

Abstract
Histiocytosis-lymphadenopathy plus syndrome (H syndrome) is caused by mutations in the SLC29A3 gene that result in histiocytic infiltration of numerous organs. Patients suffering from this disorder can be easily mistaken for similar conditions such as Muckle–Wells syndrome. We present a 9.5-year-old boy, who is the offspring of a consanguineous marriage. He suffered from sensorineural hearing loss, dark hyperpigmented indurated dry areas on the medial thighs sparing the knees with hypertrichosis on the affected areas, and areas of hypopigmentation on the abdomen. The patient displayed mild dysmorphism including frontal bossing, synophrys, bilateral proptosis (with normal thyroid function), thick eyebrows, flat nose, long philtrum, and pectus excavatum. Formal intelligence testing showed that he was a slow learner. Laboratory findings included elevated serum amyloid-A, erythrocyte sedimentation rate, and total proteins in urine tests. Complete blood count showed mild microcytic hypochromic anemia. The molecular analysis was crucial to confirm the provisional clinical diagnosis. H syndrome is a rare autoinflammatory syndrome with pleiotropic manifestations that affect many organs and can be mistaken for other conditions. Our patient's description may expand the phenotype of H syndrome, as areas of hypopigmentation were observed on the abdomen. Molecular analysis of SLC29A3 -related diseases is essential to highlight the variability and increase the awareness of H syndrome aiming for early diagnosis and proper treatment.
Keywords: H syndrome, SLC29A3 gene , hyperpigmentation, autoinflammatory syndrome
Introduction
Histiocytosis-lymphadenopathy plus syndrome (H Syndrome; MIM: 602782) is an autosomal recessive disorder characterized by cutaneous hyperpigmentation, hypertrichosis, and induration with multiple systems affection. The prevalence is about <1/1,000,000, with approximately 100 patients reported in the literature, some of them are Arabian descent. 1 Molho-Pessach et al 2 named this syndrome based on its most common clinical features. These features include hyperpigmentation, hypertrichosis, hepatosplenomegaly, hearing loss, heart anomalies, hypogonadism, and hyperglycemia. In addition, patients may present with short stature, lymphadenopathy, microcytic anemia, and flexion contractures of the proximal interphalangeal joints with camptodactyly and hallux valgus. Skin lesions involving the lower limbs particularly the medial sides of the thighs are pathognomonic to H syndrome. Histopathological findings can include epidermal hyperplasia with an increase in the basal pigmentation. There are also dermal infiltrates by histiocytes, lymphocytes, and plasma cells with hemosiderin depositions or calcifications. 3 4 5
The H syndrome is caused by homozygous or compound heterozygous mutations in the SLC29A3 gene (OMIM: 612373). This gene is located on chromosome 10q22 and encodes for human equilibrative nucleoside transporter 3 (hENT3). The hENT3 is a member of the equilibrative nucleoside transporter (ENT) family which consists of 475 amino acids and is localized in the endosomes, lysosomes, and mitochondria. This transporter helps with passive sodium-independent transportation of nucleotides, nucleobases, and nucleotide analogs across the lysosomal membrane to the cytoplasm. In addition, it helps transportation across the inner mitochondrial membranes. This maintains the cytoplasmic pool of nucleosides required for different cellular pathways. 6 The SLC29A3 mutation may hinder the nucleoside transportation leading to intracellular nucleosides accumulation. 7 H syndrome may result from missense, nonsense, compound, or deletion mutation of SLC29A3 , which may partially account for its large interfamilial variability. 8
Muckle–Wells syndrome (MWS; MIM: 191900) is an autosomal dominant autoinmmune disease whose clinical features overlap with H syndrome. Patients with MWS, however, mainly present with severe inflammatory symptoms, including fever, rash, conjunctivitis, headache, arthralgia/arthritis, amyloidosis, progressive sensorineural hearing loss, and renal failure. MWS is caused by a mutation in the NLRP3 gene that encodes for the protein cryopyrin/ NALP3 . The NLRP3 activates intracellular caspase 1, and mutations can lead to excessive production of interleukin-1 (IL-1). Excess IL-1 leads to systemic inflammatory symptoms. 9
We present a patient with H syndrome presenting with novel findings of amyloidosis and some hypopigmented areas. To the best of our knowledge, neither of these clinical findings has been described previously in association with H syndrome.
The documentation of this case report conforms to the Declaration of Helsinki protocols, was approved by the Ethical Research Committee of the National Research Centre, and informed consent was given by the child's parent for publication of photographs and a description of the child's presentation.
Case Presentation
History
A 9.5-year-old boy was referred with dark hyperpigmented areas on both lower limbs and hearing loss from the outpatient clinic of New Children's Hospital to the Department of Clinical Genetics, National Research Centre of Egypt. He was the offspring of healthy consanguineous parents. The pregnancy and delivery were unremarkable. There was no family history of birth defects or spontaneous abortions. Our patient's birth weight was 2.4 kg (<3rd percentile), and his psychomotor development was normal. At 2 months, he developed peptic ulcer, and endoscopy revealed gastritis and multiple gastric erosions causing acute abdomen. At 3 months, dark pigmented skin lesions developed over his body and disappeared by 12 months. At 4 years, he developed progressive mixed conductive and severe sensorineural hearing loss and was treated with tympanostomy and hearing aids. He also suffered from periodic fever and recurrent joint pains.
Examination
At 9.5 years of age, the patient's weight, height, and head circumference were 23 kg (standard deviation [SD] = − 1), 120.5 cm (SD = − 2.1), and 52.5 cm (SD = 0.01), respectively. The patient displayed mild dysmorphism, including frontal bossing, synophrys, bilateral proptosis (with normal thyroid function), thick eyebrows, flat nose, long philtrum ( Fig. 1A and B ), and pectus excavatum. Examination of the hands revealed clinodactyly of fifth finger in the right hand ( Fig. 1C ). He also had dark hyperpigmented indurated dry areas on the medial thighs, sparing the knees, with hypertrichosis on the affected areas ( Fig. 1D ). Areas of hypopigmentation were present on the abdomen ( Fig. 1E ). No abnormality was detected on cardiac, chest, neurologic, and genitourinary examination. Stanford Binet test for intelligent quotient was 72 (slow learner). Laboratory findings were significant for elevated serum amyloid A (145 mg/L [normal: up to 6.4 mg/L]), elevated erythrocyte sedimentation rate (ESR; 52 mm at the first hour, 100 mm at the second hour), elevated total proteins in urine (245 mg/24 h [normal: 25–150 mg/24 h]), and complete blood count showed mild microcytic hypochromic anemia. Liver enzymes, blood glucose, echocardiogram, magnetic resonance imaging of the brain, and radiographic skeletal survey were normal. Abdominal ultrasound revealed mild hepatosplenomegaly. Auditory brain response revealed bilateral profound hearing loss with very poor speech discrimination.
Fig. 1.
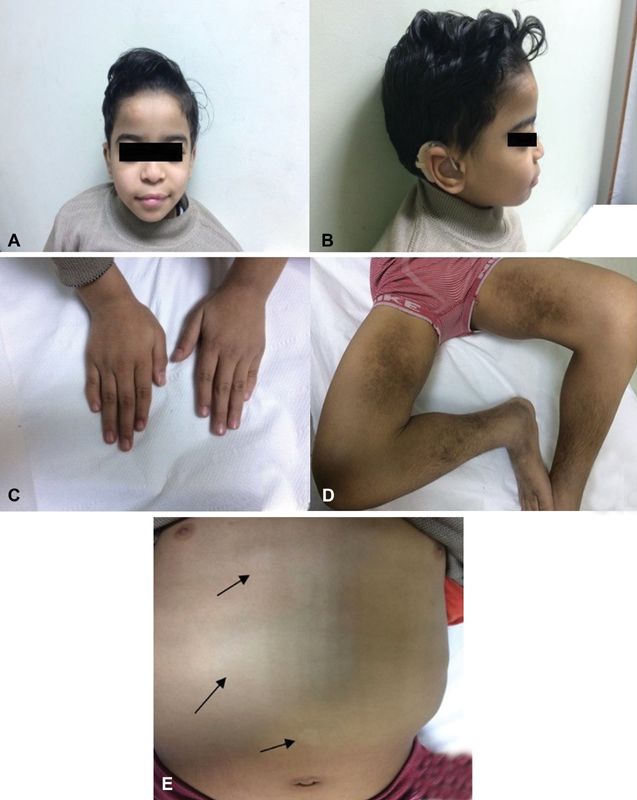
( A, B ) Frontal bossing, synophrys, bilateral proptosis, thick eye brows, flat nose, long philtrum. ( C ) Clinodactyly of the right fifth digit. ( D ) Bilateral dark hyperpigmented indurated dry areas on the medial thigh sparing the knees with hypertrichosis on the affected areas. ( E ) Areas of hypopigmentation on the abdomen.
The patient was provisionally diagnosed with MWS based on periodic fever, sensorineural hearing loss, elevated serum amyloid A, and elevated total proteins in urine.
Molecular Diagnosis
Whole exome sequencing (WES; Centogene laboratory) of the complete coding region of the SLC29A3 gene revealed a homozygous mutation in SLC29A3 c.1309G > A p.(Gly437Arg) in our patient ( Fig. 2 ). This finding confirmed H syndrome diagnosis for our patient, accompanied by amyloidosis and some hypopigmented areas.
Fig. 2.
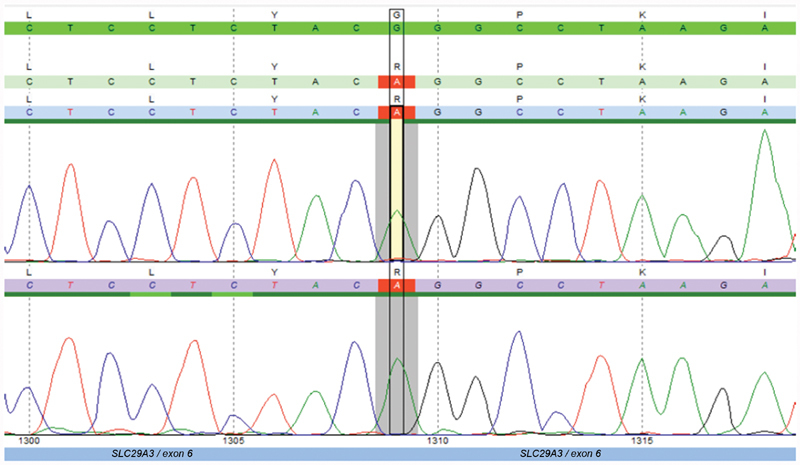
Sanger sequencing chromatogram confirmation of WES showing homozygous mutation in SLC29A3 c.1309G > A p.(Gly437Arg).
Discussion
H syndrome and MWS are two disorders with different clinical courses and pathogeneses. The reported case was initially diagnosed as MWS based on the presence of periodic fever, recurrent joint pains, sensorineural hearing loss, and amyloidosis. Other symptoms were not associated with MWS, such as skin hyperpigmentation involving the lower limbs—especially the inner thighs—with hypertrichosis. Positive consanguinity suggested an autosomal recessive disorder, and later investigations revealed hepatosplenomegaly and microcytic hypochromic anemia.
Molho-Pessach et al 2 described H syndrome in 10 patients from 6 consanguineous Arab families who presented with hyperpigmentation, hypertrichosis, and indurated cutaneous patches in the middle and lower parts of their bodies. In addition, these patients suffered from sensorineural hearing loss and hepatosplenomegaly. The clinical findings of our patient were in agreement with the previously described cases. Bolze et al 10 noted that sensorineural hearing loss and hepatosplenomegaly occur in half of the patients with H syndrome. Many studies reported that the characteristic hyperpigmentation and hypertrichosis are the most prevalent clinical manifestations among H syndrome patients and should be considered as pathognomonic clinical signs of H syndrome, 1 4 but Bloom et al 5 could only diagnose their patients with H syndrome by WES. They suggested that diagnostic difficulty of H syndrome was due to the relatively recent description of the syndrome and the small number of patients previously published. Likewise, there are few patients in Egypt diagnosed with H syndrome which we feel is due to the overlapping clinical manifestations of this rare syndrome. It is worth mentioning that a study done by El-Darouti et al 11 reported six patients diagnosed clinically with MWS who had skin lesions similar to those of H syndrome. Molho-Pessach and Zlotogorski 12 suggested that these patients may have H syndrome, owing to the hyperpigmented hypertrichotic skin plaques, and the autosomal recessive pattern of inheritance reported. The report by El-Darouti et al 11 illustrates the difficulty in diagnosing both disorders and the value in molecular diagnosis. In a patient with hepatosplenomegaly, hypertrichosis, heart anomalies, hearing loss, hypogonadism, and short stature, H syndrome should be considered in the differential diagnosis. 13
The areas of hypopigmentation present on the abdomen and the elevated serum amyloid are seemingly unreported findings in the setting of H syndrome that may contribute to the understanding of this rare condition. There were reports of H syndrome patients with recurrent fever, some of which are accompanied by joint inflammation. 8 14 15 16 SLC29A3 gene is widely expressed in various organs and regulates the inflammatory cascade. Consequently, the inflammatory process could explain the elevated serum amyloid and total proteins in urine observed in our patient. These findings may indicate amyloid deposition, a potential marker of chronic inflammation that may lead to renal damage manifesting as proteinuria, nephrotic syndrome, or derangement in renal function. 17 Molho-Pessach et al 8 found chronic elevation of inflammatory markers. Previous studies reported poor response of H syndrome patients to agents that directed against IL-1 or tumor necrosis factor-α (TNF-α), such as anakinra, canakinumab, and adalimumab. These studies also reported partial relief on colchicine or nonsteroidal anti-inflammatory drugs. 4 15 Our patient did not improve on colchicine treatment. Biological markers of chronic inflammation observed in our patient, such as anemia and increased ESR, were also reported by Elbarbary et al 18 and Al-Haggar et al. 7
WES revealed a homozygous mutation in the gene SLC29A3 c.1309G > A p.(Gly437Arg) in our patient. Mutations in the SLC29A3 gene were reported in many diseases, such as H syndrome, pigmented hypertrichotic dermatosis with insulin-dependent diabetes, Faisalabad histiocytosis, and Rosai–Dorfman disease (sinus histiocytosis with massive lymphadenopathy). Therefore, it had been suggested that these diseases should be regarded as one spectrum disorder ( SLC29A3 -related disorders) characterized as a monogenic autoinflammatory syndrome. 4 10 18 19
Conclusion
H syndrome is a rare autoimmune syndrome with pleiotropic manifestations affecting many systems and is often mistaken for other autoimmune disorders. It is a very rare genetic disorder and difficult to diagnose clinically. Our patient's findings may expand the phenotype. Areas of hypopigmentation on the abdomen and elevated serum amyloid level in blood seen in our patient are seemingly unreported in the literature in the context of H syndrome. Studies are required to define the phenotype–genotype correlation of SLC29A3 -related disorders and increase the awareness of H syndrome to facilitate early diagnosis and proper treatment.
Acknowledgments
We thank the family of patient for participating in this research. We also thank Centogene for performing the molecular analysis.
Footnotes
Conflict of Interest None declared.
References
- 1.Awake P P, Penmetcha L C, Fonseca A, Jawalkar P P. A tale of H Syndrome with typical radiographic findings. Indian J Dermatol. 2018;63(02):172–175. doi: 10.4103/ijd.IJD_573_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molho-Pessach V, Agha Z, Aamar S et al. The H syndrome: a genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol. 2008;59(01):79–85. doi: 10.1016/j.jaad.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 3.Molho-Pessach V, Lerer I, Abeliovich D et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet. 2008;83(04):529–534. doi: 10.1016/j.ajhg.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molho-Pessach V, Ramot Y, Camille F et al. H syndrome: the first 79 patients. J Am Acad Dermatol. 2014;70(01):80–88. doi: 10.1016/j.jaad.2013.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Bloom J L, Lin C, Imundo L et al. H syndrome: 5 new cases from the United States with novel features and responses to therapy. Pediatr Rheumatol Online J. 2017;15(01):76. doi: 10.1186/s12969-017-0204-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang N, Jun A H, Bhutia Y D, Kannan N, Unadkat J D, Govindarajan R. Human equilibrative nucleoside transporter-3 (hENT3) spectrum disorder mutations impair nucleoside transport, protein localization, and stability. J Biol Chem. 2010;285(36):28343–28352. doi: 10.1074/jbc.M110.109199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Haggar M, Salem N, Wahba Y et al. Novel homozygous SLC29A3 mutations among two unrelated Egyptian families with spectral features of H-syndrome. Pediatr Diabetes. 2015;16(04):305–316. doi: 10.1111/pedi.12160. [DOI] [PubMed] [Google Scholar]
- 8.Molho-Pessach V, Varma M, Godbole K, Kamath N, Zlotogorski A. H syndrome--four new patients from India. Indian J Dermatol Venereol Leprol. 2014;80(06):579. doi: 10.4103/0378-6323.144229. [DOI] [PubMed] [Google Scholar]
- 9.Kuemmerle-Deschner J B, Koitschev A, Tyrrell P N et al. Early detection of sensorineural hearing loss in Muckle-Wells-syndrome. Pediatr Rheumatol Online J. 2015;13(01):43. doi: 10.1186/s12969-015-0041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bolze A, Abhyankar A, Grant A V et al. A mild form of SLC29A3 disorder: a frameshift deletion leads to the paradoxical translation of an otherwise noncoding mRNA splice variant. PLoS One. 2012;7(01):e29708. doi: 10.1371/journal.pone.0029708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.El-Darouti M A, Marzouk S A, Abdel-Halim M R. Muckle-Wells syndrome: report of six cases with hyperpigmented sclerodermoid skin lesions. Int J Dermatol. 2006;45(03):239–244. doi: 10.1111/j.1365-4632.2004.02466.x. [DOI] [PubMed] [Google Scholar]
- 12.Molho-Pessach V, Zlotogorski A. H syndrome and Muckle-Wells syndrome. J Am Acad Dermatol. 2009;61(02):365. doi: 10.1016/j.jaad.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 13.Doviner V, Maly A, Ne'eman Z et al. H syndrome: recently defined genodermatosis with distinct histologic features. A morphological, histochemical, immunohistochemical, and ultrastructural study of 10 cases. Am J Dermatopathol. 2010;32(02):118–128. doi: 10.1097/DAD.0b013e3181b28572. [DOI] [PubMed] [Google Scholar]
- 14.Kismet E, Köseoglu V, Atay A A, Deveci S, Demirkaya E, Tuncer K. Sinus histiocytosis with massive lymphadenopathy in three brothers. Pediatr Int. 2005;47(04):473–476. doi: 10.1111/j.1442-200x.2005.02096.x. [DOI] [PubMed] [Google Scholar]
- 15.Melki I, Lambot K, Jonard L et al. Mutation in the SLC29A3 gene: a new cause of a monogenic, autoinflammatory condition. Pediatrics. 2013;131(04):e1308–e1313. doi: 10.1542/peds.2012-2255. [DOI] [PubMed] [Google Scholar]
- 16.Senniappan S, Hughes M, Shah Pet al. Pigmentary hypertrichosis and non-autoimmune insulin-dependent diabetes mellitus (PHID) syndrome is associated with severe chronic inflammation and cardiomyopathy, and represents a new monogenic autoinflammatory syndrome J Pediatr Endocrinol Metab 201326(9,10):877–882. [DOI] [PubMed] [Google Scholar]
- 17.Lane T, Gillmore J D, Wechalekar A D, Hawkins P N, Lachmann H J. Therapeutic blockade of interleukin-6 by tocilizumab in the management of AA amyloidosis and chronic inflammatory disorders: a case series and review of the literature. Clin Exp Rheumatol. 2015;33(06) 94:S46–S53. [PubMed] [Google Scholar]
- 18.Elbarbary N S, Tjora E, Molnes J et al. An Egyptian family with H syndrome due to a novel mutation in SLC29A3 illustrating overlapping features with pigmented hypertrichotic dermatosis with insulin-dependent diabetes and Faisalabad histiocytosis. Pediatr Diabetes. 2013;14(06):466–472. doi: 10.1111/j.1399-5448.2012.00925.x. [DOI] [PubMed] [Google Scholar]
- 19.Morgan N V, Morris M R, Cangul H et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010;6(02):e1000833. doi: 10.1371/journal.pgen.1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]