

Abstract
We report the design, synthesis, and characterization of a short peptide trapped in a π-helix configuration. This high-energy conformation was nucleated by a preorganized π-turn, which was obtained by replacing an N-terminal intramolecular main chain i and i + 5 hydrogen bond with a carbon-carbon bond. Our studies highlight the nucleation parameter as a key factor contributing to the relative instability of the π-helix and allow us to estimate fundamental helix-coil transition parameters for this conformation.
Classical analyses of protein structure predict several hydrogen-bonded helical configurations, including the R-, 310-, and π-helices (1–3). These configurations differ from each other in the number of atoms in the hydrogen-bonded helix turn. The R-helix is characterized by a 13-member intramolecular hydrogen bond between the i and i + 4 residues; the 310-helix contains a 10-member hydrogen bond between the i and i + 3 residues, and the π-helix features a 16-member hydrogen bond between the i and i + 5 residues (Figure 1). The α-helix is a ubiquitous element of protein structure and function, while the 310-helix is found at the ends of R-helices (4). By contrast, the third member of the protein helix family, the π-helix, is more rare (5). The relative infrequency of the π-helix has been attributed to the instability of this conformation, especially as compared to the R-helix. Molecular dynamics simulations have implicated both 310- and π-helices as high-energy folding intermediates of the R-helix (4, 6–9). We sought to trap the metastable π-helix and estimate its fundamental folding parameters. Detailed characterization the π-helix should aid in theoretical and experimental studies of protein folding and in defining the role of this motif in proteins.
FIGURE 1:
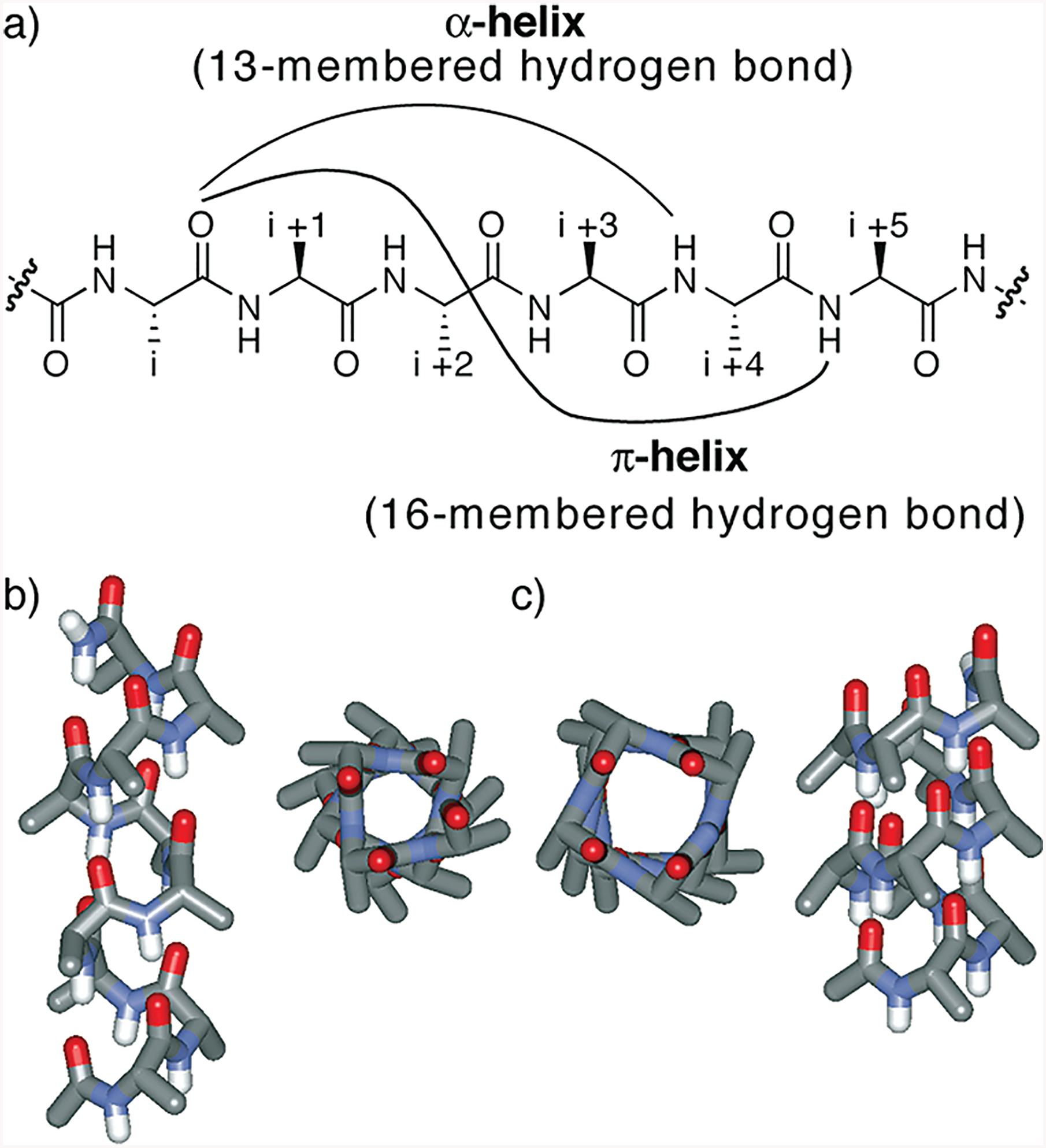
(a) Hydrogen bonding patterns that describe the α- and π-configurations in peptides. (b and c) Idealized α- and π-helices from Ala10, respectively.
Here we report the design, synthesis, and characterization of a short helix nucleated by a preorganized π-turn. The π-turn was prepared via replacement of an N-terminal main chain i and i + 5 hydrogen bond with a carbon-carbon bond (10, 11). As the π-helix is stable under only unique environments in a few proteins, it has not been possible to isolate and characterize this fundamental protein secondary structure (12). Our studies highlight the nucleation parameter as a key factor contributing to the relative instability and scarcity of the π-helix in proteins and allow us to assign a circular dichroism spectrum to this conformation. Importantly, our results suggest that the propagation constant for the π-helix is not as disfavored relative to the R-helix as expected; thus, the π-helix represents a likely intermediate in the helix-coil transition.
Three reasons have been invoked to explain the relative instability of the π-helix (5, 12): (i) poor sterics, since the dihedral angles required for each amino acid residue to adopt a π-helix conformation are slightly unfavorable (13); (ii) the idealized π-helix structure would feature a 1 Å hole leading to loss of van der Waals interactions (2); and (iii) four residues need to be organized to nucleate a π-turn, an energetically unfavorable proposition relative to 310- or α-helix (14, 15).
Inspection of the Protein Data Bank suggests that helices containing i and i + 5 hydrogen bonds, indicative of the π-helical conformation, may be more prevalent than originally believed (5, 16); a report about the key functional role played by a conformationally labile π-helix in human ferrochelatase highlights the potential role of this metastable conformation in protein structure and function (17). These studies also suggest that additional stabilizing interactions, such as metal complexation, may be necessary to stabilize π-helices in proteins (18). Considering that stabilizing moieties are often required to access even the more favored R-helical conformation in short peptides (19), stabilization of short peptides into the unstable π-helical conformation has remained a daunting challenge despite some ingenious efforts (12).
Treatments of the helix-coil transition in peptides emphasize the requirement to nucleate a series of consecutive amino acids into the helical orientation as an unfavorable step in helix formation (20, 21). Preorganization of amino acid residues is expected to overcome the intrinsic nucleation barrier and initiate helix formation. On the basis of classical theories, we have developed a general strategy for the nucleation of short peptides in α-helical conformations (10,22). In an R-helix, a hydrogen bond between the C = O group of amino acid residue i and the NH group of amino acid residue i + 4 nucleates the helical structure. To mimic the C = O · H-N hydrogen bond as closely as possible and to preorganize the R-turn, we envisioned a covalent bond of the type CdX-Y-N, where X and Y would be part of residues i and i + 4, respectively (Figure 2a). In our method, the covalent bond between the residues i and i + 4 is a carbon-carbon bond derived from a ring-closing metathesis reaction (Figure 2b) (11, 23). We have shown that this hydrogen bond surrogate (HBS)1 approach affords stable R-helices from a variety of short peptide sequences (10). High-resolution NMR and crystal structures of short HBS R-helices have been reported (10, 24). The crystal structure unambiguously shows that the alkene-based macrocycle faithfully reproduces the conformation of a prenucleated α-turn.
FIGURE 2:
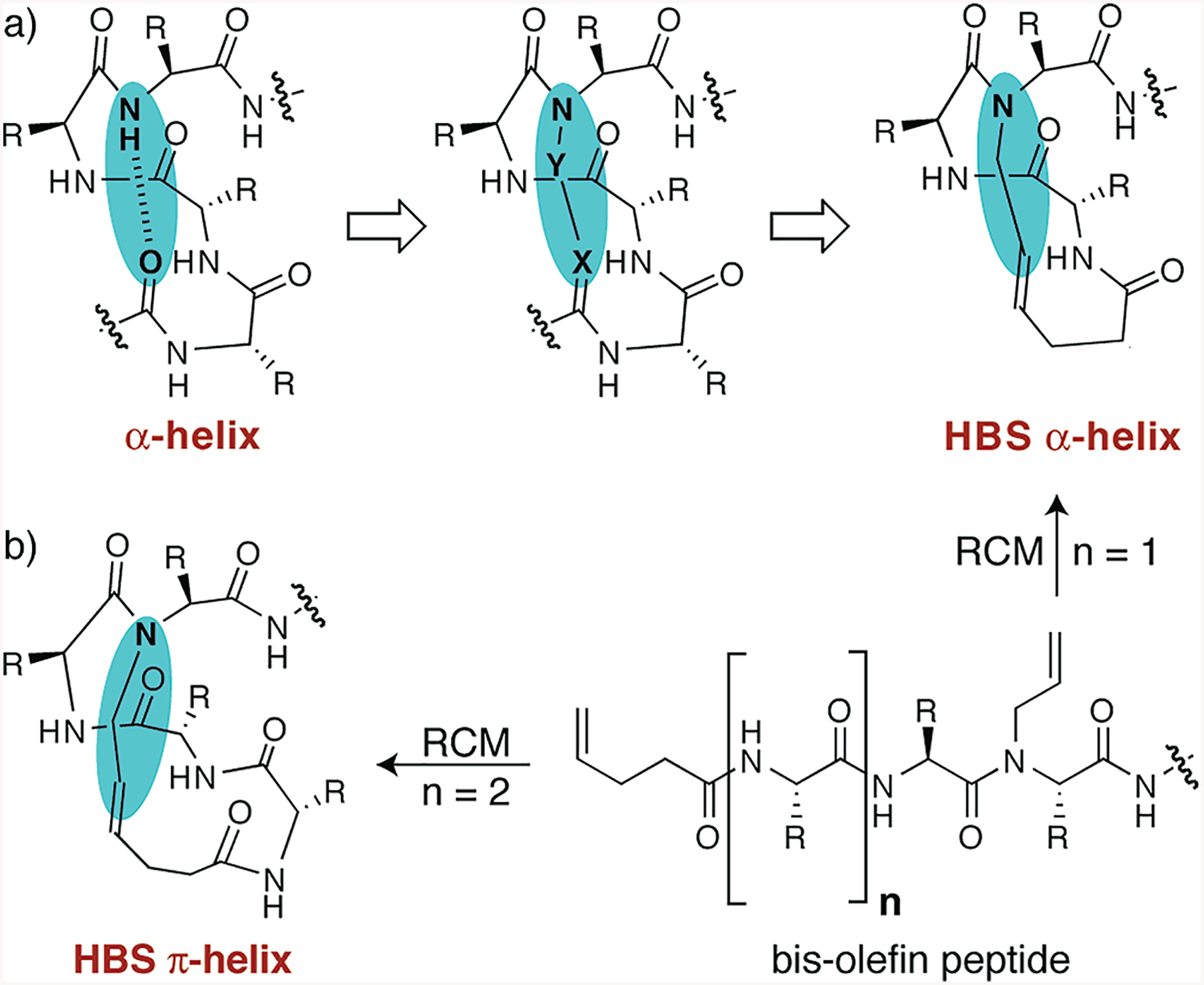
(a) Nucleation of short R-helices by replacement of an N-terminal i and i + 4 hydrogen bond (C=O· ·H−N) with a covalent link (C=X−Y−N). The hydrogen bond surrogate (HBS)-based helices contain a carbon-carbon bond derived from an olefin metathesis reaction. (b) Synthesis of HBS α- and π-helices from a bis-olefin peptide by the ring-closing metathesis (RCM) reaction. R is the amino acid side chain.
Formation of stable helical structures from disordered polypeptides depends on the nucleation and propagation parameters (20, 21, 25). The nucleation parameter is the equilibrium constant for the organization of three consecutive amino acid residues in an R-turn. The value for the nucleation constant is typically very low (10−3-10−4) in unconstrained peptides, disfavoring R-helix formation in short chains (26, 27). The aim of our hydrogen bond surrogate strategy is to prepay the penalty required for the nucleation of a helix and artificially set the σ to a value of g1 (28). As mentioned earlier, formation of the π-helix requires organization of four consecutive residues as opposed to three in an R-helix; this extra residue would render the nucleation process extremely difficult in π-helices (14, 15). Theoretical considerations suggest that the nucleation problem for a π-helix coupled with the requirement for each residue to adopt disfavored dihedral angles renders the formation of a π-helix in short peptides highly unlikely (6, 9, 14). The success of the HBS strategy for stabilization of the R-helices suggested a promising approach for nucleation of the intractable π-helical configuration.
MATERIALS AND METHODS
Peptide Synthesis.
All HBS helices (1–3) and linear peptides (4 and 5) were synthesized and purified as described previously (10, 11, 23); details are included in the Supporting Information.
Circular Dichroism.
CD spectra were recorded on AVIV 202SF CD spectrometer equipped with a temperature conroller using 1 mm length cells and a scan speed of 5 nm/min. The spectra were averaged over 10 scans with the baseline subtracted from buffer scans identical to those for the peptide samples. Samples were prepared in 2 mM potassium phosphate buffer (pH 6.0) containing 20% trifluoroethanol with a final peptide concentration of 50 μM. The concentrations of unfolded peptides were determined by the UV absorption of tyrosine residue at 275 nm in 6.0 M aqueous guanidinium chloride.
NMR.
NMR experiments were performed as described previously in 50 mM potassium phosphate buffer (pH 6.0) containing 20% trifluoroethanol (10). Spectra were recorded on a Varian INOVA 600 apparatus over a temperature range from 0 to 60 °C in 10 °C increments as indicated. NMR spectra were collected by using 64000 real data points and 32 scans averaged over a spectral width of 6400 Hz by using a one-dimensional Watergate or presaturation pulse sequence. Coupling constants were read directly from the peak splitting of amide protons or α-protons with simultaneous decoupling of β -protons. No window function except line broadening (lb = 0.1–0.5) was applied to the original free induction decay before Fourier transformation. The coupling constants were derived directly from the deconvolution function provided in VNMR version 6.3 (Varian). Two-dimensional (2D) rotating-frame Overhauser effect spectroscopy-NOESY measurements were carried out over a temperature range from 0 to 60 °C in 10 °C increments by using a mixing time of 250 or 450 ms. The resulting 2D data sets were processed with VNMR version 6.3
NOE-Restrained Dynamics.
The solution structure of HBS π-helix 3 was computed using a typical simulated annealing molecular dynamics protocol followed by energy minimization in XPLOR-NIH (29, 30). The protein force field (protein.par) was applied to three different starting conformations: π-helix ((φ = −55°; ψ = −70°), extended strand (φ = −180°; ψ = −180°), and α-helix (φ = −60°; ψ = −40°). A total of 1000 initial conformers were obtained (for each starting conformation) using 27 medium and long-range, 74 sequential, and 55 intraresidue constraints (Table 4 of the Supporting Information). Refined structures were generated and collected until the ensemble’s rmsd reached a plateau value. This occurred within 20 structures for each of the starting conformations. The 20 lowest-energy structures from different starting conformations exhibit a minimal overall deviation (Figure 10 and Table 5 of the Supporting Information). On the basis of the NOE intensities from a 200 ms ROESY experiment, the NOE-derived interproton distance restraints were classified into four distance ranges with upper limits of 3, 4, 5, and 6 Å corresponding to strong, medium, weak, and very weak NOE cross-peak intensities, respectively. These distances were employed using a soft square well function with force constants of 50 kcal mol−1 Å−2 and a maximum force value of 1000 kcal mol−1 Å−2. Pseudoatom corrections were applied as necessary (Table 4 of the Supporting Information). Hydrogen bonding restraints were not included in any part of the structure calculations. The 3JNH-CHR coupling constants for all residues except A3 (due to its lack of amide hydrogen) were measured, and the φ angles were set to −55°and allowed to rotate ±30°(31, 32). An implicit solvent was used for the simulated annealing and energy minimization using a distance-dependent dielectric and no cutoffs for electrostatic interactions. Structures were analyzed with Pymol (33), MOLMOL (34), and InsightII (35).
RESULTS
Synthesis of a HBS π-helix requires a 16-member macrocycle between residues i and i + 5 that can be readily achieved through our synthetic strategy (Figure 2b and the Supporting Information) (11). Examination of the π-turns in the Protein Data Bank (5) and theoretical studies (8) on alanine-rich peptides suggest that some amino acid residues (such as asparagine and glutamine) may have a stronger propensity for the π-conformation than others. However, we reasoned that to fully evaluate the success of our nucleation strategy, we should utilize an unbiased peptide sequence. Accordingly, we sought to convert a stable and well-characterized HBS α-helix to an HBS π-helix by enlarging the 13-member nucleation macrocycle in the HBS R-helix by one residue to a 16-member nucleation macrocycle (Figure 2b).
We have previously described the structure and stability of HBS R-helix 1, with a sequence derived from a naturally occurring α-helix (10). To test the viability of the hydrogen bond surrogate approach for stabilizing a π-configuration, we inserted an alanine residue into the macrocycle which yielded HBS π-helix 2 (Table 1, and Figure 6 of the Supporting Information). HBS R-helix 1 contains arginine 4 and glutamic acid 8 residues positioned for a potential i-i + 4 salt bridge. The i-i + 4 salt bridges are known to stabilize α-helices but are also properly spaced to form within a π-helix (36, 37); an i-i + 5 salt bridge should favor a π-helix over an α-helix (38). HBS π-helix 2 contains a 16-member nucleation macrocycle with arginine 5 and glutamic acid 9 residues positioned to form a potential i-i + 4 salt bridge. To gauge the effect of this side chain ionic interaction on the stability of the π-helical conformation, we prepared HBS π-helix 3 by exchanging two appropriate residues at the C-terminus of the peptide sequence. Peptide 3 features a 16-member HBS macrocycle with the arginine 5 and glutamic acid 10 residues positioned for a potential i-i + 5 salt bridge. We expected 3 to form a more stable π-helix than 2 because of the potentially superior ionic side chain interaction in 3 (37). We also prepared and studied linear peptides 4 and 5, featuring potential i-i + 4 and i-i + 5 salt bridges, respectively, as controls. The complete set of sequences and structures is described in Table 1.
Table 1:
Design and Sequences of Peptides and the Experimentally Observed Conformation
| compound | sequencea | composition | experimental resultb |
|---|---|---|---|
| 1 | 13-member HBS ring; i−i + 4 salt bridgec | α-helix | |
| 2 | 16-member HBS ring; i−i + 4 salt bridgec | multiple conformations | |
| 3 | 16-member HBS ring; i−i + 5 salt bridgec | π-helix | |
| 4 | AcQVARQLAEIY-NH2 | unconstrained peptide; i−i + 4 salt bridgec | unstructured |
| 5 | AcQVAARQLAIEY-NH2 | unconstrained peptide; i−i + 5 salt bridgec | unstructured |
X denotes the pentenoic acid residue in the HBS macrocycle.
Experimental results as determined by NMR and circular dichroism spectroscopies.
Potential ionic interaction between the arginine and glutamic acid side chains.
Table 1 also summarizes the results we obtained for compounds 1–5 from examination of extensive NMR and circular dichroism spectra. We began by determining the conformation of individual peptides in 0–20% trifluoroethanol (TFE) in phosphate buffer (pH 6.0) at −5 °C with NOESY and TOCSY experiments. Aggregation of peptides in purely aqueous solutions necessitated addition of an organic cosolvent (10). TFE is known to stabilize helical conformations in peptides and should help us to trap the difficult π-conformation (39, 40). We considered a compound to be unstructured under the experimental conditions if its NOESY spectrum contained very few or no NH-NH cross-peaks. Unconstrained peptides 4 and 5 provided such featureless NOESY spectra (data not shown). CD spectroscopy corroborated our NMR results; spectra of 4 and 5 are consistent with these peptides being unstructured or slightly helical (Figure 9 of the Supporting Information).
We were encouraged to find that HBS peptides 2 and 3 are structured according to CD spectroscopy (Figure 3). HBS helices 2 and 3 display double minima at 204 and 218 nm and maxima at 190 nm in 20% TFE/phosphate buffer. CD spectroscopy suggests that 2 and 3 are more helical in the 20% TFE solution than in aqueous phosphate buffer or organic solvents such as methanol and acetonitrile (Figure 20 of the Supporting Information). While the CD spectra are clearer in the presence of TFE, there is clear evidence of helix formation in aqueous solution. Circular dichroism spectroscopy provides compelling evidence that the hydrogen bond surrogate approach can stabilize helical structure in 2 and 3. However, as opposed to the well-characterized CD traces for R-helices, the expected CD spectrum for a π-helix is not well-established (12, 41). Consequently, CD spectroscopy may not be used to endorse these peptides as π-helices.
FIGURE 3:
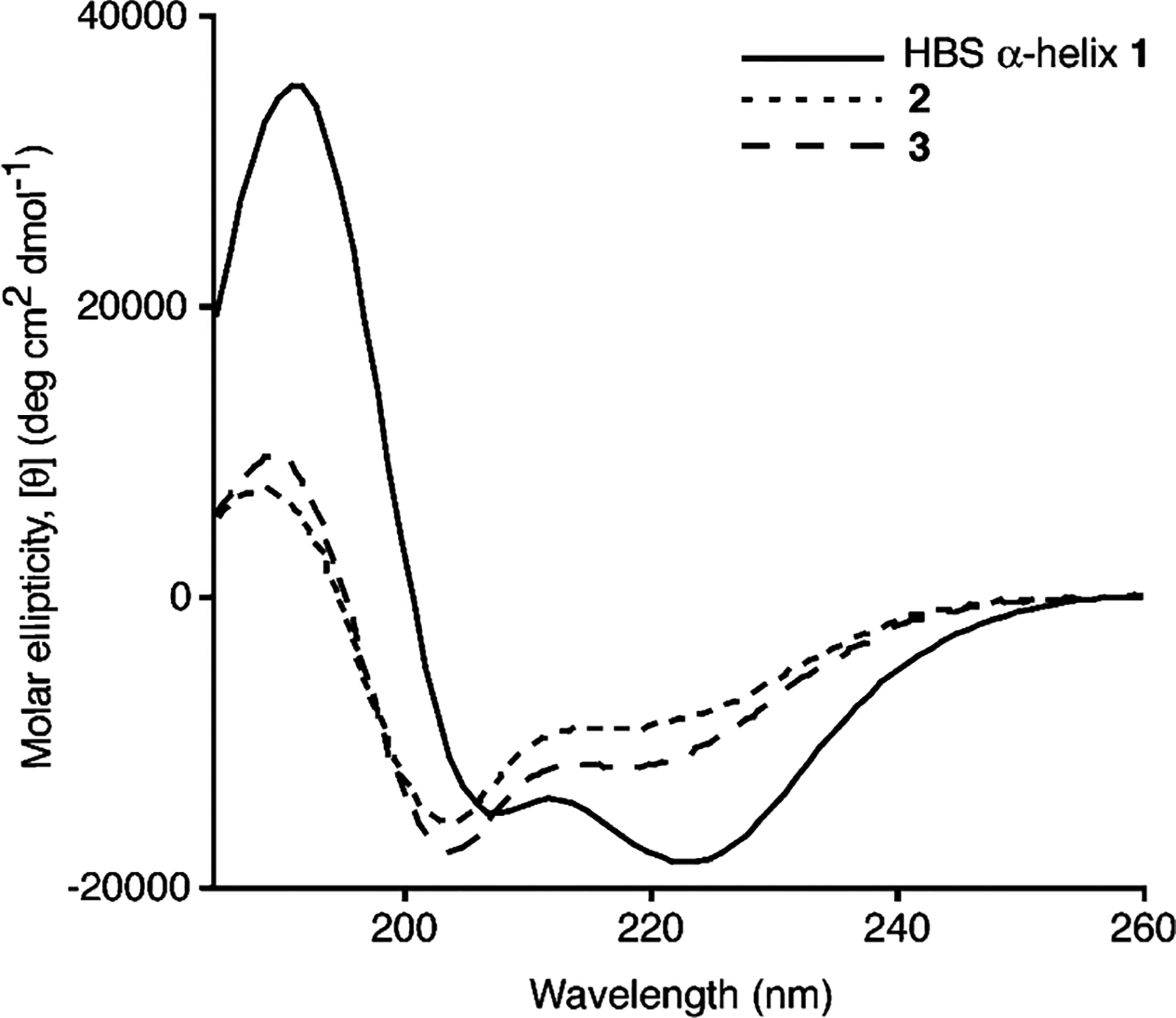
Circular dichroism spectra of HBS R-helix 1 and HBS peptides 2 and 3. The CD spectra were recorded in 20% TFE in phosphate buffer.
2D NMR studies suggest that HBS 2 exists as an interconverting mixture of two dominant conformations at −5 °C (data not shown). The complexity of the NMR spectrum of 2 precluded us from assigning a sufficient number of cross-peaks. The NOESY spectrum of 3, however, could be assigned in detail at −5 °C. A combination of COSY, TOCSY, NOESY, and ROESY spectra revealed that a single conformation predominates for HBS 3 in 20% trifluoroethanol (TFE) in phosphate buffer at −5 °C. The NOE correlation chart for 3 is shown in Figure 4a, and the representative NOESY, TOCSY, and ROESY spectra are included in the Supporting Information. Figure 4 also shows the most important short- and medium-range NOEs that would be expected for the α- and π-helices. Sequential NN (i and i + 1) NOESY cross-peaks are considered to be essential signatures of helical structure and should be observed for both the α- and π-helices. Figure 4b shows the expected NOEs for an R-helical peptide. Expected mediumrange NOEs for an α-helix include dαN(i, i + 3), dαN(i, i + 4), and dαβ (i, i + 3). The π-helix contains one extra residue in the helical turn, and the NOESY spectrum would be expected to exhibit dαN(i, i + 4), dαN(i, i + 5), and dαβ (i, i + 4) NOEs (Figure 4c).
FIGURE 4:
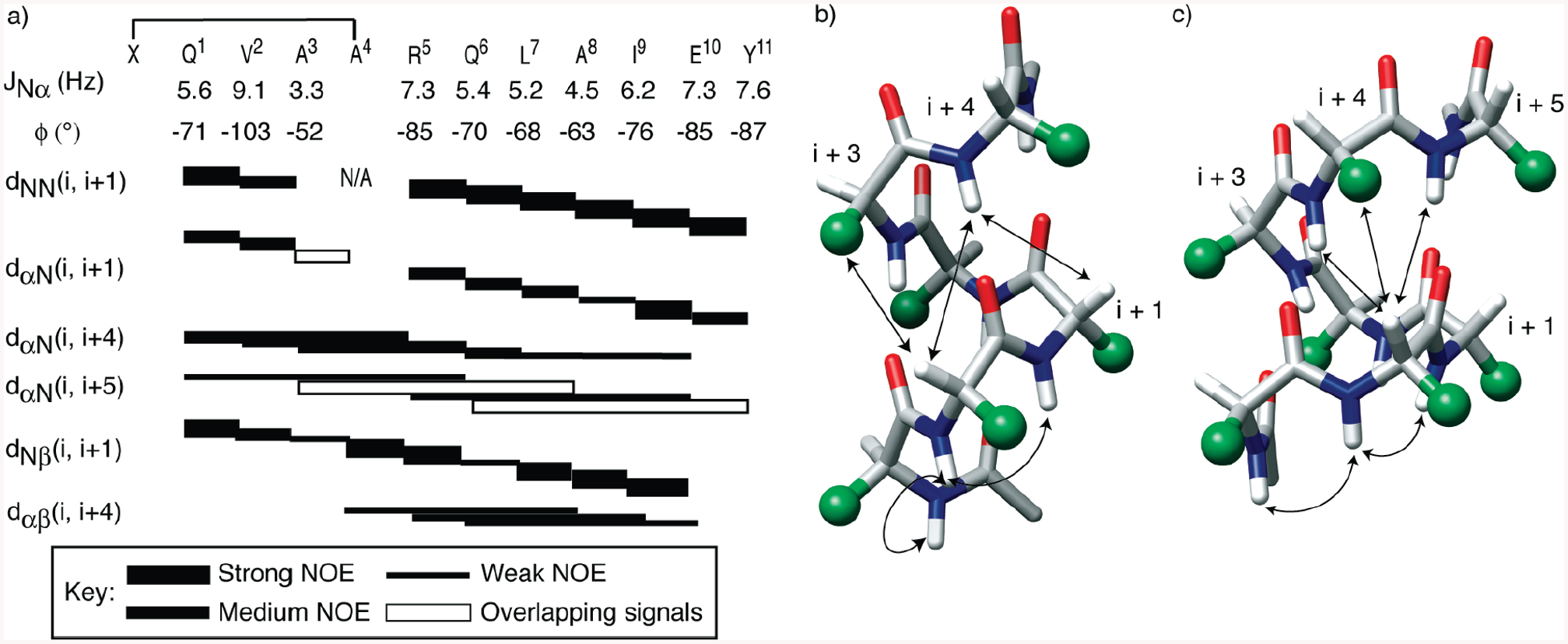
(a) Representative NOESY correlation chart for 3. The complete list of NOE assignments is included in the Supporting Information. The alanine 4 residue in 3 is N-alkylated. Filled rectangles depict the relative intensities of the NOE cross-peaks. Empty rectangles indicate NOEs that could not be unambiguously assigned because of overlapping signals. The chart also lists the 3JNH-CαH coupling constant for each residue and the calculated φ angles (31, 42). (b and c) Medium-range NOEs expected from an idealized (b) R-helix and (c) π-helix. Amino acid side chains are represented as green spheres.
Sequential NN (i and i + 1) NOESY cross-peaks were observed for HBS 3 as shown in Figure 4a; while spectral overlap prevented assignment of some cross-peaks, the NOESY spectrum reveals a series of sequential mediumrange NOEs that provide unequivocal evidence of π-helical structure. The fact that we can detect dαN(i, i + 4) and dαN(i, i + 5) but not dαN(i, i + 3) allows us to term HBS 3 a π-helix with confidence. The clear presence of several sequential dαβ (i, i + 4) cross-peaks outside the macrocycle strongly implicates a π-conformation beyond the HBS macrocycle. The sequential NN (i and i + 1) cross-peaks throughout the sequence and the helical CD signature allow us to rule out predominance of any β-turn type conformation. Since we observe NOEs involving the last residues, the helix has not started to significantly fray near the C-terminus. The complete set of NOEs observed for 3, including relative intensities of NOE cross-peaks, is listed in Table 4 of the Supporting Information.
The 3JNH-CHα coupling constant provides a measure of the φ angle and, thereby, the local conformation (42). The 3JNH-Hα values for α-helices typically range between 4 and 6 Hz (−70 < φ < −30), and a series of three or more coupling constants in this range are indicative of R-helical structure. The proposed dihedral (φ and ψ) values for model π-helices (−57° and −70°, respectively) (43, 44) are significantly different from the average dihedral values observed (−76 ± 25° and −41 ± 24°, respectively) in protein crystal structures (5) or seen in molecular dynamics calculations (−77°and −54°, respectively) (7). The calculated φ angles, derived from 1H NMR coupling constants, for HBS π-helix 3 are listed in Figure 4a (31, 42). All coupling constants and the φ angles for 3 fall in the range observed for π-helices in crystal structures with the exception of the value for valine 2. However, the valine residue is part of the macrocycle and is likely to be strained (10).
We utilized the NOE data and 3JNH-CHα coupling constants to determine the solution structure of HBS π-helix 3 using simulated annealing and the energy minimization protocol in XPLOR-NIH (29, 30). A total of 147 NOE restraints (27 medium- and long-range, 74 sequential, and 55 intraresidue) and 10 φ angle restraints were used (Figure 4a). There were no significant distance violations (Tables 4 and 5 of the Supporting Information). The ensemble of 20 conformers obtained for the peptide shows a backbone root-mean-square deviation (rmsd) of 0.33 (0.15 Å and all heavy atom rmsd of 1.03 (0.20 Å (Figure 5a, and Table 5 of the Supporting Information). The lowest-energy structure reveals a hydrogen bonding network along the backbone in an i and i + 5 configuration consistent with a well-defined π-helix (Figure 5b). Because of overlapping peaks, we could not fully delineate NOESY cross-peaks involving the hydrocarbon cross-link within the macrocycle and thus consider this region in the NMR-derived structure to be a semiqualitative model. The C-terminal glutamic acid and tyrosine residues appear flexible and show some unwinding. Interestingly, the structure suggests that the C-terminal primary amide group may form an i · i + 4 hydrogen bond and serve as an R-turn type C-cap in HBS π-helix 3 (45).
FIGURE 5:
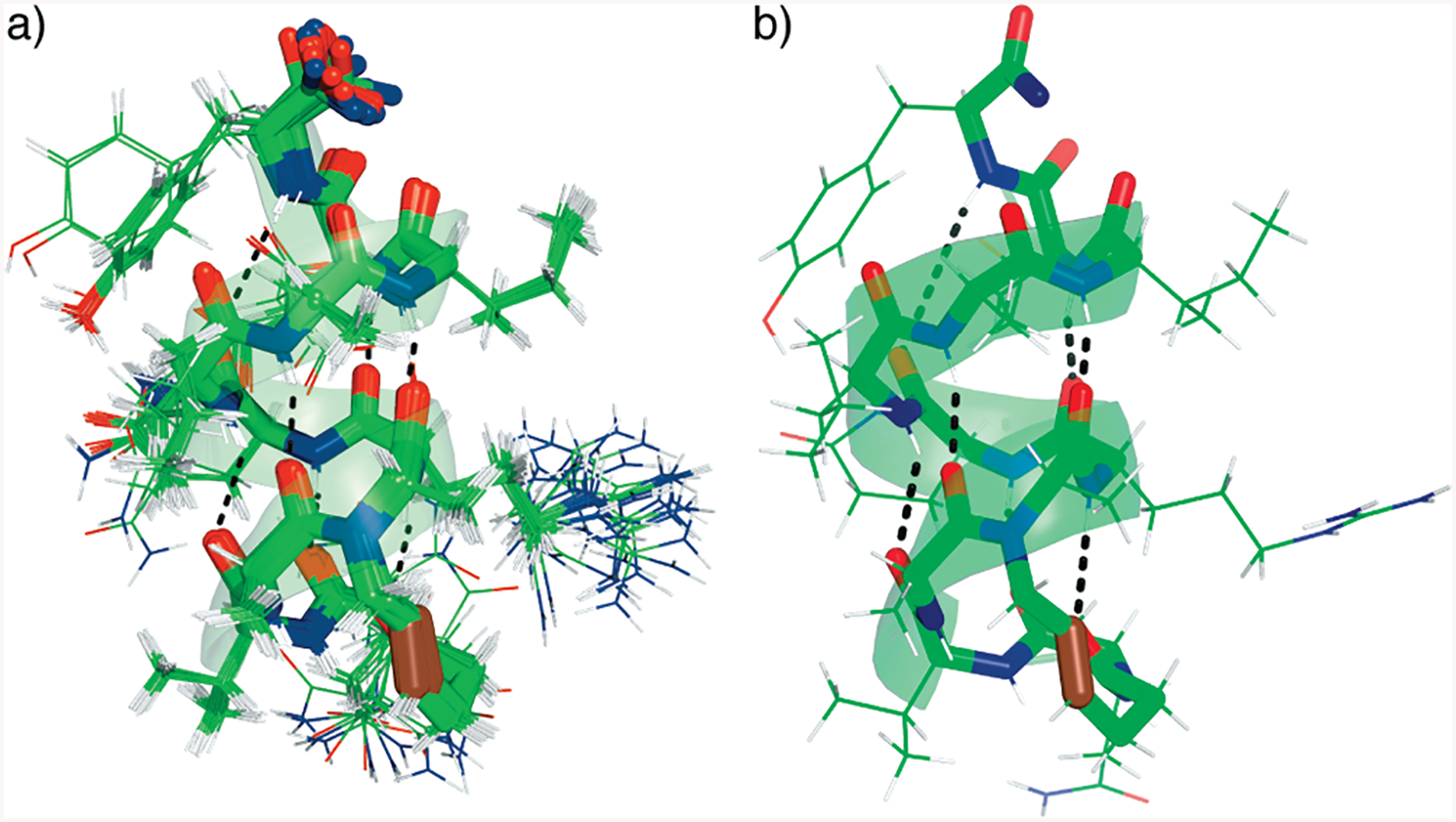
NMR-derived structures of HBS π-helix 3. (a) Twenty lowest-energy structures and (b) structure showing the hydrogen bonding pattern within the artificial π-helix. All carbon, nitrogen, and oxygen atoms are colored green, blue, and red, respectively, with the exception of the trans-alkene group which is colored maroon.
The 2D NMR studies on HBS 3 suggest that the authentic π-helix configuration predominates at −5 °C in aqueous phosphate buffer containing 20% trifluoroethanol. The contiguous dαN(i, i + 4), dαβ (i, i + 4), and dαN(i, i + 5) NOE cross-peaks provide key evidence of this structure. Nucleation of the π-helix removes a major barrier to the formation of this configuration; however, nucleation is only one of the proposed reasons for the inherent difficulty in stabilization of a π-helix. The HBS approach prepays the nucleation penalty for the formation of a π-helix; in reported studies with α-helices, we have detailed the exceptional conformational stability endowed by the HBS template on the attached peptide (10, 22, 28). However, because of the disfavored dihedral angles and the expected 1 Å hole in the middle of the helix, we expected the stability of the nucleated π-helix to be especially susceptible to the peptide sequence and the environment.
We employed several means to explore the stability of HBS π-helices. The observation that switching of a single potential i-i + 5 salt bridge in 3 to an i-i + 4 salt bridge (HBS 2) leads to a mixture of at least two interconverting conformations suggests the stabilizing role of appropriately placed side chain interactions (8, 38). We conjecture that the second conformation in 2 corresponds to an R-helical structure stabilized by the i-i + 4 salt bridge, as the π-helix has been proposed as an intermediate in the unfolding of R-helices (6, 9, 14). The similarity of the CD traces of 2 and 3 strongly suggests contributions from helical configurations (Figure 3b) (41).
We also investigated the stability of HBS π-helix 3 using thermal denaturation experiments (see the Supporting Information for details). The NMR thermal denaturation experiments suggested that a second well-defined conformation is populated as the temperature is increased from −5 to 55 °C. We evaluated TOCSY, NOESY, and ROESY spectra at regular intervals between these temperature ranges and found that a second conformation starts competing with the π-helix structure in 3 as the temperature is increased (Figures 17 and 18 of the Supporting Information). At 55 °C, we found the two conformations to be present in equal proportions. Due to chemical exchange effects in the NMR, we could not fully define this second conformation that equilibrates with the π-helix at high resolution. We observed sequential NN (i and i + 1) NOESY cross-peaks for this conformation, which implicate a helical structure. We, again, hypothesize that the second conformation corresponds to an R-helical configuration. The proposed α-helical conformation starts competing with the π-conformation at high temperatures in 3, whereas the two conformations are nearly equally populated even at −5 °C in HBS peptide 2. The additional stability of 3 potentially reflects the formation of an i-i + 5 salt bridge between the arginine 5 and glutamic acid 10 residues.
We further explored the thermal stability of HBS π-helix 3 by circular dichroism spectroscopy (Figure 19 of the Supporting Information). Interestingly, the CD spectra of 3 do not change considerably as the temperature is increased from 5 to 95 °C. This result is consistent with our hypothesis that at higher temperatures, the π-helix does not completely unravel but rather equilibrates with a second helical configuration, namely, R-helix. The CD spectra of 2 and 3 reflect a mixture of helical configurations, implying that the CD signature of the π-helix is similar to that of the α-helix.
DISCUSSION
The thermal instability of the π-helix 3 is consistent with the expectation of the π-helix as a metastable conformation. On the basis of our assumption that the nucleated π-helix competes with α-helix at high temperatures, we can evaluate the stability of a prenucleated π-helix. At −5 °C, the ratio of the two helical states is 8:2 on the basis of TOCSY cross-peak volumes, π-helix being the major conformation. The cross-peak volumes become roughly equal at 55 °C. HBS 3 contains seven residues (RQLAIEY-NH2) beyond the HBS macrocycle that would be expected to equilibrate. Using simple arguments, ΔG = −RT ln[f/(1 − f)], where f is the fractional π-helicity, we estimate the stability of the π-conformation as compared to that of the competing α-structure in 3 to be roughly 1.0 kcal/mol. The propagation constant in the π-helix can be obtained from standard Zimm–Bragg theory (21), in which the fraction helicity, f, of a peptide composed of n residues can be expressed as a function of the nucleation (σ) and propagation (s) constants. We find that the mean propagation constant for π-helix 3, sπ, is 1.13. This value is not very different from the s value in a nonnucleated α-helix (46) and implies that propagation per se is not a barrier to the formation of π-helices in proteins. This calculation assumes that the HBS π-template partially nucleates the α-conformation; more precise analysis will require experiments with additional sequences. HBS π-helix 3 was based on a naturally occurring helix because we wanted to examine the ability of the HBS strategy to nucleate a difficult conformation from an unbiased, mixed sequence. Peptide sequences that favor the π-helix conformation may lead to more stable HBS constructs (5, 38).
In summary, we have succeeded in trapping the π-helix in an unbiased sequence by replacing an N-terminal intramolecular hydrogen bond with a covalent bond. The resulting hydrogen bond surrogate helix displays NOE patterns expected of a π-helix. Our results suggest that the propagation constant for the π-helix is not disfavored relative to the R-helix as might have been expected from the unfavorable dihedral angles the individual residues need to adopt. The fundamental reason for the scarcity of the π-helices in proteins is, thus, the energetically unfavorable nucleation process. These results point to a potential role for the π-helix as an intermediate in the folding of helices.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant RO1 GM073943 (P.S.A.), equipment gants from the NSF (MRI-0116222 and CHE-0234863), and Research Facilities Improvement Grant C06 RR-16572 from the NIH.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Synthesis and characterization of peptides, circular dichroism and 2D NMR spectra, and distance restraints used for the calculation of the NMR structure of 3. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: HBS, hydrogen bond surrogate; NOE, nuclear Overhauser effect; NOESY, nuclear Overhauser enhancement spectroscopy; ROESY, rotational nuclear Overhauser effect spectroscopy; TOCSY, total correlation spectroscopy.
REFERENCES
- 1.Pauling L, Corey RB, and Branson HR (1951) The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. U.S.A 37, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Low BW, and Baybutt RB (1952) The π-Helix: A Hydrogen Bonded Configuration of the Polypeptide Chain. J. Am. Chem. Soc 74, 5806–5807. [Google Scholar]
- 3.Donohue J (1953) Hydrogen Bonded Helical Configurations of the Polypeptide Chain. Proc. Natl. Acad. Sci. U.S.A 39, 470–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bolin KA, and Millhauser GL (1999) Alpha and 3(10): The split personality of polypeptide helices. Acc. Chem. Res 32, 1027–1033. [Google Scholar]
- 5.Fodje MN, and Al-Karadaghi S (2002) Occurrence, conformational features and amino acid propensities for the π-helix. Protein Eng. 15, 353–358. [DOI] [PubMed] [Google Scholar]
- 6.Armen R, Alonso DO, and Daggett V (2003) The role of R-, 310-, and π-helix in helix→ coil transitions. Protein Sci. 12, 1145–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KH, Benson DR, and Kuczera K (2000) Transitions from R to π helix observed in molecular dynamics simulations of synthetic peptides. Biochemistry 39, 13737–13747. [DOI] [PubMed] [Google Scholar]
- 8.Shirley WA, and Brooks CL (1997) Curious structure in “canonical” alanine based peptides. Proteins: Struct., Funct., Genet 28, 59–71. [DOI] [PubMed] [Google Scholar]
- 9.Sorin EJ, Rhee YM, Shirts MR, and Pande VS (2006) The solvation interface is a determining factor in peptide conformational preferences. J. Mol. Biol 356, 248–256. [DOI] [PubMed] [Google Scholar]
- 10.Wang D, Chen K, Kulp JL III, and Arora PS (2006) Evaluation of biologically relevant short R-helices stabilized by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc 128, 9248–9256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapman RN, and Arora PS (2006) Optimized synthesis of hydrogen-bond surrogate helices: Surprising effects of microwave heating on the activity of grubbs catalysts. Org. Lett 8, 5825–5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan DM, Lynn DG, Miller-Auer H, and Meredith SC (2001) A designed Zn2+-binding amphiphilic polypeptide: Energetic consequences of π-helicity. Biochemistry 40, 14020–14029. [DOI] [PubMed] [Google Scholar]
- 13.Ramachandran GN, and Sasisekharan V (1968) Conformation of polypeptides and proteins. AdV. Protein Chem 23, 283–438. [DOI] [PubMed] [Google Scholar]
- 14.Rohl CA, and Doig AJ (1996) Models for the 310-helix/coil, π-helix/coil, and R-helix/310-helix/coil transitions in isolated peptides. Protein Sci. 5, 1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikhonin AV, and Asher SA (2006) Direct UV Raman monitoring of 310-helix and π-bulge premelting during α helix unfolding. J. Am. Chem. Soc 128, 13789–13795. [DOI] [PubMed] [Google Scholar]
- 16.Weaver TM (2000) The π-helix translates structure into function. Protein Sci. 9, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medlock AE, Dailey TA, Ross TA, Dailey HA, and Lanzilotta WN (2007) A π-helix switch selective for porphyrin deprotonation and product release in human ferrochelatase. J. Mol. Biol 373, 1006–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sazinsky MH, Dunten PW, McCormick MS, DiDonato A, and Lippard SJ (2006) X-ray structure of a hydroxylase regulatory protein complex from a hydrocarbon-oxidizing multicomponent monooxygenase, Pseudomonas sp. OX1 phenol hydroxylase. Biochemistry 45, 15392–15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andrews MJI, and Tabor AB (1999) Forming stable helical peptides using natural and artificial amino acids. Tetrahedron 55, 11711–11743. [Google Scholar]
- 20.Lifson S, and Roig A (1961) On the theory of helix-coil transitions in polypeptides. J. Chem. Phys 34, 1963–1974. [Google Scholar]
- 21.Zimm BH, and Bragg JK (1959) Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys 31, 526–535. [Google Scholar]
- 22.Chapman RN, Dimartino G, and Arora PS (2004) A highly stable short R helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc 126, 12252–12253. [DOI] [PubMed] [Google Scholar]
- 23.Dimartino G, Wang D, Chapman RN, and Arora PS (2005) Solid-phase synthesis of hydrogen-bond surrogate-derived α-helices. Org. Lett 7, 2389–2392. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Wang D, Zheng Q, Lu M, and Arora PS (2008) Atomic Structure of a Short α-Helix Stabilized by a Main Chain Hydrogen Bond Surrogate. J. Am. Chem. Soc (in press). [DOI] [PubMed] [Google Scholar]
- 25.Qian H, and Schellman JA (1992) Helix-Coil Theories: A Comparative Study for Finite Length Preferences. J. Phys. Chem 96, 3987–3994. [Google Scholar]
- 26.Yang JX, Zhao K, Gong YX, Vologodskii A, and Kallenbach NR (1998) α-Helix nucleation constant in co-polypeptides of alanine and ornithine or lysine. J. Am. Chem. Soc 120, 10646–10652. [Google Scholar]
- 27.Matheson RR, and Scheraga HA (1983) Calculation of the Zimm-Bragg Cooperativity Parameter Sigma from a Simple-Model of the Nucleation Process. Macromolecules 16, 1037–1043. [Google Scholar]
- 28.Wang D, Chen K, Dimartino G, and Arora PS (2006) Nucleation and stability of hydrogen-bond surrogate-based α-helices. Org. Biomol. Chem 4, 4074–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwieters CD, Kuszewski JJ, and Clore GM (2006) Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magon. Reson. Spectrosc 48, 47–62. [Google Scholar]
- 30.Schwieters CD, Kuszewski JJ, Tjandra N, and Clore GM (2003) The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson 160, 65–73. [DOI] [PubMed] [Google Scholar]
- 31.Karplus M (1959) Contact Electron-Spin Coupling of Nuclear Magnetic Moments. J. Chem. Phys 30, 11–15. [Google Scholar]
- 32.Pardi A, Billeter M, and Wuthrich K (1984) Calibration of the angular dependence of the amide proton-Cα proton coupling constants, 3JHNα, in a globular protein. Use of 3JHNα for identification of helical secondary structure. J. Mol. Biol 180, 741–751. [DOI] [PubMed] [Google Scholar]
- 33.DeLano WL (2003) PyMol, DeLano Scientific, San Carlos, CA. [Google Scholar]
- 34.Koradi R, Billeter M, and Wuthrich K (1996) MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics Modell 14, 51–55. [DOI] [PubMed] [Google Scholar]
- 35.Insight II, version 2000 (2000) Accelrys, San Diego, CA. [Google Scholar]
- 36.Marqusee S, and Baldwin RL (1987) Helix stabilization by Glu− ·Lys+ salt bridges in short peptides of de novo design. Proc. Natl. Acad. Sci. U.S.A 84, 8898–8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi ZS, Olson CA, Bell AJ, and Kallenbach NR (2001) Stabilization of R helix structure by polar side-chain interactions: Complex salt bridges, cation-π interactions, and C−H· · O H-bonds. Biopolymers 60, 366–380. [DOI] [PubMed] [Google Scholar]
- 38.Sudha R, Kohtani M, Breaux GA, and Jarrold MF (2004) π-Helix preference in unsolvated peptides. J. Am. Chem. Soc 126, 2777–2784. [DOI] [PubMed] [Google Scholar]
- 39.CammersGoodwin A, Allen TJ, Oslick SL, McClure KF, Lee JH, and Kemp DS (1996) Mechanism of stabilization of helical conformations of polypeptides by water containing trifluoroethanol. J. Am. Chem. Soc 118, 3082–3090. [Google Scholar]
- 40.Rajan R, and Balaram P (1996) A model for the interaction of trifluoroethanol with peptides and proteins. Int. J. Pept. Protein Res 48, 328–336. [DOI] [PubMed] [Google Scholar]
- 41.Manning MC, and Woody RW (1991) Theoretical CD Studies of Polypeptide Helices: Examination of Important Electronic and Geometric Factors. Biopolymers 31, 569–586. [DOI] [PubMed] [Google Scholar]
- 42.Wuthrich K (1986) NMR of Proteins and Nucleic Acids, Wiley, New York. [Google Scholar]
- 43.Low BW, and Grenvillewells HJ (1953) Generalized Mathematical Relationships for Polypeptide Chain Helices: The Coordinates of the Ii-Helix. Proc. Natl. Acad. Sci. U.S.A 39, 785–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramachandran GN, and Sasisekharan V (1968) Conformation of polypeptides and proteins. AdV. Protein Chem 23, 283–438. [DOI] [PubMed] [Google Scholar]
- 45.Aurora R, and Rose GD (1998) Helix capping. Protein Sci. 7, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohl CA, Chakrabartty A, and Baldwin RL (1996) Helix propagation and N-cap propensities of the amino acids measured in alanine-based peptides in 40 volume percent trifluoroethanol. Protein Sci. 5, 2623–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.