

Abstract
Two new cyclic depsipeptides named swinhopeptolides A (1) and B (2), have been isolated from the marine sponge Theonella swinhoei cf. verrucosa, collected from Papua New Guinea. They each contain 11 diverse amino acid resudues and 13-carbon polyketide moieties attached at the N-terminus. Compounds 1 and 2 each exist as two conformers in DMSO-d6 due to cis/trans isomerism of the proline residue, and their structures were successfully assigned by extensive NMR analyses complemented by chemical degradation and derivatization studies. Swinhopeptolide B (2) contains a previously undescribed 2,6,8-trimethyldeca-(2E,4E,6E)-trienoic acid moiety N-linked to a terminal serine residue. Swinhopeptolides A (1) and B (2) showed significant inhibition of the Ras/Raf signaling pathway with IC50 values of 5.8 and 8.5 μM, respectively.
Graphical Abstract
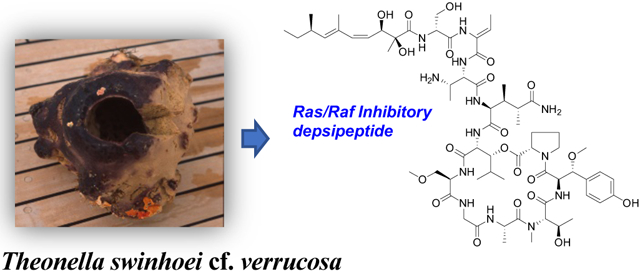
Sponge-derived cyclic depsipeptides are widely recognized for their unusual structural features and potent bioactivities. The callipeltins, isolated from a Callipelta sp. marine sponge, were the first representatives of a family of nonribosomal peptides that contain many unusual amino acids including 3,4-dimethylglutamine and β-methoxytyrosine residues, along with diverse new polyketide moieties.1–5 Related macrocyclic peptides include papuamides A−F from Theonella mirabilis, T. swinhoei, and Melophlus sp.,6–8 mirabamides A−H from Siliquariaspongia mirabilis and Stelletta clavosa,9,10 neamphamides A and B from Neamphius huxleyi and Neamphius sp.,11–13 stellettapentins A and B from Stelletta sp.,14 and stellatolides A-G from Ecionemia acervus.15 These peptides exhibit potent biological properties including anti-HIV, antifungal, and cytotoxic activities. Inhibition of HIV by the papuamides involves blocking virus entry into target cells,16 while the antifungal and cytotoxic effects of these peptides are mediated through specific binding to phosphatidylserine in cell membranes.17,18
Based on the varied biological properties reported for these metabolites and other cyclic depsipeptides, we tested a diverse array of prefractionated marine sponge extracts in a cell-based screen for materials that can impede the interaction between Ras, an ocogenic GTPase, and Raf, a serine/threonine protein kinase. These proteins are part the Ras-Raf-MEK-ERK signal transduction cascade, which is also described as the mitogen-activated protein kinase (MAPK) pathway.19,20 Ras/Raf signaling pathways are considered good potential theapeutic targets for cancer treatment as mutations in the ras and raf genes are abundant in numerous cancer types including pancreatic (95%), colorectal (45%), and lung (35%) for ras, and melanoma (60%), thyroid (39%), and ovarian (12%) for raf.21,22 While a number of small-molecule inhibitors targeting this pathway have been approved as drugs or are currently undergoing clinical trials, response rates are highly variable and therapeutic efficacy is often limited by the development of resistance.23,24 Additional therapeutic options for targeting dysfunction of Ras-Raf-MEK-ERK signaling are clearly needed.
In conjuction with an ongoing anticancer natural product discovery effort at the National Cancer Institiute,25 the extract of a Papua New Guinea collection of the sponge Theonella swinhoei cf. verrucosa was tested and showed potent activity in a cell-based anti-Ras/Raf drug screen. Bioassay-guided fractionation of the extract by sequential application of C18 flash chromatography, Sephadex LH-20 separation, and C18 HPLC provided two peptide-based metabolites. Herein, we report the structures of two cyclic depsipeptides named swinhopeptolides A (1) and B (2), which are comprised of numerous unusual amino acid residues and N-linked aliphatic polyketide moieties. Compound 2 contains a 2,6,8-trimethyldeca-(2E,4E,6E)-trienoic acid (Tdta) moiety which is the first report of this aliphatic substituent among cyclic depsipetides. These new sponge metabolites exhibited low micromolar activity in the Ras/Raf inhibition assay.
The molecular formula of swinhopeptolide A (1) was deduced to be C66H103N13O21 by HRESIMS measurements in conjunction with extensive NMR analyses. However, both the 13C and 1H NMR spectra of 1 showed a greater number of signals than expected, based on molecular weight and molecular formula considerations. Detailed examination of the NMR data (Table 1) revealed that numerous signals were paired with each other (1H NMR integration ratio of approx. 3:2 in DMSO-d6), indicating a mixture of conformational isomers. The presence of a large number of exchangeable amide NH protons (δH 9.18−6.95) and carbonyl carbons (δc 175.6−165.0) in the 1H and 13C NMR spectra of 1 indicated characteristic signals of a peptide. Detailed analysis of 2D NMR (HSQC, COSY, TOCSY, HSQC-TOCSY, HMBC, and ROESY) data led to the assignment of four common amino acid residues consisting of proline (Pro), alanine (Ala), glycine (Gly) and serine (Ser), together with the following seven nonproteinogenic amino acid residues: β-methoxytyrosine (β-OMeTyr), N-methylthreonine (N-MeThr), O-methylserine (O-MeSer), 3-hydroxyleucine (3-OHLeu), 3,4-dimethylglutamine (3,4-DiMeGln), 2,3-diaminobutanoic acid (Dab), and 2-amino-2-butenoic acid (Aba). In addition, a 2,3-dihydroxy-2,6,8-trimethyldeca-(4Z,6E)-dienoic acid (Dhtda) moiety was established. NMR assignments for the major and minor constituents 1a and 1b, respectively, are listed in Table 1.
Table 1.
1H NMR (600 MHz) and 13C NMR Data (150 MHz) of Compound 1 in DMSO-d6.
| major conformer 1a | minor conformer 1b | ||||
|---|---|---|---|---|---|
| units | position | δC, type | δH, (J in Hz) | δC, type | δH, (J in Hz) |
| Pro | 1 | 171.7, C | 170.8, C | ||
| 2 | 59.2, CH | 4.43, ma | 58.0, CH | 4.19, m | |
| 3 | 29.0, CH2 | 2.19, m | 28.4, CH2 | 2.16, m | |
| 1.89, m | 1.78, m | ||||
| 4 | 24.4, CH2 | 1.87, (2H,m) | 24.4, CH2 | 1.87, (2H,m) | |
| 5 | 46.6, CH2 | 3.53, m | 46.2, CH2 | 3.65, m | |
| 3.41, m | 3.53, m | ||||
| β-OMeTyr | 1 | 167.5, C | 168.2, C | ||
| 2 | 53.5, CH | 4.90, dd (9.2, 8.3) | 53.3, CH | 4.80, dd (9.2, 8.3) | |
| 3 | 82.9, CH | 4.30, d (9.2) | 83.6, CH | 4.15, d (9.2) | |
| 1′ | 127.3, C | 127.7, C | |||
| 2′, 6′ | 129.2, CH | 7.11, d (8.4) | 129.2, CH | 7.08, d (8.4) | |
| 3′, 5′ | 114.8, CH | 6.71, d (8.4) | 114.7, CH | 6.67, d (8.4) | |
| 4′ | 157.8, C | 157.6, C | |||
| 3-OMe | 56.2, CH3 | 3.03, s | 56.1, CH3 | 2.97, s | |
| NH | 8.23, d (8.3) | 8.42, d (8.3) | |||
| N-MeThr | 1 | 170.2, C | 169.9, C | ||
| 2 | 62.1, CH | 4.24, d (9.4) | 64.7, CH | 4.16, d (9.4) | |
| 3 | 62.5, CH | 3.69, m | 62.6, CH | 3.67, m | |
| 4 | 20.0, CH3 | 0.39, d (5.8) | 19.6, CH3 | 0.52, d (5.8) | |
| N-Me | 30.1, CH3 | 2.91, s | 28.7, CH3 | 2.65, s | |
| Ala | 1 | 171.8, C | 172.5, C | ||
| 2 | 46.0, CH | 4.43, m | 43.6, CH | 4.81, m | |
| 3 | 16.2, CH3 | 1.23, d (6.8) | 17.6, CH3 | 1.16, d (6.8) | |
| NH | 7.18, m | 8.41, d (7.8) | |||
| Gly | 1 | 168.0, C | 168.7, C | ||
| 2 | 42.5, CH2 | 4.02, m | 41.3, CH2 | 3.86, m | |
| 3.35, m | 3.52, m | ||||
| NH | 8.84, br t (5.5) | 7.93, m | |||
| O-MeSer | 1 | 170.2, C | 170.2, C | ||
| 2 | 55.9, CH | 4.27, m | 55.8, CH | 4.26, m | |
| 3 | 70.7, CH2 | 3.91, m | 70.3, CH2 | 3.90, m | |
| 3.63, m | 3.66, m | ||||
| 3-Me | 58.1, CH3 | 3.27, s | 58.2, CH3 | 3.27, s | |
| NH | 8.10, br d (4.1) | 8.10, br d (4.1) | |||
| 3-OHLeu | 1 | 170.3, C | 170.1, C | ||
| 2 | 52.8, CH | 4.76, m | 54.9, CH | 4.75, m | |
| 3 | 77.3, CH | 4.90, d (10.2) | 76.8, CH | 5.41, d (10.3) | |
| 4 | 28.3, CH | 1.96, m | 27.8, CH | 1.95, m | |
| 5 | 19.4, CH3 | 0.72, d (6.6) | 18.8, CH3 | 0.79, d (6.7) | |
| 6 | 19.5, CH3 | 0.72, d (6.6) | 18.8, CH3 | 0.79, d (6.7) | |
| NH | 8.24, br d (8.9) | 8.68, br d (8.9) | |||
| 3,4-DiMeGln | 1 | 170.2, C | 172.3, C | ||
| 2 | 56.3, CH | 4.31, dd (9.1, 6.6) | 56.9, CH | 4.17, dd (9.1, 6.6) | |
| 3 | 38.1, CH | 1.98, m | 35.1, CH | 2.10, m | |
| 4 | 40.8, CH | 2.35, m | 42.3, CH | 2.56, m | |
| 5 | 176.7, C | 178.1, C | |||
| 3-Me | 13.6, CH3 | 0.84, d (6.5) | 15.1, CH3 | 0.97, d (6.5) | |
| 4-Me | 15.9, CH3 | 1.03, d (6.8) | 14.1, CH3 | 1.12, d (6.8) | |
| NH | 8.59, br d (6.6) | 8.08, d (6.6) | |||
| 5-NH2 | 7.21, br s | 7.48, br s | |||
| 6.95, br s | 7.30, br s | ||||
| Dab | 1 | 168.7, C | 168.3, C | ||
| 2 | 53.9, CH | 4.57, t (6.6) | 54.1, CH | 4.58, t (6.6) | |
| 3 | 47.8, CH | 3.67, m | 47.7, CH | 3.66, m | |
| 4 | 14.4, CH3 | 1.11, d (6.8) | 14.6, CH3 | 1.12, d (6.8) | |
| NH | 7.98, m | 7.97, m | |||
| 3-NH2 | NDb | ND | |||
| Aba | 1 | 165.0, C | 165.1, C | ||
| 2 | 129.9, C | 130.0, C | |||
| 3 | 128.7, CH | 6.42, q (6.8) | 127.7, CH | 6.38, q (6.8) | |
| 4 | 12.9, CH3 | 1.68, d (6.8) | 12.9, CH3 | 1.68, d (6.8) | |
| NH | 9.18, br s | 9.14, br s | |||
| Ser | 1 | 170.0, C | 169.8, C | ||
| 2 | 54.7, CH | 4.37, m | 55.0, CH | 4.36, m | |
| 3 | 61.4, CH2 | 3.85, m | 61.6, CH2 | 3.84, m | |
| 3.64, m | 3.63, m | ||||
| NH | 7.93, d (7.8) | 7.92, d (7.8) | |||
| Dhtda | 1 | 175.6, C | 175.4, C | ||
| 2 | 77.0, C | 77.1, C | |||
| 3 | 71.1, CH | 4.64, d (10.2) | 70.9, CH | 4.64, d (10.2) | |
| 4 | 126.4, CH | 5.41, t (10.6) | 126.5, CH | 5.41, t (10.6) | |
| 5 | 135.9, CH | 6.03, d (12.0) | 135.8, CH | 6.03, d (12.0) | |
| 6 | 130.8, C | 130.8, C | |||
| 7 | 137.8, CH | 5.23, d (9.3) | 137.7, CH | 5.23, d (9.3) | |
| 8 | 33.6, CH | 2.32, m | 33.6, CH | 2.32, m | |
| 9 | 29.8, CH2 | 1.35, m | 29.8, CH2 | 1.35, m | |
| 1.22, m | 1.22, m | ||||
| 10 | 11.8, CH3 | 0.82, t (6.2) | 11.8, CH3 | 0.82, t (6.2) | |
| 2-Me | 22.4, CH3 | 1.13, s | 22.5, CH3 | 1.13, s | |
| 6-Me | 16.4, CH3 | 1.75, s | 16.4, CH3 | 1.75, s | |
| 8-Me | 20.5, CH3 | 0.93, d (6.5) | 20.5, CH3 | 0.93, d (6.5) | |
Coupling constant not determined due to signal broadening or overlapped signals.
Chemical shifts not determined.
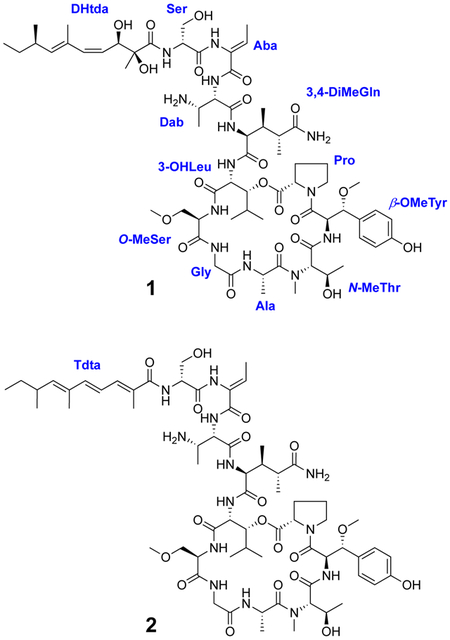
The amino acid sequence of 1 was assigned from a combination of inter-residue ROESY and HMBC correlations (Figure 1). The sequence of Ser-Aba-Dab was assigned from HMBC correlations between the Aba NH (δH 9.18)/Ser C-1 (δC 170.0) and the Dab H-2 (δH 4.57)/Aba C-1 (δC 165.0), and ROESY cross-peaks between the Aba NH/Ser H-2 (δH 4.37) and the Dab NH (δH 7.98). The Z configuration of Aba was confirmed by a ROESY correlation between the Aba H3-4 (δH 1.68) and Aba NH (δH 9.18). The next sequence of Dab-3,4-DiMeGln-3-OHLeu-O-MeSer-Gly-Ala-N-MeThr-β-OMeTyr-Pro was very similar to the corresponding sequence of papuamide A, the only difference being replacement of the C-terminal homoproline residue with a proline in 1. The connectivity of amino acids was also confirmed by both HMBC and ROESY data, with the following correlations observed: the 3,4-DiMeGln NH/Dab C-1, the O-MeSer H-2/3-OHLeu C-1, the Gly H2-2/O-MeSer C-1, the Ala H-2/Gly C-1, the N-MeThr N-Me/Ala C-1, and the Pro H2-5/β-OMeTyr C-1 from the HMBC experiment; the Dab H-2/3,4-DiMeGln NH, the 3,4-DiMeGln H-2/3-OHLeu NH, the 3-OHLeu H-2/O-MeSer NH, the Gly NH/ O-MeSer NH, the Gly NH/Ala NH, the Ala H3-3/N-MeThr N-Me, the N-MeThr H-2/β-OMeTyr NH, and the β-OMeTyr 3-OMe/Pro H2-4 from the ROESY data. The presence of an ester bond between the carbonyl group of Pro and the hydroxy group of the 3-OHLeu was suggested by the deshielded chemical shift of the 3-OHLeu oxymethine proton H-3 (δH 4.90) and confirmed by the presence of an HMBC correlation between this proton and the Pro carbonyl carbon C-1 (δC 171.7). The Dhtda moiety was linked to the N-terminus of the serine residue by HMBC correlations from the Ser H-2 (δH 4.37) and the Dhtda 2-Me (δH 1.13) protons to the carbonyl (δC 175.6) of Dhtda (Figure 1). The C-4/C-5 olefin in the Dhtda residue of 1 was assigned as Z based on a J4,5 = 12.0 Hz, while the C-6/C-7 double bond was assigned as E on the basis of ROESY correlations between H-5/H-7 and 6-Me/8-Me. These double bond configurations are the same as the Dhtda moieties of the papuamides6–8 and mirabamides.9,10 Thus, the planar structure of swinhopeptolide A (1) was elucidated.
Figure 1.
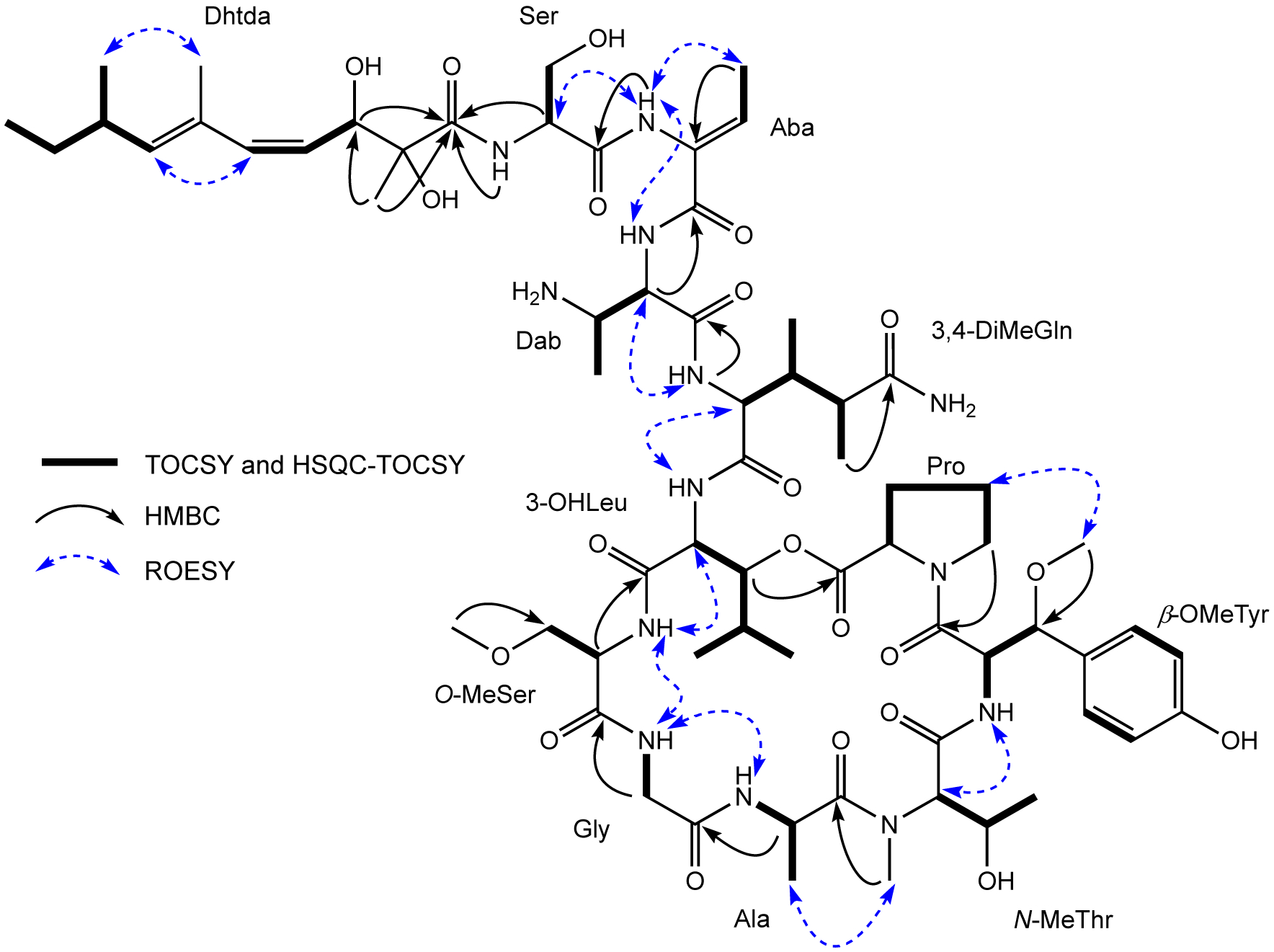
Key TOCSY, HSQC-TOCSY, HMBC, and ROESY correlations for swinhopeptolide A (1).
The absolute configurations of the amino acid constituents of 1 were determined by acid hydrolysis and application of the advanced Marfey’s method.26 The amino acids with a single stereogenic center, L-Pro, D-Ser, L-Ala and D-O-MeSer were analyzed by preparing both the L- and D-DLA derivatives, and LC-MS comparison with appropriate amino acid standards (Figure S16A–D). For the amino acids with multiple chiral centers, L-Dab, D-3-OHLeu, L-N-MeThr, and 3,4-dimethyl-L-glutamine, commercial standards were not available. Thus, their DLA derivatives were compared by LC-MS with the corresponding derivatives prepared from an authentic sample of mirabamide A,9 which contains the same nonproteinogenic amino acid residues. In addition to comparison of the LC-MS retention time of the DLA derivatives, the presence of a (2S,3S)-Dab residue in 1 was supported by the characteristic large coupling constant of J2,3 = 6.6 Hz that was observed, and by a ROESY correlation between NH and H-4. In previous NMR studies with synthetic Dab,27 the diastereomers of Dab showed distinct differences since the H-2/H-3 coupling constant in the (2R,3S)-isomer is approximately 3.0 Hz, while in the (2S,3S)-isomer it is 6.1–7.0 Hz, thus supporting the (2S,3S)-Dab assignment (Figure S17-A). Similarly, for D-3-OHLeu, a characteristic large coupling constant J2,3 = 10.2 Hz and a ROESY correlation between NH and H-4 supported the absolute configuration assignment of C-3 as R (Figure S17-B). A large coupling constant J2,3 = 9.4 Hz in L-N-MeThr, in conjunction with a ROESY correlation between H3-4 and NHβ-OMeTyr and the absence of a ROESY observed between the N-Me group and H3-4 was consistent with an R configuration at C-3 (Figure S17-C). For the 3,4-dimethyl-L-glutamine residue, a large coupling J2,3 = 9.1 Hz and the presence of ROESY correlations between 3-Me/H-2 and 4-Me/NH supported an absolute configuration assignment of S for C-3. Comparison by LC-MS of the L-DLA derivative of 3,4-DiMeGln derived from 1 with the same residue from mirabamide A, showed similar retention times. Thus, the configuration was determined as (2S, 3S, 4R)-DiMeGln in 1 (Figure S17-D), which is consistent with the configuration of all the other DiMeGln residues that have been reported from marine cyclicpeptides. Due to the decomposition of β-OMeTyr during acid hydrolysis conditions, this residue of 1 was converted to β-OMeAsp via ozonolysis and oxidative work-up, prior to acid hydrolysis and Marfey’s derivatization.2 Then, LC-MS comparison with a mirabamide A sample that was ozonized in a similar manner before hydrolysis and Marfey’s analysis, revealed (2R, 3R) as the configuration of the β-OMeTyr residue in 1 (Figure S18). The large coupling of J2,3 = 9.2 Hz observed for the β-OMeTyr residue supported this assignment.12 This completed the absolute configuration assignment for the amino acid-derived portion of swinhopeptolide A (1). The Dhtda polyketide moiety in 1 was unstable as it decomposed during acid hydrolysis, and also during various attempts of direct derivatization, so the configuration of this component could not be directly assigned. However, papuamide B contains a Dhtda side chain and its absolute configuration was assigned as 2S, 3R, 8R in a total synthesis study.8 Very close correspondence between the 1H and 13C NMR data for the Dhtda residues in both swinhopeptolide A (1) and papuamide B when measured in CD3OD, (Table S2, Supporting Information) suggest that they share the same 2S, 3R, 8R absolute configuration.
The molecular formula of 2 was deduced to be C66H101N13O19 by HRESIMS and NMR analysis. The 1H and 13C NMR spectra of 2 (Table S1) were very similar to those of 1, except the C-2 and C-3 carbinol carbons of the Dhtda moiety in 1 were replaced with a nonprotonated sp2 carbon (δC 128.7) and an sp2 methine (δC 134.4). This suggested that the amide linked 2,3-dihydroxy-2,6,8-trimethyldeca-(4Z,6E)-dienoic acid (Dhtda) residue in 1 was replaced with a 2,6,8-trimethyldeca-(2,4,6)-trienoic acid (Tdta) moiety in 2. The more shielded chemical shift of C-4 in the Tdta relative to C-4 in Dhtda (from δC 126.4 in 1 to δC 121.8 in 2), and the deshielded nature of C-5 (from δC 135.9 to δC 142.9) was consistent with a conjugated triene functionality in 2. This assignment was confirmed by TOCSY correlations that extended from H-3 (δH 7.00) to the Tdta H-5 (δH 6.56), and by HMBC correlations from the 2-Me to C-1 (δC 169.3), C-2 (δC 128.7), and C-3 (δC 134.4). The double bond configurations at C-2/C-3, C-4/C-5, and C-6/C-7 were assigned as E based on ROESY correlations between 2-Me/H-4, H-4/6-Me, H-3/H-5 and H-5/H-7 (Figure 2). Characteristic coupling constants J3,4 = 10.6 Hz and J4,5 = 15.7 Hz were consistent with the assignment of 2,6,8-trimethyldeca-(2E,4E,6E)-trienoic acid (Tdta). The absolute configuration of 2 was assigned by LC-MS analysis of the D/L-DLA derivatives of the acid hydrolysate and comparison of the retention times with those of 1 and mirabamide A. This established that all of the amino acid residues in swinhopeptolide B (2) possessed identical configurations to those in 1.
Figure 2.
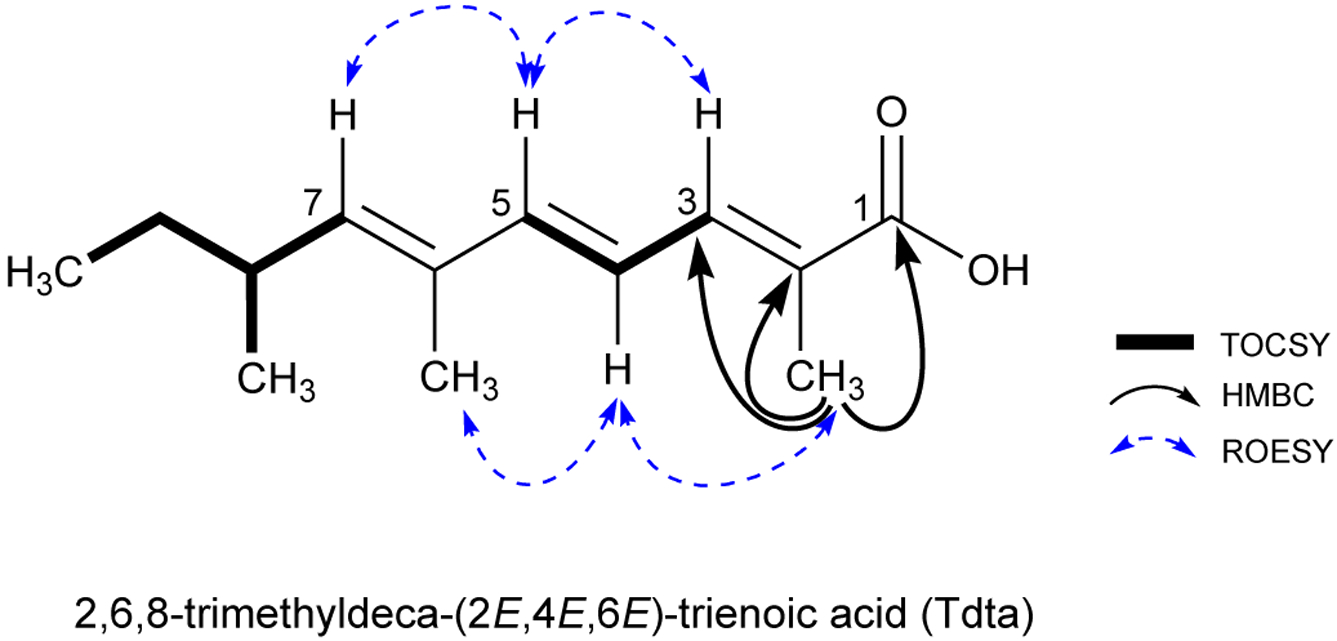
Key TOCSY, HMBC and ROESY NMR correlations for Tdta moiety in 2.
Swinhopeptolides A (1) and B (2) were tested in a cell-based Bioluminescence Resonance Energy Transfer (BRET) assay to assess their effects on Ras-Raf protein-protein interactions. This assay employs the NanoLuc luciferase attached to the full-length Raf protein as the energy donor and a HaloTag fluorescent ligand on the full-length Ras protein that acts as a fluorescent energy acceptor. When the two proteins bind together the two fluorescent reagents are brought into close spatial proximity and an acceptor signal at 618 nm is generated, but in the absence of protein-protein binding, the two reagents remain spatially separated and the donor signal at 460 nm predominates. The ration between the fluorescent signals at 460 nm and 618 nm is indicative of the degree of Ras-Raf binding. Using this assay system, swinhopeptolides A (1) and B (2) were effective inhibitors of this protein-protein interaction with IC50 values of 5.8 μM and 8.5 μM, respectively (Supporting Information).
Most efforts to suppress the Ras-Raf-MEK-ERK signal transduction cascade have targeted the active sites of the various signaling proteins. Much less attention has focused on disrupting critical protein-protein interactions such as the Ras-Raf complex. Small molecules that can effectively inhibit protein-protein interactions are relatively rare, in large part because protein-protein interaction domains are generally flat and quite large, covering 100s to 1000s of square nanometers.28,29 However, a number of natural products such as cyclosporine and rapamycin have been found to effectively block specific protein-protein interactions. The finding that swinhopeptolides A (1) and B (2) were active in the cell-based BRET assay for Ras-Raf interactions suggests that they can suppress or destabilize the Ras-Raf complex. Detailed biochemical confirmation and evaluation of these effects is warranted, but these initial results indicate that the swinhopeptolides may represent a new structural class of protein-protein interaction modulators.
EXPERIMENTAL SECTION
General Experimental Procedures.
Optical rotations were measured on a Perkin-Elmer 241 polarimeter and UV spectra were acquired in spectroscopy grade MeOH using a Varian Cary 50 UV-Vis spectrophotometer. NMR spectra were obtained with a Bruker Avance III NMR spectrometer equipped with a 3 mm cryogenic probe and operating at 600 MHz for 1H and 150 MHz for 13C. Spectra were referenced to residual solvent signals at δH 2.50 and δC 39.5 (DMSO-d6), and HMBC experiments were optimized for nJCH = 8.3 Hz. HRESIMS data were acquired on an Agilent Technology 6530 Accurate-mass Q-TOF LC/MS. HPLC separations were performed on a Varian ProStar HPLC system using a Phenomenex Luna C18 (5μ, 100 Å, 250 × 10 mm) column run with the indicated gradient.
Animal Material.
Samples of the marine sponge Theonella swinhoei cf. verrucosa were collected by scuba at a depth of 18 feet, in Papua New Guinea in 1993, and kept frozen until extraction. The collection was carried out by the Coral Reef Research Foundation under contract with the National Products Branch, U.S. National Cancer Institute. A voucher specimen (voucher ID # 0CDN1829) was deposited at the Smithsonian Institution, Washington, D. C.
Extraction and Isolation.
The frozen specimens of Theonella swinhoei (194.5 g wet wt.) were repeatedly extracted according to the standard NCI methodology outlined in McCloud30 to give 50 g of an aqueous extract. A 5.2 g aliquot of the extract was subjected to C18 reversed-phase flash column chromatography using a stepwise gradient elution with 100% H2O (fraction A, 4.2 g), 2:1 H2O-MeOH (fraction B, 267 mg), 1:2 H2O-MeOH (fraction C, 179 mg), and 100% MeOH (fraction D, 90 mg). The active fraction D was chromatographed on a Sephadex LH-20 column (25 × 510 mm), using 1:1 MeOH-CH2Cl2 as eluent, to obtain 20 fractions. The combined active fractions (13–14) was separated by semi-preparative reversed-phase HPLC (Phenomenex Luna C18 column, 10 mm × 250 mm; 2.0 mL/min, MeCN-H2O, 50:50), yielding 6 peaks rich in secondary metabolites. Purification of the sub-fractions (4 and 5) was accomplished by analytical HPLC (Phenomenex Luna C18 column, 4.6 mm × 250 mm; 0.8 mL/min, CH3CN-H2O gradient (20:80–100:0, containing 0.1% trifluoroacetic acid) to yield compounds 1 (5.1 mg) and 2 (0.9 mg), respectively, as amorphous solids.
Swinhopeptolide A (1):pale yellow, amorphous powder; −5 (c 0.1, MeOH); UV (MeOH) λmax (log ε) 203 (3.24), 229 (3.17), 276 (2.07) nm; IR (film) νmax 3300, 2936, 1740, 1670, 1453, 1204, 1137 cm−1; 1H and 13C NMR data, Table 1; HRESIMS m/z 1436.7281 [M + Na]+ (calcd for C66H103N13O21Na, 1436.7288).
Swinhopeptolide B (2): white, amorphous powder; −13 (c 0.1, MeOH); UV (MeOH) λmax (log ε)) 202 (3.05), 228 (2.52), 274 (1.81) nm; IR (film) νmax 3300, 2948, 1743, 1679, 1447, 1208, 1141cm−1; 1H and 13C NMR data, Table S1; HRESIMS m/z 1380.7416 [M + H]+ (cald for C66H102N13O19, 1380.7420).
Acid Hydrolysis of Peptides and LC-MS Analysis of D/L-DLA Derivatives.25
Compounds 1, 2, and mirabamide A (0.2 mg each) were separately dissolved in degassed 6 N HCl (0.6 mL) and heated in sealed glass vials at 110 °C for 17 h. The hydrolysates were evaporated to dryness, dissolved in H2O (50 μL) and to each sample 20 μL of 1 N NaHCO3 and 100 μL of a 1% solution in acetone of 1-fluoro-2,4-dinitrophenyl-5-L-leucinamide (L-FDLA or D/L-FDLA) was added. The mixtures were heated to 40 °C for 40 min and then cooled to room temperature, neutralized with 2 N HCl (20 μL), and evaporated to dryness. The residues were dissolved in CH3CN and then analyzed by LC-MS (Poroshell 120 EC-C18 column, 4.6 × 150 mm, 1.0 mL/min, CH3CN-H2O gradient 5:95–100:0, containing 0.1% formic acid in 60 min). An Agilent 6130 Quadrupole mass spectrometer was used for ESIMS detection (positive and negative ion mode). DLA derivatives were detected by absorption at 340 nm, and assignment was secured by ion-selective monitoring. The retention times (tR) of the D/L-DLA mixtures (with the L-DLA tR underlined) were as follows.
Swinhopeptolide A (1): L-Pro (23.10), D-Pro (24.85), m/z 408 [M-H]−; L-Ser (20.88), D-Ser (21.41), m/z 400 [M+H]+; L-Ala (22.96), D-Ala (25.47), m/z 384 [M+H]+; L-O-MeSer (22.50), D-O-MeSer (25.43), m/z 414 [M+H]+ ; L-Dab (32.25), D-Dab (34.82), (32.32, from mirabamide A), m/z 705 [M-H]− (bis DLA derivative); L-3-OHLeu (23.10), D-3-OHLeu (26.91), (26.97, from mirabamide A), m/z 440 [M-H]−; L-N-MeThr (21.31), D-N-MeThr (22.81), (21.30, from mirabamide A), m/z 426 [M-H]−; 3,4-DiMe-L-Gln (22.89), 3,4-DiMe-D-Gln (24.43), (22.25, from mirabamide A), m/z 468 [M-H]−.
Swinhopeptolide B (2): L-Pro (23.11), D-Pro (24.87), m/z 408 [M-H]−; L-Ser (20.90), D-Ser (21.45), m/z 398 [M-H]−; L-Ala (22.97), D-Ala (25.49), m/z 382 [M-H]−; L-O-MeSer (22.54), D-O-MeSer (25.48), m/z 412 [M-H]−; L-Dab (32.28), D-Dab (34.85), m/z 705 [M-H]− (bis derivative); L-3-OHLeu (22.81), D-3-OHLeu (24.31), m/z 440 [M-H]−; L-N-MeThr (21.29), D-N-MeThr (22.81), m/z 426 [M-H]−; 3,4-DiMe-L-Gln (22.49), 3,4-DiMe-D-Gln (25.51), m/z 468 [M-H]−.
Absolute Configuration of β-Methoxytyrosine.
A stream of ozone in O2 was bubbled through individual cooled solutions of compounds 1, 2, and mirabamide A (0.2 mg each) in MeOH (2 mL) at −78 °C for 5 h. Hydrogen peroxide (30%, 10 drops) was added and the reaction mixture was allowed to stand at 23 °C overnight. The solvent was removed under a stream of N2 and the resultant ozonolysis products were dissolved in degassed 6 N HCl (0.5 mL) in an evacuated glass tube and heated at 160 °C for 16 h. The solvent was removed in vacuo and the resulting material was subjected to Marfey’s derivatization as described above. The derivatized amino acids were dissolved in CH3CN and then analysis of the DLA mixtures (with the L-DLA tR underlined) were as follows: : L-β-OMeAsp (33.40), D-β-OMeAsp (34.71) for 1, L-β-OMeAsp (32.94), D-β-OMeAsp (34.74) for 2, (34.62, from mirabamide A), m/z 458 [M+H]+.
Ras-Raf Assay.
A modification of a live cell bioluminescence resonance energy transfer (BRET) analysis system for assessing Ras-Raf protein interactions was used to identify disrupters of Ras-Raf binding in cells.31 Cells expressing Ras and Raf proteins fused to BRET acceptor and donor domains, respectfully, were treated with test samples at 10 μg/mL for 4 h followed by assessment of the BRET signal. Acceptor, donor, and BRET signals were each monitored to control for protein expression levels. The same assay system was used for bioassay-guided fractionation and to estimate potency of the final purified active components.
Supplementary Material
ACKNOWLEDGMENTS
Grateful acknowledgement goes to the Natural Products Support Group (NCI at Frederick) for extraction, and A. Wamiru for Ras/Raf assay support. This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supporting Information.
The Supporting Information is available free of charge on the ACS Publications website. Experimental procedures, additional figures, and full spectroscopic data for all new compounds (PDF)
The authors declare no competing financial interest.
References
- 1.Zampella A; D’Auria MV; Gomez-Paloma L; Casapullo A; Minale L; Debitus C; Henin YJ Am. Chem. Soc 1996, 118, 6202–6209. [Google Scholar]
- 2.Zampella A; D’Orsi R; Sepe V; Casapullo A; Monti MC; D’Auria MV Org. Lett 2005, 7, 3585–3588. [DOI] [PubMed] [Google Scholar]
- 3.D’ Auria MV; Zampella A; Gomez-Paloma L; Minale L; Debitus C; Roussakis C; Le Bert V Tetrahedron 1996, 52, 9589–9596. [Google Scholar]
- 4.Zampella A; Randazzo A; Borbone N; Luciani S; Trevisi L; Debitus C; D’Auria MV Tetrahedron Lett. 2002, 43, 6163–6166. [Google Scholar]
- 5.Sepe V; D’Orsi R; Borbone N; D’Auria MV; Bifulco G; Monti MC; Catania A; Zampella A Tetrahedron 2006, 62, 833–840. [Google Scholar]
- 6.Ford PW; Gustafson KR; McKee TC; Shigematsu N; Maurizi LK; Pannel LK; Williams DE; de Silva ED; Lassota P; Allen TM; Soest RV; Andersen RJ; Boyd MR J. Am. Chem. Soc 1999, 121, 5899–5909. [Google Scholar]
- 7.Prasad P; Aalbersberg W; Feussner K-D; Van Wagoner RM Tetrahedron 2011, 67, 8529–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie W; Ding D; Zi W; Li G; Ma D Angew. Chem. Int. Ed 2008, 47, 2844–2848. [DOI] [PubMed] [Google Scholar]
- 9.Plaza A; Gustchina E; Baker HL; Kelly M; Bewley CA J. Nat. Prod 2007, 70, 1753–1760. [DOI] [PubMed] [Google Scholar]
- 10.Lu Z; Van Wagoner RM; Harper MK; Baker HL; Hooper JNA; Bewley CA; Ireland CM J. Nat. Prod 2011, 74, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oku N; Gustafson KR; Cartner LK; Wilson JA; Shigematsu N; Hess S; Pannell LK; Boyd MR; McMahon JB J. Nat. Prod 2004, 67, 1407–1411. [DOI] [PubMed] [Google Scholar]
- 12.Oku N; Krishnamoorthy R; Benson AG; Ferguson RL; Lipton MA; Phillips LR; Gustafson KR; McMahon JB J. Org. Chem 2005, 70, 6842–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamano Y; Arai M; Kobayashi M Bioorg. Med. Chem. Lett 2012, 22, 4877–4881. [DOI] [PubMed] [Google Scholar]
- 14.Shin HJ; Rashid MA; Cartner LK; Bokesch HR; Wilson JA; McMahon JB; Gustafson KR Tetrahedron Lett. 2015, 56, 4215–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martín MJ; Rodríguez-Acebes R; García-Ramos Y; Martínez V; Murcia C; Digón I;Marco I; Pelay-Gimeno M; Fernández R; Reyes F: Francesch AM; Munt S; Tulla-Puche J; Albericio F; Cuevas C J. Am. Chem. Soc 2014, 136, 6754–6762. [DOI] [PubMed] [Google Scholar]
- 16.Andjelic CD; Planelles V; Barrows LR Marine Drugs, 2008, 6, 528–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parsons AB; Lopez A; Givoni IE; Williams DE; Gray CA; Porter J; Chua G; Sopko R; Brost RL; Ho C-H; Wang J; Ketela T; Brenner C; Brill JA; Fernandez GE; Lorenz TC; Payne GS; Ishihara S; Ohya Y; Andrews B; Hughes TR; Frey BJ; Graham TR; Andersen RJ; Boone C Cell, 2006, 126, 611–625. [DOI] [PubMed] [Google Scholar]
- 18.Cassilly CD; Maddox MM; Cherian PT; Bowling JJ; Hamann MT; Lee RE; Reynolds TB PLoS One, 2016, 11, e0154932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonni A; Brunet A; West AE; Datta SR; Takasu MA; Greenberg ME Science 1999, 286, 1358–1362. [DOI] [PubMed] [Google Scholar]
- 20.Chang L; Karin M Nature 2001, 410, 37–40. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez-Medarde A; Santos E Genes Cancer 2011, 2, 344–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davies H; Bignell GR; Cox C; Stephens P; Edkins S; Clegg S; Teague J; Woffendin H; Garnett MJ; Bottomley W; Davis N; Dicks E; Ewing R; Floyd Y; Gray K; Hall S; Hawes R; Hughes J; Kosmidou V; Menzies A; Mould C; Parker A; Stevens C; Watt S; Hooper S; Wilson R; Jayatilake H; Gusterson BA; Cooper C; Shipley J; Hargrave D; Pritchard-Jones K; Maitland N; Chenevix-Trench G; Riggins GJ; Bigner DD; Palmieri G; Cossu A; Flanagan A; Nicholson A; Ho JWC; Leung SY; Yuen ST; Weber BL; Seigler HF; Darrow TL; Paterson H; Marairs R; Marshall CJ; Wooster R; Stratton MR; Futreal PA Nature 2002, 417, 949–954. [DOI] [PubMed] [Google Scholar]
- 23.Samatar AA; Poulikakos PI Nat. Rev. Drug Discov 2014, 13, 928–942. [DOI] [PubMed] [Google Scholar]
- 24.Ryan MB; Der CJ; Wang-Gillam A; Cox AD Trends Cancer 2015, 1, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.see for example:; (a) Yan P; Ritt DA; Zlotkowski K; Bokesch HR; Reinhold WC; Schneekloth JS Jr.; Morrison DK; Gustafson KR J. Nat. Prod 2018, 81, 1666–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thornburg CC; Britt JR; Evans JR; Akee RK; Whitt JA; Trinh SK; Harris MJ; Thompson JR; Ewing TL; Shipley SM; Grothaus PG; Newman DJ; Schneider JP; Grkovic T; O’Keefe BR ACS Chem. Biol 2018, 13, 2484–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tran TD; Wilson BAP; Henrich CJ; Staudt LM; Krumpe LRH; Smith EA; King J; Wendt KL; Stchigel AM; Miller AN; Cichewicz RH; O’Keefe BR; Gustafson KR J. Nat. Prod 2019, 82, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Fujii K; Ikai Y; Mayumi T; Oka H; Suzuki M; Harada K Anal. Chem 1997, 69, 3346–3352. [Google Scholar]; (b) Fujii K; Ikai Y; Mayumi T; Oka H; Suzuki M; Harada K Anal. Chem 1997, 69, 5146–5151. [Google Scholar]
- 27.(a) Han H; Yoon J; Janda KD J. Org. Chem 1998, 63, 2045–2048. [Google Scholar]; (b) Bunnage ME; Burke AJ; Davies SG; Millican NL; Nicholson RL; Roberts PM; Smith AD Org. Biomol. Chem 2003, 1, 3708–3715. [DOI] [PubMed] [Google Scholar]
- 28.Smith MC; Gestwicki JE Expert Rev. Mol. Med 2012, 14, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott DE; Bayly AR; Abell C; Skidmore J Nat. Rev. Drug Discov 2016, 15, 533–550. [DOI] [PubMed] [Google Scholar]
- 30.McCloud TG Molecules 2010, 15, 4526–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terrell EM, Durrant DE, Ritt DA, Sealover NE, Sheffels E, Spencer-Smith R, Esposito D, Zhou Y, Hancock JF, Kortum RL, Morrison DK Mol. Cell 2019, 76, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.