a variety of bacterial infections, in which excessive inflammatory signaling could be detrimental to host survival. Further work would be needed to determine whether the translational mode of regulation identified in this study plays a role in bacterially induced, RIPK1/3 kinase-dependent inflammation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Requests for further information, resources and reagents should be directed to the Corresponding Author, Alexander Poltorak (alexander.poltorak@tufts.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6J, MOLF/Ei, and Ifnar−/−(B6.129S2-Ifnar1tm1Agt/Mmjax) (Müller et al., 1994) mouse strains were obtained from The Jackson Laboratory. Ifnb−/− mice (Deonarain et al., 2003) were a gift from Dr. S. Vogel (University of Maryland). Mlkl−/− (Murphy et al., 2013) mice were generated by Dr. W. Alexander (Walter and Eliza Hall Institute) and were a gift from Dr. M. Kelliher (University of Massachusetts). Ifnb−/− Mlkl−/− double knockout mice were generated by crossing Ifnb−/− and Mlkl−/− mice to homozygosity. Similarly, RIP1 Ki Ifnb−/− mice were generated by crossing Ifnb−/− and Ripk1K45A/K45A mice to homozygosity. Akt1−/−, Akt3−/− and wild-type littermate controls (Easton et al., 2005; Mao et al., 2007) were a gift from Dr. P. Hinds (Tufts University). 4E-BP triple knockout mice were a gift from Dr. N. Sonenberg (McGill University). RIP1/3 kinase inactive mice (Ripk1K45A/K45ARipk3K51A/K51A, RIPKi) (Berger et al., 2014; Mandal et al., 2014) were gifts from Dr. A. Degterev. All genetically modified mice were fully backcrossed to the C57BL/6J background. All mice were housed in a pathogen-free facility at the Tufts University School of Medicine and experiments were performed in accordance with regulations and approval of the Tufts University Institutional Animal Care and Use Committee.
Mouse Bone Marrow Derived Macrophages (BMDMs)
Bone marrow was flushed from femurs of 6–12 week old female mice of the indicated strains above with cold RPMI. Isolated cells were pelleted and re-suspended in BMDM differentiating media (RPMI with L-glutamine, 20% FBS, 30% L-cell conditioned media, 2% Pen-Strep) and cultured for 7 days at 37°C in 5% CO2 to differentiate. Matured BMDMs were rested overnight in RPMI with L-glutamine 10% FBS, 2% Pen-Strep prior to experiments.
Human PBMC Derived Macrophages
De-identified human peripheral blood was obtained from New York Biologics and used in accordance with protocols approved by the Tufts University School of Medicine Institutional Review Board. Peripheral blood mononuclear cells were isolated by Ficoll gradient, and differentiated into macrophages over the course of 7 days in RPMI containing 20% FBS, 2% Penicillin/Streptomycin and 10 ng/ml human monocyte colony stimulating factor (M-CSF). Differentiated macrophages were rested overnight in RPMI with L-glutamine 10% FBS, 2% Penicillin/Streptomycin prior to experiments.
METHOD DETAILS
Reagents
Lipopolysaccharide (LPS) (Escherichia coli 0111:B4) was purchased from Millipore Sigma and used at 10ng/ml. zVAD.fmk was purchased from Millipore and used at 50uM. Necrostatin-1 was purchased from Millipore Sigma, and Nec-1 s was purchased from abcam, both were used at 10uM. Recombinant mouse IFNβ (12404–1) was purchased from PBL Assay Science and used at indicated concentrations. In all experiments, IFNβ pre-treatment occurred 16 hours prior to stimulation, and the IFNβ is washed away before the addition of experimental treatments. 4EGi-1 (15362) was purchased from Cayman Chemical and used at 100uM. Torin-2 (14185) was purchased from Cayman Chemical and used at 2.5uM. AKT inhibitor X (14863) was purchased from Cayman Chemical and used at 1.25uM. For all experiments, macrophages were pre-treated with 4EGi-1, Torin-2 or AKT Inhibitor X for 1 hour prior to the addition of indicated experimental treatments. Ruxolitinib (11609) and Baricitinib (16707) were purchased from Cayman Chemical and used at 10uM. Blocking antibody to mouse IFNAR (MAR1–5A3) and control IgG were purchased from BD PharMingen and used at 20ug/ml. Blocking antibody to Human IFNAR (21385–1) was purchased from PBL Assay Science and used at 20ug/ml.
Next Generation Sequencing
Total RNA was isolated from unstimulated, LPS, and LPS+zVAD stimulated B6, Ifnb−/− and RIP1 Ki BMDMs using TRIzol. A TrueSeq kit was used to make a directional cDNA library. Seventy-five bp pair-end reads from cDNA libraries were generated on MiSeq (Illumina) and aligned using TopHat2 and Cufflinks software.
Cytokine Arrays
Cell free supernatants were analyzed for relative cytokine levels using the Proteome Profiler Mouse Cytokine Array Kit, Panel A from R&D (ARY006). Arrays were visualized using a BioRad Chemidoc MP imaging system and signal intensity was quantified using Image Studio software.
Ingenuity Pathway Analysis (IPA)
Pathway analysis was performed on RNA sequencing data from LPS+zVAD stimulated bone marrow derived macrophages using Ingenuity Pathway Analysis software. Using FPKM values, a core analysis was performed, utilizing the Ingenuity Knowledge Base as a reference set. The analysis was set to look for direct and indirect relationships and to filter the data specifically for the species mouse and the cell type bone marrow derived macrophages. This expression analysis revealed the top upregulated canonical pathways as well as diseases and disorders associated with the dataset.
RNA Isolation and Analysis
5×105 BMDMs were plated on 24-well TC treated plates, cells were lysed with Trizol (Invitrogen) and RNA extraction was carried out according to the manufacturer’s instructions. Reverse transcription was performed using M-MuLV reverse transcriptase, RNase inhibitor, random primers 9, and dNTP mix (New England BioLabs) to synthesize cDNA. cDNA was analyzed for relative mRNA levels using SYBR Green (Applied Biosystems) and intron spanning primers. Gapdh was used to normalize mRNA levels. Post amplification melting curve analysis was performed to confirm primer specificity.
Quantitative PCR Primers
TNF: (F) 5′-CTGTAGCCCACGTCGTAGC-3′, (R) 5′-TTGAGATCCATGCCGTTG-3′; CXCL1: (F) 5′-TGAGCTGCGCTGTCAGTG-3′, (R) 5′-AGAAGCCAGCGTTCACCAGA-3′
IRF7: (F) 5′-CTTCAGCACTTTCTTCCGAGA-3′, (R) 5′-TGTAGTGTGGTGACCCTTGC-3′; ISG15: (F) 5′-GAGCTAGAGCCTGCAG CAAT-3′, (R) 5′-TTCTGGGCAATCTGCTTCTT-3′,
GAPDH: (F) 5′-GGAGAGTGTTTCCTCGTCCC-3′, (R) 5′-TTCCCATTCTCGGCCTTGAC-3′.
ELISA
Cell free supernatants were analyzed for mouse TNFα (DY410), mouse CXCL-1 (DY453) and human CXCL-1 (DY275) protein levels as appropriate using the DuoSet ELISA kit from R&D Systems.
Cell viability assays
BMDMs were plated in tissue culture treated, 384-well, optical bottom plates (Nunc 142761) at 25×104 cells per well in RPMI with L-glutamine 10% FBS, 2% Pen-Strep. Indicated treatments were added to cells in media containing 10 ug/mL propidium iodide (Life Technologies, P3566). The Cytation3 Imager (BioTek) was used to maintain temperature at 37°C and 5% CO2 during the experiments. Kinetic microscopy was performed by the Cytation3 Imager to quantify propidium iodide uptake every 30 minutes. Wells containing 0.1% Triton X-100 lysed cells were used as 100% cytotoxicity controls, and wells containing unstimulated cells were used as controls for baseline death overtime.
Western blotting
Whole-cell lysates were prepared by lysing cells directly in 1X Laemmli Buffer with 5% β–mercaptoethanol. Samples were boiled for 10 minutes, followed by a 10-minute incubation on ice. Protein lysates were resolved on a 10% or 15% Bis-Tris SDS gel, and transferred to a nitrocellulose membrane using the Pierce Power transfer system. Membranes were blocked with 5% BSA in TBS-T for 1 hour. Primary antibodies were diluted to 1:1000 in 1% BSA in TBS-T, and membranes were incubated with primary antibodies overnight at 4°C. Infrared secondary antibodies (680 or 700 nm) were diluted 1:30,000 in 1% BSA in TBS-T, and membranes were incubated with secondary antibodies for 45 minutes at room temperature. Membranes were imaged using an Odyssey ® CLx Imaging System, and image analysis was performed using Image Studio software. Phospho-eIF4E (Ser 209) (9741), 4E-BP1 (9452), RIPK1 (3493), GAPDH (2118), Anti-rabbit IgG DyLight 680 conjugate (5366), and Anti-rabbit IgG DyLight 800 conjugate (5151) antibodies were purchased from Cell Signaling Technology. Total MLKL antibody (MABC604) was purchased from Millipore Sigma, and phospho-MLKL antibody (ab196436) was purchased from abcam.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mean values are presented ± standard deviation. Error bars in qPCR and ELISA experiments represent the standard deviation of three independent experiments. Data from kinetic cytotoxicity experiments are representative of 3 or more experiments, and error bars represent the standard deviation between triplicate samples. Significance was determined using a Student’s two tailed t test or Two-way ANOVA as appropriate: ns (non-significant) p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
DATA AND CODE AVAILABILITY
The accession number for the RNA senquencing data reported in this paper is Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc GEO: GSE83885.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p-eIF4E (Ser 209) | Cell Signaling Technology | Cat#9741; RRID: AB_331677 |
| 4E-BP | Cell Signaling Technology | Cat#9452; RRID: AB_331692 |
| GAPDH | Cell Signaling Technology | Cat#2118; RRID: AB_561053 |
| Anti-rabbit IgG DyLight 680 conjugate | Cell Signaling Technology | Cat#5366; RRID: AB_10693812 |
| Anti-rabbit IgG DyLight 800 conjugate | Cell Signaling Technology | Cat#5151; RRID: AB_10697505 |
| Purified NA/LE Mouse anti-Mouse IFN-a/b Receptor 1 (MAR1-5A3) | BD PharMingen | Cat#561183; RRID: AB_10611858 |
| Anti-Human IFNAR2 Antibody, Clone MMHAR-2 Neutralizing (mAb) | PBL Assay Science | Cat#21385-1; RRID: AB_354167 |
| RIPK1 | Cell Signaling Technology | Cat#3493; RRID: AB_2305314 |
| MLKL | Sigma | Cat#MABC604 |
| p-MLKL | Abcam | Cat#ab196436; RRID: AB_2687465 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Lipopolysaccharide (LPS, Escherichia coli 0111:B4) | Sigma | Cat#L4391 |
| zVAD.fmk | Millipore | Cat#627610 |
| Necrostatin-1 | Sigma | Cat#N9037 |
| Necrostatin-1 s | Abcam | Cat#ab221984 |
| Recombinant mouse IFNb | PBL Assay Science | Cat#12404-1 |
| 4EGi-1 | Cayman Chemical | Cat#15362 |
| Torin-2 | Cayman Chemical | Cat#14185 |
| AKT inhibitor X | Cayman Chemical | Cat#14863 |
| Ruxolitinib | Cayman Chemical | Cat#11609 |
| Baricitinib | Cayman Chemical | Cat#16707 |
| Critical Commercial Assays | ||
| Proteome Profiler Mouse Cytokine Array Kit, Panel A | R&D | Cat#ARY006 |
| TNFa DuoSet ELISA Kit | R&D | Cat#DY410 |
| CXCL-1 DuoSet ELISA Kit | R&D | Cat#DY453 |
| Deposited Data | ||
| RNA sequencing data from B6, Ifnb−/− and Rip1K45A/K45A BMDMs stimulated with LPS and LPS/zVAD | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE83885 | |
| Experimental Models: Cell Lines | ||
| Primary Mouse Bone Marrow Derived Macrophages | N/A | N/A |
| Human PBMC derived Macrophages (blood) | New York Biologies | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J Mouse | Jackson Laboratories | 000664 |
| MOLF/Ei Mouse | Jackson Laboratories | 000550 |
| Ifnar−/−(B6.129S2-Ifnar1tm1Agt/Mmjax) Mouse | Jackson Laboratories | 3204-JAX |
| Ifnb−/−Mouse | Dr. S Vogel | N/A |
| Mlkl−/− Mouse | Dr. W. Alexander, Dr. M. Kelliher | N/A |
| Ifnb−/− Mlkl−/− Mouse | N/A | N/A |
| Ifnb−/− Rip1K45A/K45A Mouse | N/A | N/A |
| Akt1−/−, Akt3−/− Mouse | Dr. P. Hinds | N/A |
| 4E-BP Triple KO Mouse | Dr. N. Sonenberg | N/A |
| Ripk1K45A/K45ARipk3K51A/K51A (RIPK1/3 Kinase inactive) Mouse | Dr. A. Degterev | N/A |
| Oligonucleotides | ||
| TNF: (F) 5′-CTGTAGCCCACGTCGTAGC-3′ qRT-PCR primer | Sigma | N/A |
| TNF: (R) 5′-TTGAGATCCATGCCGTTG-3′ qRT-PCR primer | Sigma | N/A |
| CXCL1: (F) 5′-TGAGCTGCGCTGTCAGTG-3′ qRT-PCR primer | Sigma | N/A |
| CXCL1: (R) 5′-AGAAGCCAGCGTTCACCAGA-3′ qRT-PCR primer | Sigma | N/A |
| IRF7: (F) 5′-CTTCAGCACTTTCTTCCGAGA-3′ qRT-PCR primer | Sigma | N/A |
| IRF7(R) 5′-TGTAGTGTGGTGACCCTTGC-3′ qRT-PCR primer | Sigma | N/A |
| ISG15: (F) 5′-GAGCTAGAGCCTGCAGCAAT-3′ qRT-PCR primer | Sigma | N/A |
| ISG15(R) 5′-TTCTGGGCAATCTGCTTCTT-3′ qRT-PCR primer | Sigma | N/A |
| GAPDH: (F) 5′-GGAGAGTGTTTCCTCGTCCC-3′ qRT-PCR primer | Sigma | N/A |
| GAPDH(R) 5′-TTCCCATTCTCGGCCTTGAC-3′ qRT-PCR primer | Sigma | N/A |
Highlights.
RIPK1/3 kinase activity promotes cytokine translation in mouse and human macrophages
IFN signaling inhibits RIPK1/3-driven initiation of cap-dependent translation
eIF4E-binding protein 4E-BP regulates RIPK1/3-kinase-dependent cytokine translation
ACKNOWLEDGMENTS
We thank Dr. Katherine Fitzgerald, Dr. Stefanie Vogel, Dr. Michelle Kelliher, Dr. Warren Alexander, and Dr. Philip Hinds for sharing various mouse strains used in this study. Additionally, we thank the Eshe Fund for support of Tufts core facilities and equipment. This work was supported by NIH grants AI056234, AI126050, and AI135369, Russian Science Fund Project 15-15-00100 (RNA-seq) to A.P., and NIAID Grant T32-AI-007077 to the Immunology Graduate Program.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.073.
REFERENCES
- Abt MC, Osborne LC, Monticelli LA, Doering TA, Alenghat T, Sonnenberg GF, Paley MA, Antenus M, Williams KL, Erikson J, et al. (2012). Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 37, 158–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn D, and Prince A (2017). Participation of necroptosis in the host response to acute bacterial pneumonia. J. Innate Immun 9, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, and Barber GN (2012). STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. U S A 109, 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battistini A (2009). Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J. Interferon Cytokine Res. 29, 765–780. [DOI] [PubMed] [Google Scholar]
- Berger SB, Kasparcova V, Hoffman S, Swift B, Dare L, Schaeffer M, Capriotti C, Cook M, Finger J, Hughes-Earle A, et al. (2014). Cutting edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol 192, 5476–5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunn GJ, Fadden P, Haystead TA, and Lawrence JC Jr. (1997). The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J. Biol. Chem. 272, 32547–32550. [DOI] [PubMed] [Google Scholar]
- Chee AV, and Roizman B (2004). Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 78, 4185–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, and Chan FK (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, and Yuan J (2012). A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 3, e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalet A, Gatti E, and Pierre P (2015). Integration of PKR-dependent translation inhibition with innate immunity is required for a coordinated anti-viral response. FEBS Lett. 589, 1539–1545. [DOI] [PubMed] [Google Scholar]
- Deonarain R, Verma A, Porter ACG, Gewert DR, Platanias LC, and Fish EN (2003). Critical roles for IFN-beta in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor alpha. Proc. Natl. Acad. Sci. U S A 100, 13453–13458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, et al. (2005). Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol 25, 1869–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz KM, Neidermyer WJ, Tan YJ, Whelan SPJ, and Kagan JC (2018). STING-dependent translation inhibition restricts RNA virus replication. Proc. Natl. Acad. Sci. U S A 115, E2058–E2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, and Kocher O (1999). Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene 240, 157–163. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, and Hay N (1998). 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 12, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, and Sonenberg N (1999). eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem 68, 913–963. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Gygi SP, Niedzwiecka A, Miron M, Burley SK, Polakiewicz RD, Wyslouch-Cieszynska A, Aebersold R, and Sonenberg N (2001). Hierarchical phosphorylation of the translation inhibitor 4E-BP1. Genes Dev. 15, 2852–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, and Orihuela CJ (2015). Pore-forming toxins induce macrophage necroptosis during acute bacterial pneumonia. PLoS Pathog. 11, e1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Juarbe N, Bradley KM, Shenoy AT, Gilley RP, Reyes LF, Hinojosa CA, Restrepo MI, Dube PH, Bergman MA, and Orihuela CJ (2017). Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 24, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, and Wang X (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111. [DOI] [PubMed] [Google Scholar]
- He S, Liang Y, Shao F, and Wang X (2011). Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. USA 108, 20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, and Goeddel DV (1996). TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4, 387–396. [DOI] [PubMed] [Google Scholar]
- Ingram JP, Thapa RJ, Fisher A, Tummers B, Zhang T, Yin C, Rodriguez DA, Guo H, Lane R, Williams R, et al. (2019). ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J. Immunol 203, 1348–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, Sehon CA, Marquis RW, Bertin J, and Mocarski ES (2013). Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem 288, 31268–31279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Joe CO, and Choi EJ (2001). Role of receptor-interacting protein in tumor necrosis factor-alpha -dependent MEKK1 activation. J. Biol. Chem 276, 27064–27070. [DOI] [PubMed] [Google Scholar]
- Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, and Prince A (2015). Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 11, e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitur K, Wachtel S, Brown A, Wickersham M, Paulino F, Peñaloza HF, Soong G, Bueno S, Parker D, and Prince A (2016). Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 16, 2219–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, and Kanneganti T (2018). ZBP1: innate sensor regulating cell death and inflammation. Trends Immunol. 39, 123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Man SM, Malireddi RKS, Karki R, Kesavardhana S, Place DE, Neale G, Vogel P, and Kanneganti T-D (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol 1, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Shank J, Cusson N, and Kelliher MA (2004). The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J. Biol. Chem. 279, 33185–33191. [DOI] [PubMed] [Google Scholar]
- Li S, Zhang L, Yao Q, Li L, Dong N, Rong J, Gao W, Ding X, Sun L, Chen X, et al. (2013). Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 501, 242–246. [DOI] [PubMed] [Google Scholar]
- Li MMH, MacDonald MR, and Rice CM (2015). To translate, or not to translate: viral and host mRNA regulation by interferon-stimulated genes. Trends Cell Biol. 25, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, and Green DR (2014). Necroptosis. N. Engl. J. Med 370, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Qiu J, Liang M, Golinski J, van Leyen K, Jung JE, You Z, Lo EH, Degterev A, and Whalen MJ (2014). Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 5, e1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, et al. (2014). RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakoa I, Horie K, Gaitanaris GA, Fidanza V, Ludwig T, Wiest DL, et al. (2007). Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J. Immunol 178, 5443–5453. [DOI] [PubMed] [Google Scholar]
- McComb S, Cessford E, Alturki NA, Joseph J, Shutinoski B, and Startek JB (2014). Type-I interferon signaling through ISGF3 complex is required for sustained Rip3 activation and necroptosis in macrophages. Proc. Natl. Acad. Sci. U S A 111, 3206–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Ahuja R, Osafo-Addo AD, Barrows D, Kettenbach A, Skidan I, Teng X, Cuny GD, Gerber S, and Degterev A (2013). Akt regulates TNFα synthesis downstream of RIP1 kinase activation during necroptosis. PLoS ONE 8, e56576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, and Tschopp J (2004). RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κ B activation. Nat. Immunol. 5, 503–507. [DOI] [PubMed] [Google Scholar]
- Micheau O, and Tschopp J (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190. [DOI] [PubMed] [Google Scholar]
- Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, Gross JD, Degterev A, Yuan J, Chorev M, et al. (2007). Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 128, 257–267. [DOI] [PubMed] [Google Scholar]
- Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K, Raj T, Ye CJ, Chevrier N, Zhang SY, Feng T, et al. ; Immunological Genome Project Consortium (2016). Parsing the interferon transcriptional network and its disease associations. Cell 164, 564–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller U, Steinhoff U, Reis LFL, Hemmi S, Pavlovic J, Zinkernagel RM, and Aguet M (1994). Functional role of type I and type II interferons in antiviral defense. Science 264, 1918–1921. [DOI] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. [DOI] [PubMed] [Google Scholar]
- Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, Finger JN, Polykratis A, Gough PJ, Bertin J, et al. (2016). RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by Toll-like receptor 4. Immunity 45, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orozco S, and Oberst A (2017). RIPK3 in cell death and inflammation: the good, the bad, and the ugly. Immunol. Rev. 277, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, and Vandenabeele P (2015). Necroptosis and its role in inflammation. Nature 517, 311–320. [DOI] [PubMed] [Google Scholar]
- Pearson JS, Giogha C, Muhlen S, Nachbur U, Pham CLL, Zhang Y, Hildebrand JM, Oates CV, Lung TWF, Ingle D, et al. (2017). EspL is a bacterial cysteine protease effector that cleaves RHIM proteins to block necroptosis and inflammation. Nat. Microbiol. 2, 16258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin F, and Sonenberg N (2013). Mechanism of Translation Initiation in Eukaryotes (Madame Curie Bioscience Database). [Google Scholar]
- Reynaud JM, Kim DY, Atasheva S, Rasalouskaya A, White JP, Diamond MS, Weaver SC, Frolova EI, and Frolov I (2015). IFIT1 differentially interferes with translation and replication of alphavirus genomes and promotes induction of type I interferon. PLoS Pathog. 11, e1004863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, and Sad S (2012). Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol. 13, 954–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh D, Najjar M, Zelic M, Shah S, Nogusa S, Polykratis A, Paczosa MK, Gough PJ, Bertin J, Whalen M, et al. (2017). Kinase activities of RIPK1 and RIPK3 can direct IFN-β synthesis induced by lipopolysaccharide. J. Immunol. 198, 4435–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarhan J, Liu BC, Muendlein HI, Weindel CG, Smirnova I, Tang AY, Ilyukha V, Sorokin M, Buzdin A, Fitzgerald KA, and Poltorak A (2018). Constitutive interferon signaling maintains critical threshold of MLKL expression to license necroptosis. Cell Death Differ. 26, 332–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer SA, Smirnova II, Kurbatova I, Bagina U, Churova M, Fowler T, Roy AL, Degterev A, and Poltorak A (2014). Toll-like receptor-mediated down-regulation of the deubiquitinase cylindromatosis (CYLD) protects macrophages from necroptosis in wild-derived mice. J. Biol. Chem. 289, 14422–14433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutinoski B, Alturki NA, Rijal D, Bertin J, Gough PJ, Schlossmacher MG, and Sad S (2016). K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 23, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silke J, Rickard JA, and Gerlic M (2015). The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 16, 689–697. [DOI] [PubMed] [Google Scholar]
- Spriggs KA, Bushell M, and Willis AE (2010). Translational regulation of gene expression during conditions of cell stress. Mol. Cell 40, 228–237. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, and Wang X (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surpris G, Chan J, Thompson M, Ilyukha V, Liu BC, Atianand M, Sharma S, Volkova T, Smirnova I, Fitzgerald KA, and Poltorak A (2015). Cutting edge : novel Tmem173 allele reveals importance of STING N terminus in trafficking and type I IFN production. J. Immunol. 196, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfay MZ, Yin J, Gardner CL, Khoretonenko MV, Korneeva NL, Rhoads RE, Ryman KD, and Klimstra WB (2008). Alpha/beta interferon inhibits cap-dependent translation of viral but not cellular mRNA by a PKR-independent mechanism. J. Virol. 82, 2620–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa RJ, Nogusa S, Chen P, Maki JL, Lerro A, Andrake M, Rall GF, Degterev A, and Balachandran S (2013). Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. U S A 110, E3109–E3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Moraga I, Levin D, Krutzik PO, Podoplelova Y, Trejo A, Lee C, Yarden G, Vleck SE, Glenn JS, et al. (2011). Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 146, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting AT, Pimentel-Muiños FX, and Seed B (1996). RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 15, 6189–6196. [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2008). Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, and Mocarski ES (2012). DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Shubina M, and Balachandran S (2017). RIPK3-driven cell death during virus infections. Immunol. Rev 277, 90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilcek J (2006). Fifty years of interferon research: aiming at a moving target. Immunity 25, 343–348. [DOI] [PubMed] [Google Scholar]
- Vogel SN, Hansen CT, and Rosenstreich DL (1979). Characterization of a congenitally LPS-resistant, athymic mouse strain. J. Immunol 122, 619–622. [PubMed] [Google Scholar]
- Wallach D, Kovalenko A, and Kang TB (2011). ‘Necrosome’-induced inflammation: must cells die for it? Trends Immunol. 32, 505–509. [DOI] [PubMed] [Google Scholar]
- Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, and Wang X (2014). Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Okabe Y, Kawane K, Fukuyama H, and Nagata S (2005). Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat. Immunol 6, 49–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA senquencing data reported in this paper is Gene Expression Omnibus: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc GEO: GSE83885.