

Abstract
Background
Dopamine replacement therapy is an established treatment for motor symptoms of Parkinson's disease, but its long-term use is often limited by the eventual development of motor complications, including levodopa-induced dyskinesia. Genetic background, particularly polymorphisms of dopamine metabolism genes, may affect the occurrence of dyskinesia in Parkinson's disease patients.
Methods
We investigated polymorphisms of dopamine metabolism genes, including catechol-O-methyltransferase, monoamine oxidase B, dopamine beta-hydroxylasedopamine, dopamine receptors D1, D2, and D3, and dopamine transporter, in 110 patients with Parkinson's disease. Cox proportional hazards regression was used to detect associations between genotypes and levodopa-induced dyskinesia.
Results
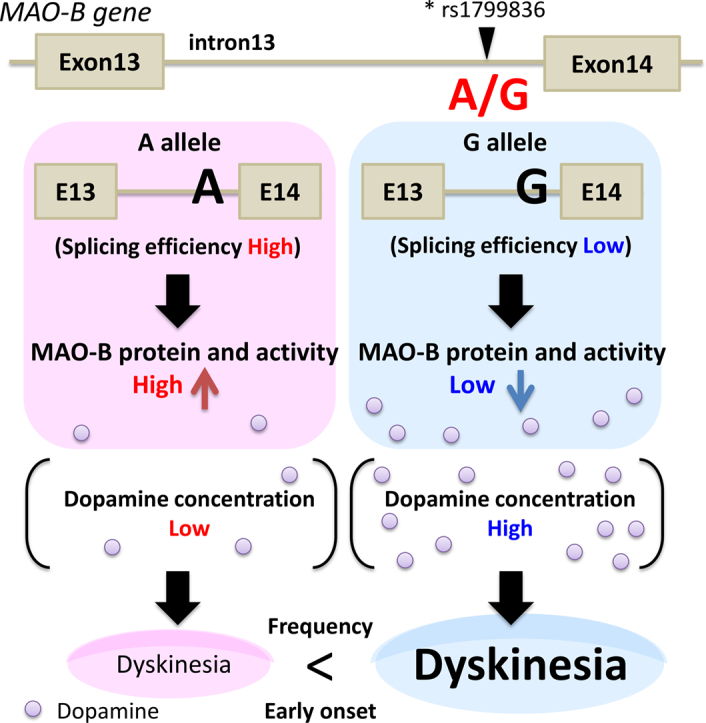
Monoamine oxidase B rs1799836 was the only polymorphism correlated with risk of dyskinesia. Patients with an AG or GG genotype were more likely to have dyskinesia than those with an AA genotype (adjusted hazard ratio, 3.41; 95% confidence interval, 1.28–9.10). Also, Kaplan-Meier curves demonstrated that patients with an AG or GG genotype developed dyskinesia earlier than those with an AA genotype (log-rank test, p = .004).
Conclusions
In Parkinson's disease patients, the monoamine oxidase B rs1799836 G allele is associated with a greater likelihood of developing dyskinesia than the A allele, possibly due to its association with lower monoamine oxidase B activity in the brain. Thus, detection of monoamine oxidase B polymorphisms may be useful for determining the optimal dosing of antiparkinson medications.
Keywords: Parkinson's disease, MAO-B polymorphism, Dyskinesia, Dopamine metabolism genes
Graphical abstract
Highlights
-
•
Average dose of L-dopa induced dyskinesia in Parkinson's disease was over 400 mg/day.
-
•
MAO-B rs1799836 G allele increases risk of L-dopa induced dyskinesia in PD.
-
•
50% of PD with MAO-B G allele developed L-dopa induced dyskinesia within 8 years.
1. Introduction
Parkinson's disease (PD) results from the aggregation of alpha-synuclein, which forms Lewy bodies and eventually leads to degeneration of dopaminergic neurons in the substantia nigra pars compacta [1]. Both genetic and environment factors are involved in the pathophysiology of PD. In addition, mitochondrial dysfunction and oxidative stress within the substantia nigra are believed to promote neuronal degeneration in PD [1]. The dopaminergic system is a major contributor to the motor symptoms of PD, which include bradykinesia, rigidity, and tremor at rest [2] .
Dopamine replacement therapy is the most effective treatment for PD, although its effectiveness changes over the protracted course of treatment and involves motor complications including delayed-on, dyskinesia, and wearing-off [3]. The severity of these motor complications varies widely among patients. One of the most common motor complications, levodopa-induced dyskinesia (LID), can be a major obstacle to increasing the dose of dopaminergic medications, and the symptom itself can impair patients' activities of daily living. Preclinical and clinical findings suggest that pulsatile stimulation of striatal postsynaptic dopamine receptors (DRs) and degeneration of pre- and post-synapses may be a key mechanism underlying LID [3,4]. Additionally, several lines of evidence have suggested that the large increase in synaptic dopamine concentration play a role in the emergence of LID [5]. However, the genetic factors contributing to the pathogenesis of LID remain unclear.
Age at disease onset, sex (female), duration of disease, length of treatment, and daily levodopa dose is suggested as a risk to develop LID [6]. In addition to these factors, alterations in dopaminergic metabolism have also been investigated, including changes in DRs, the dopamine transporter (DAT) [7], and dopamine degradation via catechoal-O-methyltransferase (COMT) and monoamine oxidase (MAO) [8]. In the present study, to address genetic factors contributing to the development of LID, we examined associations between dopamine metabolism gene polymorphisms and the development of dyskinesia.
2. Methods
2.1. Patients
A total of 110 Asian patients with PD were recruited from as subjects for this study at the Chiba University Hospital; one patient was Taiwanese, and the others were Japanese. It was noted that we excluded patients who have familial history about PD, who have PARK2 mutations, who were not taking antiparkinson drugs or whose disease duration was 3 years or less in recruiting process. Patients who had undergone deep brain stimulation were also excluded. All patients were diagnosed according to the United Kingdom Parkinson's Disease Society Brain Bank criteria [9]. The ethics committee of Chiba University approved this study. Study details were explained to participants by neurologists with over 10 years of experience, and all participants provided written informed consent.
Patient profiles, clinical findings, treatments, and outcomes were retrospectively reviewed from medical records. Levodopa-induced dyskinesia was determined by neurologists who have over fifteen years of experience based on either direct observation or by patients response to questioning. To evaluate the role of dopaminergic therapy, daily levodopa equivalent dose (LED) was calculated [10].
2.2. Genomic DNA extraction
DNA was isolated from peripheral blood using a MagNA Pure Compact System (Roche Diagnostics, Penzberg, Germany) according to the manufacturer's protocol. DNA samples were measured using a NanoDrop 1000 spectrophotometer (Coleman Technologies, Orlando, FL), diluted to 3 ng/μL, and stored at 4 °C.
2.3. Polymorphism selection
We analyzed nine polymorphisms over seven dopamine metabolism genes, including COMT, MAO-B, dopamine beta-hydroxylasedopamine (DBH), DRD1, DRD2, DRD3, and DAT. These polymorphisms were selected from a previous study based on (1) minor genotype frequencies of samples >1% and (2) their strong associations with biological measures [11]. Polymorphism genotyping was carried out using high resolution melt (HRM) analysis and confirmed by direct sequencing for the following polymorphism: COMT rs4680; MAO-B rs1799836; DBH rs1611115; DRD1 rs4532; DRD2 rs1799732, rs1800497, and rs1079597; and DRD3 rs6280. Polymorphism genotyping for DAT rs28363170 was performed by fragment analysis. The genotype frequency of dopaminergic polymorphisms in the present study was confirmed against Japanese genotype frequency data from the NCBI database (https://www.ncbi.nlm.nih.gov) or previous studies [[12], [13], [14], [15], [16], [17], [18], [19], [20], [21]] (Supplementary Table 1).
2.4. HRM analysis
Primers were designed by LightCycler Probe Design Software 2.0 (Roche Diagnostics). HRM polymerase chain reaction (HRM-PCR) was performed on a LightCycler 480 Instrument (Roche Diagnostics). The reaction mixture contained 3 ng genomic DNA and either 0.7 μM of each primer with Type-it HRM-PCR reagents (Qiagen, Hilden, Germany) or 0.2 μM of each primer with Multiplex PCR Master Mix (Qiagen) and PCR-grade water, adjusted to a total volume of 20 μL. For PCR cycles, the initial denaturation step was 95 °C for 10 min followed by 50 cycles of 95 °C for 10 s, touchdown annealing from 66 °C to 56 °C (1 °C/cycle), and a final step at 72 °C for 10 s. After amplification, products were heated to 95 °C for 1 min and then cooled to 40 °C for 1 min to favor heteroduplex formation. For the melting step, the temperature was raised from 40 °C to 95 °C with a ramp rate of 0.02 °C/s and 25 acquisitions/°C. HRM curve analysis was performed using Light Cycler 480 Gene Scanning Software (version 1.5) to detect nucleotide variation [22].
2.5. PCR and fragment analysis
The amplification reaction was performed in a mixture containing 50–100 ng genomic DNA, 1 μM of each primer, 5% DMSO, Multiplex PCR Master Mix (Qiagen), and PCR-grade water, adjusted to a total volume of 20 μL. PCR was performed with initial denaturation at 95 °C for 15 min followed by 40 cycles of 94 °C for 30 s, annealing at 66 °C for 90 s, and a final step at 72 °C for 90 s. PCR products were subjected to fragment analysis as previously described [23].
2.6. Direct sequencing
PCR products generated after HRM were column-purified according to standard protocols. Using BigDye terminator v.3.1, a cycle sequencing reaction was performed with an initial step at 96 °C for 10 min followed by heat denaturation as 96 °C for 10 s, annealing at 50 °C for 5 s, and extension at 60 °C for 1 min. After products were purified by BigDye Xterminator (Life Technologies, Carlsbad, CA), sequence detection was performed using the Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA).
2.7. Measurement of MAO-B activity
Platelets samples were obtained from 21 patients with PD (12 patients with MAO-B AA(A) genotypes and 9 patients with MAO-B AG or GG (G) genotypes). Determination of MAO-B was performed in platelets using the MAO-Glo Assay (Promega, Germany). MAO-Glo Assay System Protein lysates were diluted to 1.0 mg/mL using lysis buffer. Twenty-five microliters of the diluted lysate was incubated with 25 μL of MAO substrate solution (1:250 dilution of provided MAO substrate to measure MAO-B activity) (Promega) for 50 min at 37 °C. Fifty microliters of luciferin detection reagent was then added and luminescence was measured.
2.8. Western blotting analysis
Peripheral blood samples were obtained from 33 patients with PD (16 patients with MAO-B AA(A) genotypes and 17 patients with MAO-B AG or GG (G) genotypes). Whole blood was centrifuged for 10 min with 200 ×g at 4 °C. Obtained platelet rich plasma (PRP) was centrifuged for 10 min with 5000 ×g at 4 °Cto separate platelets as precipitates. The samples were sonicated and diluted to 1.0 mg/mL using RIPA buffer. The proteins (10 μg/per lane) were separated on a 7.5% SDS-polyacrylamide gel, transferred to the nitrocellulose membrane and detected using Anti-Monoamine Oxidase B antibody (abcam, ab175136). Densitometric measurements of the Western blot band images were performed using image J software.
2.9. Statistical analysis
Association between single nucleotide polymorphisms and the development of LID was analyzed using Fisher's exact tests and odds ratios (ORs). Kruskal-Wallis tests were used to analyze interactions between genotype, sex, age at disease onset, disease duration, duration of antiparkinson drug therapy, LED, use of selegiline, and levodopa dose and to perform comparisons between patients with and without dyskinesia. The correlation between MAO-B genotype and time to develop LID from disease or treatment onset was analyzed by Kaplan-Meier curves and log-rank tests. Statistical significance in MAO-B enzyme activities analysis and immunoblotting was ascertained by Student's t-test. Fisher's exact tests, ORs, Kruskal-Wallis tests, Kaplan-Meier curves and Student's t-test were performed using SPSS statistics (version 22; IBM, Inc., Chicago, IL).
3. Results
3.1. Patient characteristics
Clinical characteristics of PD patients are summarized in Table 1. Of the 110 enrolled patients, 37 (34%) developed LID. There were no significant differences in treatment with selegiline between patients with and without LID. By contrast, there were significant differences in sex, age at onset, disease duration, duration of anti-parkinson drug therapy, total LED, daily levodopa dose and Hoehn and Yahr stage between patients with and without LID. The average levodopa dose among patients with LID was over 400 mg/day, consistent with previous reports [6] .
Table 1.
PD patient characteristics, overall and with or without LID.
| Characteristics | Overall | No LID | LID | p-value |
|---|---|---|---|---|
| Patients, no. (%) | 110 | 73 (66) | 37 (34) | |
| Male, no. (%) | 54 (49) | 42 | 12 | 0.016 |
| Age at disease onset, mean ± SD, y | 61.0 ± 11.8 | 63.0 ± 12.0 | 58.0 ± 10.5 | 0.026 |
| Disease duration, mean ± SD, y | 11 ± 5.1 | 9.0 ± 4.3 | 14.0 ± 5.1 | < 0.0001 |
| Duration of anti-parkinson drug therapy, mean ± SD, y | 8.0 ± 5.0 | 7.0 ± 4.1 | 12.0 ± 5.0 | < 0.0001 |
| Total LED,a mean ± SD, mg/day | 604.0 ± 412.9 | 505.0 ± 408.9 | 799.0 ± 345.6 | < 0.0001 |
| Levodopa dose, mean ± SD, mg/day | 411.4 ± 386.5 | 368.0 ± 456.9 | 497.0 ± 144.7 | < 0.0001 |
| Treatment with selegiline, no. (%) | 52 (47) | 36 | 16 | 0.686 |
| Hoehn and Yahr, no. (%) | < 0.0001b | |||
| Stage 1–2 | 36 (33) | 34 | 2 | |
| Stage 3–5 | 74 (67) | 39 | 35 |
LED, levodopa equivalent dose; LID, levodopa-induced dyskinesia.
Calculated by summing the LEDs for levodopa alone or in combination with dopamine agonists.
Hoehn and Yahr stage 1–2 vs. 3–5.
3.2. Associations between genotype, LID, and treatment duration
Associations between each polymorphism and the presence of LID were analyzed. The polymorphism identified as being significantly correlated with LID was MAO-B rs1799836 (Table 2). No significant differences in the frequencies of the other eight polymorphisms were detected between patients with and without LID (Supplementary Table 2).
Table 2.
Association between the MAO-B rs1799836 polymorphism and the development of LID in PD patients.
| Polymorphism | sNo LID | LID | p-value | OR (95% CI) |
|---|---|---|---|---|
| MAO-B rs1799836 | 0.011 | |||
| AA (A) | 64 | 25 | ||
| AG | 8 | 7 | ||
| GG (G) | 1 | 5 | ||
| AA (A) vs. AG + GG (G) | 64 vs. 9 | 25vs. 12 | 0.019 | 3.41 (1.28–9.10) |
| A vs. G (count of allele) | 95 vs. 9 | 46 vs. 16 | 0.006 | 3.67 (1.50–8.93) |
LID, levodopa-induced dyskinesia; MAO-B, monoamine oxidase B; OR, odds ratio; CI, confidence interval
Clinical characteristics of patients with respect to MAO-B genotype are shown in Supplementary Table 3. The minor allele frequencies of MAO-B in our study were comparable to those of healthy Japanese individuals in the NCBI database (Supplementary Table 1). There were no significant differences in sex, age at disease onset, disease duration, duration of anti-parkinsonian drug usage, total LED, levodopa dose, treatment with selegiline or Hoehn and Yahr stage among the three genotypes. Table 2 shows ORs with 95% CIs for the occurrence of LID according to MAO-B genotype. Patients with G allele of MAO-B genotype (AG + GG (G)) showed significant increased risk of developing LID. In addition, the presence of the G allele (count of allele) was significantly associated with an increased risk of developing LID.
To further determine the relationship between LID and duration of disease and duration of treatment depending on MAO-B genotype, we performed Kaplan-Meier analysis. Kaplan-Meier curves for disease duration without LID were significantly different between the two MAO-B genotypes (AA (A) vs. AG + GG (G); Fig. 1A). Patients with an AA (A) genotype developed LID 2 to 25 years after disease onset. By contrast, patients with an AG or GG (G) genotype developed LID 4 to 20 years after disease onset. Of the patients with an AA (A) genotype, 50% developed LID within 25 years, whereas 50% of those with AG or GG (G) genotypes developed LID within 8 years. Patients with an AG or GG (G) genotype were at a significantly higher risk for developing LID. We found similar significant associations between genotype and duration of treatment (Fig. 1B).
Fig. 1.

Kaplan-Meier curves showing association between MAO-B polymorphism and increased LID risk for (A) disease duration and (B) treatment duration. Kaplan-Meier curves showing association between MAO-B polymorphism and increased LID risk without selegiline treatment for (C) disease duration and (D) treatment duration. Number of patients in each groups is indicated.
3.3. Association between MAO-B genotype and LID depending on MAO-B inhibitor treatment
In the present study, this MAO-B inhibitor was prescribed to 52 patients (47%). Mean daily selegiline dose was 4.1 ± 1.9 mg, with a minimum of 2.5 mg and maximum of 10 mg. Four patients discontinued treatment with selegiline due to camptocormia and hallucination.
To assess influence of MAO-B inhibitor on LID, we performed Kaplan-Meier analysis in patients treated without selegiline, the only approved MAO-B inhibitor in Japan. Among AA genotype, fourty-nine patients treated without selegiline, of which 13 developed LID. Nine patients with an AG + GG (G) genotype treated without selegiline, of which eight developed LID. Among patients without selegiline, the duration of disease (Fig. 1C) and duration of treatment (Fig. 1D) until the development of LID was significantly different between patients with an AA or AG + GG genotype; patients with an AG + GG genotype developed LID earlier than patients with an AA genotype.
3.3.1. Reduction of MAO-B enzyme activity and protein expressions in patients with MAO-B G allele
To determine which MAO-B genotypes affect MAO-B enzyme activity and protein levels, we performed measurement of MAO-B enzyme activity and immunoblotting. Platelet MAO-B enzyme activity was significantly higher in patients with an AA (A) genotype than patients with an AG or GG (G) genotype (Fig. 2A). Similar trends were observed in immunoblotting analysis (Fig. 2B and Supplementary Fig. 1).
Fig. 2.

MAO-B enzyme activeties and protein expression were decreased in patients with MAO-B rs1799836 G allele polymorphism. (A) MAO-B enzyme activeties are reduced in patients with G allele. (B) Densitometry of Western blotting revealed that MAO-B are expressed to a less extent in patients with G allele.
4. Discussion
Using retrospective clinical data from 110 Asian patients with PD, we performed correlation analysis between disease parameters and polymorphisms in genes related to dopamine metabolism, including COMT, MAO-B, DBH, DR, and DAT. Of the nine polymorphisms, only MAO-B rs1799836 correlated with the risk of developing dyskinesia. More specifically, we determined that PD patients carrying the G allele (AG or GG) exhibited an approximately 3-fold increase in the occurrence of dyskinesia. The relationship between MAO-B allele and PD risk has not been confirmed in Western populations [24,25].
Our results indicate that MAO-B allele may affect the risk of developing LID. Similarly, the MAO-B AG genotype was shown to have trends in correlation with an increased risk of dyskinesia in Chinese study [8,26]. MAO activity is highly disposed in the human striatum and hypothalamus. MAO-B is the main form found in the basal ganglia, serotonergic neurons (including the dorsal raphe nucleus), and astrocytes [27]. The MAO-B rs1799836 polymorphism influences the enzymatic activity of MAO-B; the A allele may lead to elevated MAO-B activity, whereas the G allele may lead to lower MAO-B activity [13]. In this study, both MAO-B enzyme activity and protein levels were higher in patients with an AA (A) genotype. Our findings confirm that A allele of the MAO-B rs1799836 polymorphism involve high MAO-B enzyme activity and expression of MAO-B proteins. In patients receiving long-term levodopa therapy, the concentration of dopamine in the synaptic cleft is no longer appropriately regulated. Patients with low MAO-B activity (G allele) would likely have even higher synaptic dopamine levels in the striatal synaptic cleft. Our finding that the MAO-B G allele is correlated with the development of LID suggests that lower MAO-B activity may have functional consequences in terms of levodopa metabolism, eventually resulting in increased risk of developing LID. MAO-B polymorphism may possibly correlate with the development of PD. We found similar proportions of PD patients with each MAO-B genotype compared with those of Japanese individuals in the NCBI database. In this context, it was reported that the A allele is a risk factor for developing PD in the Japanese population [14]. As MAO catabolism of dopamine produces reactive oxygen species as a byproduct, oxidative stress levels and cell death in the striatum and substantia nigra could depend on the MAO-B allele. We speculate that high MAO-B activity (A allele) could produce more oxidative damage, leading to increased cell death. This could be a contributing factor for individuals with the MAO-B A allele and a higher risk of developing PD. Nevertheless, it is necessary to investigate the relationship between A allele and PD prevalence using more large number of patients.
Although previous studies report an association between dyskinesia risk and the low-activity genotype of COMT rs4680 (Val158Met) polymorphism [12], we were unable to detect a correlation between dyskinesia risk and other dopaminergic polymorphisms, including COMT (Supplementary Table 1). In a previous study, PD patients with low COMT activity (A allele) tended to develop dyskinesia, although no statistically significant differences were observed [28]. COMT metabolizes dopamine mainly in the cortex but not in the striatum [29] and contributes to cognitive function rather than motor function. On the other hand, MAO-B participates in dopamine degradation in the basal ganglia. Thus, aberrant hyperkinesia induced by higher synaptic dopamine level may well explained by low striatal MAO-B activity.
A recent study [6] shows that factors predictive of dyskinesia are, in rank order: young age at disease onset, high levodopa dose, low body weight, treatment allocation, female gender, and a more severe score on part 2 of the Unified Parkinson's Disease Rating Scale. In line with this finding, the MAO-B gene is located on the X chromosome, and thus it is possible that sex differences are related to this risk factor for dyskinesia development. In this study, we detected age at disease onset, sex, or Hoehn and Yahr stage as risk factors. These results supported that the characteristics of our study cohort were similar to that of general patients with Parkinson's disease.
The duration of disease and duration of treatment were independently assessed in our survival analyses. Considering disease duration, patients with a MAO-B G allele exhibited dyskinesia earlier. However, treatment duration did not significantly differ between genotypes despite the strong correlation between disease duration and treatment duration. In a previous study examining a large cohort of PD patients in Ghana, a sub-Saharan African country in which access to medication is limited and initiation of levodopa therapy often delays by many years after disease onset, multivariate analysis showed that disease duration and daily levodopa dose, but not disease duration at the initiation of levodopa treatment, were associated with motor complications [30]. Together, the previous and present results indicate that, regardless of the time at which levodopa is initiated, disease duration is a major factor contributing to the development of dyskinesia.
The following are the supplementary data related to this article.
A western blot showing decreased MAO-B expression in platelets derived from patients with MAO-B rs1799836 G allele polymorphism.
Supplementary material
Contributors
SK, SS, MB, SK, FN, and TT conceived of and designed the experiments. SH, YY, TY, and MB collected and assembled the data. SK, TI, KS, SI, MC, AN, XA, AA, GY and MS performed experiments and analyzed the data. SK, MB, SH, SS, and TT wrote the paper. TT takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This work was supported by Grants-in-Aid for Advanced Research and Development Programs for Medical Innovation, Scientific Research (B) #19H03708, (C) #19K07635, #17K09774, Young Scientists (B) #15K19475, the Takeda Science Foundation, Naito Foundation, Novartis Foundation, Uehara Memorial Foundation, and GSK Japan Research Grant 2018.
Patient consent
Obtained.
Ethics approval
The ethics committee of Chiba University approved this study. (application numbers192).
Declaration of Competing Interest
None.
Acknowledgements
We thank E. Utsuno and Y. Uchigaki for assistance with specimen collection,T. Suichi and K. Himuro for assistance with data collection, and S. Takahashi for assistance with data analysis.
References
- 1.Farrer M.J. Genetics of Parkinson's disease: paradigm shifts and future prospects. Nat. Rev. Genet. 2006;7(4):306–318. doi: 10.1038/nrg1831. doi: 10.1038/nrg1831[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 2.Hely M.A., Reid W.G., Adena M.A., Halliday G.M., Morris J.G. The Sydney multicenter study of Parkinson's disease: the inevitability of dementia at 20 years. Mov. Dis. 2008;23(6):837–844. doi: 10.1002/mds.21956. doi: 10.1002/mds.21956[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 3.Calabresi P., Di Filippo M., Ghiglieri V., Tambasco N., Picconi B. Levodopa-induced dyskinesias in patients with Parkinson's disease: filling the bench-to-bedside gap. Lancet Neurol. 2010;9(11):1106–1117. doi: 10.1016/S1474-4422(10)70218-0. doi: 10.1016/S1474-4422(10)70218-0[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 4.Jenner P. Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci. 2008;9(9):665–677. doi: 10.1038/nrn2471. doi: 10.1038/nrn2471[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 5.de la Fuente-Fernandez R., Sossi V., Huang Z. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson's disease: implications for dyskinesias. Brain J. Neurol. 2004;127(Pt 12):2747–2754. doi: 10.1093/brain/awh290. doi: 10.1093/brain/awh290[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 6.Warren Olanow C., Kieburtz K., Rascol O. Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson's disease. Mov. Dis. 2013;28(8):1064–1071. doi: 10.1002/mds.25364. doi: 10.1002/mds.25364[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 7.Kaiser R., Hofer A., Grapengiesser A. L -dopa-induced adverse effects in PD and dopamine transporter gene polymorphism. Neurology. 2003;60(11):1750–1755. doi: 10.1212/01.wnl.0000068009.32067.a1. [DOI] [PubMed] [Google Scholar]
- 8.Hao H., Shao M., An J. Association of Catechol-O-Methyltransferase and monoamine oxidase B gene polymorphisms with motor complications in parkinson's disease in a Chinese population. Parkinsonism Relat. Disord. 2014;20(10):1041–1045. doi: 10.1016/j.parkreldis.2014.06.021. doi: 10.1016/j.parkreldis.2014.06.021[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 9.Hughes A.J., Daniel S.E., Lees A.J. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology. 2001;57(8):1497–1499. doi: 10.1212/wnl.57.8.1497. [DOI] [PubMed] [Google Scholar]
- 10.Tomlinson C.L., Stowe R., Patel S., Rick C., Gray R., Clarke C.E. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov. Dis. 2010;25(15):2649–2653. doi: 10.1002/mds.23429. doi: 10.1002/mds.23429[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 11.Pearson-Fuhrhop K.M., Dunn E.C., Mortero S. Dopamine genetic risk score predicts depressive symptoms in healthy adults and adults with depression. PLoS One. 2014;9(5):e93772. doi: 10.1371/journal.pone.0093772. doi: 10.1371/journal.pone.0093772[published Online First: Epub Date] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Lau L.M., Verbaan D., Marinus J., Heutink P., van Hilten J.J. Catechol-O-methyltransferase Val158Met and the risk of dyskinesias in Parkinson's disease. Mov. Dis. 2012;27(1):132–135. doi: 10.1002/mds.23805. doi: 10.1002/mds.23805[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 13.Balciuniene J., Emilsson L., Oreland L., Pettersson U., Jazin E. Investigation of the functional effect of monoamine oxidase polymorphisms in human brain. Hum. Genet. 2002;110(1):1–7. doi: 10.1007/s00439-001-0652-8. doi: 10.1007/s00439-001-0652-8[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 14.Kiyohara C., Miyake Y., Koyanagi M. Genetic polymorphisms involved in dopaminergic neurotransmission and risk for Parkinson's disease in a Japanese population. BMC Neurol. 2011;11:89. doi: 10.1186/1471-2377-11-89. doi: 10.1186/1471-2377-11-89[published Online First: Epub Date] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zabetian C.P., Anderson G.M., Buxbaum S.G. A quantitative-trait analysis of human plasma-dopamine beta-hydroxylase activity: evidence for a major functional polymorphism at the DBH locus. Am. J. Hum. Genet. 2001;68(2):515–522. doi: 10.1086/318198. doi: 10.1086/318198[published Online First: Epub Date] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Comings D.E., Gade R., Wu S. Studies of the potential role of the dopamine D1 receptor gene in addictive behaviors. Mol. Psychiatry. 1997;2(1):44–56. doi: 10.1038/sj.mp.4000207. [DOI] [PubMed] [Google Scholar]
- 17.Ohara K., Nagai M., Tani K., Nakamura Y., Ino A., Ohara K. Functional polymorphism of —141C Ins/Del in the dopamine D2 receptor gene promoter and schizophrenia. Psychiatry Res. 1998;81(2):117–123. doi: 10.1016/s0165-1781(98)00092-4. [DOI] [PubMed] [Google Scholar]
- 18.Jonsson E.G., Nothen M.M., Grunhage F. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol. Psychiatry. 1999;4(3):290–296. doi: 10.1038/sj.mp.4000532. [DOI] [PubMed] [Google Scholar]
- 19.Savitz J., Hodgkinson C.A., Martin-Soelch C. The functional DRD3 Ser9Gly polymorphism (rs6280) is pleiotropic, affecting reward as well as movement. PLoS One. 2013;8(1):e54108. doi: 10.1371/journal.pone.0054108. doi: 10.1371/journal.pone.0054108[published Online First: Epub Date] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue-Murayama M., Adachi S., Mishima N. Variation of variable number of tandem repeat sequences in the 3′-untranslated region of primate dopamine transporter genes that affects reporter gene expression. Neurosci. Lett. 2002;334(3):206–210. doi: 10.1016/s0304-3940(02)01125-4. [DOI] [PubMed] [Google Scholar]
- 21.Fuke S., Suo S., Takahashi N., Koike H., Sasagawa N., Ishiura S. The VNTR polymorphism of the human dopamine transporter (DAT1) gene affects gene expression. Pharm. J. 2001;1(2):152–156. doi: 10.1038/sj.tpj.6500026. [DOI] [PubMed] [Google Scholar]
- 22.Obul J., Itoga S., Abliz M. High-resolution melting analyses for gene scanning of APC, MLH1, MSH2, and MSH6 associated with hereditary colorectal cancer. Gene. Test. Mol. Biomark. 2012;16(5):406–411. doi: 10.1089/gtmb.2011.0166. doi: 10.1089/gtmb.2011.0166[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 23.Ishige T., Sawai S., Itoga S. Pentanucleotide repeat-primed PCR for genetic diagnosis of spinocerebellar ataxia type 31. J. Hum. Genet. 2012;57(12):807–808. doi: 10.1038/jhg.2012.112. doi: 10.1038/jhg.2012.112[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 24.Costa P., Checkoway H., Levy D. Association of a polymorphism in intron 13 of the monoamine oxidase B gene with Parkinson's disease. Am. J. Med. Genet. 1997;74(2):154–156. doi: 10.1002/(sici)1096-8628(19970418)74:2<154::aid-ajmg7>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]
- 25.Ho S.L., Kapadi A.L., Ramsden D.B., Williams A.C. An allelic association study of monoamine oxidase B in Parkinson's disease. Ann. Neurol. 1995;37(3):403–405. doi: 10.1002/ana.410370318. doi: 10.1002/ana.410370318[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 26.Sampaio T.F., Dos Santos E.U.D., de Lima G.D.C. MAO-B and COMT Genetic Variations Associated With Levodopa Treatment Response in Patients With Parkinson's Disease. J. Clin. Pharmacol. 2018;58(7):920–926. doi: 10.1002/jcph.1096. [published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 27.Youdim M.B., Edmondson D., Tipton K.F. The therapeutic potential of monoamine oxidase inhibitors. Nat. Rev. Neurosci. 2006;7(4):295–309. doi: 10.1038/nrn1883. doi: 10.1038/nrn1883[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 28.Watanabe M., Harada S., Nakamura T. Association between catechol-O-methyltransferase gene polymorphisms and wearing-off and dyskinesia in Parkinson's disease. Neuropsychobiology. 2003;48(4):190–193. doi: 10.1159/000074637. doi: 10.1159/000074637[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 29.Meyer-Lindenberg A., Kohn P.D., Kolachana B. Midbrain dopamine and prefrontal function in humans: interaction and modulation by COMT genotype. Nat. Neurosci. 2005;8(5):594–596. doi: 10.1038/nn1438. doi: 10.1038/nn1438[published Online First: Epub Date] [DOI] [PubMed] [Google Scholar]
- 30.Cilia R., Akpalu A., Sarfo F.S. The modern pre-levodopa era of Parkinson's disease: insights into motor complications from sub-Saharan Africa. Brain J. Neurol. 2014;137(Pt 10):2731–2742. doi: 10.1093/brain/awu195. doi: 10.1093/brain/awu195[published Online First: Epub Date] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A western blot showing decreased MAO-B expression in platelets derived from patients with MAO-B rs1799836 G allele polymorphism.
Supplementary material