

Abstract
PD-1, a T cell checkpoint receptor and target of cancer immunotherapy, is also expressed on myeloid cells. The role of myeloid-specific vs. T cell-specific PD-1 ablation on anti-tumor immunity has remained unclear because most studies have used either PD-1 blocking antibodies or complete PD-1 KO mice. We generated a conditional allele, which allowed myeloid-specific (PD-1f/fLysMcre) or T cell-specific (PD-1f/fCD4cre) targeting of Pdcd1 gene. Compared to T cell-specific PD-1 ablation, myeloid cell-specific PD-1 ablation more effectively decreased tumor growth. We found that granulocyte/macrophage progenitors (GMP), which accumulate during cancer-driven emergency myelopoiesis and give rise to myeloid-derived suppressor cells (MDSC), express PD-1. In tumor-bearing PD-1f/fLysMcre but not PD-1f/fCD4cre mice, accumulation of GMP and MDSC was prevented, while systemic output of effector myeloid cells was increased. Myeloid cell-specific PD-1 ablation induced an increase of T effector memory (TEM) cells with improved functionality, and mediated anti-tumor protection despite preserved PD-1 expression in T cells. In PD-1-deficient myeloid progenitors, growth factors driving emergency myelopoiesis induced increased metabolic intermediates of glycolysis, pentose phosphate pathway and TCA cycle but, most prominently, elevated cholesterol. As cholesterol is required for differentiation of inflammatory macrophages and DC, and promotes antigen presenting function, our findings indicate that metabolic reprogramming of emergency myelopoiesis and differentiation of effector myeloid cells might be a key mechanism of anti-tumor immunity mediated by PD-1 blockade.
One sentence summary:
PD-1 ablation regulates metabolism-driven lineage fate commitment of myeloid progenitors and differentiation of effector myeloid cells
Introduction
PD-1 is a major inhibitor of T cell responses expressed on activated T cells. It is also expressed on NK, B, Treg, T follicular helper (TFH) and myeloid cells (1). The current model supports that a key mechanism dampening anti-tumor immune responses is the upregulation of PD-1 ligands in cancer cells and antigen presenting cells (APC) of the tumor microenvironment (TME), which mediate ligation of PD-1 on tumor-infiltrating CD8+ T-cells, leading to the development of T incapable of generating anti-tumor responses (2). Therapeutic targeting of the PD-1 pathway with antibodies blocking the PD-1 receptor or its ligands induces expansion of oligoclonal CD8+ TILs that recognize tumor neoantigens (3). Thus, in the context of cancer, PD-1 is considered a major inhibitor of T effector (TEFF) cells, whereas on APC and cancer cells, emphasis has been placed on the expression of PD-1 ligands. PD-L1 expression in the TME is often a pre-requisite for patient enrolment to clinical trials involving blockade of the PD-1 pathway. However, responses do not always correlate with PD-L1 expression and remains incompletely understood how the components of the PD-1: PD-L1/2 pathway suppress anti-tumor immunity.
Recent studies indicated that PD-1 can be induced by TLR signaling in macrophages (MΦ), and negatively correlates with M1 polarization (4). PD-1 expression in macrophages plays a pathologic role by suppressing the innate inflammatory response to sepsis (5) and inhibiting M.tubeculosis phagocytosis in active tuberculosis (6). Our knowledge about the function of PD-1 on myeloid cells in the context of cancer is very limited. However, similarly to its role in infections, PD-1 expression inversely correlates with M1 polarization and phagocytic potency of tumor-associated MΦ (TAM) against tumor (7, 8). The mechanisms of PD-1 expression in myeloid cells and the role of PD-1-expressing myeloid cells in tumor immunity remain unknown.
The rapid increase in myeloid cell output in response to immunologic stress is known as emergency myelopoiesis. Terminally differentiated myeloid cells are essential innate immune cells and are required for the activation of adaptive immunity. Strong activation signals mediated by pathogen-associated molecular patterns (PAMP) or danger-associated molecular patterns (DAMP) molecules lead to a transient expansion and subsequent differentiation of myeloid progenitors to mature monocytes and granulocytes to protect the host. In contrast, during emergency myelopoiesis mediated by continuous low-level stimulation mediated by cancer-derived factors and cytokines, bone marrow common myeloid progenitors (CMP) but predominantly granulocyte/macrophage progenitors (GMP) undergo modest expansion with hindered differentiation and a fraction of myeloid cells with immunosuppressive and tumor-promoting properties, named myeloid-derived suppressor cells (MDSC), accumulates. MDSC suppress CD8+ T cells responses by various mechanisms (9). In the mouse, MDSC consist of two major subsets, CD11b+Ly6ChiLy6G− (thereafter named CD11b+Ly6C+) monocytic (M-MDSC) and CD11b+Ly6CloLy6G+ (thereafter named CD11b+Ly6G+) polymorphonuclear (PMN-MDSC) (10). These cells have similar morphology and phenotype to normal monocytes and neutrophils but distinct genomic and biochemical profiles (9). In humans, in addition to M-MDSC and PMN-MDSC, a small subset of early stage MDSC (eMDSC) has been identified (10).
Although PMN-MDSC represent the major subset of circulating MDSC, they are less immunosuppressive than M-MDSC when assessed on per cell basis (11–13). Current views support the two-signal requirement for MDSC function. The first signal controls MDSC generation, whereas the second signal controls MDSC activation, depends on cues provided by the TME and promotes MDSC differentiation to TAM (14). Proinflammatory cytokines and ER stress response in the TME contribute to pathologic myeloid cell activation that manifests as weak phagocytic activity, increased production of reactive oxygen species (ROS) and nitric oxide (NO) and expression of arginase-1 (ARG1), and convert myeloid cells to MDSC (9). MDSC are associated with poor outcomes in many cancer types in patients and negatively correlate with response to chemotherapy, immunotherapy and cancer vaccines (15–19).
In the present study, we examined how PD-1 regulates the response of myeloid progenitors to cancer-driven emergency myelopoiesis and its implications on anti-tumor immunity. We determined that myeloid progenitors, which expand during cancer-driven emergency myelopoiesis, express PD-1 and PD-L1. PD-L1 was constitutively expressed on CMP and GMP, whereas PD-1 expression displayed a striking increase on GMP that arose during tumor-driven emergency myelopoiesis. PD-1 was also expressed on tumor-infiltrating myeloid cells, including M-MDSC and PMN-MDSC, CD11b+F4/80+ MΦ and CD11c+MHCII+ dendritic cells (DC) in tumor bearing mice, and on MDSC in patients with refractory lymphoma. Ablation of PD-1 signaling in PD-1 KO mice prevented GMP accumulation and MDSC generation and resulted in increase of Ly6Chi effector monocytes, MΦ and DC. We generated mice with conditional targeting of the Pdcd1 gene (PD-1f/f) and selectively eliminated PD-1 in myeloid cells or T cells. Compared to T cell-specific ablation of PD-1, myeloid-specific PD-1 ablation more effectively decreased tumor growth in various tumor models. At a cellular level, only myeloid-specific PD-1 ablation skewed the myeloid cell fate commitment from MDSC to effector Ly6Chi monocytes, MΦ and DC and induced TEM cells with improved functionality. Our findings reveal a previously unidentified role of the PD-1 pathway and suggest that skewing of myeloid cell fate during emergency myelopoiesis and differentiation to effector antigen presenting cells, thereby reprogramming T cell responses, might be a key mechanism by which PD-1 blockade mediates anti-tumor function.
Results
PD-1 is expressed in myeloid cells during cancer-mediated emergency myelopoiesis.
For our studies, we selected the murine B16-F10 melanoma tumor model because it has been informative in dissecting mechanisms of resistance to checkpoint immunotherapy (20). First, we examined whether B16-F10 induces tumor-driven emergency myelopoiesis similarly to the MC17-51 fibrosarcoma, a mouse tumor model well-established to induce cancer-driven emergency myelopoiesis (21). We assessed expansion of myeloid progenitors in the bone marrow and increase of CD11b+CD45+ myeloid cells in the spleen and tumor (Supplementary Fig. S1 and S2). Both tumor types induced increase of myeloid progenitors in the bone marrow and systemic increase of CD45+CD11b+ myeloid cells (Supplementary Fig. S3), providing evidence that B16-F10 melanoma is an appropriate tumor model to study tumor-driven emergency myelopoiesis and its consequences in tumor immunity. In the spleen of non tumor-bearing mice, few myeloid cells constitutively expressed very low levels of PD-L1 whereas PD-1 was very low to undetectable (Fig. 1A, B). In B16-F10 tumor-bearing mice, expression of PD-1 and PD-L1 was upregulated on myeloid cells of the spleen (Fig. 1C–F). PD-1 and PD-L1 were also expressed on myeloid cells at the tumor site (Fig. 1G–I). Notably, all subsets of myeloid cells expanding in tumor-bearing mice including M-MDSC, PMN-MDSC, CD11b+F4/80+ MΦ and CD11c+MHCII+ DC, expressed PD-1 (Fig. 1D, G). Kinetics studies of PD-1 expression on myeloid cells in the spleen of tumor-bearing mice showed a gradual increase overtime (Fig.1 J–M).
Figure 1: PD-1 and PD-L1 are expressed on myeloid cells that expand in tumor-bearing mice.
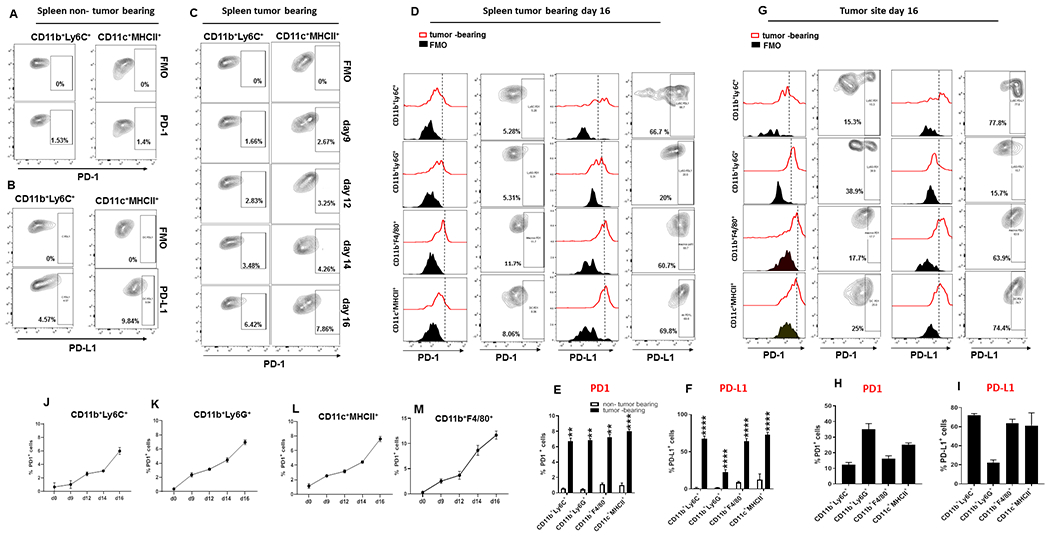
(A-B) Expression of PD-1 and PD-L1 on CD11b+Ly6C+ monocytes and CD11c+MHCII+ DC in the spleen of non-tumor bearing C57BL/6 mice. (C) C57BL/6 mice were inoculated with B16-F10 mouse melanoma and at the indicated time points, expression of PD-1 was examined by flow cytometry in the spleen after gating on the indicated myeloid populations; contour plots depicting % positive cells are shown. On day 16 after tumor inoculation, expression of PD-1 and PD-L1 was assessed in the spleen (D) and the tumor site (G) after gating on the indicated myeloid populations. (D, G) FACS histograms and contour plots depicting % positive cells, and bar graphs (E, F, H, I) of mean ± SEM positive cells. Results are representative of 12 independent experiments with 6 mice per group. (J-M) Kinetics of PD-1 upregulation on CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+ and CD11c+MHCII+ of the spleen after tumor inoculation.
Because myeloid cells that give rise to MDSC and TAM are generated from myeloid progenitors in the bone marrow during tumor-driven emergency myelopoiesis, we examined PD-1 and PD-L1 expression in these myeloid progenitors. In non-tumor bearing mice, PD-1 was detected at very low levels on GMP (Fig. 2A) whereas PD-L1 was constitutively expressed in CMP but mostly on GMP (Fig. 2B). In tumor-bearing mice, PD-L1 was upregulated in CMP and GMP and its expression levels remained elevated during all assessed time points (Fig. 2F–J). Strikingly, PD-1 expression was induced on CMP but more prominently GMP (Fig. 2C–I). Kinetics studies showed that PD-1 expression on GMP peaked early after tumor inoculation (Fig. 2C, E, I), at a time point when tumor growth was not yet measurable. Thus, induction of PD-1 expression in myeloid progenitors is an early event during tumor development.
Figure 2: PD-1 and PD-L1 is expressed on CMP and GMP myeloid progenitors during cancer-driven emergency myelopoiesis.
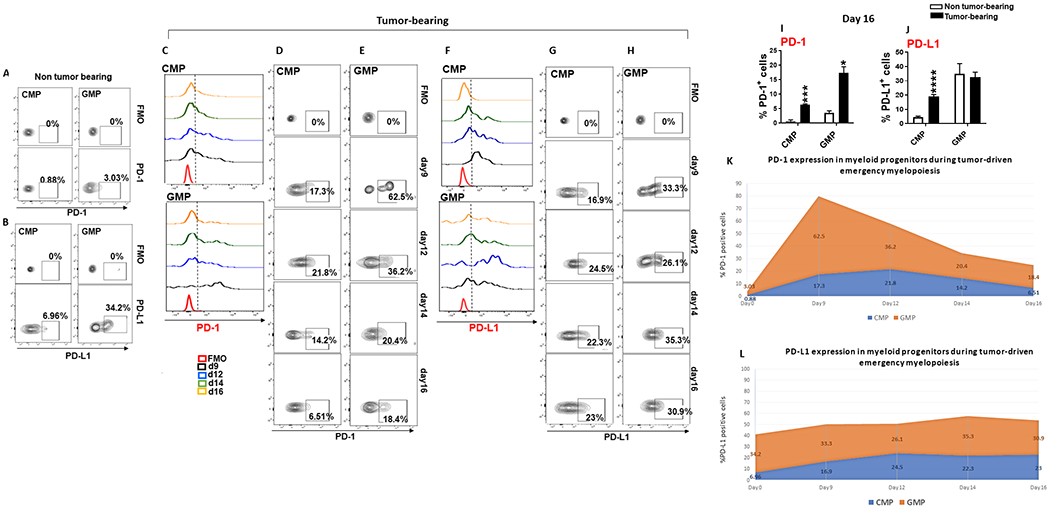
(A-B) Expression of PD-1 and PD-L1 on CMP and GMP of non-tumor bearing mice. (C-J) C57BL/6 mice were inoculated with B16-F10 mouse melanoma and expression of PD-1 and PD-L1 on CMP and GMP was examined on days 9, 12, 14 and 16 after implantation. FACS histograms (C, F), contour plots (D, E, G, H) indicating % positive cells and bar graphs of mean ± SEM positive cells (I, J) are shown. Results are representative of 4 independent experiments with 6 mice per group. (K, L) Kinetics of PD-1 (K) and PD-L1 (L) expression on CMP (blue) and GMP (orange) during tumor-driven emergency myelopoiesis. Results are representative of four separate experiments with 6 mice per group.
To determine whether PD-1 expression on GMP was mediated by growth factors regulating emergency myelopoiesis, we cultured bone marrow cells from non-tumor bearing mice with G-CSF, GM-CSF and the TLR4 ligand LPS. PD-1 that was constitutively expressed at low levels in GMP, was upregulated by culture with each of these factors (Supplementary Fig. S4A), consistent with our findings that PD-1 expression was rapidly induced on GMP of tumor bearing mice in vivo (Fig. 2C, E, I). Quantitative PCR in purified Linneg bone marrow cells showed that PD-1 mRNA was constitutively expressed in myeloid progenitors and was upregulated by culture with G-CSF or GM-CSF (Supplementary Fig. S4B). Together these in vivo and in vitro studies provide evidence that PD-1 expression on myeloid progenitors is regulated by a direct cell-intrinsic effect of factors driving cancer-mediated emergency myelopoiesis.
To examine weather PD-1 was expressed in MDSC in humans, we used samples from healthy donors and patients with malignant non-Hodgkin’s lymphoma (NHL) (Supplementary Fig. S5 and Fig. S6). A high level of PD-1-expressing M-MDSC was detected in the peripheral blood of three patients with treatment-refractory NHL but not in two patients who responded to treatment or five healthy donors (Supplementary Fig. S6). These results show that PD-1 expression is detected in human MDSC and serve as a paradigm suggesting that PD-1 expression in MDSC of cancer patients might be a clinically relevant event.
PD-1 ablation alters emergency myelopoiesis and the profile of myeloid cell output.
To examine whether PD-1 might have an active role in tumor-induced stress myelopoiesis, we used PD-1 deficient (PD-1−/−) mice. PD-1 deletion, which resulted in decreased tumor growth (Fig. 3A, B), significantly altered tumor-induced stress myelopoiesis (Fig. 3C–E). Although accumulation of CMP was comparable, accumulation of GMP was significantly diminished in PD-1−/− mice (Fig. 3C, D), indicating that GMP might be a key target on which PD-1 mediated its effects on myeloid progenitors (Fig. 3E). Kinetics studies showed sustained GMP expansion in WT tumor-bearing mice. In contrast, in PD-1−/− tumor-bearing mice, GMP displayed a rapid expansion and subsequent decline (Supplementary Fig. S7). In parallel, in PD-1−/− mice there was an increase of differentiated CD11b+Ly6Chi monocytic cells in the tumor (Fig. 3H) but also in the spleen and the small intestine, which also displayed an increase in CD11c+MHCII+ DC (Fig. 3F and G). Moreover, at these sites, there was a significant increase of the CD11b+Ly6C+/CD11b+Ly6G+ ratio (Fig. 3I–K) indicating a shift of myelopoiesis output toward monocytic lineage dominance. Notably, these Ly6Chi monocytes, CD11b+F4/80+ MΦ and CD11c+MHCII+ DC in PD-1−/− tumor-bearing mice expressed IRF8, and all myeloid subsets had elevated expression of the retinoic acid receptor-related orphan receptor gamma (RORC or RORγ) (Fig. 3L–N and Supplementary Fig. S8). Similar results were observed in two additional tumor models, the MC38 colon adenocarcinoma and the MC17-51 fibrosarcoma model (Supplementary Fig. S9) both of which induced cancer-driven emergency myelopoiesis (Supplementary Fig. S3).
Figure 3. PD-1 ablation alters emergency myelopoiesis and the profile of myeloid cell output.
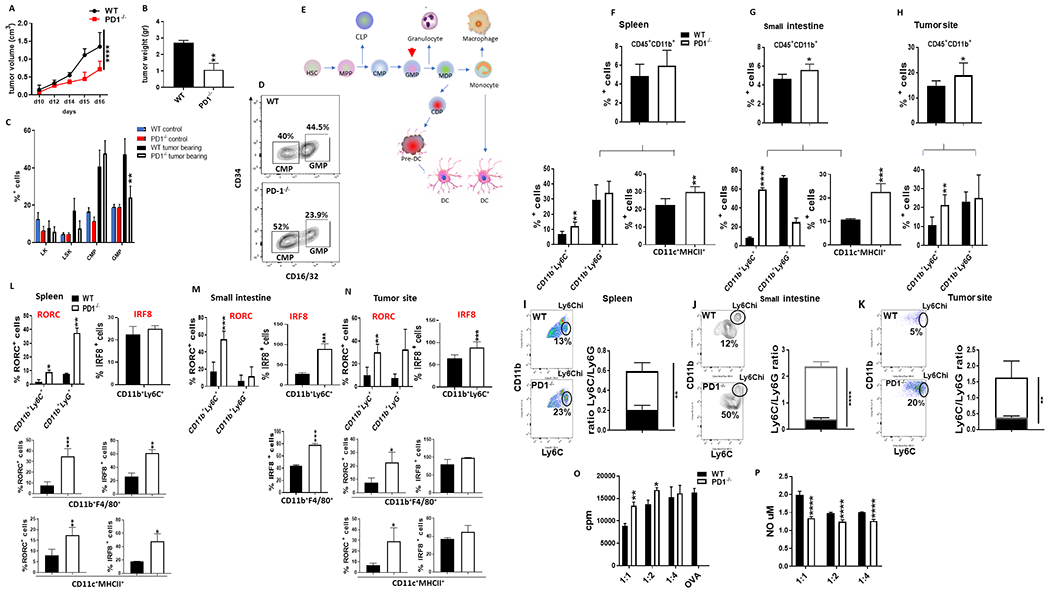
(A, B) WT and PD-1−/− mice were inoculated with B16-F10 melanoma and tumor size was monitored daily (A). Mice were euthanized on day 16 and tumor weight was measured (B). Data shown are the mean ± SEM of 6 mice/group and are representative of six independent experiments. (C) Mean percentages ± SEM of LK, LSK, CMP and GMP in the bone marrow of non tumor-bearing and tumor-bearing WT and PD-1−/− mice. GMP in PD-1−/− mice were significantly lower compared to GMP in WT mice (** p< 0.01). (D) Representative contour plots of FACS analysis for CMP and GMP in the bone marrow of tumor-bearing WT and PD-1−/− mice. (E) Schematic presentation of myeloid lineage differentiation. Arrowhead indicates GMP, the key target population of PD-1 during emergency myelopoiesis. (F-H) Mean percentages of CD45+CD11b+, CD11b+Ly6C+, CD11b+Ly6G+ and CD11c+MHCII+ in the spleen (F), small intestine (G), and B16-F10 site (H) of tumor-bearing WT and PD-1−/− mice. (I-K) Representative plots of FACS analysis for CD11b+Ly6Chi and CD11b+Ly6C+/ CD11b+Ly6G+ ratio in the spleen (I), small intestine (J) and B16-F10 site (K). (L-N) Mean percentages ± SEM of RORC and IRF8 expressing CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+ and CD11c+MHCII+ myeloid cells within the CD45+CD11b+ gate in spleen (L), small intestine (M), and B16-F10 site (N). Data from one representative experiment out of three independent experiments with 6 mice per group is shown. (O-P) Diminished suppressive activity (O) and NO production (P) of CD11b+Ly6C+ cells isolated from PD-1−/− tumor bearing mice. CD11b+Ly6C+ cells were isolated from tumor-bearing WT and PD-1−/− mice and cultured at various ratios with OTI splenocytes stimulated with OVA257-264. Data show the mean ± SEM of one representative of two experiments (*p<0.05, **p<0.01, ***p<0.001). HSC, hematopoietic stem cells; CMP, common myeloid progenitor; GMP, granulocyte and macrophage progenitor.
IRF8 regulates myeloid cell fate to monocyte/macrophage and DC differentiation vs. granulocyte differentiation (22, 23), explaining the increase of CD11b+Ly6C+/CD11b+Ly6G+ ratio that we observed in tumor-bearing PD-1 KO mice. IRF8 is designated as one of the “terminal selectors” that control the induction and maintenance of the terminally differentiated state of these myeloid cells (22, 23). Moreover, IRF8 shifts the fate of myeloid cells away from immature MDSC, which are characterized by a restriction in IRF8 expression (24, 25). Retinoid related orphan nuclear receptors are required for myelopoiesis and are mediators of the inflammatory response of effector Ly6Chi monocytes and macrophages (21, 26) but can also be expressed by MDSC (21). For these reasons, we examined the functional properties of CD11b+Ly6C+ cells in PD-1−/− tumor bearing mice. A key mechanism by which CD11b+Ly6C+ M-MDSC mediate suppression of T cell responses involves the production of nitric oxide (NO) (27). We assessed the immunosuppressive function and found diminished NO production and diminished suppressor capacity of CD11b+Ly6C+ myeloid cells isolated from tumor-bearing PD-1−/− mice compared to their counterparts isolated from tumor-bearing WT control mice (Fig. 3O–P). Thus, PD-1 ablation switches the fate and function of myeloid cells away from immunosuppressive MDSC and promotes the generation of differentiated monocytes, MΦ and DC. Importantly, the expansion of CD11b+Ly6Chi monocytes, the increase of the CD11b+Ly6C+/CD11b+Ly6G+ ratio and the upregulation of RORC in myeloid cells of the spleen of PD-1−/− mice were already observed on day 9 after tumor inoculation, when tumors were not yet measurable, and on day 12, when tumors in WT and PD-1−/− mice had comparable size (Supplementary Fig. S10). These results indicate that the effects of PD-1 ablation on the myeloid compartment of PD-1−/− tumor-bearing mice preceded the differences in tumor growth.
To determine the potential therapeutic relevance of these findings we examined whether changes in the myeloid compartment might be detected during treatment with PD-1 blocking antibody. Compared to the control treatment group, mice receiving anti-PD-1 antibody (Supplementary Fig. 11A) had diminished accumulation of GMP in the bone marrow (Supplementary Fig. S11B), and increased expansion of Ly6C+ monocytes and DC in the tumor site (Supplementary Fig. S11D) with effector features characterized by expression of RORC, IRF8 and IFNγ (Supplementary Fig. S11E, F, G, I). In contrast, cells expressing IL-4Rα, a marker of MDSC (10, 28), were significantly decreased (Supplementary Fig. 11H). Thus, treatment with anti-PD-1 blocking antibody promotes the differentiation of myeloid cells with effector features, while suppressing expansion of MDSC in tumor-bearing mice.
Myeloid-specific PD-1 ablation is the key driver of anti-tumor immunity.
To determine if these changes on myeloid cell fate in PD-1−/− mice were mediated by myeloid cell-intrinsic effects of PD-1 ablation or by the effects of PD-1neg T cells on myeloid cells, we generated mice with conditional targeting of Pdcd1 gene (PD-1f/f) (Supplementary Fig. S12A) and crossed them with mice expressing Cre recombinase under the control of the lysozyme (LysM) promoter to induce selective ablation of the Pdcd1 gene in myeloid cells (PD-1f/fLysMcre) or with mice expressing Cre recombinase under the control of the CD4 promoter to induce selective ablation of the Pdcd1 gene in T cells (PD-1f/fCD4cre) (Supplementary Fig. S12B, C). In PD-1f/fLysMcre mice, tumor growth was significantly diminished (Fig. 4A and B) indicating that despite the preserved PD-1 expression in T cells, myeloid-specific PD-1 ablation in PD-1f/fLysMcre mice was sufficient to inhibit tumor growth. Tumor-driven emergency myelopoiesis was selectively affected in PD-1f/fLysMcre mice. Although myeloid-specific PD-1 ablation resulted in expansion of CMP, accumulation of GMP was prevented (Fig. 4C). In contrast, no change on cancer-driven emergency myelopoiesis was detected in PD-1f/fCD4cre mice, which had comparable expansion of CMP and GMP to PD-1f/f control mice (Fig. 5A).
Figure 4. Myeloid-specific PD-1 ablation is the driver of altered tumor-driven emergency myelopoiesis, inflammatory myeloid cell differentiation and anti-tumor immunity.
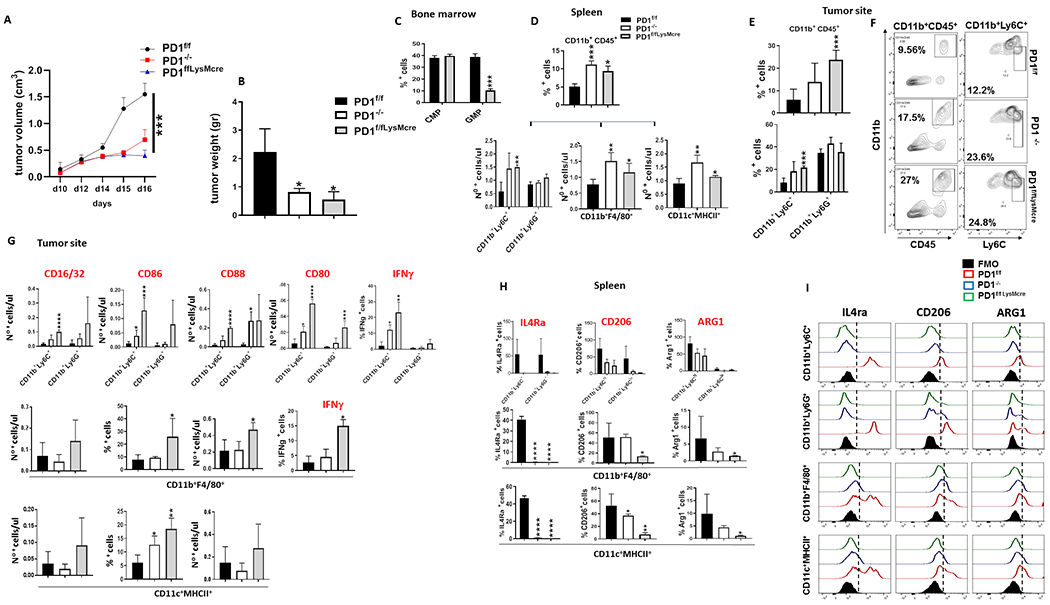
(A, B) PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice were inoculated with B16-F10 melanoma and tumor size was monitored daily (A). After mice were euthanized, tumor weight was measured (B). (C) Mean percentages ± SEM of CMP and GMP in the bone marrow of tumor-bearing PD-1f/f and PD-1f/fLysMcre mice (D) Mean percentages ± SEM of CD11b+CD45+ cells and CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+ and CD11c+MHCII+ myeloid subsets in the spleen of tumor-bearing mice. (E) Mean percentages ± SEM of CD11b+CD45+, CD11b+Ly6C+ and CD11b+Ly6G+ cells and (F) representative contour plots of FACS analysis for CD11b+CD45+ and CD11b+Ly6C+ cells at the tumor site in PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice. (G) Mean percentages ± SEM of CD16/CD32+, CD86+, CD88+ and CD80+ cells and IFN-γ-expressing myeloid cell subsets within the CD45+CD11b+ gate in B16-F10 tumors from PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice. (H) Mean percentages ± SEM and (I) FACS histograms of IL-4Ra, CD206 and ARG1 expression in CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+ and CD11c+MHCII+ myeloid cells within the CD11b+CD45+ gate in the spleen of tumor-bearing PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice. Data from one representative of three independent experiments, with 6 mice per group are shown in all panels (* p< 0.05, ** p< 0.01, *** p< 0.001).
Figure 5. T cell-specific PD-1 ablation has no impact on tumor-driven emergency myelopoiesis and profile of myeloid cell output and provides minimal protection against tumor growth.
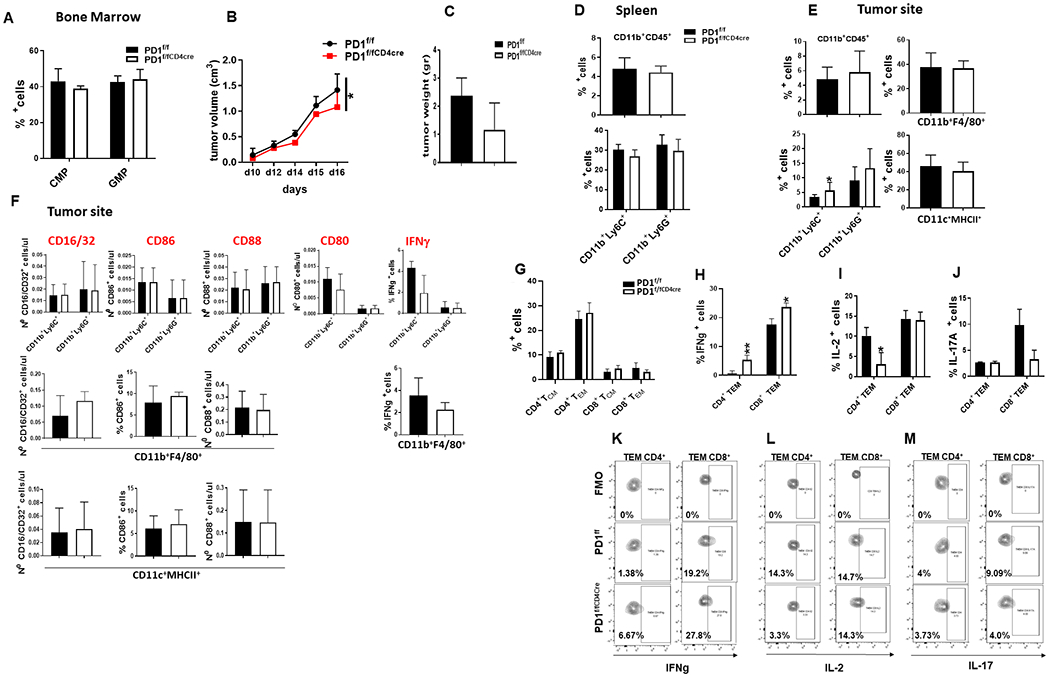
PD-1f/f and PD-1f/fCD4cre mice were inoculated with B16-F10 melanoma. (A) On day 16, mice were euthanized and bone marrow CMP and GMP were examined by flow cytometry. Mean percentages ± SEM of CMP or GMP are shown. (B-C) Tumor size was assessed evert other day from inoculation (B). On the day of euthanasia, tumor weight was measured (C). (D) Mean percentages ± SEM of CD11b+CD45+ cells and CD11b+Ly6C+ and CD11b+Ly6G+ populations within the CD11+CD45+ gate in the spleen. (E) Mean percentages ± SEM of CD11b+CD45+ cells and CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+ and CD11c+MHCII+ cells within the CD11b+CD45+ gate in the tumor site. (F) Mean percentages ± SEM of CD16/CD32+, CD86+, CD88+, CD80+ and IFN-γ expression in the indicated myeloid subsets (CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F4/80+, CD11c+MHCII+) within the CD11b+CD45+ gate in the tumor site. (G-J) Mean percentages ± SEM of CD4+ and CD8+TCM and TEM (G), as well as IFNγ, IL-2, and IL-17 (H-J) expression in CD4+ and CD8+ TEM and TCM at the tumor site, and respective contour plots (K-M). Results from one representative of two independent experiments with 6 mice per group are shown. (*p< 0.05, **p< 0.01, ***p< 0.001).
Myeloid-specific PD-1 ablation in PD-1f/fLysMcre mice shifted the differentiation of CD11b+Ly6C+ and CD11b+Ly6G+ myeloid subsets and increased the CD11b+Ly6C+/CD11b+Ly6G+ ratio in the spleen and tumor site as in PD-1−/− mice (Fig. 4D–F)), but also resulted in a strikingly different immunological profile of CD11b+Ly6C+ monocytic myeloid cells consistent with effector myeloid function as indicated by the expression of effector myeloid cell markers including CD80, CD86, CD16/32 (FcRII/III) and CD88 (C5aR) (Fig. 4G). Consistent with the improved function of myeloid cells, PD-1f/fLysMcre mice also had higher levels of IFNγ-expressing CD11b+Ly6Chi monocytes and CD11b+F4/80+ macrophages (Fig. 4G and Supplementary Fig. S13A, B) and increase of IRF8+ and RORC+ CD11b+Ly6Chi monocytes (Supplementary Fig. S13C, D). In contrast, cells expressing IL-4Ra, CD206, and ARG1, which are markers of MDSC, immunosuppressive neutrophils and tolerogenic DC (29–33) were diminished (Fig. 4H, I). Thus, myeloid-intrinsic PD-1 ablation skews the fate of myeloid cells away from immunosuppressive MDSC, promotes the differentiation of functional effector monocytes, macrophages, and DC, and has a decisive role in systemic anti-tumor immunity despite PD-1 expression in T cells.
We studied anti-tumor responses in mice with T cell-specific PD-1 ablation and found that PD-1f/fCD4cre mice had diminished anti-tumor protection (Fig. 5B, C). Consistent with the causative role of myeloid cell-specific PD-1 targeting in the differentiation and function of myeloid cells, T cell-specific PD-1 ablation did not induce expansion of CD11b+CD45+ leukocytes, CD11b+F4/80+ macrophages and CD11c+MHCII+ DC, increase of CD11b+Ly6C+/CD11b+Ly6G+ ratio (Fig. 5D, E) or immunological features of functional effector myeloid cells (Fig. 5F) in PD-1f/fCD4cre tumor-bearing mice, compared to control tumor-bearing mice. Moreover, despite PD-1 ablation, tumor-bearing PD-1f/fCD4cre mice did not have quantitative differences in tumor infiltrating TEM cells compared to control tumor-bearing mice (Fig. 5G) or features of enhanced effector function as determined by assessment of cytokine producing cells (Fig. 5H–M).
Similar outcomes to those observed with B16-F10 tumor in the differentiation of myeloid cells toward myeloid effectors vs. MDSC were obtained when PD-1f/fLysMCre and PD-f/fCD4Cre mice were inoculated with MC38 colon adenocarcinoma cells (Fig. 6B–I). Moreover, PD-1f/fLysMcre but not PD-1f/f CD4cre mice inoculated with MC38 had functional differences in tumor infiltrating TEM and TCM cells compared to control tumor-bearing mice (Fig. 6J–L). Notably, in the context of this highly immunogenic tumor, PD-1 ablation in myeloid cells resulted in complete tumor eradication, whereas mice with PD-1 ablation in T cells showed progressive tumor growth (Fig. 6A). Together these results suggest that by preventing the differentiation of effector myeloid cells and promoting generation of MDSC, myeloid-specific PD-1 expression has a decisive role on T cell function. Thus, although PD-1 is an inhibitor of T cell responses (2, 34, 35), ablation of PD-1 signaling in myeloid cells is an indispensable requirement for induction of systemic anti-tumor immunity in vivo.
Figure 6. T cell-specific PD-1 ablation provides diminished protection against tumor growth compared to myeloid-specific PD-1 ablation.

(A) PD-1f/f, PD-1f/fCD4cre and PD-1f/fLysMcre mice were inoculated with MC38 colon adenocarcinoma and tumor size was monitored daily. Mice were euthanized on day 21 and mean percentages ± SEM of CD45+CD11b+ cells and CD11b+Ly6C+, CD11b+Ly6G+ and CD11b+F4/80+ and CD11c+MHCCII+ myeloid subsets in the spleen (B), and tumor site (C) were determined. (D) Mean percentages ± SEM of RORC- and IRF8-expressing CD11b+Ly6C+, CD11b+Ly6G+, CD11b+F/480+ and CD11c+MHCII+ myeloid cells, and (E) mean percentages ± SEM of Arg1, IL-4Ra, CD88 and CD80 cells within the same myeloid subsets in the spleen. (F, G) Representative flow cytometry plots for RORC and IRF8 expression. (H) Mean percentages ± SEM and (I) representative flow cytometry plots of IFN-γ and Arg1-expressing CD11b+Ly6C+ and CD11b+Ly6G+ myeloid cells at the tumor site. (J-L) Mean percentages ± SEM of CD4+ and CD8+TCM and TEM cells (J) and IFN-γ-expressing CD4+ and CD8+ TEM and TCM at the tumor site (K) and respective contour plots (L). Data from one representative of three experiments with 6 mice per group (*p< 0.05, ** p< 0.01, *** p <0.001).
To further investigate the direct effects of PD-1 on myeloid cell fate in the absence of T cells, we used RAG2 KO mice (lacking mature T cells and B cells). Treatment of RAG2 KO tumor-bearing mice with anti-PD-1 blocking antibody resulted in decreased accumulation of GMP during tumor-driven emergency myelopoiesis (Supplementary Fig. S14A), myeloid cell expansion in the spleen and tumor site (Supplementary Fig. S14B, C), and enhanced generation of effector myeloid cells (Supplementary Fig. S14D–G ), providing evidence that blockade of PD-1-mediated signals skews myeloid lineage fate to myeloid effector cells in a myeloid cell-intrinsic and T cell-independent manner. In RAG2 KO mice treated with anti-PD-1 antibody, despite the absence of T cells, a decrease of tumor growth was also observed (Supplementary Fig. S14H–I), suggesting that ablation of PD-1 signaling promotes myeloid-specific mechanisms that induce tumor suppression, one of which might involve increased phagocytosis (8).
PD-1 ablation alters the signaling responses of myeloid cells to factors of emergency myelopoiesis.
To understand mechanisms that might be responsible for the significant differences of myeloid cell fate commitment induced by myeloid-specific PD-1 targeting, we examined whether PD-1 deficient bone marrow myeloid progenitors might have distinct signaling responses to the key hematopoietic growth factors that mediate cancer-driven emergency myelopoiesis, which also induced PD-1 expression in GMP during in vitro culture. To avoid any potential impact of bone marrow-residing PD-1−/− T cells or mature myeloid cells on the signaling responses of myeloid progenitors we used Linneg bone marrow from PD-1f/fLysMcre mice because LysM-cre is expressed in CMP and GMP (36) allowing us to take advantage of the selective deletion of PD-1 in these myeloid progenitors. PD-1 deficient GMP (Supplementary Fig. S15) had enhanced activation of Erk1/2, mTORC1, and STAT1 in response to G-CSF, a main mediator of emergency myelopoiesis (37, 38). These results are striking because each of these signaling targets has a decisive role in the differentiation and maturation of myeloid cells while preventing the generation of immature immunosuppressive MDSC (39–42). These findings indicate that PD-1 might affect the differentiation of myeloid cells by regulating the fine tuning of signaling responses of myeloid progenitors to hematopoietic growth factors that induce myeloid cell differentiation and lineage fate determination during emergency myelopoiesis.
PD-1 ablation alters the metabolic program of myeloid progenitors and activates cholesterol synthesis.
Metabolism has a decisive role in the fate of hematopoietic and myeloid precursors. Stemness and pluripotency are regulated by maintenance of glycolysis (43). Switch from glycolysis to mitochondrial metabolism and activation of oxidative phosphorylation (OXPHOS) and TCA cycle are associated with differentiation (44). This is initiated by glycolysis-mediated mitochondrial biogenesis and epigenetic regulation of gene expression (43). The structural remodeling of the mitochondrial architecture during differentiation is characterized by increased replication of mitochondrial DNA to support production of TCA cycle enzymes and electron transport chain subunits, linking mitochondrial metabolism to differentiation (45).
We examined whether PD-1 ablation, which promoted the differentiation of myeloid cells in response to tumor-mediated emergency myelopoiesis, might affect the metabolic properties of myeloid precursors. Linneg bone marrow myeloid precursors were cultured with the cytokines G-CSF/GM-CSF/IL-6 that drive tumor-mediated emergency myelopoiesis in cocktail (Fig. 7A, B) or individually (Fig. 7C, D). HSC differentiation was documented by decrease of Linneg, which was more prominent in the cultures of PD-1 deficient bone marrow cells, and coincided with increase of CD45+CD11b+ cells (Fig. 7A, B). Ly6C+ monocytic cells dominated in the PD-1f/fLysMcre cultures whereas Ly6G+ granulocytes were decreasing compared to PD-1f/f control cultures (Fig. 7C, D) providing evidence for a cell-intrinsic mechanism of PD-1 deficient myeloid precursors for monocytic lineage commitment. Glucose uptake, but more prominently mitochondrial biogenesis, were elevated in PD-1 deficient CMP and GMP (Fig. 7E, F). Bioenergetics studies showed that PD-1 deficient cells developed robust mitochondrial activity (Fig. 7G) and increase of OCR/ECAR ratio during culture (Fig. 7H), indicating that mitochondrial metabolism progressively dominated over glycolysis. This bioenergetic profile is consistent with metabolism-driven enhanced differentiation of hematopoietic and myeloid precursors (45, 46).
Figure 7. Myeloid-specific PD-1 ablation reprograms myeloid cell signalling and metabolism and induces cholesterol synthesis.

(A, B) Linneg BM from PD-1f/f and PD-1f/fLysMcre mice was cultured with GM-CSF, G-CSF and IL-6 for the indicated time intervals. Mean percentages ± SEM of CD11b+CD45+ (A) and Linneg cells (B) are shown. (C, D) Bone marrow cells purified as in (A, B) were cultured with the indicated growth factors and mean percentages ± SEM of CD11b+Ly6C+ and CD11b+Ly6G+ cells were examined after 48 hours of culture. (E-H) Bone marrow cells were prepared and cultured as in (A, B) and at 48 hours of culture glucose uptake was assessed by 2-NBDG (E) and mitochondrial biogenesis was assessed by MitoGreen staining and flow cytometry (F). (G) At 24, 48 and 72 hrs of culture oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by Seahorse extracellular flux analyser and mitostress responses at each time point of culture were examined. (H) OCR/ECAR ratio was measured at these time points and the increase of OCR/ECAR ratio during stimulation was calculated. (I) BM Linneg cells from PD-1f/f and PD-1f/fLysMcre mice were cultured with G-CSF and GM-CSF for 48 hours and metabolite analysis was performed by mass spectrometry. Unsupervised hierarchical clustering heat map of the top 50 metabolites is shown. (J) At 24, 48 and 72 hours of culture with G-CSF and GM-CSF, mRNA was extracted and analysed for the expression of the indicated genes by qPCR. Results of 48-hr culture are shown and are presented as the fold increase over the mRNA level expressed by PD-1f/f cells. Results are from one of three independent experiments. (K-M) At 24 hrs of culture with GM-CSF, G-CSF or IL-6, content of neutral lipid droplets, including triglycerides and cholesterol esters, was assessed by flow cytometry using BODIPY 493/503. Mean percentages ± SEM (K) of BODIPY 493/503 positive cells within the CD11b+CD45+ gate, representative contour plots (L) and histograms of FACS analysis (M) are shown. (N) PD-1f/f and PD-1f/fLysMcre DC were differentiated in the presence of B16-F10 tumor supernatant and content of neutral lipids was assessed. Mean percentage ± SEM of BODIPY 493/503 positive DC within the CD45+CD11b+ gate is shown. Results are representative of three experiments.
We performed unbiased global metabolite analysis to determine whether PD-1 deficient myeloid precursors developed a distinct metabolic program. Compared to control, PD-1 deficient cells had elevated metabolic intermediates of glycolysis and pentose phosphate pathway (PPP), acetyl-coA, and the TCA cycle metabolites citrate and α-ketoglutarate, but the most prominent difference was the elevated cholesterol (Fig. 7I, Supplementary Fig. S16, Supplementary Fig. S17 and Table S1). Abundant cytosolic acetyl-coA can be utilized for fatty acid and cholesterol biosynthesis (Supplementary Fig. S17) (43). Moreover, mTORC1 activates de novo cholesterol synthesis via SREBP1, which regulates transcription of enzymes involved in cholesterol synthesis (47, 48). Because acetyl-coA was elevated (Fig. 7I and Supplementary Fig. S17) and mTORC1 activation was enhanced in PD-1 deficient myeloid progenitors in response to growth factors driving emergency myelopoiesis (Supplementary Fig. S15), we examined whether activation of the mevalonate pathway that induces cholesterol synthesis (Supplementary Fig. S18A) might be involved. In PD-1 deficient myeloid progenitors cultured with growth factors of emergency myelopoiesis, mRNA of genes regulating cholesterol synthesis and uptake was increased, mRNA of genes promoting cholesterol metabolism was decreased (Fig. 7J, and Supplementary Fig. S18B), whereas cellular cholesterol and neutral lipid content was elevated (Fig. 7K–M). PD-1-deficient DC, differentiated in vitro in the presence of B16-F10 tumor supernatant, also had a significant increase of cholesterol and neutral lipids compared to similarly differentiated DC from control mice (Fig. 7N). Consistent with these in vitro findings, glucose uptake and content of cholesterol and neutral lipids were elevated in GMP of tumor-bearing PD-1 KO mice compared to control mice at day 7 or day 9 after tumor inoculation, respectively, when tumors were not yet detectable or tumors in WT and PD1 mice had equal size (Supplementary Fig. S19). Thus, features associated with metabolism-driven differentiation of myeloid progenitors are enhanced early in tumor-bearing PD-1 KO mice.
In addition to cholesterol synthesis, mevalonate also leads to the synthesis of isoprenoids, including geranylgeranyl pyrophosphate (GGPP) (Supplementary Fig. S17), which is required for protein geranylgeranylation catalyzed by geranylgeranyltransferase (GGTase) and has an active role in the upregulation of RORC expression (49). Our metabolite analysis showed increased GGPP (Fig. 7I), providing a mechanistic explanation for the upregulation of RORC in PD-1-deficient myeloid cells. Cholesterol accumulation is associated with skewing of hematopoiesis toward myeloid lineage and monocytosis, induces a proinflammatory program in monocytes/macrophages and DC, and amplifies TLR signaling (50–52). Together these results unravel a previously unidentified role of PD-1 targeting in regulating myeloid lineage fate commitment and proinflammatory differentiation of monocytes, macrophages, and DC during tumor-driven emergency myelopoiesis, through metabolic reprogramming.
Myeloid-specific PD-1 ablation induces improved T cell functionality.
Previously, it was determined that monocyte/macrophage terminal differentiation is controlled by the combined actions of retinoid receptors and the nuclear receptor PPARγ, which is regulated by cholesterol and promotes gene expression and lipid metabolic processes, leading to terminal macrophage differentiation (26, 53). Because our in vitro studies showed that PD-1 deficient myeloid progenitors developed a distinct metabolic program with elevated cholesterol metabolism, we examined whether PD-1 ablation might alter expression of PPARγ in addition to RORC. We found that expression of PPARγ was elevated in CD11b+Ly6C+ monocytic cells and MΦ isolated from tumors of PD-1−/− and PD-1f/fLysMcre mice (Fig. 8A, B, C). Since PD-1 deficient myeloid progenitors developed robust mitochondrial activity during culture in vitro (Fig. 7G, H), and PPARγ is involved in mitochondrial function (53), we examined whether myeloid cells in tumor-bearing mice have improved mitochondrial metabolism, a feature that has an important role in supporting anti-tumor function of other immune cells (54). Monocytes, MΦ, and DC isolated from tumor of PD-1−/− and PD-1f/fLysMcre mice had increased mitochondrial membrane potential, compared to myeloid cells from control tumor-bearing mice, consistent with enhanced mitochondrial metabolism (Fig. 8D–G).
Figure 8. PD-1 ablation induces enhanced mitochondrial metabolism of myeloid cells in tumor-bearing mice and improved T cell function.
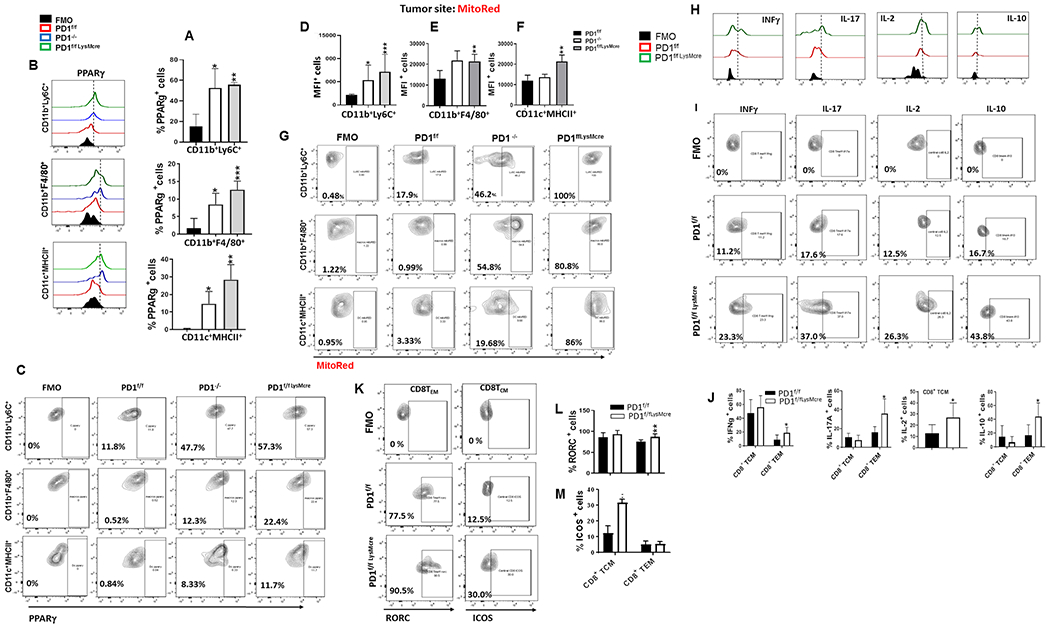
(A-C) Expression of PPARγ in myeloid cells at the B16-F10 site in PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice was examined by flow cytometry. Mean percentages ± SEM (A), representative histograms (B) and contour plots (C) of PPARγ-expressing CD11b+Ly6C+, CD11b+F4/80+ and CD11c+MHCII+ subsets. (D-G) Mitochondrial metabolic activity of myeloid cells at the B16-F10 tumor site in PD-1f/f, PD-1f/fLysMcre and PD-1−/− mice was examined by assessing mitochondrial membrane potential using MitoRed. MFI ± SEM of MitoRed positive CD11b+Ly6C+, CD11b+F4/80+, and CD11c+MHCII+ subsets within the CD45+CD11b+ gate (D-F) and representative plots of FACS analysis (G) are shown. (H-L) In parallel, expression of IFN-γ, IL-17A, IL-2, IL-10, RORC and ICOS in CD8+ TCM and TEM isolated from B16-F10-bearing PD-1f/f and PD-1f/fLysMcre mice was assessed by flow cytometry. Representative histograms (H), contour plots (I, K), and mean percentages ± SEM (J, L, M) within the CD44hiCD62Lhi gate (for TCM) and CD44hiCD62lo gate (for TEM) cells are shown. Data are from one representative of four independent experiments. (* p< 0.05, ** p< 0.01, *** p< 0.001).
We investigated whether these significant immunometabolic changes of myeloid cells, induced by myeloid-specific PD-1 targeting, affected immunological properties of T cells that have key roles in their anti-tumor function. Compared to control PD-1f/f tumor-bearing mice, PD-1f/fLysMcre tumor-bearing mice had no quantitative differences in CD4+ or CD8+ TEM and central memory (TCM) cells (Supplementary Fig. S20A) but had significant functional differences. There was an increase of IFNγ-, IL-17-, and IL-10-producing CD8+ TEM cells and IL-2-producing CD8+ TCM cells (Fig. 8H–J). ICOS and LAG3 were elevated in T cells from PD-1f/fLysMcre tumor-bearing mice but CTLA4, Tim3, CD160 and PD-1/PD-L1 were comparable in T cells from PD-1f/f and PD-1f/fLysMcre tumor-bearing mice (Fig. 8K–M and Supplementary Fig. S20B). These findings are significant because IL-17 producing Th17/Tc17 cells have enhanced anti-tumor function and mediate durable tumor growth inhibition (55). Moreover, T cells with a “hybrid” phenotype producing both IFN-γ and IL-17 might have superior anti-tumor properties by combining the enhanced effector function of Th1/Tc1 and the longevity and stemness of Th17/Tc17 cells (56). Indeed, in our studies these properties of TEM cells correlated with improved anti-tumor function in PD-1f/fLysMcre mice.
To examine experimentally whether PD-1 deficient myeloid cells differentiated in tumor-bearing mice in vivo have improved capacity of inducing antigen-specific T cell responses, we assessed responses of the same primary CD4+ or CD8+ T cells to antigen-loaded DCs isolated from PD-1−/− or control mice bearing B16-F10 tumors (Supplementary Fig. S21A). DCs isolated from the spleen of tumor-bearing WT and PD-1−/− mice were pulsed with ovalbumin and co-cultured with ovalbumin-specific CD4+ or CD8+ T cells from OTI or OTII TCR-transgenic mice. DCs from tumor-bearing PD-1−/− mice had superior ability to induce OTI and OTII T cell proliferation and IFN-γ expression (Supplementary Fig. S21B, C).
Together our data provide evidence that myeloid cell-intrinsic PD-1 ablation induces potent anti-tumor immunity by decreasing accumulation of MDSC and promoting proinflammatory and effector monocytic/macrophage and DC differentiation, thereby leading to enhanced effector T cell responses.
Discussion
Our present studies reveal a previously unidentified role of the PD-1 pathway in regulating lineage fate commitment and function of myeloid cells that arise from tumor-driven emergency myelopoiesis. These outcomes are mediated by myeloid-intrinsic effects of PD-1 ablation leading to altered signaling and metabolic reprogramming of myeloid progenitors characterized by enhanced differentiation and elevated cholesterol synthesis. Consequently, the accumulation of immature immunosuppressive and tumor promoting MDSC is diminished and the output of differentiated, inflammatory effector monocytes, MΦ, and DC is enhanced. These immunometabolic changes of myeloid cells promote differentiation of TEM cells and systemic anti-tumor immunity in vivo, in spite of preserved PD-1 expression in T cells.
We found that PD-1 deficient myeloid progenitors had enhanced activation of Erk1/2 and mTORC1 in response to G-CSF. These results indicate that Erk1/2 and mTORC1, a downstream mediator of PI3K/Akt signaling, which are major targets of PD-1 in T cells (2), are subjected to PD-1-mediated inhibition in myeloid cells. These results are revealing because Erk1/2 phosphorylation subverts MDSC-mediated suppression by inducing M-MDSCs differentiation to APC (39). Erk and PI3K regulate glycolysis in response to G-CSF (57). PI3K/Akt/mTORC1 signaling is critical in myeloid lineage commitment. Expression of constitutively active Akt in CD34+ cells induces enhanced monocyte and neutrophil development whereas a dominant negative Akt has the opposite effect (58). mTORC1 is necessary for the transition of hematopoietic cells from a quiescent state to a prepared “alert” state in response to injury-induced systemic signals (59), for G-CSF-mediated differentiation of myeloid progenitors (40), and for M-CSF-mediated monocyte/macrophage generation (41). mTORC1 stimulates translation initiation through phosphorylation of 4E-BP and ribosomal S6 kinases and has a decisive role in the expression of glucose transporters and enzymes of glycolysis and PPP (47). Consistent with these, our studies showed that PD-1-deficient myeloid progenitors had elevated expression of glycolysis and PPP intermediates after culture with emergency cytokines in vitro and enhanced monocytic differentiation in tumor-bearing mice in vivo. Together our findings indicate that PD-1 might affect the differentiation of myeloid cells by regulating the fine tuning of signaling responses of myeloid progenitors to hematopoietic growth factors that induce myeloid cell differentiation and lineage fate determination during emergency myelopoiesis. Further studies will identify how receptor-proximal signaling events mediated by hematopoietic growth factors are targeted by PD-1 in a manner comparable to PD-1-mediated targeting of signaling pathways in T cells (2, 34, 35).
Our metabolite analysis showed that a striking difference of PD-1 deficient myeloid progenitors was the increased expression of mevalonate metabolism enzymes and the elevated cholesterol. Notably, mTORC1 activates SREBP1, which induces transcription of enzymes involved in fatty acid and cholesterol synthesis (48) thereby leading to glycolysis-regulated activation of the mevalonate pathway. Our signaling studies showing enhanced mTORC1 activation and our metabolic studies showing enhanced mitochondrial metabolism and increased cholesterol content in PD-1 deficient myeloid cells, provide a mechanistic link between the altered differentiation of PD-1 deficient myeloid progenitors and the altered immunophenotypic and functional program of PD-1 deficient monocytes, MΦ and DC in tumor bearing mice. Cholesterol drives myeloid cell expansion and differentiation of macrophages and DC (50, 51, 60) and promotes antigen presenting function (61). These properties are consistent with the metabolic profile and the increased cholesterol of PD-1-deficient myeloid progenitors, the inflammatory and effector features of differentiated monocytes, MΦ and DC, and the enhanced T effector cell activation in tumor-bearing mice with myeloid-specific PD-1 ablation that we identified in our studies. By such mechanism, PD-1 might centrally regulate anti-tumor immunity, independently of the expression of PD-1 and its ligands in the tumor microenvironment. Notably, our studies showed that PD-1 expression on myeloid progenitors is an early event during tumor-mediated emergency myelopoiesis, and indicate that PD-1 blockade at early stages of cancer might have a decisive effect on anti-tumor immunity by preventing MDSC generation from myeloid progenitors and inducing the systemic output of effector myeloid cells that drive anti-tumor T cell responses.
In addition to its expression in myeloid progenitors in the bone marrow, we found that PD-1 is expressed in all myeloid subsets including M-MDSC, PMN-MDSC, CD11b+F4/80+ MΦ and CD11c+MHCII+ DC, in the tumor and the spleen of tumor bearing mice, albeit at different levels. This difference might be related to gradient of tumor-derived factors responsible for PD-1 induction, such as G-CSF and GM-CSF that we found to induce PD-1 transcription in myeloid progenitors. This possibility would be consistent with the gradual upregulation of PD-1 expression in splenic myeloid cells, determined by our kinetics studies, which correlates with tumor growth that might be responsible for the increase of systemic levels of tumor-derived soluble factors that induce PD-1. Other cues of the tumor microenvironment known to mediate the activation step of MDSC (14) might also be responsible for the induction of higher PD-1 expression level in the tumor vs. the splenic myeloid cells. Our findings unravel a previously unidentified role of PD-1 in myeloid cell fate commitment during emergency myelopoiesis, a process that is involved not only in anti-tumor immunity but also in the control of pathogen-induced innate immune responses and sterile inflammation (62).
An additional important finding of our studies is that the nuclear receptors RORC and PPARγ are upregulated in myeloid cells by PD-1 ablation. RORs were initially considered retinoic acid receptors, but were subsequently identified as sterol ligands. RORC not only is induced by sterols and isoprenoid intermediates (49) but also serves as the high affinity receptor of the cholesterol precursor desmosterol (63, 64), a metabolic intermediate of cholesterol synthesis via the mevalonate pathway that regulates inflammatory responses of myeloid cells (52, 60). Desmosterol, as well as sterol sulfates, function as endogenous RORC agonists and induce expression of RORC target genes (63, 64). Our studies showed that, in addition to cholesterol, the mevalonate metabolism product GGPP that has an active role in the upregulation of RORC expression (49) was elevated in PD-1-deficient myeloid cells providing a mechanistic basis for our finding of the elevated RORC expression. Retinoid receptors and PPARγ together regulate monocyte/macrophage terminal differentiation (26). Although initially thought to be involved in proinflammatory macrophage differentiation, it was subsequently understood that PPARγ predominantly promotes macrophage-mediated resolution of inflammation by inducing expression of the nuclear receptor LXR and the scavenger receptor CD36, thereby regulating tissue remodeling (65). PPARγ also regulates macrophage-mediated tissue remodeling by efferocytosis and production of pro-resolving cytokines (66), which can suppress cancer growth (67). The combined actions of RORC and PPARγ induced by myeloid-specific PD-1 ablation might be involved in the anti-tumor function by promoting both proinflammatory and tissue remodeling properties of myeloid cells. Future studies will dissect the specific role of each of these nuclear receptors on the anti-tumor immunity induced by myeloid cell-specific ablation of PD-1.
In conclusion, our results provide multiple levels of evidence that myeloid-specific PD-1 targeting mediates myeloid cell-intrinsic effects, which have a decisive role on systemic anti-tumor responses. This might be a key mechanism by which PD-1 blockade induces anti-tumor function. Recapitulating this immunometabolic program of myeloid cells will improve the outcome of cancer immunotherapy.
Materials and Methods
Animals
All procedures were in accordance with National Institutes of Health Guidelines for the Care and Use of Animals and approved by the Institutional Animal Care and Use Committee (IACUC) at Beth Israel Deaconess Medical Center (Boston MA). C57BL/6 Wild-type (WT) mice were purchased from Charles River (Franklin, MA). PD-1−/− mice were kindly provided by Dr. Tasuku Honjo, Kyoto University, Japan. PD-1−/− mice (B6.Cg-Pdcd1tm1.1Shr/J) were also purchased from The Jackson Laboratory (Bar Harbor, Maine). All the studies requiring the use of PD-1−/− mice were performed with both PD-1-deficient strains and resulted in similar outcomes. Pdcd1 flox/flox (PD-1f/f) mice on a C57BL/6 background were generated by Ozgene (Australia) using goGermline technology. Briefly, a targeting vector was prepared containing LoxP sites in introns 1 and 3, closely flanking exons 2 and 3 respectively, of the Pdcd1 (Supplementary Fig. S12A). The genomic 5’ and 3’ arms of homology and the Floxed genomic region were generated by PCR amplification of C57BL/6 genomic DNA. An Frt-PGK-NeoR-Frt selection cassette was placed immediately 5’ of the LoxP site in intron 3. Homologous recombination of the targeting vector was carried out by electroporation of ES cells and clones were selected for neomycin resistance. Correctly targeted ES clones were identified by Southern blot RFLP analysis and microinjected into goGermline blastocysts to generate germline chimeras. Following germline transmission, the FRT-PGK-NeoR-FRT cassette was deleted by mating to a transgenic line containing FLP recombinase. The Flp gene was removed by segregation in subsequent crosses. PD-1f/f mice were mated with LysMcre mice (B6.129P2- Lyz2tm1(cre)Ifo/J) or CD4cre mice (B6.Cg-Tg(Cd4-cre)1Cwi/BfluJ), obtained from The Jackson Laboratory. Selective ablation of PD-1 protein expression in T cells vs. myeloid cells in each strain was confirmed by flow cytometry (Supplementary Fig. S12B, C). Rag2 deficient mice (B6(Cg)-Rag2tm1.1Cgn/J), OTI TCR transgenic mice (C57BL/6- Tg(TcraTcrb)1100Mjb/J) and OTII TCR transgenic mice (B6.Cg-Tg(TcraTcrb)425Cbn/J) were purchased from Jackson Laboratory (Bar Harbor, Maine).
Tumor cell lines and tumor experiments
MC17-51 and B16-F10 cell lines were purchased from ATCC. B16-F10 cell line was subcloned and subclones with intermediate growth rate were selected for use. MC38 cell line was purchased from Kerafast. For tumor implantation, 105 murine fibrosarcoma (MC17-51) were injected intramuscularly in the left hind limb whereas 2.5x105 murine colon carcinoma (MC38) or 5x105 murine melanoma (B16-F10) cells were injected subcutaneously in the left flank. Tumor growth was monitored daily with a caliper fitted with a Vernier scale, starting from day 9. Tumor volume was calculated based on 3 perpendicular measurements. At day 15 -16 for F10-B16 tumors, at day 12-14 for MC17-51 tumors and at day 15-21 for MC38 after tumor inoculation, mice were euthanized and tumor, spleen, small intestine and bone marrow were harvested. 8-12 weeks old male mice were used for MC17-51 inoculations and 8-12 weeks old male or female mice for MC38 and B16-F10 inoculations. For studies at various time points after tumor implantation, a large cohort of mice of each strain was used for simultaneous tumor inoculation, and at the indicated times, equal number of mice were euthanized and assessment of the indicated endpoints was performed. For treatment with anti-PD-1 blocking antibody, 250 μg of either anti- PD-1 (clone RMP1-14, BioXCell) or IgG2a control (clone 2A3, BioXCell) diluted in sterile PBS were administered intraperitoneally in a volume of 100 μl per dose on days 9, 12 and 15 after tumor inoculation.
Statistics
Statistical significance for comparison between two groups was determined by two-tailed Student’s t test or Mann-Whitney U test. Statistical significance for comparison among three or more groups was determined by ANOVA. (*p value < 0.05; **p value < 0.01; ***p value < 0.001).
Supplementary Material
Table S5. Raw data in Excel spreadsheet.
Fig. S1. Gating strategy of hematopoietic and myeloid precursors in the bone marrow.
Fig. S2. Gating strategy of myeloid subsets in the spleen and tumor site.
Fig. S3. Cancer-induced emergency myelopoiesis in three different mouse tumor models.
Fig. S4. PD-1 expression is induced on myeloid progenitors by emergency cytokines.
Fig. S5. Gating strategy for identification of MDSC in human blood samples.
Fig. S6. PD-1 expression in human MDSC.
Fig. S7. PD-1 ablation alters tumor-driven emergency myelopoiesis.
Fig. S8. PD-1 ablation induces expression of RORC and IRF8 in myeloid cells expanding in response to tumor-driven emergency myelopoiesis.
Fig. S9. PD-1 ablation induces expression of RORC and IRF8 in myeloid cells expanding in mice bearing MC38 or MC17–51 tumors.
Fig. S10. PD-1 ablation increases the output of RORChi effector-like myeloid cells at early stages of tumor growth.
Fig. S11. Therapeutic targeting of PD-1 increases effector features of myeloid cells and decreases tumor growth.
Fig. S12. Myeloid-specific and T cell-specific PD-1 deletion.
Fig. S13. Myeloid-specific PD-1 ablation promotes expansion of IRF8hi and RORChi monocytes and IFN-γ−producing monocytes and macrophages in the tumor site.
Fig. S14. Tumor-induced emergency myelopoiesis and myeloid effector differentiation in Rag2 deficient mice treated with PD-1 Ab.
Fig. S15. PD-1 ablation reduces the threshold of growth factor-mediated signalling in GMP.
Fig. S16. Myeloid-specific PD-1 ablation induces a distinct metabolic profile, characterized by elevated cholesterol.
Fig. S17. Metabolic pathways linking glycolysis to PPP, fatty acid and cholesterol synthesis.
Fig. S18. Schematic presentation of the mevalonate pathway.
Fig. S19. Increase of glucose uptake and neutral lipid content in PD-1 deficient myeloid progenitors early after tumor implantation.
Fig. S20. Myeloid-specific PD-1 deletion alters the immunological profile of CD8+ TEM cells.
Fig. S21. PD-1 ablation enhances antigen presentation ex vivo by tumor-matured DC.
Table S1. List of significantly different metabolites.
Table S2. List of antibodies used for surface staining.
Table S3. List of antibodies used for intracellular staining.
Table S4. List of antibodies used for phenotype of human MDSC.
Acknowledgements
Funding: This work was supported by NIH grants CA183605, CA183605S1 and AI098129-01 and by the DoD grant PC140571.
Footnotes
Competing interests: Vassiliki A. Boussiotis has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis and Dako. The authors report no other competing interests related to this work.
References and Notes
- 1.Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T, A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nature immunology 14, 1212–1218 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Boussiotis VA, Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. The New England journal of medicine 375, 1767–1778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rizvi NA et al. , Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, N.Y 348, 124–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen W, Wang J, Jia L, Liu J, Tian Y, Attenuation of the programmed cell death-1 pathway increases the M1 polarization of macrophages induced by zymosan. Cell Death Dis 7, e2115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang X et al. , PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proceedings of the National Academy of Sciences of the United States of America 106, 6303–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen L et al. , PD-1/PD-L pathway inhibits M.tb-specific CD4(+) T-cell functions and phagocytosis of macrophages in active tuberculosis. Sci Rep 6, 38362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qorraj M et al. , The PD-1/PD-L1 axis contributes to immune metabolic dysfunctions of monocytes in chronic lymphocytic leukemia. Leukemia 31, 470–478 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Gordon SR et al. , PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumor immunity. Nature 545, 495–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V, Coordinated regulation of myeloid cells by tumours. Nature reviews 12, 253–268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bronte V et al. , Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature communications 7, 12150 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolcetti L et al. , Hierarchy of immunosuppressive strength among myeloid-derived suppressor cell subsets is determined by GM-CSF. European journal of immunology 40, 22–35 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Youn JI, Nagaraj S, Collazo M, Gabrilovich DI, Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181, 5791–5802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Movahedi K et al. , Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111, 4233–4244 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Kumar V, Patel S, Tcyganov E, Gabrilovich DI, The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends in immunology 37, 208–220 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng PH et al. , CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. American journal of respiratory and critical care medicine 186, 1025–1036 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D, Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother 62, 909–918 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer C et al. , Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother 63, 247–257 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Azzaoui I et al. , T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 128, 1081–1092 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Weber R et al. , Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Frontiers in immunology 9, 1310 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pan D et al. , A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (New York, N.Y 359, 770–775 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Strauss L et al. , RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer cell 28, 253–269 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Sichien D et al. , IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 45, 626–640 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Yanez A, Ng MY, Hassanzadeh-Kiabi N, Goodridge HS, IRF8 acts in lineage-committed rather than oligopotent progenitors to control neutrophil vs monocyte production. Blood 125, 1452–1459 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Netherby CS et al. , The Granulocyte Progenitor Stage Is a Key Target of IRF8-Mediated Regulation of Myeloid-Derived Suppressor Cell Production. J Immunol 198, 4129–4139 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Waight JD et al. , Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. The Journal of clinical investigation 123, 4464–4478 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szanto A, Nagy L, Retinoids potentiate peroxisome proliferator-activated receptor gamma action in differentiation, gene expression, and lipid metabolic processes in developing myeloid cells. Molecular pharmacology 67, 1935–1943 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Raber PL et al. , Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. International journal of cancer 134, 2853–2864 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandruzzato S et al. , IL4Ralpha+ myeloid-derived suppressor cell expansion in cancer patients. J Immunol 182, 6562–6568 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Bronte V et al. , IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 170, 270–278 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Giallongo C et al. , Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with polymorphonuclear leukocytes (PMNs) in chronic myeloid leukemia patients. PloS one 9, e101848 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zou JM et al. , IL-35 induces N2 phenotype of neutrophils to promote tumor growth. Oncotarget 8, 33501–33514 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woytschak J et al. , Type 2 Interleukin-4 Receptor Signaling in Neutrophils Antagonizes Their Expansion and Migration during Infection and Inflammation. Immunity 45, 172–184 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Mondanelli G et al. , A Relay Pathway between Arginine and Tryptophan Metabolism Confers Immunosuppressive Properties on Dendritic Cells. Immunity 46, 233–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parry RV et al. , CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Molecular and cellular biology 25, 9543–9553 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patsoukis N et al. , Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Science signaling 5, ra46 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye M et al. , Hematopoietic stem cells expressing the myeloid lysozyme gene retain long-term, multilineage repopulation potential. Immunity 19, 689–699 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Lieschke GJ et al. , Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 84, 1737–1746 (1994). [PubMed] [Google Scholar]
- 38.Liu F, Wu HY, Wesselschmidt R, Kornaga T, Link DC, Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity 5, 491–501 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Albeituni SH et al. , Yeast-Derived Particulate beta-Glucan Treatment Subverts the Suppression of Myeloid-Derived Suppressor Cells (MDSC) by Inducing Polymorphonuclear MDSC Apoptosis and Monocytic MDSC Differentiation to APC in Cancer. J Immunol 196, 2167–2180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li D et al. , Identification of key regulatory pathways of myeloid differentiation using an mESC-based karyotypically normal cell model. Blood 120, 4712–4719 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karmaus PWF et al. , Critical roles of mTORC1 signaling and metabolic reprogramming for M-CSF-mediated myelopoiesis. The Journal of experimental medicine 214, 2629–2647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coccia EM et al. , STAT1 activation during monocyte to macrophage maturation: role of adhesion molecules. International immunology 11, 1075–1083 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Moussaieff A et al. , Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell metabolism 21, 392–402 (2015). [DOI] [PubMed] [Google Scholar]
- 44.TeSlaa T et al. , alpha-Ketoglutarate Accelerates the Initial Differentiation of Primed Human Pluripotent Stem Cells. Cell metabolism 24, 485–493 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu WM et al. , Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell stem cell 12, 62–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ito K, Ito K, Hematopoietic stem cell fate through metabolic control. Exp Hematol 64, 1–11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Duvel K et al. , Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Molecular cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Porstmann T et al. , PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24, 6465–6481 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Kagami S et al. , Protein geranylgeranylation regulates the balance between Th17 cells and Foxp3+ regulatory T cells. International immunology 21, 679–689 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Murphy AJ et al. , ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. The Journal of clinical investigation 121, 4138–4149 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westerterp M et al. , Cholesterol Accumulation in Dendritic Cells Links the Inflammasome to Acquired Immunity. Cell metabolism 25, 1294–1304 e1296 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tall AR, Yvan-Charvet L, Cholesterol, inflammation and innate immunity. Nature reviews 15, 104–116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM, PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93, 241–252 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Scharping NE et al. , The Tumor Microenvironment Represses T Cell Mitochondrial Biogenesis to Drive Intratumoral T Cell Metabolic Insufficiency and Dysfunction. Immunity 45, 374–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowers JS et al. , Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight 2, e90772 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chatterjee S et al. , CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell metabolism 27, 85–100 e108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee AW, States DJ, Colony-stimulating factor-1 requires PI3-kinase-mediated metabolism for proliferation and survival in myeloid cells. Cell death and differentiation 13, 1900–1914 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Buitenhuis M et al. , Protein kinase B (c-akt) regulates hematopoietic lineage choice decisions during myelopoiesis. Blood 111, 112–121 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Rodgers JT et al. , mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert). Nature 510, 393–396 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spann NJ et al. , Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 151, 138–152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson HA, Roche PA, MHC class II association with lipid rafts on the antigen presenting cell surface. Biochimica et biophysica acta 1853, 775–780 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.King KY, Goodell MA, Inflammatory modulation of HSCs: viewing the HSC as a foundation for the immune response. Nature reviews 11, 685–692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu X et al. , Sterol metabolism controls T(H)17 differentiation by generating endogenous RORgamma agonists. Nature chemical biology 11, 141–147 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Santori FR et al. , Identification of natural RORgamma ligands that regulate the development of lymphoid cells. Cell metabolism 21, 286–297 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chawla A et al. , A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Molecular cell 7, 161–171 (2001). [DOI] [PubMed] [Google Scholar]
- 66.Yoon YS et al. , PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol 8, 1031–1046 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sulciner ML et al. , Resolvins suppress tumor growth and enhance cancer therapy. The Journal of experimental medicine 215, 115–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zanoni I et al. , CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature 460, 264–268 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Weigmann B et al. , Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc 2, 2307–2311 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Dumitru CA, Moses K, Trellakis S, Lang S, Brandau S, Neutrophils and granulocytic myeloid-derived suppressor cells: immunophenotyping, cell biology and clinical relevance in human oncology. Cancer Immunol Immunother 61, 1155–1167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yuan M, Breitkopf SB, Yang X, Asara JM, A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 7, 872–881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S5. Raw data in Excel spreadsheet.
Fig. S1. Gating strategy of hematopoietic and myeloid precursors in the bone marrow.
Fig. S2. Gating strategy of myeloid subsets in the spleen and tumor site.
Fig. S3. Cancer-induced emergency myelopoiesis in three different mouse tumor models.
Fig. S4. PD-1 expression is induced on myeloid progenitors by emergency cytokines.
Fig. S5. Gating strategy for identification of MDSC in human blood samples.
Fig. S6. PD-1 expression in human MDSC.
Fig. S7. PD-1 ablation alters tumor-driven emergency myelopoiesis.
Fig. S8. PD-1 ablation induces expression of RORC and IRF8 in myeloid cells expanding in response to tumor-driven emergency myelopoiesis.
Fig. S9. PD-1 ablation induces expression of RORC and IRF8 in myeloid cells expanding in mice bearing MC38 or MC17–51 tumors.
Fig. S10. PD-1 ablation increases the output of RORChi effector-like myeloid cells at early stages of tumor growth.
Fig. S11. Therapeutic targeting of PD-1 increases effector features of myeloid cells and decreases tumor growth.
Fig. S12. Myeloid-specific and T cell-specific PD-1 deletion.
Fig. S13. Myeloid-specific PD-1 ablation promotes expansion of IRF8hi and RORChi monocytes and IFN-γ−producing monocytes and macrophages in the tumor site.
Fig. S14. Tumor-induced emergency myelopoiesis and myeloid effector differentiation in Rag2 deficient mice treated with PD-1 Ab.
Fig. S15. PD-1 ablation reduces the threshold of growth factor-mediated signalling in GMP.
Fig. S16. Myeloid-specific PD-1 ablation induces a distinct metabolic profile, characterized by elevated cholesterol.
Fig. S17. Metabolic pathways linking glycolysis to PPP, fatty acid and cholesterol synthesis.
Fig. S18. Schematic presentation of the mevalonate pathway.
Fig. S19. Increase of glucose uptake and neutral lipid content in PD-1 deficient myeloid progenitors early after tumor implantation.
Fig. S20. Myeloid-specific PD-1 deletion alters the immunological profile of CD8+ TEM cells.
Fig. S21. PD-1 ablation enhances antigen presentation ex vivo by tumor-matured DC.
Table S1. List of significantly different metabolites.
Table S2. List of antibodies used for surface staining.
Table S3. List of antibodies used for intracellular staining.
Table S4. List of antibodies used for phenotype of human MDSC.