

Abstract
Mucolipidosis II α/beta (MLII) is an autosomal recessive disease in which a gene mutation leads to improper targeting of lysosomal enzymes with an end result of accumulation of lysosomes in the mitochondria resulting in a dysfunctional mitochondria. 1 Leigh syndrome (LS) is a rare progressive neurodegenerative disorder associated with dysfunctional mitochondria and oxidative phosphorylation. 4 Both disease processes typically present in infancy. 3 7 Herein, we present a case of an infant diagnosed with both mucolipidosis II and Leigh syndrome. Genetic analysis in this case revealed two mutations (NDUFA12 c.178C > T p.Arg60* and GNPTAB c.732_733delAA) on the long arm of chromosome 12 as the etiology of MLII and LS in this neonate, respectively. We are unaware of any previously published cases of the presence of these two diseases occurring in the same patient. The complex clinical presentation of this case led to a delay in the diagnosis, and we believe that the clinical phenotypes of these two conditions were likely worsened. The genetic alterations presented in this case occurred as a result of mutations on chromosome 12. We suggest further investigation into the potential overlap in the pathophysiology, specifically the inheritance pattern, linkage disequilibrium, mitochondrial–lysosomal interaction, or crosstalk contributing to both diseases.
Keywords: mucolipidosis II, I cell disease, Leigh syndrome
Introduction
Mucolipidosis II α/beta (MLII), Online Mendelian Inheritance in Man (OMIM) 252500, is a progressively debilitating lysosomal storage disease (LSD) caused by the mutation in GNPTAB 1 and is inherited in an autosomal recessive pattern. The mutation results in improper tagging and transportation of lysosomal enzymes resulting in toxic accumulation of lysosomal substrates in the mitochondria. 2 MLII affects approximately 1 in 100,000 to 400,000 live births. 3 Clinical features, which may be present at birth, include skeletal abnormalities and coarse facial features. 2
Leigh syndrome (LS; OMIM 256000) is a mitochondrial disease first reported by Denis Leigh in 1951 in a 7-month-old infant. 4 5 Although it typically presents in infancy and early childhood, cases of adult-onset LS have been described in literatures. 6 LS affects approximately 1 in 40,000 live births. 7 The most common inheritance pattern is autosomal recessive, but over 75 gene mutations have been associated with LS. Most mutations affect nuclear DNA, although 20% of the cases have mutations in mitochondrial DNA. 7 The mutations affect mitochondrial energy production through oxidative phosphorylation. LS is characterized by focal, bilateral lesions in one or more areas of the central nervous system, commonly in the brainstem and basal ganglia leading to progressive neurological impairment. 8
Case Report
We present a term female neonate with facial dysmorphisms, short limbs, persistent thrombocytopenia, direct hyperbilirubinemia, and poor feeding. The pregnancy was complicated by intrauterine growth restriction and suspected fetal skeletal dysplasia on prenatal ultrasound. Prenatal serology was unremarkable. Prenatal genetic testing was declined by the family. The neonate was born to a 26-year-old G2P2 Caucasian mother at 37 weeks of gestation by cesarean section due to nonreassuring fetal heart tones. Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. The family was of Mennonite (German Baptist) descent and denied consanguinity. The patient had a healthy older sister.
Vital signs were normal for age. Birth weight, head circumference, and length were all less than the 3rd percentile and consistent with symmetric growth restriction. The physical examination showed large ears with increased folding anteriorly, a long philtrum, a small mouth with prominent alveolar ridge, epicanthal folds, jaundice and scattered petechiae on the trunk, short upper and lower extremities with bowing of the tibia and fibula bilaterally, supinated ankles, and mild generalized hypertonia. Flexion wrist contractures were present with clenched fists. Thumbs were held between the second and third fingers, bilaterally.
Laboratory testing revealed elevated alkaline phosphatase (ALP) with a peak serum ALP of 2596U/L and a low serum vitamin D 25-OH of 17 ng/mL. Direct hyperbilirubinemia was present with a direct bilirubin of 3.4 mg/dL and a total bilirubin of 13.2 mg/dL. Her stools were acholic but an abdominal ultrasound was normal. Complete blood counts showed persistent severe thrombocytopenia with a platelet count of 41K but no signs of overt bleeding. Congenital infection screen was negative. A head ultrasound was performed and found to be normal, with no evidence of intracranial hemorrhage or calcifications. An echocardiogram showed a patent ductus arteriosus and patent foramen ovale. A chest X-ray ( Fig. 1 ) showed hypoinflated lungs as well as dysplastic bones throughout the thorax and visualized upper extremities. A skeletal survey ( Fig. 2 ) demonstrated marked osteopenia, foreshortened long bones with thickened diaphyses, irregular “raggedy” metaphyses, and no wormian bones. Genetic testing (microarray, achondroplasia panel, and FGFR3 mutation) was performed. Based on physical exam and laboratory analysis, the differential diagnosis included Mucolipidosis II, congenital TORCH infection, isolated rhizomelia, Trisomy 18, and osteogenesis imperfecta. On careful evaluation of the family history and pedigree, it was discovered that the patient's maternal cousin died at 12 months of age with concurrent diagnoses of MLII and LS.
Fig. 1.

Chest X-ray showing hypoinflated lungs, dysplastic bones throughout the thorax, and visualized upper extremities.
Fig. 2.

Long bones X-rays showing osteopenia with significant shortening of the long bones, thickening of the diaphyses, and irregular metaphyses.
The patient was treated in our neonatal intensive care unit for 27 days by a multispecialty team. During that time, she remained stable on room air but failed the car seat test multiple times necessitating discharge with home oxygen for travel. She had slow weight gain and poor oral feeding and was discharged with a combination of nasogastric tube and oral feeding. Following discharge, she was re-admitted to the hospital three times due to acute hypoxemic respiratory failure and was ultimately discharged to home with hospice care. The patient died at 12 months and 29 days of age.
Microarray testing revealed homozygous mutations at 12q22 and 12q23.2. The variant at 12q22 (c.178C > T p.Arg60*) in the NDUFA gene is a pathogenic mutation predicted to lead to abnormal mitochondrial oxidative phosphorylation resulting in LS. The variant at 12q23.2 (c.732_733delAA) in the GNPTAB gene is a pathogenic variant which disrupts lysosomal enzyme transport resulting in MLII. Genetic testing of the parents was deemed not necessary because the NDUFA12 and GNPTAB variants have been well documented in the plain community (Amish and Mennonite). It was presumed that one allele of the family haplotype in the region of chromosome 12 contained both of these pathogenic variants which were coinherited by the infant. Of note, the older sibling of the infant was completely asymptomatic ( Fig. 3 ).
Fig. 3.

Pedigree depicting the biological relationship and inheritance pattern of the genetic mutation c.178C > T of the NDUFA12 gene resulting in Leigh syndrome and the c.732_733delAA mutation of the GNPTAB gene, causing mucolipidosis II α/beta. The patient in the presented case is indicated by an arrow.
Discussion
MLII is due to a mutation in the GNPTAB gene which encodes for the enzyme GlcNAc-1-PT. 9 This mutation leads to incorrectly synthesized mannose 6-phosphate (M6P), a lysosomal catabolic enzyme marker that aides in proper transportation of lysosomes. In cases of defective M6P, the enzymes are transported extracellularly into intercellular space, resulting in a deficiency of lysosomal enzymes and thus accumulation of waste material in lysosomes resulting in inclusion cells. 9
LS may occur as a result of many gene mutations. Regardless of the specific gene mutation, the most common defect is an abnormal oxidative phosphorylation process most commonly due to gene disruptions or deficiency of complex I, NADH: ubiquinone oxidoreductase. However, other common gene mutations resulting in LS have been found in complex IV and V, termed cytochrome c oxidase and adenosine triphosphate (ATP) synthase protein complex, respectively. The impaired process of oxidative phosphorylation causes a deficiency of ATP resulting in cellular death with a significant impact on regions of high energy consumption such as the brain, heart, and muscles leading to the clinical features of LS. 7 The specific mutation (NDUFA12 c.178C > T p.Arg60*) noted in our patient is extremely rare and has only been reported once. 10 This particular mutation was reported to result in a premature stop codon resulting in a loss of function involved in complex I activity of the mitochondrial respiratory chain. 10
It is possible that in patients with only complex I disease, this NDUFA12 variant may be amenable to supportive therapy as it decreases the activity of complex I but does not destroy the complex. 10 11 It is very likely that the NDUFA12 is a gene required in the last few steps in the completion of complex I or participates in the stability of complex I once it has been synthesized and should be investigated in future studies. 10
Diagnosis of MLII and LS is usually made through microarray or whole exome sequencing, but targeted testing can be done in certain populations (such as the Mennonite), which is rapid and inexpensive. Phenotypic signs should be appreciated by physicians as markers for potential further genomic work-up ( Table 1 ). Overlapping features between MLII and LS include psychomotor delay and low muscle tone. 8 9 Additional signs of MLII include joint contractures, skeletal abnormalities, coarse facial features, gingival hyperplasia, and cardiomegaly. 9 As mentioned previously, our patient exhibited diffuse skeletal dysplasia consistent with MLII with significantly high ALP levels. Bone pathologic examination in MLII suggests that the high serum ALP is likely from abnormal osteoblasts, osteocytes, fibroblasts, and chrondroblasts. 12 Ataxia, weakness, vision loss, seizures, and dysphagia are other clinical features associated with LS. 5 Our patient exhibited dysphagia. It is uncommon to see other features of LS at the age of diagnosis. A patient presenting with both diseases may prove to be difficult to diagnose as the diseases have both shared features and characteristics that are unique to each pathology. Thus, while one diagnosis may be suspected and confirmed, physicians should not halt the investigative process if not all symptoms can be explained by one confirmed diagnosis. Further testing may therefore be warranted in such cases.
Table 1. Comparison of the phenotype exhibited by the presented case and the phenotypes from reported cases with the same mutations for mucolipidosis II α/beta (MLII) 9 and Leigh syndrome (LS) 10 .
| Mutation | Pathology | Phenotype | |||
|---|---|---|---|---|---|
| Facial dysmorphia | Neurological complications | Other | |||
| Our patient | GNPTAB c.732_733delAA | Mucolipidosis II alpha/beta | Large anterior folded ears, long philtrum, small mouth with prominent alveolar ridge, epicanthal folds | Mild generalized hypertonia | Failure to thrive, short limbs, wrist contractures, bowing of tibia and fibula |
| NDUFA12 c.178C > T | Leigh syndrome | ||||
| Reported case 9 | GNPTAB | Mucolipidosis II alpha/beta | Metopic prominence, coarse facial features, depressed nasal bridge, shallow orbits, prominent mouth with thickened alveolar ridges, gingival hypertrophy | Range of neuromotor deficiencies (unable to sit by themselves, some able to walk without assistance), hypotonia, limited verbal skills, cognitive impairment | Premature statural growth cessation, short femurs, bowed limbs, mitral and aortic valve thickening |
| Reported case 10 | NDUFA12 homozygous c.178C> T | Leigh syndrome | None reported | Loss of motor function beginning at 2 years and wheel chair bound at 10 years, dystonia, severe muscular dystrophy, hypotonia | Growth retardation: height and weight in 3rd percentile, hypertrichosis, scoliosis |
Work-up of an infant with suspected MLII or LS necessitates a thorough history and physical examination. Such patients benefit from multidisciplinary team management. Establishing care with a primary care provider familiar with these conditions is essential for long-term management. No cure is currently available for MLII or LS. However, supportive therapy including physical, occupational, and speech therapy is available. Nutritional support is necessary as poor weight gain is common in both diseases. Recurrent respiratory infections and cardiopulmonary insufficiency are common and the most common cause of death in cases of MLII. 13 MRI to evaluate for cervical instability and spinal stenosis should be performed at least bi-annually. Newer therapies under investigation include treatment of MLII by bone marrow or hematopoietic stem cell transplantation with no current published outcome data. 14 15
Families with affected children should be informed of the poor prognosis associated with these disorders. In cases of MLII, most children die in early childhood or typically by age 10. 9 13 15 Outcome in LS is dependent on whether there is partial or complete enzyme deficiency with the former living an average of 6 to 7 years and the latter living up to the first few years of life. 16
Conclusion
Herein, we present a case of a term neonate with skeletal dysplasia, growth restriction, and dysmorphic features with genetic testing confirming two genetic mutations causing MLII and LS. To our knowledge, this is the first published report of MLII and LS in the same patient. Recently, mitochondrial dysfunction has been implicated in the pathogenesis of LSD. 8 17 Scientists speculate that common signaling pathways and “crosstalk” could occur between mitochondria and lysosomes. 8 18 19 20 The basis of these mitochondrial–lysosomal interactions warrants exploration. Additionally, the two mutations presented in this case occurred on the long arm of chromosome 12 within nearby loci ( Fig. 4 ). It is possible that “homozygosity mapping” played a role as genes are not inherited individually but in blocks of DNA. In our patient, although we do not have the parents' genetic testing, one allele of the family haplotype (father's and mother's) in the region of chromosome 12 that contains both of these pathogenic variant of genes was likely co-inherited by their offspring. The mutation of NDUFA12 gene has been reported only in one other instance as a cause of LS, 10 and its proximity to the GNPTAB gene resulting in MLII requires additional investigation, particularly in the Mennonite population who likely harbor both variants.
Fig. 4.
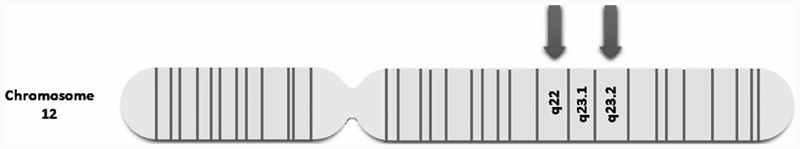
Diagram of chromosome 12. The first arrow at 12q22 marks the c.178C > T mutation of the NDUFA12 gene causing Leigh syndrome. The second arrow at 12q23.2 marks the c.732_733delAA mutation of the GNPTAB gene, causing mucolipidosis II α/beta.
Acknowledgments
The authors thank the family for providing the written consent and permission to publish this case and are grateful to Karlla Brigatti, PhD (Clinic for Special Children, Strasburg, PA) and Holmes Morton, MD (Central Pennsylvania Clinic, A Medical Home for Special Children & Adults, Belleville, PA), for their help in reviewing this manuscript.
Funding Statement
Funding None.
Conflict of Interest None declared.
Authors' Contributions
R.R.S. and U.C.E. drafted the initial and reviewed the final version of the manuscript. C.N.O.-M., S.J.B., and K.M.G. reviewed and edited the final manuscript. All the authors reviewed and accepted the final manuscript for publication.
References
- 1.Cury G K, Matte U, Artigalás O et al. Mucolipidosis II and III alpha/beta in Brazil: analysis of the GNPTAB gene. Gene. 2013;524(01):59–64. doi: 10.1016/j.gene.2013.03.105. [DOI] [PubMed] [Google Scholar]
- 2.Wraith J E. Amsterdam, Netherlands: Elsevier Health Sciences; 2013. Mucopolysaccharidoses and mucolipidoses; pp. 1723–1729. [DOI] [PubMed] [Google Scholar]
- 3.Genetics Home Reference: Mucolipidosis II alpha/beta. U.S.National Library of Medicine, National Institutes of HealthAvailable at:https://ghr.nlm.nih.gov/condition/mucolipidosis-ii-alpha-beta. Accessed December 11, 2018
- 4.Pincus J H. Subacute necrotizing encephalomyelopathy (Leigh's disease): a consideration of clinical features and etiology. Dev Med Child Neurol. 1972;14(01):87–101. doi: 10.1111/j.1469-8749.1972.tb02563.x. [DOI] [PubMed] [Google Scholar]
- 5.Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14(03):216–221. doi: 10.1136/jnnp.14.3.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McKelvie P, Infeld B, Marotta R, Chin J, Thorburn D, Collins S. Late-adult onset Leigh syndrome. J Clin Neurosci. 2012;19(02):195–202. doi: 10.1016/j.jocn.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Genetics Home Reference: Leigh Syndrome.U.S. National Library of Medicine, National Institutes of HealthAvailable at:https://ghr.nlm.nih.gov/condition/leigh-syndrome. Accessed December 11, 2018
- 8.Lake N J, Bird M J, Isohanni P, Paetau A. Leigh syndrome: neuropathology and pathogenesis. J Neuropathol Exp Neurol. 2015;74(06):482–492. doi: 10.1097/NEN.0000000000000195. [DOI] [PubMed] [Google Scholar]
- 9.Cathey S S, Leroy J G, Wood T et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010;47(01):38–48. doi: 10.1136/jmg.2009.067736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ostergaard E, Rodenburg R JT, van den Brand M et al. Respiratory chain complex I deficiency due to NDUFA12 mutations as a new cause of Leigh syndrome. J Med Genet. 2011;48(11):737–740. doi: 10.1136/jmg.2011.088856. [DOI] [PubMed] [Google Scholar]
- 11.Chinnery P F.Mitochondrial disorder overview. In: Pagon RA, Adam MP, Ardinger HH, et al., eds. GeneReviews [Internet] Seattle, WA: University of Washington; 2000[Updated 2014]. Available at:https://www.ncbi.nlm.nih.gov/books/NBK1224/#mt-overview. Accessed May 7, 2019 [Google Scholar]
- 12.Pazzaglia U E, Beluffi G, Bianchi E, Castello A, Coci A, Marchi A. Study of the bone pathology in early mucolipidosis II (I-cell disease) Eur J Pediatr. 1989;148(06):553–557. doi: 10.1007/BF00441557. [DOI] [PubMed] [Google Scholar]
- 13.Leroy J G, Cathey S, Friez M J.Mucolipidosis II Seattle, WA: University of Washington; 2008[Updated 2012]. Available at:https://www.ncbi.nlm.nih.gov/books/NBK1828/. Accessed April 7, 2019 [Google Scholar]
- 14.Kurobane I, Inoue S, Gotoh Y et al. Biochemical improvement after treatment by bone marrow transplantation in I-cell disease. Tohoku J Exp Med. 1986;150(01):63–68. doi: 10.1620/tjem.150.63. [DOI] [PubMed] [Google Scholar]
- 15.Lund T C, Cathey S S, Miller W P et al. Outcomes after hematopoietic stem cell transplantation for children with I-cell disease. Biol Blood Marrow Transplant. 2014;20(11):1847–1851. doi: 10.1016/j.bbmt.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inclusion-Cell (I-Cell) Disease.(Mucolipidosis Type II) Follow-Up: Complications, Prognosis, Patient Education. Inclusion-Cell (I-Cell) Disease (Mucolipidosis Type II) Follow-Up: Complications, Prognosis, Patient EducationAvailable at:https://emedicine.medscape.com/article/945460-followup. Published January 4, 2019. Accessed April 5, 2019
- 17.Leigh's Disease Information Page.National Institute of Neurological Disorders and StrokeAvailable at:https://www.ninds.nih.gov/Disorders/All-Disorders/Leighs-Disease-Information-Page. Accessed April 5, 2019
- 18.Plotegher N, Duchen M R. Mitochondrial dysfunction and neurodegeneration in lysosomal storage disorders. Trends Mol Med. 2017;23(02):116–134. doi: 10.1016/j.molmed.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 19.de Pablo-Latorre R, Saide A, Polishhuck E V, Nusco E, Fraldi A, Ballabio A. Impaired parkin-mediated mitochondrial targeting to autophagosomes differentially contributes to tissue pathology in lysosomal storage diseases. Hum Mol Genet. 2012;21(08):1770–1781. doi: 10.1093/hmg/ddr610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Audano M, Schneider A, Mitro N. Mitochondria, lysosomes, and dysfunction: their meaning in neurodegeneration. J Neurochem. 2018;147(03):291–309. doi: 10.1111/jnc.14471. [DOI] [PubMed] [Google Scholar]