

Abstract
The pool of beta cell-specific CD8+ T-cells in type 1 diabetes (T1D) sustains an autoreactive potential despite having access to a constant source of antigen. To investigate the long-lived nature of these cells, we established a DNA methylation-based T cell “multipotency index” and found that beta cell-specific CD8+ T-cells retained a stem-like epigenetic multipotency score. Single cell ATAC-seq analysis confirmed the co-existence of naive and effector-associated epigenetic programs in individual beta cell-specific CD8+ T-cells. Assessment of beta cell-specific CD8+ T-cell anatomical distribution and the establishment of stem-associated epigenetic programs revealed that self-reactive CD8+ T-cells isolated from murine lymphoid tissue retained developmentally plastic phenotypic and epigenetic profiles relative to the same cells isolated from the pancreas. Collectively, these data provide new insight into the longevity of beta cell-specific CD8+ T cell responses, and document the utility of this novel methylation-based multipotency index for investigating human and mouse CD8+ T-cell differentiation.
Introduction
Self-reactive T cells play an important role in the development of a wide spectrum of life-long immunopathologies1, 2, 3. Often considered a quintessential autoimmune disease, type 1 diabetes (T1D) is distinguished by self-reactive T cell-mediated destruction of insulin-secreting beta cells in the pancreatic islets of Langerhans4, 5,6, 7, 8. Importantly, the cytolytic capacity among beta cell-specific T-cells can be preserved for long periods of time as pancreatic islet transplant recipients have a predisposition to undergo a rapid antigen-specific effector CD8+ T cell response. The extraordinarily long-lived effector capacity of these cells raises questions regarding the mechanisms that preserve this quality throughout the life of the individual9. Based on the phenotypic characterization of beta cell-specific CD8+ T cells found in circulation, recent studies reported a correlation between disease severity and a T cell phenotype associated with limited homeostatic proliferation (namely, effector-memory or Tem)10. However, another study reported that the majority of beta cell-specific CD8+ T cells possess a less differentiated stem-cell memory (Tscm) phenotype11. The discrepancy between these phenotypic analyses raises many unresolved questions about the differentiation status of these cells. Hence, there is a critical need to more broadly investigate the mechanisms that contribute to reinforcing effector and memory T cell-associated properties of beta cell-specific CD8+ T cells.
Broadly, epigenetic modifications, which include histone modifications and DNA methylation, influence gene expression patterns without altering the underlying DNA sequence12, 13. By providing a mechanism to heritably propagate acquired gene expression programs in a dividing population of cells, epigenetic modifications can be utilized to reinforce cell fate decisions. Our group and others recently demonstrated a causal relationship between epigenetic programming and the maintenance of effector and memory-associated functions during T cell homeostasis to sustain long-lived immunity14, 15, 16, 17. During the development of long-lived memory CD8+ T cells, activated naive antigen-specific CD8+ T cells transition through the effector stage of differentiation enabling a subset of cells to acquire effector-associated programs prior to their continued development into memory CD8+ T cells16, 18. The transient exposure to effector-promoting signals imparts memory T cells with long-lived effector-associated gene expression that endow memory T cells with a heightened ability to recall effector functions while retaining the naive-like capacity to develop into other memory and effector cell types. Importantly, the blend of naive and effector properties among memory CD8+ T cells is reflected by their epigenetic profiles being similar to both naive and effector T cells19.
Here, we applied the concept that changes in DNA methylation reinforce CD8+ T cell fate decisions and investigated the relationship between epigenetic programs and the longevity of human autoreactive T cell responses during T1D. Characterization of MHC class I tetramer+ beta cell-specific CD8+ T cells isolated from the circulation of type 1 diabetics revealed that this pool of autoreactive T cells is imparted with epigenetic programs associated with both naive and effector-associated properties. Indeed single cell ATAC-seq confirmed that beta cell-specific CD8+ T cells exhibit transcriptionally permissive regions consistent with both naive and effector stages of differentiation. Consistent with the results from our human self-reactive T cell epigenetic analyses, mouse beta cell-specific CD8+ T cells isolated from lymphoid tissues away from the source of antigen retain a stem-like epigenetic state. Collectively, the results presented here indicate that beta cell-specific CD8+ T cells can acquire a hybrid of naive and effector associated epigenetic programs and provide a mechanism to explain how the stem-like state of the cells can sustain the autoreactive immune response.
Results
Beta cell-specific CD8+ T cells acquire Tscm-like epigenetic programming
In order to fully contextualize the differentiation-associated programs among beta cell-specific T cells, we first created an epigenetic atlas of human CD8+ T cell differentiation. To establish a broad spectrum of human CD8+ T cell differentiation-associated epigenetic profiles, we isolated naive, short-lived, and long-lived memory CD8+ T cells from healthy donors to generate whole-genome bisulfite sequencing (WGBS) DNA methylation profiles (Fig. 1a, top panel). These polyclonal CD8+ T cell subsets cover a developmental spectrum ranging from less differentiated (naive and Tscm) to more differentiated CD8+ T cells (Tem)14. In addition to the polyclonal CD8+ T cell subsets, we also analyzed HIV-specific CD8+ T cells from infected individuals to characterize DNA methylation profiles of cells that have undergone differentiation in the setting of chronic antigen exposure. The HIV-specific CD8+ T cells exhibited phenotypic properties similar to Tem cells but also expressed high levels of the inhibitory receptor, PD-1 (Fig. 1a, bottom panel), which is epigenetically reinforced even following therapeutic reduction in viral load20. Taken together, these data indicate that the HIV-specific T cells are representative of a chronically stimulated population of cells and provide a benchmark for terminal differentiation in our analysis.
Figure 1: Generation of the human CD8+ T cell DNA methylation landscape for assessment of beta cell -specific CD8+ T cell differentiation.

(A) Representative FACS plots showing expression of PD-1 expression among true naive and memory CD8+ T cells subsets from healthy adult subjects (Upper panel) and cell surface expression of CCR7, CD45RO, and PD-1 among HIV-specific CD8+ T cells from HIV-ART patients (Lower Panel). (B) PCA of methylation status of the total CpG sites in CD8+ T cells showing 63.5% of principal component 1 (PC1). Principal component 2 = 4% of variance (not shown). Naive CD8+ T cells from HIV patients (n=3), T1D patient (n=5), and healthy adult donors [HD] (n=4), T1D Tetramer+ (n=5), HD Tscm (n=3), Tcm (n=3), Tem (n=3), and HIV Tetramer+ (n=4). (mean +/− SEM). (C) Venn diagram showing the number of DMRs in T1D-specific CD8+ T cells genomes relative to Tcm, Tem, Tscm, and HIV-specific CD8+ T cell genome. The number of demethylated regions was calculated based on >or= 30%methylation between the two population. The number of methylated regions was calculated based on <or= - 30%methylation between the two population. (D) Normalized plots of CpG methylation at sites surrounding and within DMRs of stemness programs including transcription factors (BATF and TOX), and the de novo DNA methyl transferase enzyme (DNMT3A). Red and blue lines depict methylated and unmethylated CpG sites, respectively.
Having generated whole-genome nucleotide-resolution methylation profiles of three or more biological replicates for each of these T cell populations, we next performed an unsupervised principal component analysis (PCA) of the methylation status across the most variable 3000 CpG sites in the genome. Notably, PC1 explained >60% of the variance among all samples (Supplementary Fig. 1). The greatest segregation was among the naive compared to Tem and HIV-specific CD8+ T cells, consistent with these subsets providing a lower and upper bounds on the developmental spectrum, respectively. Furthermore, this analysis revealed a notable clustering of naive CD8+ T cells isolated from both HIV-infected and healthy individuals. Lastly, the PCA placed the most developmentally plastic memory T cell population, Tscm, in an area of this “epigenetic spectrum” that is approximately equidistant between naive and chronically stimulated virus-specific T cells (Fig. 1b). Broadly, this analysis documents the linkage between DNA methylation programming and the putative multipotent capacity of human memory CD8+ T cells as well as provides a framework to further characterize beta cell-specific CD8+ T cells.
With an epigenetic atlas of human T cell differentiation established, we proceeded to generate whole-genome DNA methylation profiles of beta cell-specific CD8+ T cells from a well-defined cohort of T1D patients with a range in disease duration spanning 1 to 20 years (Supplementary Fig. 1a bottom table). After generating five independent whole-genome DNA methylation profiles of beta cell-specific CD8+ T cells, we again performed PCA of all the whole-genome DNA methylation profiles. Beta cell-specific T cell populations from all five participants, regardless of disease duration, clustered most closely to the Tscm CD8+ T cells (Fig. 1b, Supplementary Fig. 2a, 2b, & 2c). Notably, this analysis revealed that the self-reactive T cells isolated from the circulation of T1D patients were strikingly segregated from T cells that had progressed to a more terminally differentiated state (i.e., chronically stimulated HIV-specific CD8+ T cells). Thus, despite T cell activation with lasting access to their cognate antigen, beta cell-specific CD8+ T cells in the circulation of T1D do not acquire an epigenetic program associated with prolonged TCR engagement. Rather, beta cell-specific CD8+ T cells acquire an epigenetic program associated with the multipotent capacity of Tscm CD8+ T cells.
To further define the DNA methylation programs associated with the developmental status of self-reactive CD8+ T cells, we performed a pairwise comparison of gene-associated differentially methylated regions (DMRs) between beta cell-specific CD8+ T cells and each individual CD8+ T cell subset in addition to HIV-specific CD8+ T cells. Notably, Tscm and beta cell-specific CD8+ T cells had the most similar DNA methylation profiles with only thirty-seven DMR-associated genes (Fig. 1c). Further inspection of other DMR pairwise comparisons demonstrated a significantly greater number of differences in gene-associated DNA methylation programs between the T1D self-reactive CD8+ T cells and Tcm, Tem, and HIV-specific T cell populations. Previously, self-reactive T cells were often described as sharing features with naive CD8+ T cells and exhibiting a similar CD45RA+ CCR7+ phenotype (Supplementary Fig. 2a). Therefore, we proceeded to interrogate potential differences among the naive and beta cell-specific DNA methylation profiles. We identified more than 1000 regions that were differentially methylated between the naive and self-reactive T cells, encompassing both de novo DNA methylation and demethylation events (Supplementary Fig. 2d, bottom panel). Most (~95%) of these DMRs were located at or near genes (Supplementary Fig. 2d, top panel). Broadly, these data document methylation programs that distinguish self-reactive CD8+ T cells from CD8+ T cells in other differentiation states and highlight the epigenetic similarity between beta cell-specific CD8+ T cells and Tscm.
The marked overlap in DNA methylation programming among Tscm and self-reactive CD8+ T cells prompted us to further characterize methylation programs that are specifically shared between these populations that delineate these cells from other differentiated populations (Fig. 1d, Supplementary Fig. 2b – 2e). Shared methylation programs included regions located within TOX, DNMT3A, and BATF loci. Importantly, each of these transcription factors is critically involved in the terminal differentiation of T cells16, 21, 22, 23. Specifically, the TOX locus, a transcription factor recently shown to play a critical role in the survival of exhausted T cells23, 24, was demethylated in Tem and HIV-specific CD8+ T cells and remained methylated in both Tscm and the beta cell-specific CD8+ T cells. Additionally, consistent with its critical role in regulating the self-renewal capacity of embryonic and hematopoietic stem cells25, 26, 27, we observed that the DNMT3a internal promoter was significantly methylated in more differentiated T cell populations and remained unmethylated in the Tscm and self-reactive T cells. Notably, expression of DNMT3a isoform 2 has been historically associated with stem cells25. Because of the unmethylated status of the DNMT3a internal promoter, we cross referenced the previously defined Dnmt3a targeted loci in murine T cells16 with our existing Tem and HIV-specific CD8+ T cell DMR-associated gene list. Indeed, many of the genes targeted for Dnmt3a-mediated methylation in terminally differentiated mouse T cells were also differentially methylated among the self-reactive versus chronically stimulated virus-specific CD8+ T cells (Supplementary Fig. 3). Lastly, the gene body of B cell activating factor (BATF), a transcription factor involved in the self-renewal of hematopoietic stem cells, was highly methylated in beta cell-specific and Tscm CD8+ T cells but unmethylated in the more differentiated CD8+ T cells (Fig. 1d). Collectively, these data indicate that differentiation of beta cell-specific CD8+ T cells is coupled to an epigenetic rewiring of the cell that may preserve its multipotent capacity.
Development of a human DNA methylation-based T cell multipotency index to assess CD8+ T cell plasticity
To more broadly characterize the “multipotency-associated” epigenetic status of beta cell-specific T cells, we next compared our datasets to a recently reported human stem-cell epigenetic index. Derived from a machine learning algorithm that interrogated gene expression and epigenetic signatures of bona fide stem-cells, this index consists of 219 CpG sites used to assign a “stemness” hierarchy among stem and tumor cells28. From this index, only 45 CpG sites were differentially methylated between human CD8+ T cells across differentiation states (Supplementary Fig 4a). Therefore, to better define a CD8+ T cell-specific multipotency index, we utilized the same machine learning approach with naive and HIV-specific CD8+ T cells as training data sets to identify a set of 245 CpG sites whose methylation status can delineate the developmental hierarchy among CD8+ T cells (Fig. 2a & Supplementary Table 1). Dendogram representation of these data show that the beta cell-specific and Tscm CD8+ T cells were nearest to the naive CD8+ T cells, with both groups being segregated from the HIV-specific and polyclonal Tem CD8+ T cells. This developmental hierarchy was then normalized on a scale of 0 to 1 to generate a human T cell “multipotency index.” Previously defined methylation profiles of CD8+ T cell subsets (Tem, Tcm, and Tscm) were used to validate this tool. As expected, Tem had the lowest score followed by Tcm and Tscm which is consistent with their respective levels of differentiation (with lower scores indicative of a more terminally differentiated state). Importantly, beta cell-specific CD8+ T cells were found to have the highest (most naive-like) multipotency score (Fig. 2b). Collectively, these data indicate that beta cell-specific CD8+ T cells acquire an epigenetic program that is associated with retention of a stem-like differentiation state. Therefore, given that the duration of disease for several of the individuals used for establishing the beta cell-specific CD8+ T cell methylation profiles spanned several years, these data suggest that long-term pathology may be coupled to the ability of these stem-like, self-reactive CD8+ T cells to retain the potential to mount an effector response.
Figure 2: Novel human multipotency index predicts beta cell-specific CD8+ T cells to retain a degree of developmental plasticity comparable to Tscm.
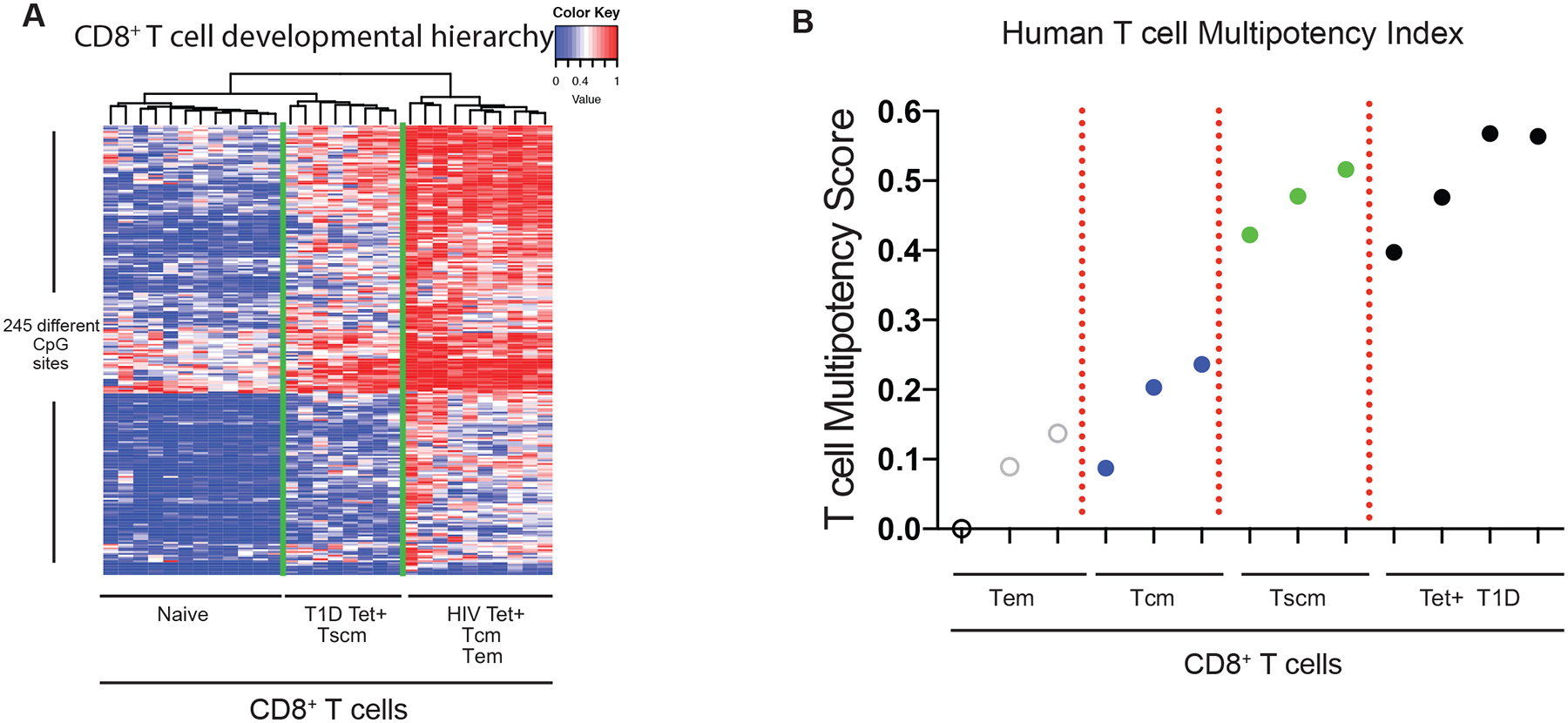
(A) Heat map showing the methylation status of 245 CpG sites of HIV-specific, T1D-specific, naive, Tem, Tcm, and Tscm CD8+ T cells used to define the human T cell multipotency index. The CpG sites were identified from a machine learning algorithm using naive and HIV-specific CD8+ T cells as the training data sets. Red and blue intensity depict methylated and unmethylated CpG sites. (B) A normalized human T cell multipotency score (0–1) was generated (see methods) based on the newly identified CpG sites that delineate the developmental potential of human T cells. Weight assignment for the methylation status at the 245 CpG sites is provided in supplemental table 1. The normalized multipotency index was applied to T1D-specific, Tem, Tcm, and Tscm CD8+ T cell methylomes.
Beta cell-specific CD8+ T cells acquire effector-associated epigenetic programs
Because progressive destruction of pancreatic islet cells relies largely on the effector functions of T cells8, 29, we next interrogated the beta cell-specific T cells for effector-associated DNA methylation programs. While our initial pairwise analysis of gene-associated DMRs documented a similarity between Tscm and self-reactive CD8+ T cell methylation profiles, it is important to note that the beta cell-specific T cells also acquired a core set of DNA methylation programs that are present in all memory T cell subsets. Indeed, >900 of the beta cell-specific CD8+ T cell-associated DMRs that delineate the self-reactive T cells from naive CD8+ T cells were also present in the Tem and/or HIV-specific CD8+ T cells (Supplementary Fig. 2d). These DMRs were primarily enriched in the 5’ distal regions, suggesting an association with transcriptional regulatory regions (Supplementary Fig. 2b). Core programs included demethylation of loci associated with effector responses such as Perforin (PRF1), GZMK, and IFNG. These loci were predominately unmethylated in beta cell-specific CD8+ T cells relative to loci in naive CD8+ T cells suggesting that they had undergone a differentiation program that enables an effector response (Fig. 3). In addition to demethylation of effector-associated loci, we observed differences in the methylation status at the loci of the transcription factors for Eomesodermin (EOMES), T-bet (TBX21), and T cell-specific transcription factor 7 (TCF7, encoding TCF1 protein), all of which have crucial roles in the differentiation of effector and memory CD8+ T cells (Fig. 3). Thus, while beta cell-specific CD8+ T cells retain many epigenetic features of a less differentiated cell, the modification of effector-associated loci indicates that the cells undergo some degree of effector differentiation.
Figure 3: Self-reactive human CD8+ T cells acquire effector-associated epigenetic programs.
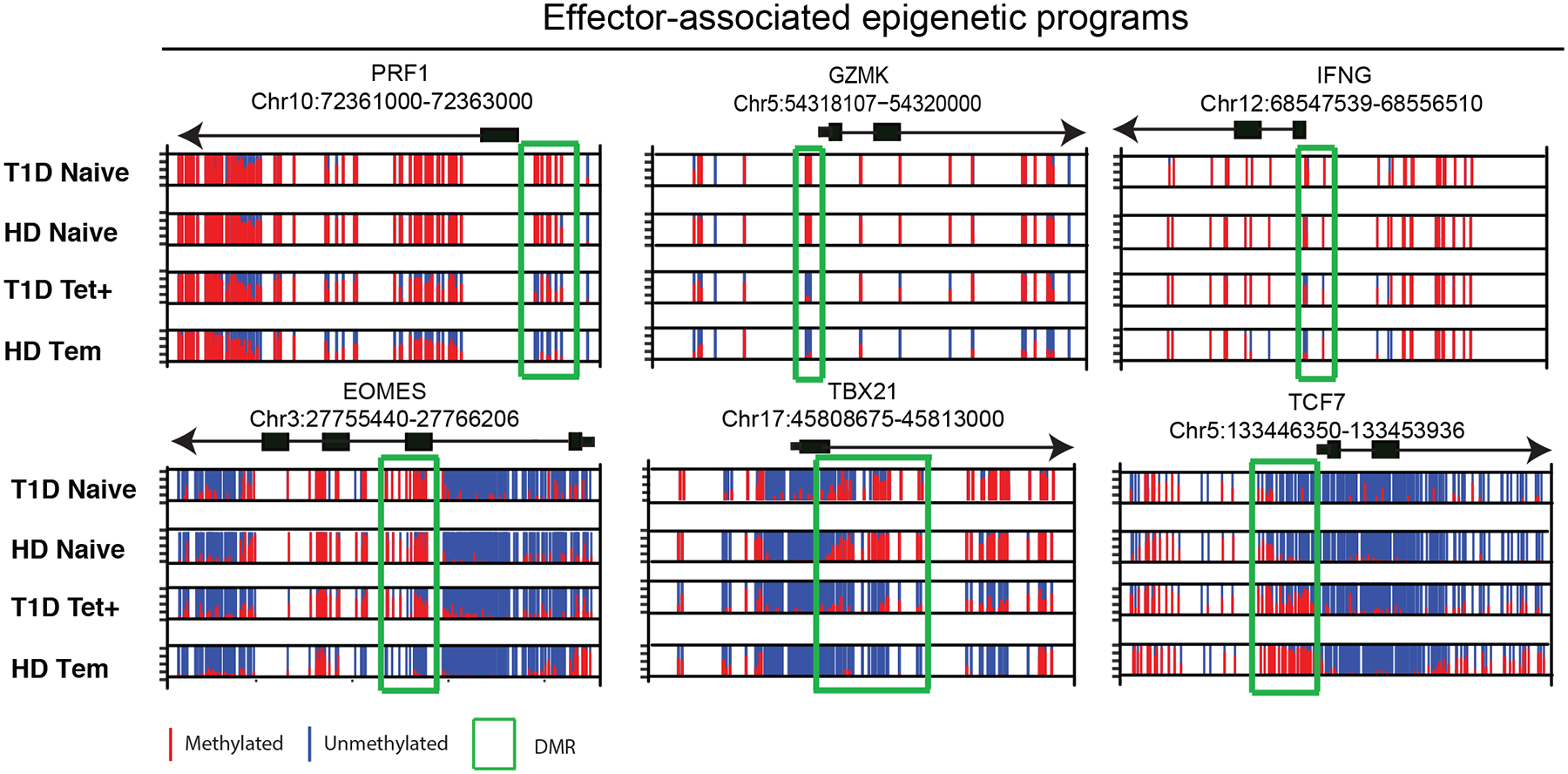
Normalized plots of CpG methylation at sites surrounding and within DMRs of effector molecules (PRF1, GZMK, and IFNg) and transcription factors (Eomes, TBX21 and Tcf7) obtained from WGBS analysis. Red and blue lines depict methylated and unmethylated CpG sites, respectively.
Naive and effector-associated epigenetic programs co-exist in individual beta cell-specific CD8+ T cells
Our DNA methylation data broadly suggest that beta cell-specific CD8+ T cells exhibit both naive and effector epigenetic programs. However, because our whole-genome DNA methylation profiling approach was performed on a pool of cells, we cannot fully resolve whether the hybrid profiles are truly co-existing in individual cells or if these data result from our analyses being performed on a heterogeneous population of cells that have distinct naive and effector-associated epigenetic programs. To further investigate the epigenetic programs acquired in individual beta cell-specific CD8+ T cells, we performed single cell ATAC-seq (scATAC-seq). Tetramer positive beta cell-specific CD8+ T cells, as well as donor-matched naive and Tem CD8+ T cells, were sorted from three T1D donors and scATAC-seq profiling was performed. t-SNE analysis of individual cell chromatin accessibility profiles for all samples broadly documented that a large portion of the beta cell-specific CD8+ T cells resemble a transition state between the naive and Tem subsets (Fig. 4a), which is similar to our results from the DNA methylation profiling studies shown in Fig. 1b. A subpopulation of beta cell-specific CD8+ T cells (highlighted in green) exhibits hybrid properties of both naive and Tem subsets. To confirm that the accessibility profiles indeed delineate the naive and effector-like states, we examined the accessibility of the CCR7 locus which is known to acquire epigenetically repressive modifications in Tem CD8+ T cells14. Indeed, all naive CD8+ T cells had high levels of chromatin accessibility at the CCR7 locus whereas this locus was largely inaccessible among the pool of Tem CD8+ T cells (Fig. 4b). Furthermore, interrogation of the CCR7 locus revealed that a population of beta cell-specific CD8+ T cells had an accessibility profile that suggested a transitional state between both the naive and Tem subsets. We next proceeded to interrogate the accessibility profiles of individual effector and naive-associated loci among the beta cell-specific CD8+ T cells. Importantly, we observed distinct subsets of tetramer+ beta cell-specific CD8+ T cells based on these profiles. Notably, one subset of tetramer+ CD8+ T cells was indistinguishable from naive CD8+ T cells. However, a separate population of tetramer+ CD8+ T cells segregated from the majority of both naive and Tem CD8+ T cells. Focusing on this subset of beta cell-specific CD8+ T cells, we examined several genes that were representative of the hybrid naive and effector state observed in the DNA methylation data set. These loci included the stem-associated transcriptional regulators TCF7, LEF1, and DNMT3A, effector/activation-associated transcription factors Tbet and Tox, and effector molecules, IFNG, PRF1, and GZMK. Notably, this cluster of beta cell-specific CD8+ T cells was found to have accessible chromatin at the loci of the transcriptional activators of the effector/chronic stimulation T response (TBX21 and TOX) and the accompanying effector molecules (IFNG and GZMK) (Fig. 4c & 4d). Quite strikingly, this same cluster of cells retained accessibility at the stem-associated loci of LEF1 and DNMT3A (Fig. 4c & 4d). These data document the co-existence of both naive and effector-associated epigenetic programs among individual beta cell-specific CD8+ T cells. Although many of the beta cell-specific CD8+ T cells overlap with either the naive and Tem, the existence of a hybrid tetramer+ cell population further supports the idea that the sustained autoreactive state of these T cells may be preserved by a long-lived population of cells that retain effector potential. These data further demonstrate the utility of the epigenetic-based multipotency index (DNA methylation profiling) to predict the developmental plasticity of human T cells.
Figure 4: Single cell ATACseq profiling identifies naive and effector epigenetic programming within individual beta cell-specific CD8+ T cells.
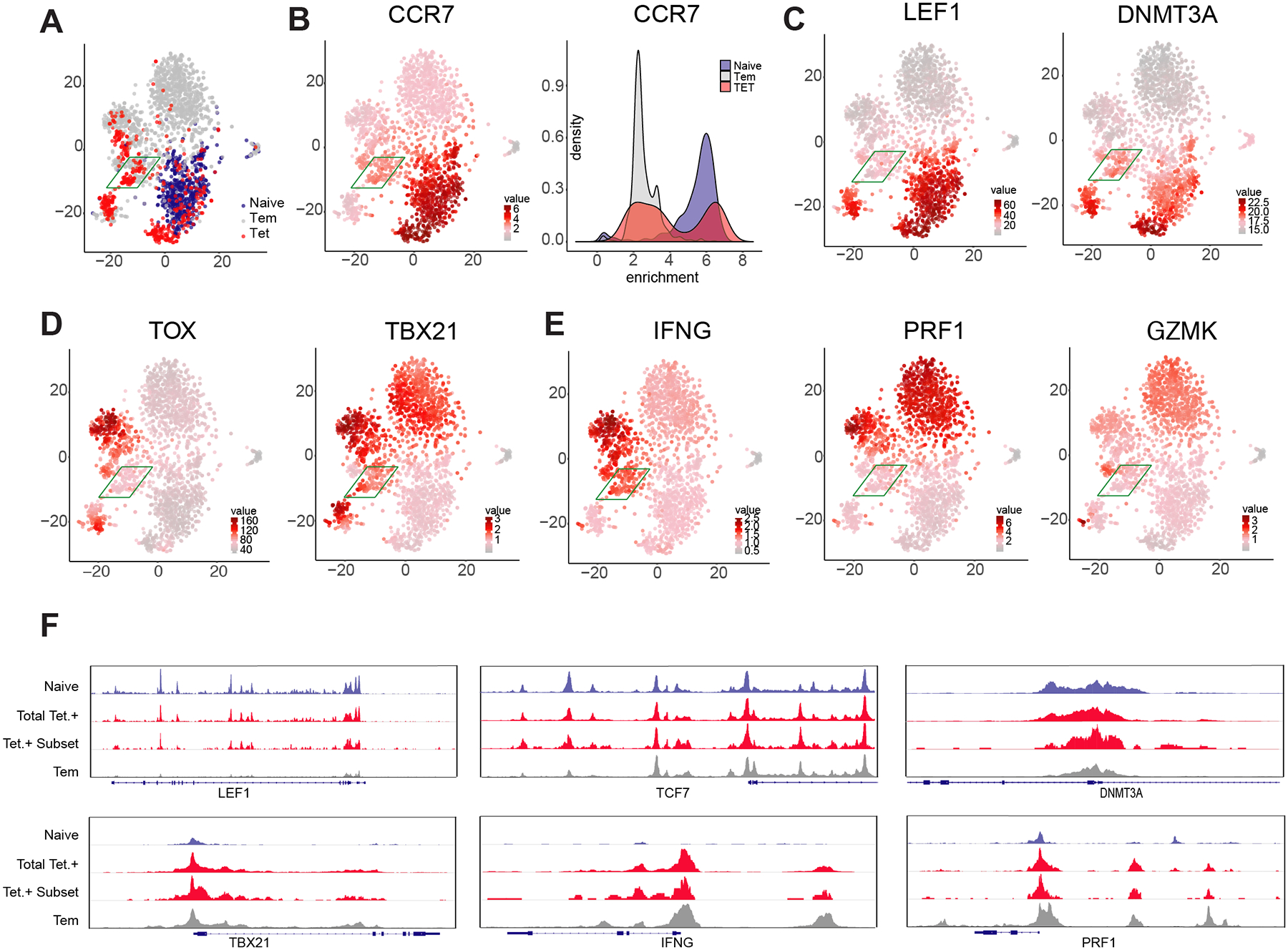
(A) t-SNE analysis of individual cell chromatin accessibility profiles for all (aggregated) Tetramer+ CD8+ T cells and representative naive and Tem populations from three donors. For all cells, heatmap for the chromatin accessibility profile of CCR7 (B), LEF1 and DNMT3a (C) TOX and TBX21 (D), IFNG, PRF1, and GZMK (E). (F) Composite (N = 3) ATACseq profiles showing accessibility peaks across the loci of LEF1, TCF7, DNMT3a, TBX21, IFNg, and PRF1 loci for naive, Tem, and beta cell-specific CD8+ T cells (Tetramer +).
Beta cell-specific CD8+ T cells maintain stemness-associated epigenetic programs during in vitro expansion
Our data suggest that the longevity of the effector T cell response in T1D patients is coupled to the ability of self-reactive CD8+ T cells to preserve their effector capacity during prolonged stimulation while also maintaining a developmentally plastic state of differentiation. To test the stability of multipotency-associated programs, we proceeded to determine if these programs were retained during ex vivo antigen-driven expansion of beta cell-specific CD8+ T cells. Peripheral blood mononuclear cells (PBMCs) were obtained from a T1D patient and labeled with cell trace violet (CTV), a dye to track cell proliferation. The CTV-labeled cells were then cultured with a mixture of peptides recognized by T1D self-reactive CD8+ T cells. After 14–24 days of ex vivo culture, tetramer positive CD8+ T cells that underwent several rounds of proliferation were phenotypically characterized and FACS-purified for subsequent DNA methylation analyses (Fig. 5a). Notably, the divided population of cells expressed high levels of CD95 and CD45RA with downregulated CCR7 (Fig. 5a), a phenotype often ascribed to an effector response. We next proceeded to determine if these cells retained the multipotency-associated epigenetic programs using DMRs at TOX and DNMT3A loci as surrogates of such programs. Importantly, targeted methylation profiling of these loci revealed that these programs remained unchanged during ex vivo antigen-dependent proliferation of beta cell-specific CD8+ T cells: the TOX locus remained partially methylated in beta cell-specific CD8+ T cells compared to mostly unmethylated Tem, while the DNMT3A locus remained partially methylated compared to the mostly methylated Tem (Fig. 5b). These data collectively demonstrate that despite having a significant proportion of cells exhibiting a Tem phenotype, beta cell-specific CD8+ T cells maintain multipotency-associated epigenetic programming after undergoing extensive antigen-dependent proliferation.
Figure 5: Stemness-associated DNA methylation programs are maintained during in vitro antigen-driven proliferation of human T1D-specific CD8+ T cells.
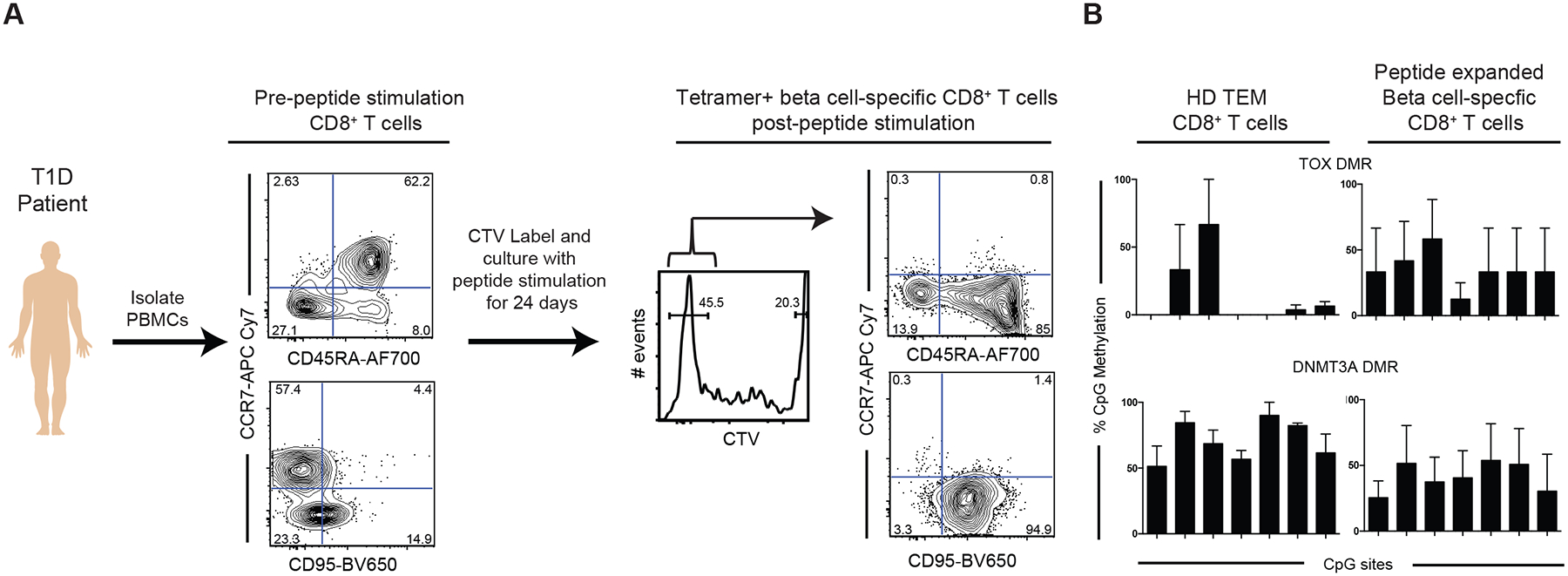
(A) Experimental setup for in vitro stimulation of total PBMCs from T1D patients. PBMCs were labeled with cell proliferation dye (cell trace violet-CTV) and subsequently maintained in culture in the presence of mixture of peptides specific for T1D-specific CD8+ T cells for 14–24days. Phenotypic analyses of beta cell- specific CD8+ T cells prior to and after 14–24 days of peptide stimulation. Antigen-driven expansion of beta-cell specific CD8+ T cells results in down regulation of CCR7 and upregulation of CD95. (B) Representative bisulfite sequencing analysis and bar graph showing % CpG methylation for individual CpG sites of stemness programs DNMT3A (CpG sites 12–18 shown) and TOX (CpG sites 4–10 shown) from beta cell-specific CD8+ T cells post peptide stimulation relative to the methylation status of bona fide effector memory CD8+ T cells N = 2.
Stem-associated epigenetic programs are enriched in lymphoid-homing beta cell-specific CD8+ T cells
Given that human beta cell-specific CD8+ T cells isolated from the peripheral blood of individuals with T1D retain stem-associated DNA methylation programs following in vitro expansion, we sought to investigate whether the developmentally plastic state of self-reactive CD8+ T cells was also preserved in cells that traffic to the source of the antigen. To overcome the challenge of examining patient beta cell-specific T cells from various anatomical locations, we utilized an established murine model that allowed us to examine an endogenous immune response in a site-specific manner30. Tetramer+ beta cell-specific CD8+ T cells were sorted from spleen, pancreatic lymph node, and pancreas of non-obese diabetic (NOD) mice and whole-genome DNA methylation profiling was performed. Notably, the phenotype of the beta cell-specific CD8+ T cells isolated from the lymphoid tissues versus the pancreas were strikingly distinct. Lymphoid residing beta cell-specific CD8+ T cells retained the phenotype of a multipotent memory T cell depicted by higher expression of CD127 and CD62L and lower PD-1 expression. Conversely, beta cell-specific CD8+ T cells isolated directly from the pancreas exhibited an effector phenotype with low expression of CD127 and CD62L and higher expression of PD-1 (Fig. 6a and 6b). Importantly, the phenotypic differences associated with the anatomical location were also consistent with the cell’s differentiation-associated epigenetic status. For instance, beta cell-specific CD8+ T cells isolated from the pancreas possess an effector-like methylation pattern at the Tcf7 locus whereas the same antigen-specific CD8+ T cells isolated from the spleen have a naive-like pattern (Fig. 6c). A similar pattern is seen at the Ifng locus with antigen-specific CD8+ T cells isolated from the pancreas exhibiting an effector-like methylation pattern compared to the more naive-like pattern in the spleen-derived antigen-specific T cells. While distinct methylation patterns are observed between the spleen- and pancreas-derived beta cell-specific CD8 T cells at several key loci, differences between the two cell populations were not uniformly observed at all gene loci. Examination of other relevant loci such as Batf, Eomes, Tbet (Tbx21), and Gzmk revealed epigenetic programming most consistent with an effector-like cell population. Collectively, these data are consistent with our findings from human beta cell-specific CD8+ T cells isolated from the peripheral blood of type 1 diabetics indicating that both murine and human beta cell-specific CD8+ T cells in the periphery retain a hybrid of naive and effector epigenetic programs. Further, these results indicate that the beta cell-specific CD8+ T cells may lose their naive-like state as they traffic to the site of antigen.
Figure 6: Lymphoid-homing murine beta cell-specific CD8+ T cells retain phenotypic and epigenetic programs indicating developmental plasticity.

(A) Representative FACS plots showing the phenotype of tetramer positive beta cell-specific CD8+ T cells isolated from murine spleen, pancreatic lymph node, and pancreas. Phenotypic characterization includes cell surface expression of CD44, PD-1, CD127, and CD62L. N = 3. (B) Summary graph from three different mice of tetramer positive beta cell-specific CD8+ T cells isolated from spleen, pancreatic lymph node, and pancreas (n=3). (C) Normalized plots of CpG methylation at sites surrounding and within DMRs of effector molecules (Ifng, Gzmk) and transcription factors (Tcf7, Batf, Eomes, and TBX21) obtained from WGBS analysis. Red and blue lines depict methylated and unmethylated CpG sites, respectively.
Differentiation status of beta cell-specific CD8+ T cells is dependent upon the site of antigen exposure
To further resolve whether a plasticity-associated epigenetic program delineates lymphoid-residing versus pancreas-residing beta cell-specific CD8+ T cells, we developed a murine-version of the CD8+ T cell multipotency index and applied it to the antigen-specific CD8+ T cell whole-genome DNA methylation data sets. This index utilized the same machine learning approach as the human multipotency index but was derived using training data sets with murine naive and LCMV-specific exhausted CD8+ T cells. This approach identified 177 CpG sites whose methylation status can delineate the developmental hierarchy among murine CD8+ T cells (Fig. 7a, Supplementary Table 2). Application of this murine multipotency index to our beta cell-specific CD8+ T cells documented that pancreas-derived CD8+ T cells have the lowest multipotency index whereas spleen-derived and pancreatic lymph node-derived CD8+ T cells have a multipotency index comparable to long-lived LCMV-specific memory T cells (Fig. 7b). Broadly, these data are consistent with our results from human beta cell-specific CD8+ T cells demonstrating that the circulating autoreactive T cells acquire an epigenetic program associated with retention of a less differentiated state. Beta cell-specific CD8+ T cells derived from murine pancreas have a lower multipotency score than Tem indicating they are more terminally differentiated, a cell fate consistent with their effector-like phenotype and epigenetic programs. Taken together, these data indicate that preservation of the auto-reactive potential of beta cell-specific CD8+ T cells is coupled to the acquisition of an epigenetic program the preserves a multipotent developmental potential in both mouse and human T1D. In addition to providing new insight into the disease etiology of T1D, our studies also highlight the general applicability of the methylation-based multipotency index to interrogate the developmental plasticity of CD8+ T cells derived from both humans and mice. Moreover, this novel tool may be broadly applied to interrogate the differentiation status of T cells from a wide range of disease settings as well as therapeutic modalities that utilize T cells in adoptive transfer settings.
Figure 7: Novel murine multipotency index predicts terminal differentiation of beta cell-specific CD8+ T cells isolated from the pancreas.
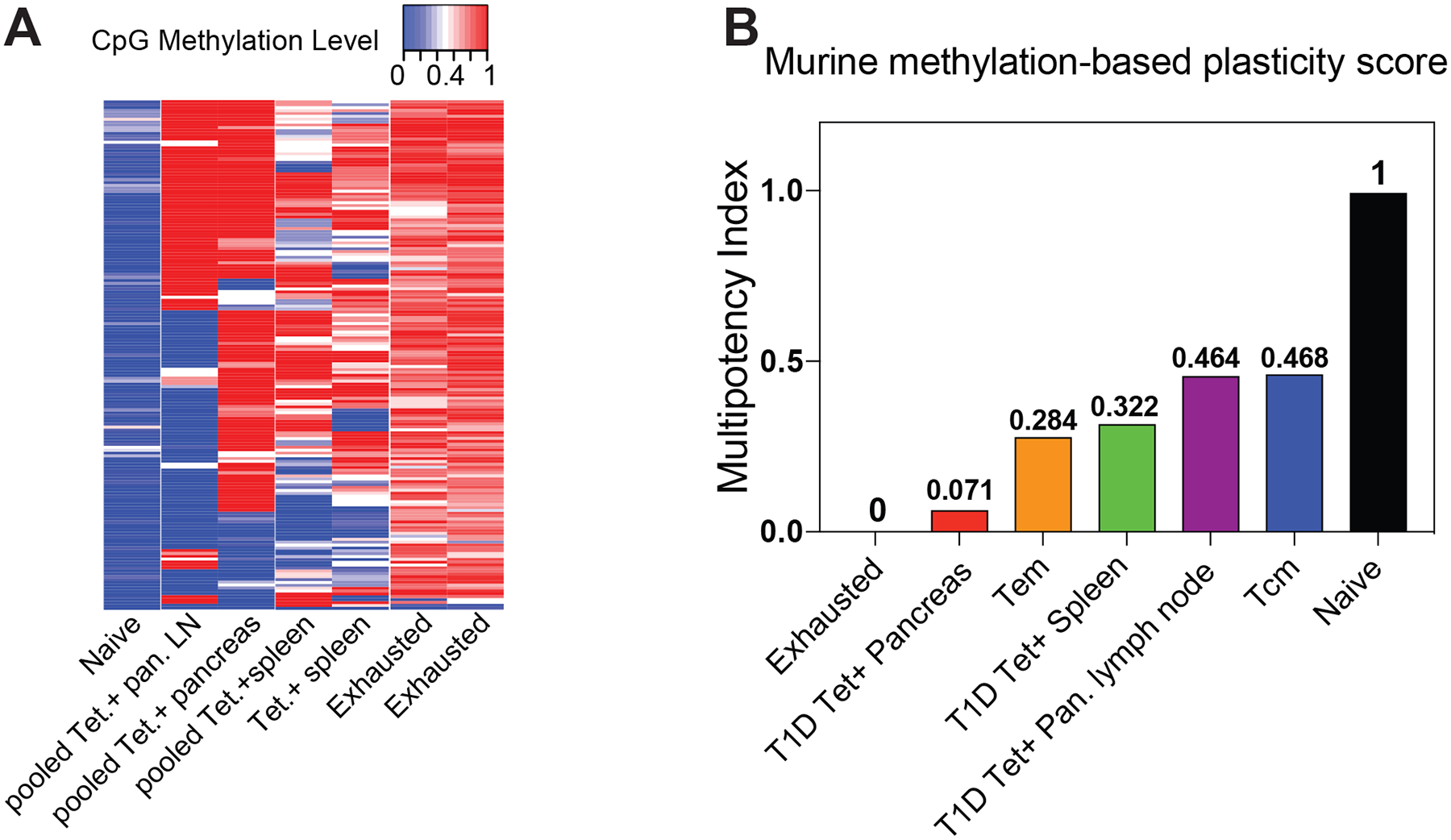
(A) Heat map showing the methylation status of 177 CpG sites of exhausted, lymphatic-derived, pancreas-derived, and naive CD8+ T cells used to define the murine T cell multipotency index. The CpG sites were identified from a machine learning algorithm using naive and LCMV-specific exhausted CD8+ T cells as the training data sets. Red and blue intensity depict methylated and unmethylated CpG sites. (B) A murine T cell multipotency score was generated as described in methods based on newly identified CpG sites that delineate the developmental potential of murine T cells. The multipotency score was derived from a machine learning algorithm using naive and LCMV-specific exhausted CD8+ T cell methylomes as the training datasets. LCMV-specific CD8+ T cell methylomes were used as test datasets for the multipotency index. The multipotency score was based on weight assignment for methylation status at 177 CpG sites.
Undeniably, there are likely to be many contributing factors that enable self-reactive T cells to resist progression to a more terminally differentiated state, yet several lines of evidence suggest that the low binding affinity of T1D autoantigens limit the stimulation that these T cells experience31, 32, 33. Consistent with this idea, we observed that LCMV-specific CD8+ T cells responding to a chronic low-affinity altered peptide ligand resist acquisition of DNA methylation programs found in T cells undergoing terminal differentiation during their response to the high-affinity ligand (Supplementary Fig. 5). These data support the idea that TCR signal strength impacts the epigenetic programs a T cell acquires in a chronic antigen environment. Thus, the combination of signal strength and duration are two factors that likely contribute to the preservation of the circulating beta cell-specific CD8+ T cells stem-like differentiation status.
Discussion
In the current study, our results reveal that beta cell-specific CD8+ T cells acquire epigenetic programs that are coupled to preservation of the autoreactive T cell’s ability to sustain a long-term effector response. For the first time, we show that a hybrid of naive and effector epigenetic programs coexist in the same self-reactive CD8+ T cells. Collectively, these data help reconcile the seemingly contradictory naive-like phenotype of beta cell-specific CD8+ T cells with results from other studies that document the effector functionality (e.g., expression of granzymes and perforin) of self-reactive CD8+ T cells. Based on our observation that circulating beta cell-specific CD8+ T cells resist acquisition of DNA methylation programs found in terminally differentiated T cells, these data raise the possibility that the antigen-specific cells found away from the source of antigen may provide a pool of self-renewing stem-like cells that can maintain an effector response. Our extension of this finding to a murine model of T1D further demonstrated that lymphoid-derived beta cell-specific CD8+ T cells were enriched in long-lived memory-associated phenotypic and epigenetic programs relative to pancreas-derived beta cell-specific CD8+ T cells. Broadly, these data suggest that it is indeed possible to induce and epigenetic program associated with terminal differentiation among autoreactive T cells.
Generally, the severity and life-long nature of T1D is intimately coupled to a failure in establishing central and/or peripheral tolerance among self-reactive T cells. Our data show that beta cell-specific CD8+ T cells infiltrating the pancreas, the source of antigen, acquire a terminally differentiated multipotency score and are methylated at gene loci related to T cell stemness such as Tcf7. These results suggest that beta cell-specific CD8+ T cells have the capacity to acquire an epigenetic program associated with restricted fate-potential and raise the possibility that therapeutic approaches that promote tolerance can be reinforced by epigenetic mechanisms. Further supporting the link between the mechanisms that preserve T cell stemness and pancreatic islet destruction, a recent publication describes that the rate of T1D disease progression is inversely related to the establishment of an exhaustion phenotype among beta cell-specific CD8+ T cells34. To attenuate the harmful nature of the beta cell-specific CD8+ T cells, significant efforts have been mounted to develop new therapeutic strategies to induce tolerance among these autoreactive CD8+ T cells. One such strategy utilized in a clinical trial (AbATE) attempted to therapeutically induce a tolerized state among T cells by treating individuals with an anti-CD3 monoclonal antibody. Although some patients in this trial exhibited a partial response, the delay in disease progression was ultimately transient35, 36. Similar to the AbATE study, anti-CD3 treatment administered to at-risk individuals prior to the onset of T1D was also shown to delay disease progression37. These data further reinforce the idea that the initial development of beta cell-specific CD8+ T cells imparts the cells with a capacity to evade a tolerizing fate38. Therefore, as opposed to generally targeting tolerization of established self-reactive T cells, the epigenetic data from beta cell-specific CD8+ T cells that we present here suggest a more successful strategy may involve inducing a tolerized state through an epigenetic reprogramming of the stem-like subset of the autoreactive T cells. Further, our results also highlight the potential utility of using epigenetic programs to track, even predict, the tolerized status of the T cells. Thus, development of the methylation-based T cell multipotency index might serve as a diagnostic tool for assessing long-term tolerance induction.
Beyond our investigation into the disease etiology of T1D, the novel bioinformatic tools that we have established to analyze beta cell-specific T cells has broad implications for understanding the role of methylation programming in T cell differentiation and establishment of long-lived memory T cells. Although prior studies examining changes in histone modifications during in vitro memory differentiation of murine CD8+ T cells have documented a lineage relationship between naive and memory CD8+ T cells39, here we have established an epigenetic atlas based on DNA methylation of the endogenous human T cell subsets. Our DNA methylation-based T cell multipotency index provides further documentation that progressive differentiation of human T cells is coupled to changes in DNA methylation. By using PD-1+ HIV-specific CD8+ T cells from a well-defined cohort of long-term infected individuals, we were able to better define the spectrum of epigenetic reprogramming events that result in the progressive loss of T cell developmental plasticity in humans. In addition to expanding our understanding of human memory T cell differentiation, this multipotency index can be applied toward improving T cell-based therapeutic approaches. As the field moves forward with development of novel strategies to induce tolerance among self-reactive T cells, the epigenetic atlas of T cell differentiation provided here will serve as a framework for assessing the long-lived impact these tolerance-focused strategies have on the differentiation status of CD8+ T cells. In addition to utilizing these tools to evaluate T cell tolerance, our novel multipotency index may be used as a resource to guide studies that aim to stably modify T cell function for an array of therapeutic purposes.
Methods
Isolation of human CD8+ T cells:
This study was conducted with approval from the Institutional Review Boards (IRBs) of St. Jude Children’s Research Hospital, Benaroya Institute (BRI), University of San Francisco (UCSF), and Case Western Reserve University (CWRU) and informed consent was obtained. For adult donors with T1D, PBMCs were previously collected and archived through the BRI Registry and Repository Sample Bank ID is IRB07109. For healthy adult donors, PBMCs were collected through the St. Jude Blood Bank, and samples for WGBS were collected under IRB protocol XPD15–086. PBMCs were purified from a platelet- apheresis blood unit using a density gradient. In brief, blood was diluted 1:2.5 using sterile Dulbecco’s PBS (Thermo Fisher Scientific). The diluted blood was then overlayed with Ficoll- Paque PLUS medium (GE Healthcare) to a final dilution of 1:2.5 (Ficoll/diluted blood). The gradient was centrifuged at 400 xg with no brake for 20 min at room temperature. The PBMC interphase layer was collected, washed with 2% FBS/1 mM EDTA PBS buffer, and centrifuged at 400 xg for 5 min. Total CD8+ T cells were enriched from PBMCs with the EasySep human CD8+ negative-selection kit (STEMCELL Technologies). All donors provided informed consent for collection of the blood samples used for all analyses. Cryopreserved PBMC samples were obtained from HIV-infected participants enrolled in the San Francisco-based SCOPE cohort who were infected for at least two years and on suppressive ART for at least two years with HIV viral load < 40 copies/mL. In case of T1D and HIV patients, the same approach has been used to collect PBMCs then tetramer/pentamer staining was performed to identify beta cell-specific and HIV-specific CD8+ T cells.
Isolation and flow cytometric analysis of naive and memory CD8+ T cell subsets:
After enrichment of CD8+ T cells, naive and memory CD8+ T cell subsets were sorted using the following markers, as previously described40, 41. Naive CD8+ T cells were phenotyped as live CD8+, CCR7+, CD45RO−, CD45RA+, and CD95− cells. CD8+ Tem cells were phenotyped as live CD8+, CCR7−, and CD45RO+ cells. TCM cells were phenotyped as live CD8+, CCR7+, and CD45RO+ cells. Tscm cells were phenotyped as live CD8+, CCR7+, CD45RO−, and CD95+ cells. Sorted cells were checked for purity (i.e., samples were considered pure if >90% of the cells had the desired phenotype). HIV-specific CD8+ T cells were identified by staining with MHC Class I tetramers (made in the laboratory of RPS) or pentamers (Proimmune, Oxford, United Kingdom) and sorted on a FACS Aria.
HLA-A2 Tetramer assembly:
Peptides representing previously characterized immunodominant beta cell epitopes INS B 10–18, PPI 15–24, GAD 114–122, IA2 797–805, IGRP 265–273, ZnT8 186–194 were synthesized by Genscript (Piscataway, NJ, USA). HLA-A2 monomers (refolded to contain each peptide of interest) were obtained through the National Institutes of Health (NIH) Tetramer Core Facility (Atlanta, GA, USA). PE and APC multimers were prepared utilizing a stepwise addition of fluophore labeled streptavidin slightly modified from the NIH Tetramer Core protocol. Briefly, 60 μl of each monomer (0.2 μg/μl) diluted 1:10 in PBS was multimerized by adding 1.2 μl of the appropriate PE or APC-labelled streptavidin 0.2mg/ml(Thermo Fisher Scientific, Waltham, MA, USA) for 10 min at 4°C 6 times. Multimers were stored at 4°C and used within 4 weeks.
Ex Vivo CD8+ T-Cell sorting:
Samples from HLA-A2–positive subjects were dual stained with a mixture containing a pool of PE and APC tetramers, PBMCs (20 ×10^6) were stained simultaneously with 3 μl each in 200 μl of PBS supplemented with 2% BSA and incubated for 15 min at 37°C. Cells were then incubated with 30 μl each of anti-PE and anti-APC magnetic beads (Miltenyi Biotec, San Diego, CA, USA) and enriched on a MS-sized magnetic column according to the manufacturer’s instructions. After enrichment, flow-through cells were reserved for bulk memory and naive CD8+ T-cell sorting described in the following section. Both enriched and flowthrough cells samples were washed and then stained for 30 min at 4°C with 5 μl each:anti- CD8+ BV605(Biolegend), AF700 CD45RA(BD Bioscience), APC-Cy7 CCR7(Biolegend), PerCP- eFluor 710 TIGIT(eBioscience), PE-Cy7 PD-1(Biolegend), BV650 CD95(Biolegend), BV421 KLRG1(Biolegend), 2.5 μl each of anti-CD4/CD14/CD16/CD20/CD40-FITC (dump channel; eBioscience, Waltham, MA, USA), and 1:10,000 syTOX green viability dye(Thermo Fisher). CD8+ cells were sorted using a FACSAria II(BD Biosciences) into lysis buffer from an Allprep DNA RNA mini prep kit (Qiagen) and processed for DNA and RNA according to the manufacturer’s instructions.
In vitro CD8+ T cell ex vivo expansion:
Isolated PBMCs were labeled with Cell Trace Violet-CTV (Life Technologies) at a final concentration of 1μM. CTV-labeled cells were maintained in culture in RPMI containing 10% FBS, penicillin-streptomycin containing mixture of 6 pooled peptides at a final concentration total of 100ug/ml. After 14–24d of incubation at 37°C and 5% CO2, adding medium and interleukin (IL)-2 as needed starting on day 7 cultures were collected for sorting. To visualize responses, cells were stained with tetramers and surface markers as described previously, and undivided (CTV++) and divided cells(CTV+/−) tetramer+ CD8+ T-cells were sorted into lysis buffer. To define the phenotypic characteristic features of the expanded cells, we used cell surface markers CD95, CCR7, and CD45RA.
Isolation and phenotypic analysis of mouse antigen-specific CD8+ T cells:
We adoptively transferred ~2000 congenically distinct naive P14 CD8+ T cells (CD45.1/1+) into C57BL/6 mice (CD45.1/2+). One day later, we infected the mice with different strains of lymphocytic choriomeningitis virus (LCMV) separately. Acute LCMV infection was performed by i.p. injection of 2×10^5 PFU Armstrong strain per mouse, while chronic LCMV infections were performed by i.v. injection of 2×10^6 PFU LCMV per mouse using either Clone 13 strain or C6 strain that has a mutated GP33 epitope with lower TCR-binding affinity to P14 cells42. After 4 or 8 weeks of LCMV infections we harvested the spleens and FACS-purified P14 cells from splenocytes. P14 cells were also phenotypically analyzed by Flow Cytometry after surface staining using monoclonal antibodies for CD45.1 (clone A20), CD45.2 (clone 104), CD8+ (clone 53–6.7), PD- 1(clone J43), and Klrg1 (clone 2F1).
Genomic methylation analysis:
DNA was extracted from the sorted cells by using a DNA- extraction kit (QIAGEN) and then bisulfite treated using an EZ DNA methylation kit (Zymo Research), which converts all unmethylated cytosines to uracils. Loci-specific PCR was performed using the primers described below. Individual clones of DNA were sequenced using the previously described blue/white bacterial cloning and screening approach14 Whole geneome bisulfie sequencing was performed as previously described. Briefly, bisulfite modified DNA-sequencing libraries were generated using the EpiGnome kit (Epicentre) per the manufacturer’s instructions. Bisulfite-modified DNA libraries were sequenced using an Illumina HiSeq system14. Sequencing data were aligned to the HG19 genome by using BSMAP software43. Differentially methylation analysis of CpG methylation among the datasets was determined with a Bayesian hierarchical model to detect regional methylation differences with at least three CpG sites44. To perform loci-specific methylation analysis, bisulfite modified DNA was PCR amplified with locus-specific primers. The PCR amplicon was cloned into a pGEMT easy vector (Promega) and then transformed into XL10-Gold ultracompetent bacteria (Agilent Technologies). Bacterial colonies were selected using a blue/white X-gal selection system after overnight growth, the cloning vector was then purified from individual colonies, and the genomic insert was sequenced. After bisulfite treatment, the methylated CpGs were detected as cytosines in the sequence, and unmethylated CpGs were detected as thymines in the sequence by using QUMA software45.
Human and mouse primer sequences for loci specific bisulfite sequencing:
Forward Human TOX: 5’-AGTAAGGTTTTTTTTTAACAATAGG-3’, Reverse Human TOX : 5’- CAATAAAATCATTCTAAAAAATAACAAC −3’ Forward Human DNMT3A: GAAGGTGTATTGAAGTGTGG-3’ Reverse Human DNMT3A: 5’- CCAAAAAAAAAACCCAACCCA-3’
Forward Mouse TOX:5’-GTGTAAGTTATTGTGATTCTGATTGTG-3’ Reverse Mouse TOX 5’- CTTTAACTACCCTCTCTAAATTAAAAAACC-3’ Forward Mouse DNMT3A: 5’- GGTTTTTGGATAGAGTGGGGATA-3’ Reverse Mouse DNMT3A: 5- CAAAAACTACCAAACVCATCAAACC-3’
T-cell multipotency index:
To identify the methylation state of the CpG sites associated with the T cell multipotent potential, a supervised analysis was performed between the methylomes from two naive and four HIV-specific CD8+ T cells (methylation difference >=0.4 and FDR<= 0.01). This analysis results in identification of 245 CpGs sites that were hypomethylated in naive HIV CD8+ T cells compared to HIV-specific CD8+ T cells. Each set of the CpGs was then used as an input to the one-class logistic regression to calculate the multipotency signature using just the HIV naive samples (Training data sets)28, 46. Once the signature was obtained, it was then applied to the beta cell-specific, HIV-specific, Naive, Tscm, Tem, and Tcm CD8+ T cell methylomes (Test data sets). The score was calculated as the dot product between the DNA methylation value and the signature. The score was subsequently converted to the [0, 1] range. Data sets with multipotency indices closer to 1 were more similar to naive cells.
Isolation of murine beta cell-specific CD8+ T cells from NOD mice:
Female, NOD mice, 8–14 weeks of age were euthanized by CO2 asphyxiation, followed by cervical dislocation. Prior to euthanasia, urine glucose was tested to exclude diabetic mice. Cells from the pancreas were harvested by perfusing the pancreas with 3 ml of collagenase IV (Gibco 17104019) (410 units/ml in Hanks’ balanced salt solution (HBSS) and 10% fetal bovine serum (FBS)) into the common bile duct using a 30G needle. The excised pancreas was incubated for 20 minutes at 37°C in 3–5 ml of collagenase, passed through a 40 um cell strainer, and centrifuged at 220 rcf for 3 minutes. Cells were then washed twice in 10 ml of HBSS + 10% FBS and centrifuged at 600 rcf for 3 minutes and resuspended in complete RPMI. For sorting, cells were washed in PBS twice and stained with the following antibodies: KLRG1 FITC (2F1; eBioscience 11-5893-82), CD62L Percp-cy5.5 (MEL-14; eBioscience 45-0621-82), CD8a PE-cy7 (53–6.7; BioLegend 100722), CD127 BV421 (A7R34; BioLegend 135024),Viability-Zombie Aqua (BioLegend 423101), CD44 APC (IM7; eBioscience 17-0441-82), CD4 BV711 (RM4-5; BioLegend 100550), PD-1 APC-750/Fire (29F.1A12; BioLegend 135240), and mouse NRP-V7 mimotope tetramer PE (KYNKANVFL/H-2Kd; NIH tetramer facility)30. Cells were sorted on viability dye-negative, CD4−CD8+, and tetramer+CD44+ on a Sony ICyt Synergy (Sony Biotechnology, Inc.). DNA was extracted from FACS purified cells and used for whole-genome DNA methylation profiling as described above.
Single cell ATAC (Assay for Transposase Accessible Chromatin) seq:
Nuclei were isolated from FACS purified tetramer+ beta cell-specific CD8+ T cells and polyclonal Tem and naive CD8+ T cells from T1D donors using the Low Cell Input Nuclei Isolation protocol from 10x Genomics. The transposition reaction was then performed on the bulk nuclei. The transposed nuclei were partitioned into nanoliter-scale barcoded Gel Beads-in-emulsion (GEMs) after running the loaded Chromium microfluidic Chip E on the Chromium Controller, and the transposed DNA was uniquely indexed and barcoded for each individual nucleus per manufacturer’s instructions (10x Genomics). Libraries were generated and sequenced using Illumina Hiseq system following the manufacturer’s protocols. For each sample, the single cell ATAC data were mapped to hg38 using the Cell Ranger ATAC pipeline. We then used SnapATAC (https://github.com/r3fang/SnapATAC) to combine multiple samples, i.e. the fragment.tsv files generated by Cell Ranger ATAC pipeline, and performed the clustering analysis.
Data were analyzed using Prism 6 software. Statistical significance was determined using the two-tailed unpaired Mann-Whitney test to compare two to three experiments.
Supplementary Material
Acknowledgments:
We would like to thank Dr. Pranay Dogra for processing samples for methylation profiling. This work was supported by the National Institutes of Health (1R01AI114442 to BY, Immune Tolerance Network UM1AI109565 to GTN, EJ, & BY, LRP to CZ), the American Lebanese Syrian Associated Charities (ALSAC to BY), and Assisi foundation (to BY).
The data that support the findings of this study are available from the corresponding author upon request. Whole genome and ATACseq data files are available at the NCBI gene expression omnibus (GEO).
Footnotes
Code availability:
All described code is publicly available.
Competing interests
The authors declare no competing interests
REFERENCES
- 1.Sun D et al. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J Immunol 166, 7579–7587 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Huseby ES et al. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J Exp Med 194, 669–676 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vizler C, Bercovici N, Cornet A, Cambouris C & Liblau RS Role of autoreactive CD8+ T cells in organ-specific autoimmune diseases: insight from transgenic mouse models. Immunol Rev 169, 81–92 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Coppieters KT et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209, 51–60 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willcox A, Richardson SJ, Bone AJ, Foulis AK & Morgan NG Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 155, 173–181 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kronenberg D et al. Circulating preproinsulin signal peptide-specific CD8 T cells restricted by the susceptibility molecule HLA-A24 are expanded at onset of type 1 diabetes and kill beta-cells. Diabetes 61, 1752–1759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skowera A et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest 118, 3390–3402 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knight RR et al. Human beta-cell killing by autoreactive preproinsulin-specific CD8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes 62, 205–213 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckner JH & Nepom GT Obstacles and opportunities for targeting the effector T cell response in type 1 diabetes. J Autoimmun 71, 44–50 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yeo L et al. Autoreactive T effector memory differentiation mirrors beta cell function in type 1 diabetes. J Clin Invest 128, 3460–3474 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skowera A et al. beta-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes 64, 916–925 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allis CD & Jenuwein T The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Portela A & Esteller M Epigenetic modifications and human disease. Nat Biotechnol 28, 1057–1068 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Abdelsamed HA et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med 214, 1593–1606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pace L et al. The epigenetic control of stemness in CD8(+) T cell fate commitment. Science 359, 177–186 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Youngblood B et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 552, 404–409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghoneim HE et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell 170, 142–157 e119 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akondy RS et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henning AN, Roychoudhuri R & Restifo NP Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol 18, 340–356 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Youngblood B et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol 191, 540–544 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee W, Kim HS, Hwang SS & Lee GR The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp Mol Med 49, e393 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liao J, Humphrey SE, Poston S & Taparowsky EJ Batf promotes growth arrest and terminal differentiation of mouse myeloid leukemia cells. Mol Cancer Res 9, 350–363 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Page N et al. Expression of the DNA-Binding Factor TOX Promotes the Encephalitogenic Potential of Microbe-Induced Autoreactive CD8(+) T Cells. Immunity 48, 937–950 e938 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alfei F et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 571, 265–269 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Chen T, Ueda Y, Xie S & Li E A novel Dnmt3a isoform produced from an alternative promoter localizes to euchromatin and its expression correlates with active de novo methylation. J Biol Chem 277, 38746–38754 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Manzo M et al. Isoform-specific localization of DNMT3A regulates DNA methylation fidelity at bivalent CpG islands. EMBO J 36, 3421–3434 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tadokoro Y, Ema H, Okano M, Li E & Nakauchi H De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 204, 715–722 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malta TM et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 173, 338–354 e315 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burrack AL, Martinov T & Fife BT T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front Endocrinol (Lausanne) 8, 343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garyu JW et al. Characterization of Diabetogenic CD8+ T Cells: IMMUNE THERAPY WITH METABOLIC BLOCKADE. J Biol Chem 291, 11230–11240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J et al. Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci U S A 111, 14840–14845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levisetti MG, Suri A, Petzold SJ & Unanue ER The insulin-specific T cells of nonobese diabetic mice recognize a weak MHC-binding segment in more than one form. J Immunol 178, 6051–6057 (2007). [DOI] [PubMed] [Google Scholar]
- 33.James EA & Kwok WW Low-affinity major histocompatibility complex-binding peptides in type 1 diabetes. Diabetes 57, 1788–1789 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wiedeman AE et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. The Journal of clinical investigation 130, 480–490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daifotis AG, Koenig S, Chatenoud L & Herold KC Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin Immunol 149, 268–278 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Herold KC et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 346, 1692–1698 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Herold KC et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med 381, 603–613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richards DM, Kyewski B & Feuerer M Re-examining the Nature and Function of Self-Reactive T cells. Trends Immunol 37, 114–125 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Crompton JG et al. Lineage relationship of CD8+ T cell subsets is revealed by progressive changes in the epigenetic landscape. Cell Mol Immunol (2015). [DOI] [PMC free article] [PubMed]
- 40.Gattinoni L et al. A human memory T cell subset with stem cell-like properties. Nat Med 17, 1290–1297 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lugli E et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc 8, 33–42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Utzschneider DT et al. High antigen levels induce an exhausted phenotype in a chronic infection without impairing T cell expansion and survival. J Exp Med 213, 1819–1834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xi Y & Li W BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10, 232 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic Acids Res 43, e141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumaki Y, Oda M & Okano M QUMA: quantification tool for methylation analysis. Nucleic Acids Res 36, W170–175 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sokolov A, Carlin DE, Paull EO, Baertsch R & Stuart JM Pathway-Based Genomics Prediction using Generalized Elastic Net. PLoS Comput Biol 12, e1004790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.