

Introduction: the Erythron
Recently, the red blood cell (RBC) series, from progenitor cells to mature erythrocytes, has, collectively, been termed the Erythron. The erythron comprises RBCs at all stages of development and is the organ (primarily composed of anucleated cells in suspension) responsible for oxygen (O2) transport from lungs to tissue1. This role is newly appreciated to include active (by RBCs) vasoregulation that links regional blood flow to O2 availability in the lung and to consumption in the periphery2. A considerable portion of our nutritional and energy budget is devoted to maintaining a robust RBC population (20–30 trillion cells circulate in the average adult - approximately 85% of the cells in the body are RBCs.); 1.4 million RBCs are released into the circulation per second, replacing ~ 1% of the circulating mass per day. Mature RBCs have a life span of ~ 4 months, the majority of which is spent traversing the microcirculation. It is estimated that RBCs travel approximately 400 km during this interval, having made 170,000 circuits through the vascular tree. Circulating RBCs demonstrate unique physiology and are adapted to withstand significant biomechanical and biochemical stress. As RBCs age, energy and antioxidant systems fail; key proteins (including hemoglobin (Hb) and lipids) suffer oxidative injury, negatively impacting performance (rheology, adhesion, gas transport, vascular signaling). Such cells acquire marks of senescence and are cleared by the spleen or undergo eryptosis (a process unique to RBCs, similar to apoptosis). Of importance, this process may be accelerated in the course of critical illness and thereby, by limiting O2 delivery, influence organ failure progression and outcome.
Moreover, it is essential to note that in the setting of insufficient O2 delivery, blood flow (rather than content) is the focus of O2 delivery regulation: O2 content is relatively fixed, whereas flow is modulated by several orders of magnitude. Thus, blood flow volume and distribution are the physiologic parameters most actively regulated to maintain coupling between O2 delivery and demand. Specifically, the trapping, processing and delivery of nitric oxide (NO) by RBCs has emerged as a conserved mechanism through which regional blood flow is linked to biochemical cues of perfusion sufficiency. By coordinating vascular signaling in a fashion that links O2 and NO flux, RBCs couple vessel caliber (and thus blood flow) to O2 need in tissue. Malfunction of this signaling system is implicated in a wide array of pathophysiologies and may be explanatory in part for the dysoxia frequently encountered in the critical care setting.
Capture and Release of Oxygen by RBCs
Hemoglobin (Hb) is formed of 2 α and 2 β polypeptide chains each carrying a heme prosthetic group, comprised of a porphyrin ring bearing a ferrous atom that can reversibly bind an oxygen (O2) molecule. In the deoxygenated state, the Hb tetramer is electrostatically held in a tense (T) conformation. Binding of the first O2 molecule leads to mechanical disruption of these bonds, an increase in free energy and transition to the relaxed (R) conformation. Each successive O2 captured by T-state Hb shifts the Hb tetramer closer to the R state, which has an estimated 500-fold increase in O2 affinity3. This concept of thermodynamically coupled “cooperativity” in O2 binding was first described by Bohr4 and explains the sigmoidal appearance of the O2-Hb binding curve, also known as the oxy-Hemoglobin dissociation curve (ODC) (Figure 1). Moreover, understanding of allosteric influence of protein function by ‘heterotropic effectors’ (e.g. For Hb, O2, which binds to the ‘active’ site (heme) is the homotropic ligand and all other molecules influencing the Hb~O2 binding relationship are termed heterotropic effectors.) was first achieved following description of the variation in Hb~O2 affinity5. In addition to the homotropic effects of ligand binding on quaternary conformational changes (e.g. cooperativity), primary ligand binding affinity (O2) is also affected by multiple heterotropic effectors of significant physiologic relevance. The major heterotropic effectors that influence Hb O2 affinity are hydrogen ion (H+), chloride ion (Cl−), carbon dioxide (CO2) and 2,3-diphosphoglycerate (DPG)3.
Figure 1.
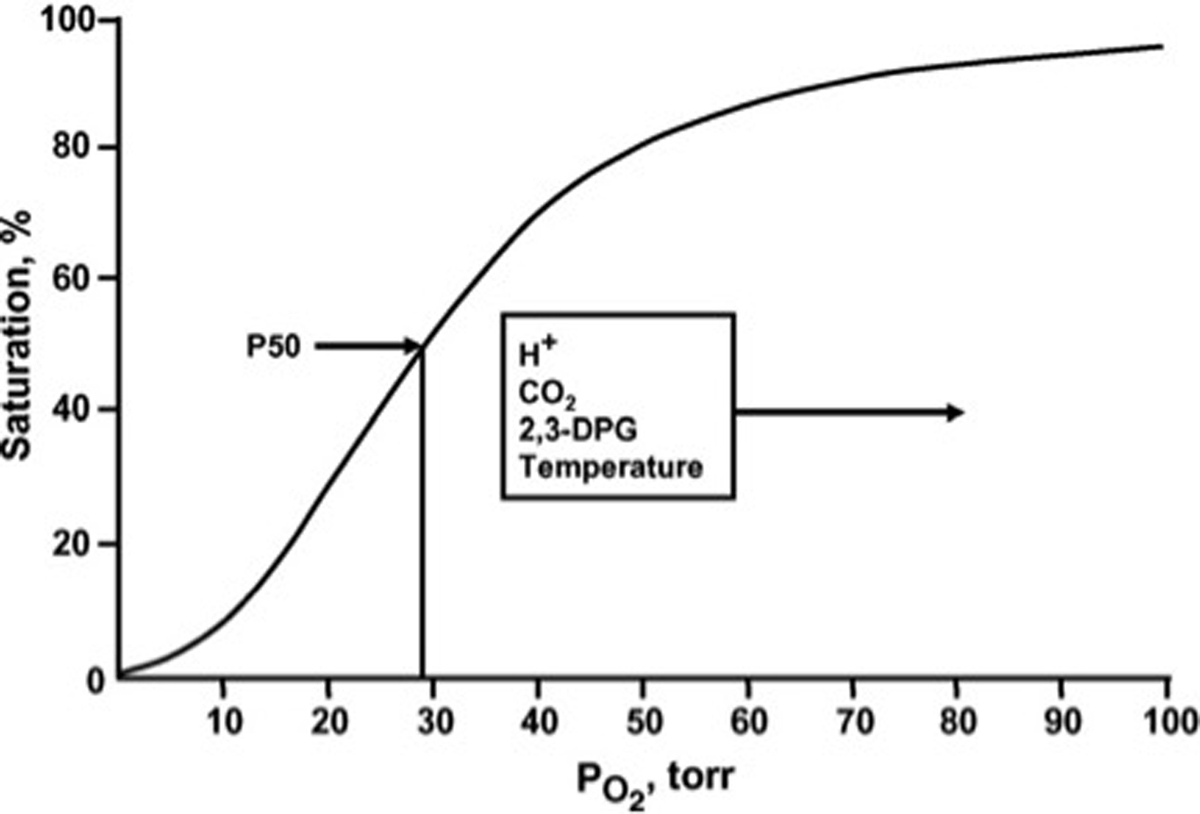
The normal whole blood oxygen equilibrium curve (OEC). P50 is the pO2 at which hemoglobin is half-saturated with O2. The principal effectors that alter the position and shape of the curve under physiological conditions are indicated.
From Winslow RM. The role of hemoglobin oxygen affinity in oxygen transport at high altitude. Respir Physiol Neurobiol 2007; 158:121–127; with permission.
P50, the oxygen tension at which 50% of Hb binding sites are saturated, is used as a standard means to quantify change in Hb~O2 affinity and is inversely related to the binding affinity of Hb for O26. Elevated levels of H+, Cl− and CO2 reduce O2 binding affinity (e.g. raise P50). This allosteric shift in O2 affinity, called the Bohr effect7, arises from the interactions among the above heterotropic effectors bound to different sites on hemoglobin – all of which serve to stabilize the low energy, low affinity, T-state Hb conformation8. This effect is achieved by complex interactions amongst carbonic anhydrase (CA) and the B3 membrane protein (also known as anion exchange protein 1, AE1). Specifically, CA generates H+ and HCO3− from CO2 encountered in the microcirculation; HCO3− then exchanges for Cl− across the RBC membrane through AE1. As a consequence, extra erythrocytic CO2 is converted into intra-erythrocytic HCl by the CA-AE1 complex, thus acidifying RBC cytoplasm and raising p50 (lowering affinity, also termed ‘right’ shifting the ODC). Additionally, through the Haldane effect, CO2 more directly lowers O2 affinity (by binding to the N-terminus of the globin chains to form a carbamino, further stabilizing T-state Hb); carbamino formation also releases another hydrogen ion (further reinforcing the ‘right shift’ in ODC)3 (Figure 2). This set of reactions is reversed in the alkaline (and low CO2) milieu in the pulmonary circulation, leading to increased Hb~O2 binding affinity (lower P50). In sum, this physiology vastly improves O2 transport efficiency by enhancing gas capture in the lung and release to tissue – and does so in proportion to perfusion sufficiency (in the setting of perfusion lack, acidosis and hypercpanea improve O2 release). Of note, this tightly regulated modulation of O2 affinity may become impaired in the setting of critical illness9–12 and may, in part explain the dysoxia commonly observed in this setting.
Figure 2.
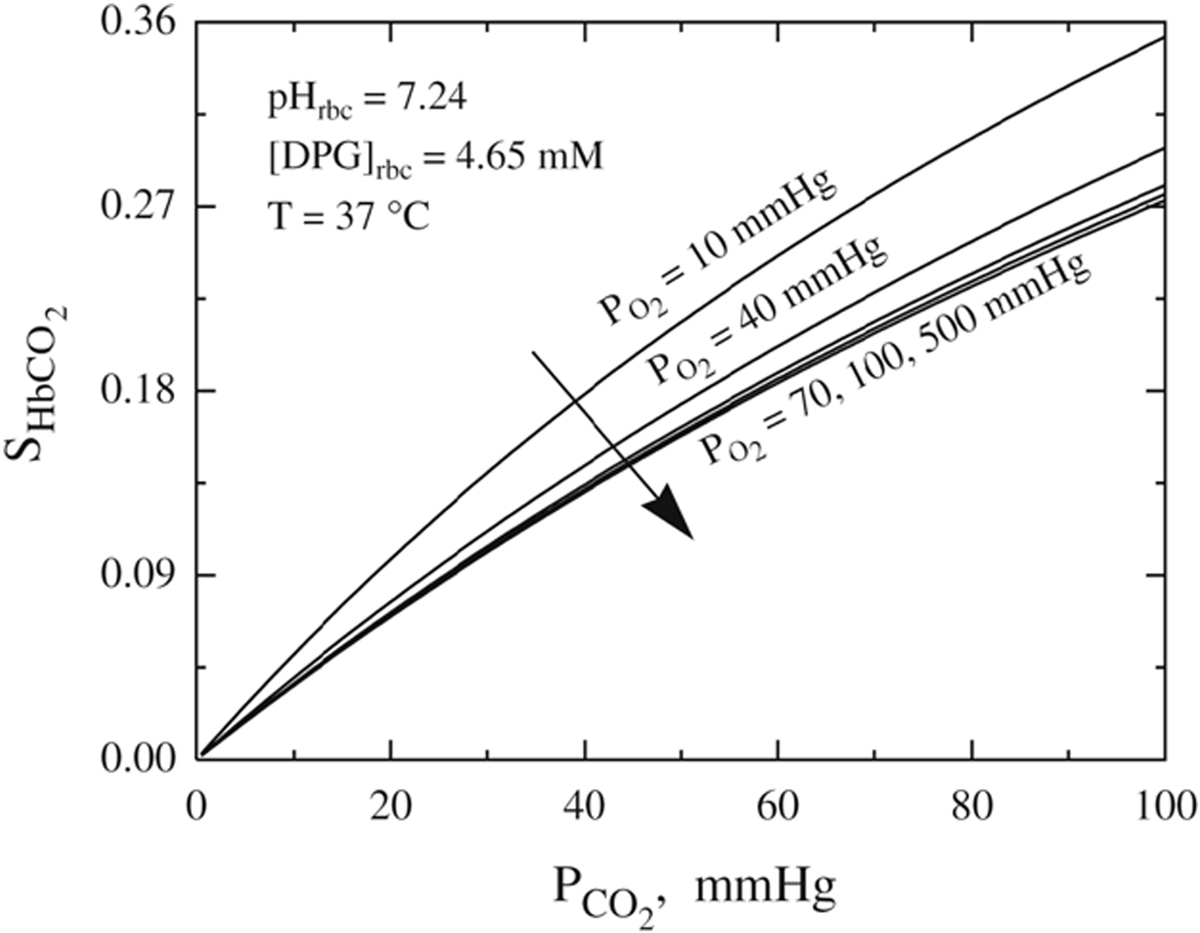
The quantitative behavior of the Carbaminohemoglobin (HbCO2) dissociation curves at various oxygen tension levels.
From Dash RK, Bassingthwaighte JB. Erratum to: Blood HbO2 and HbCO2 dissociation curves at varied O2, CO2, pH, 2,3-DPG and temperature levels. Annals of biomedical engineering 2010; 38:1683–1701; with permission.
Less acute modulation of P50 is achieved by DPG, a glycolytic intermediate that binds in an electrically charged pocket between the β chains of hemoglobin, which stabilizes the T conformation, decreasing O2 affinity and elevating P50. DPG binding also releases protons, lowering intracellular pH and further reinforcing the Bohr effect. DPG in RBCs increases whenever O2 availability is diminished (as in hypoxia or anemia) or when glycolytic flux is stimulated13. Lastly, temperature significantly influences Hb~O2 affinity. As body temperature increases, affinity lessens (P50 increases, ODC shifts right); the reverse happens in hypothermia. This feature is of physiological importance during heavy exercise, fever or induced hypothermia. It should be noted that clinical co-oximetry results and blood gas values are reported at 37°C and not at true in vivo temperature and can lead to either under or over estimation of true HbSO2% values and blood O2 tension14.
RBC Biophysical factors Influencing tissue perfusion
Blood Rheology
Disease-based variation in blood fluidity has been recognized since the early 20th century15 and there is substantive evidence that this property strongly influences tissue perfusion16. Plasma is a newtonian fluid (viscosity is independent of shear rate); its viscosity is closely related to protein content and in critical illness, physiologically significant changes in viscosity may vary with concentration of acute phase reactants. Whole blood, however, is considered a non-newtonian suspension (fluidity cannot be described by a single viscosity value); whole blood fluidity is determined by combined rheological properties of plasma and the cellular components.
The cellular components of blood, particularly RBCs, influence blood viscosity as a function of both number and deformability. RBC concentration in plasma (hematocrit) has an exponential relationship with viscosity and meaningfully diminishing tissue perfusion when Hct exceeds ~ 60–65. RBC deformability, or behavior under shear stress, also strongly influences blood fluidity. Normal RBCs behave like fluid drops under most conditions, are highly deformable under shear and orient with flow streamlines. However, during inflammatory stress, RBC tend to aggregate into linear arrays like a stack of coins (rouleaux); fibrinogen and other acute phase reactants in plasma stabilize such aggregates, significantly increasing blood viscosity. Such a change in viscosity most impacts O2 delivery during low flow (e.g. low shear) states (such as in critical illness) in the microcirculation17. RBC biomechanics and aggregation impact blood viscosity, strongly influencing the volume and distribution of O2 delivery (again more so, in the low-shear microcirculation, or when vessel tone is abnormal)18. This hemorheologic physiology is perturbed by oxidative stress (common in critical illness)19 and in sepsis20. This has been attributed to increased intracellular 2,3-DPG concentration21, intracellular free Ca2+22 and decreased intra-erythrocytic ATP with subsequent decreased sialic acid content in RBC membranes23. Both increased direct contact between RBCs and WBCs and reactive oxygen species released during sepsis have also been shown to alter RBC membrane properties24.
RBC aggregation and adhesion
As noted above, in the absence of shear, RBCs suspended in autologous plasma stack in large aggregates, known as rouleaux. Acute phase reactants especially fibrinogen, C-reactive protein, serum amyloid A, haptoglobin and ceruloplasmin have been shown to increase RBC aggregation25. Pathophysiological conditions as sepsis and ischemia-reperfusion injury have been shown to alter RBC surface proteins and increase RBC “aggregability”19. Activated white blood cells (WBC) are also thought to cause structural changes in the RBC glycocalyx and increase RBC aggregability26.
Under normal conditions, RBC adherence to endothelial cells (EC) is insignificant and RBC deformability permits efficient passage through the microcirculation. Again, under normal conditions, enhanced EC adherence plays a role in the removal of senescent RBCs in the spleen. However, during critical illness, RBC~endothelial interactions are altered by RBC injuries associated with sepsis27,28 and/or oxidative stress19. This is more prominent, with ‘activated’ endothelium, as frequently occurs in critical illness)29,30.
Such RBC~endothelial aggregates create a physiologically significant increase in apparent blood viscosity18. Moreover, RBC adhesion directly damages the endothelium31,32 and augments leukocyte adhesion33–35 further impairing apparent viscosity and microcirculatory flow. This phenomenon is commonly appreciated in the pathophysiology of vaso-occlusive crises in sickle cell disease patients, malaria, diabetic vasculopathy, polycythemia vera and central retinal vein thrombosis, but may be more widespread than originally appreciated.
RBC Deformability
Tissue deformation can be defined as the relative displacement of specific points within a cell or structure. Mature RBCs are biconcave disks ranging from 2–8 μm in thickness, which act like droplets that deform reversibly under the shear encountered during circulatory transit18. Unique RBC geometry and deformability arises from (a) cytoplasmic viscosity and (b) specific interactions between the plasma membrane and underlying protein skeleton23 (Figure 3). Cytoplasmic viscosity is mainly determined by hemoglobin concentration, which varies with intra-erythrocytic hydration, which is actively regulated by ATP-dependent cation pumps36. The integral transmembrane membrane proteins AE-1 (AKA B3) and glycophorins are reversibly anchored to a submembrane filamentous protein mesh comprised of spectrin, actin, and protein 4.1. Linear extensibility of this mesh defines the limits of RBC deformability37. Maintenance of membrane-mesh interactions and robust RBC mechanical behavior is dependent on ATP-dependent ion pumps as well as support from NADPH-dependent antioxidant systems36. The sole energy source in RBCs is anaerobic glycolysis, which is discussed in detail below. RBC geometric and mechanical alterations secondary to impaired metabolism (leading to RBC dehydration, elevated intra-erythrocytic calcium and ATP/NADPH depletion) is a well-described consequence in blood stored for prolonged periods38 and in RBCs subjected to significant metabolic stress during critical illness39,40.
Figure 3.
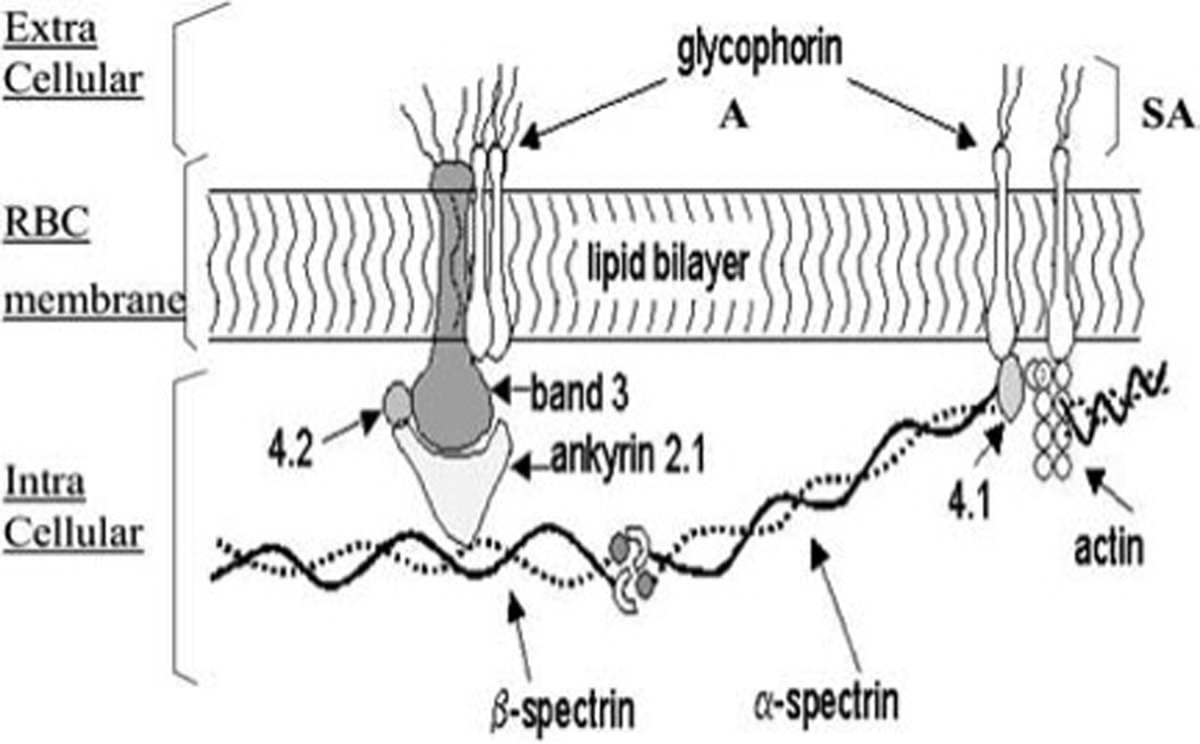
The RBC membrane is composed of a phospholipid membrane bilayer and transmembrane proteins including glycophorin A and Band 3 proteins. Glycophorin A is the major sialoglycoprotein of the RBC. Sialic acid (SA) bound to glycophorin A is responsible for the negative charge of the RBC membrane. The intracellular compartment (IC) is constituted by spectrin (α and β subunits), actin, protein 4.1, and ankyrin.
From: Piagnerelli M, et al. Red blood cell rheology in sepsis. Intensive Care Med. 2003; 29(7):1052–1061; with permission.
Regulation of Blood Flow Distribution by RBCs
Microcirculatory blood flow is physiologically regulated to instantaneously match O2 delivery to metabolic demand. This extraordinarily sensitive programmed response to tissue hypo-perfusion is termed hypoxic vasodilation (HVD)41. This process involves the detection of point-to-point variations in arteriolar O2 content42 with the subsequent initiation of signaling mechanism(s) capable of immediate modulation of vascular tone (Figure 4).
Figure 4.
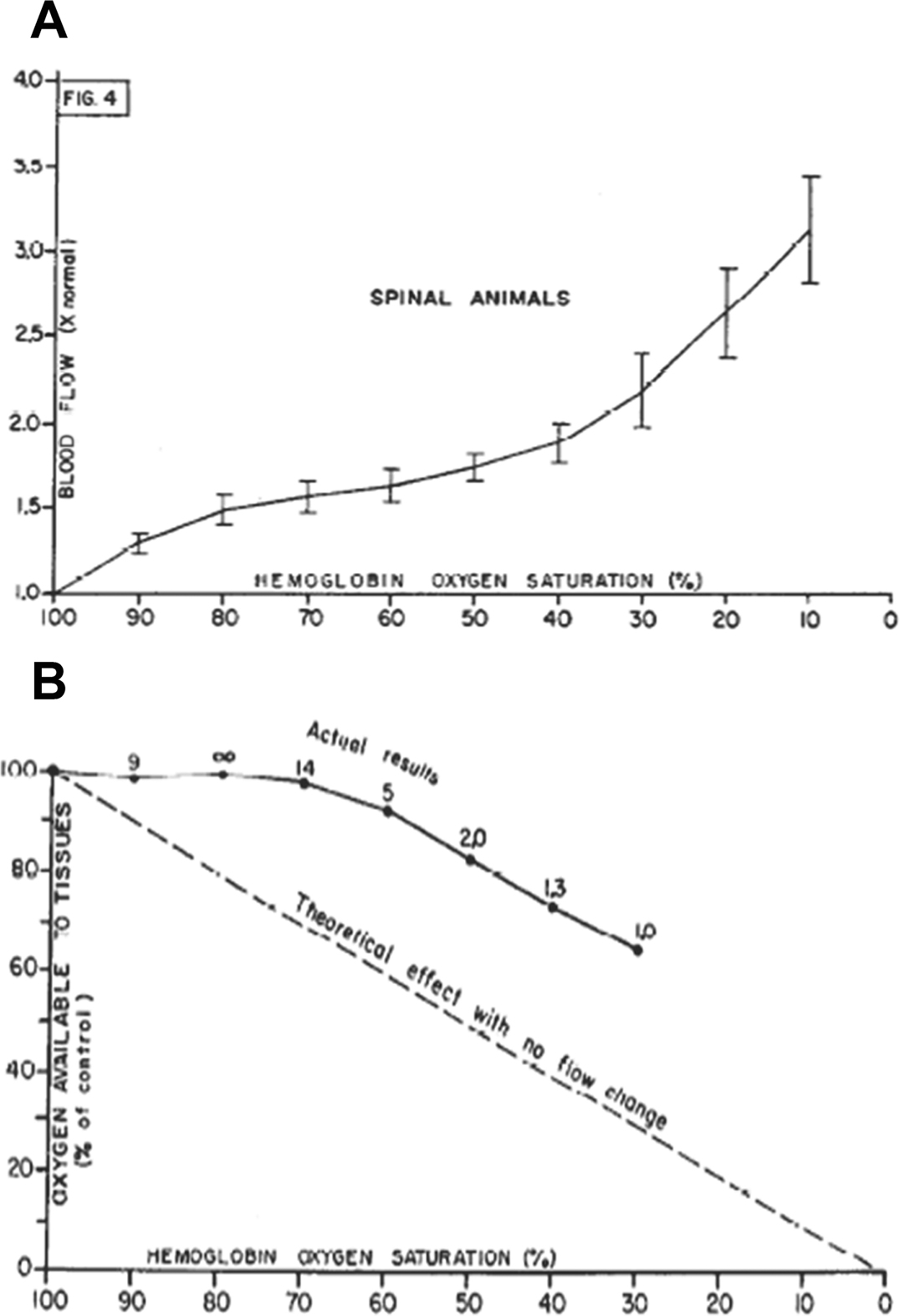
Local vascular reflexes support maintenance of O2 delivery to tissue in the setting of progressive hypoxia. In a classic paper41, Guyton demonstrated regional autoregulation of systemic blood flow in normal dogs (following spinal anesthesia) by observing variation in blood flow during constant pressure blood perfusion of the femoral artery, while reducing the hemoglobin oxygen saturation (Hb SO2%) from 100% to 0% in the perfusing blood. (A) Stepwise reduction in Hb SO2% caused a progressive increase in blood flow through the leg. (B) These data demonstrate that autoregulation of blood flow occurs at a local level and this regulation serves to improve oxygen supply when blood oxygen content falls. In addition, effects on blood flow were replicated by injecting partially deoxygenated versus oxygenated red blood cells into the artery, demonstrating that effects could be elicited during arteriovenous transit (<1 s).
From Ross JM, Fairchild HM, Weldy J, Guyton AC. Autoregulation of blood flow by oxygen lack. Am J Physiol. 1962;202:21–24; with permission.
Over 30 years ago, intracellular RBC Hb was identified as a potential circulating O2 sensor, following identification that in severe hypoxia, O2 content was more important than partial pressure of O2 (PO2) in the maintenance of regional O2 supply43. It was later demonstrated in vivo that Hb O2 saturation (HbSO2) was independent of plasma or tissue PO2, but was directly correlated with blood flow44. These findings implicated a role for RBCs in the regulation of O2 supply, given the following evidence: (1) the Hb molecule within the RBC is the only component in the O2 transport pathway directly influenced by O2 content, and (2) the level of O2 content of the RBC at a particular point in the circulation is linked to the level of O2 utilization45.
With the vascular O2 sensor identified, the mechanism involved in mediating the vasoactive response has remained in debate. To date, three HbSO2 dependent RBC derived signaling mechanisms have been proposed, the first two linked to the vasoactive effector NO, and the third to RBC adenosine triphosphate (ATP) : (1) formation and export of S-nitrosothiols, ‘catalyzed’ by Hb (SNOHb hypothesis)46–48; (2) reduction of nitrite (NO2−) to NO by deoxygenated Hb (nitrite hypothesis)49; and (3) hypoxia responsive release of ATP (ATP hypothesis)45,50. Each of these hypotheses will be addressed further, below.
Role of RBC~NO interactions in vasoregulation
Interest in the free radical NO began with the identification of EDRF (endothelium derived relaxing factor), first reported in 198051, which resolved the apparent paradox as to why acetylcholine, an agent known to be a vasodilator in vivo often caused vasoconstriction in vitro. Experiments performed with dissected segments of rabbit thoracic aorta mounted on a force transducer, demonstrated that handling of the tissue in a fashion that preserved endothelium always resulted in acetylcholine having relaxant properties. However, removal of the endothelium eradicated this action51,52. Identification of EDRF consequently led to a race to discover its chemical identity. It was not until seven years later that two groups simultaneously published definitive studies characterizing and identifying EDRF as NO53,54. However, the means by which NO exerted its physiological effects remained unknown and effort focused upon identifying the “NO receptor(s)”. This effort characterized the ‘classical’ signaling pathway for NO via soluble guanylate cyclase (sGC) and cyclic guanosine 3’,5’-monophosphate (cGMP) that appeared to clarify the means by which NO achieves its myriad effects55. Over time, however, it is now appreciated that this pathway has little to do with the vasoregulation that governs regional blood flow distribution.
In terms of the HVD response (which underlies blood flow regulation) it is essential to appreciate that endothelium-derived NO plays no direct role in this reflex44,56. Because of O2 substrate limitation, NO production by eNOS is most likely attenuated by hypoxia57,58. In fact, NO derived from eNOS46 (and perhaps other NOS isoforms59 and/or nitrite60) is taken up by RBCs, transported, and subsequently dispensed in proportion to regional O2 gradients to effect HVD at a time and place remote from the original site of NO synthesis. This key process enables RBCs to instantaneously modulate vascular tone in concert with cues of perfusion insufficiency, including hypoxia, hypercarbia, and acidosis46,47.
Metabolism of endothelium derived NO by RBCs – Historical view
In the original NO paradigm, NO derived from endothelial nitric oxide synthase (eNOS) was felt to play a purely paracrine role in the circulation, acting within the vicinity of its release61. Its metabolic fate was explained by the diffusion of the “gas” in solution and its terminal reactions (1) in vascular smooth muscle cells with the ferrous heme iron (Fe2+) of soluble guanylate cyclase (sGC)62, and (2) in the vessel lumen, with the heme group (Fe2+) of oxyHb (the resultant oxidation reaction forming MetHb and nitrate), or deoxyHb (the resultant addition reaction forming iron nitrosyl Hb; HbNO), or in plasma with dissolved O2 (the resultant autoxidation reaction)63, and/or O2 derived free radicals including superoxide (O2−), hydrogen peroxide (H2O2), or hydroxyl radicals (OH−). Several “barriers” were presumed to retard NO diffusing into the blood vessel lumen to react avidly with the abundance of Hb, including the RBC membrane, the submembrane protein matrix, an unstirred layer around the RBC64,65, in addition to laminar blood flow66. These barriers were thought to limit these luminal reactions, thus allowing the local concentration of NO adjacent to endothelial cells to increase sufficiently to provide a diffusional gradient for NO to activate the underlying vascular smooth muscle sGC. Reactions of NO in the bloodstream were assumed only to scavenge/inactivate NO via the formation of metabolites unable to activate sGC62.
Metabolism of endothelium derived NO by RBCs – Modern view
A much broader biological chemistry of endothelial NO has been elucidated67–69. Most notable is the covalent binding of NO+ to cysteine thiols, forming S-nitrosothiols (SNO). This paradigm developed following the discovery that endogenously produced NO circulated in human plasma primarily complexed to the protein albumin (S-nitrosoalbumin70), which transformed the understanding of blood borne NO signaling. SNO proteins thus offered a means to conserve NO bioactivity, allowing the storage, transport, and potential release of NO remote from its location of synthesis71. The SNO hypothesis was extended to include a reactive thiol of Hb (Cysß93) that was demonstrated to undergo S-nitrosylation and sustain bioactivity under oxygenated conditions and NO release under low O2 conditions (see HbSNO hypothesis)46.
In this SNO paradigm, the NO radical must be oxidized to an NO+ (nitrosonium) equivalent, which can then be passed between thiols in peptides and proteins preserving NO bioactivity67,68. S-nitrosylation then is akin to protein phosphorylation in terms of regulating protein function. SNO biochemistry offers NO a far broader signaling repertoire and has enabled awareness that the heme in sGC is not the sole, or even the principal, target of NO generated by endothelium. A wide array of alternative sGC (cyclic guanosine monophosphate)-independent reactions following endothelial NOS (eNOS) activation have been identified69,72.
Processing and export of S-nitrosothiols by RBCs
Hb S-nitrosylation (HbSNO), which has been characterized by both mass spectrometry73 and X-ray crystallography74, provides an explanation as to how NO circumvents terminal reactions with Hb, enabling RBCs to conserve NO bioactivity and transport it throughout the circulation46,47 (Figure 5). The formation and export of NO groups by Hb is governed by the transition in Hb conformation that occurs in the course of O2 loading/unloading during arterio-venous (A-V) transit. This is due to conformational dependent change in reactivity of the Cysβ93 residue toward NO, which is higher in the R (oxygenated) Hb state and lower in the T (deoxygenated) Hb state46,47.
Figure 5.
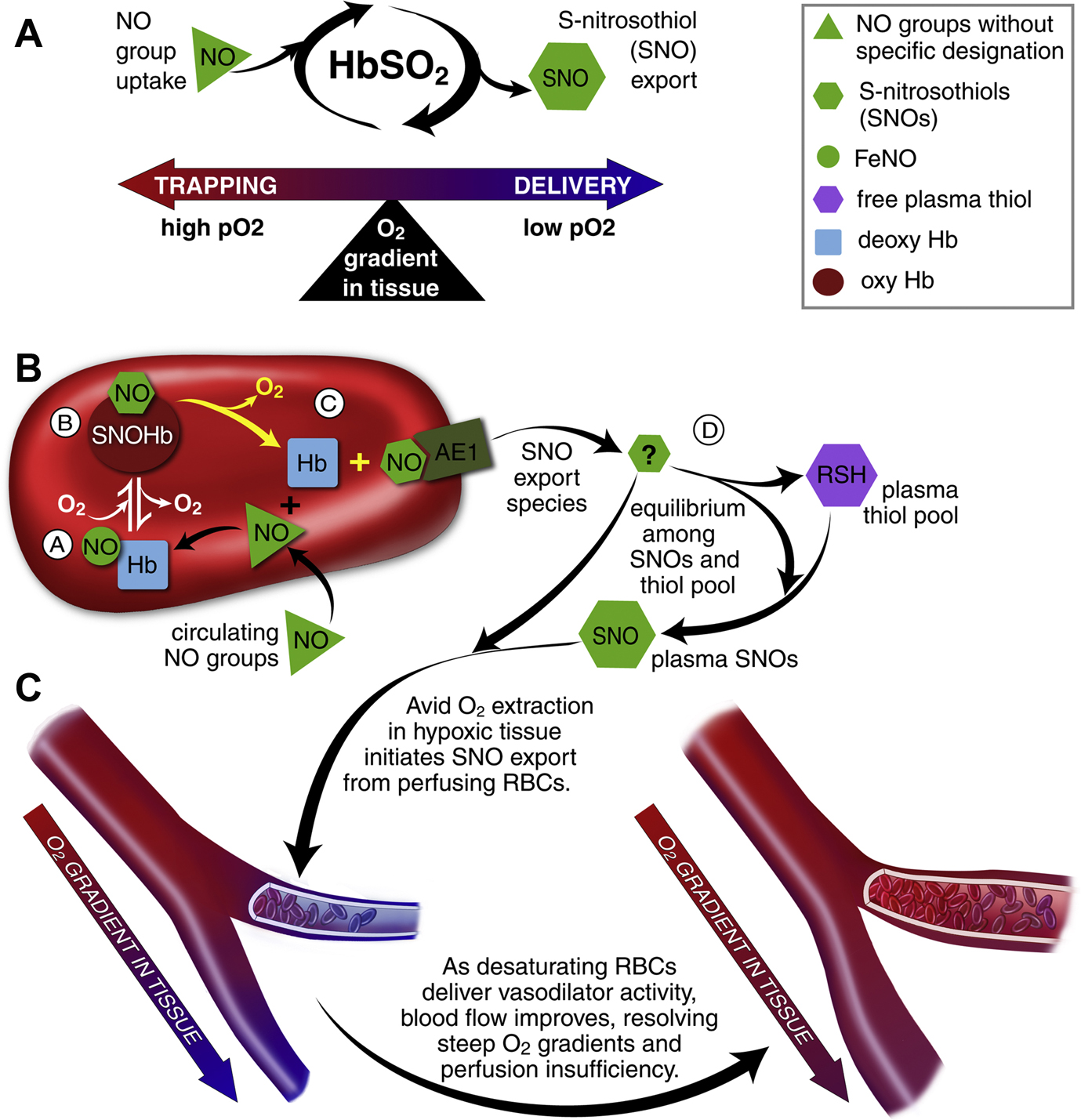
RBCs transduce regional O2 gradients in tissue to control nitric oxide (NO) bioactivity in plasma by trapping or delivering NO groups as a function of hemoglobin (Hb) O2 saturation. (A) In this fashion, circulating NO groups are processed by Hb into the highly vasoactive (thiol-based) NO congener, S-nitrosothiol (SNO). By exporting SNOs as a function of Hb deoxygenation, RBCs precisely dispense vasodilator bioactivity in direct proportion to regional blood flow lack. (B) O2 delivery homeostasis requires biochemical coupling of vessel tone to environmental cues that matches perfusion sufficiency to metabolic demand. Because oxy- and deoxy-Hb process NO differently (see text), allosteric transitions in Hb conformation afford context-responsive (O2-coupled) control of NO bioavailability, thereby linking the sensor and effector arms of this system. Specifically, Hb conformation governs the equilibria among deoxy-HbFeNO (A; NO sink), SNO-oxy-Hb (B; NO store), and acceptor thiols including the membrane protein SNO-AE-1 (C; bioactive NO source). Direct SNO export from RBCs or S-transnitrosylation from RBCs to plasma thiols (D) or to endothelial cells directly (not shown) yields vasoactive SNOs, which influence resistance vessel caliber and close this signaling loop. Thus, RBCs either trap (A) or export (D) NO groups to optimize blood flow. (C) NO processing in RBCs (A and B) couples vessel tone to tissue PO2; this system subserves hypoxic vasodilation in the arterial periphery and thereby calibrates blood flow to regional tissue hypoxia.
From Doctor A, Stamler JS. NO Transport in Blood: A third gas in the respiratory cycle. In: Comprehensive Physiology: Respiratory Physiology. Wagner P and Hlastala M, Ed’s. American Physiological Society. Compr Physiol 1:541–568, 2011; with permission.
In a tightly regulated fashion, Hb captures and binds NO at its β-hemes and then passes the NO group from the heme to a thiol (Cys-ß93-SNO)60,75. Transfer of NO between heme and thiol requires heme-redox coupled activation of the NO group, which is controlled by its allosteric transition across the lung76. Once in R state the Cys-ß93-SNO is protected through confinement to a hydrophobic pocket74. NO group export from Cys-β93-SNO occurs when steep O2 gradients are encountered in the periphery (HVD). The R to T state conformational transition that occurs on Cys-β93-SNO deoxygenation (or oxidation) results in a shift in the location of the β-chain from its hydrophobic niche toward the aqueous cytoplasmic solvent74. This allows the Cys-β93-SNO to be “chemically available” for transfer to target thiol containing proteins, including those associated with the RBC membrane protein AE-1 (Band 3)77 and extra-erythrocytic thiols78,79. Resultant plasma or other cellular SNOs, then become vasoactive at low nM concentrations)46,47. Importantly all NO transfers in this process involve NO+ 46,48, which protects bioactivity from Fe2+ heme recapture and/or inactivation. S-nitrosothiols are the only known endogenous NO compounds that retain bioactivity in the presence of Hb46,79,80.
Extensive evidence supports SNO-Hb biology, whereby RBCs exert graded vasodilator and vasoconstrictor responses across the physiological microcirculatory O2 gradient. RBCs dilate pre-constricted aortic rings at low PO2 (1% O2), while constricting at high PO2 (95% O2)47,80–82. The vasodilatory response at low O2 is enhanced following the addition of NO (or SNO) to RBCs, commensurate with SNO-Hb formation46,77,80,83. Additionally, the vasodilatory response is enhanced in the presence of extra cellular free thiol80, occurs in the absence of endothelium48,80 (which is consistent with in vivo observation that HVD is endothelium independent84), and transpires in the time frame of circulatory transit, as confirmed by measurements of A-V gradients in SNO-Hb46,78,81,82.
In addition to these ex vivo experiments, numerous groups have also demonstrated bioactivity of inhaled NO, commensurate with SNO-Hb formation85–89.
Metabolism of Nitrite by RBCs
Nitrite (NO2−), formed mainly via hydration reactions involving N-oxides, was long viewed as an inactive oxidation product of NO metabolism. More recently it has been proposed as circulating pool of bioactive NO90. Some have suggested that the reduction of nitrite by deoxyHb may serve as the RBC derived signaling mechanism regulating HVD91. However, this hypothesis has two major shortcomings in terms of known NO chemistry/biochemistry and HVD physiology. Firstly, to influence vascular tone, the NO radical produced from NO2− must escape RBCs at low O2 tension in order to elicit a vasodilatory response. Experimental evidence, however, unambiguously refutes the possibility of NO escaping RBCs as an authentic radical, especially given the proximity, high concentration, and rapid reaction kinetics (107M−1s−1) of authentic NO with deoxyHb. The only plausible reconciliation of this would be that bioactivity from this reaction may derive from heme captured NO (HbFe2+NO) being further converted into SNO-Hb60,75, as HbFe2+NO itself acts as a vasoconstrictor rather than vasodilator92. The second shortcoming relates to the fact that the NO2− reductase activity of deoxyHb is purportedly symmetrical across the physiological O2 gradient93,94, with maximal activity occurring at the P50 of Hb (~ 27 mmHg)93,95. This reaction profile does not match the HVD response, which increases in a steadily graded fashion as PO2 falls in the physiological range from 100 mmHg down to approximately 5 mmHg (HbSO2 ~ 1–2%)41,44. If RBC based vasoactivity were maximal at Hb’s P50, then blood flow would be diverted away from regions with PO2 below 27 mmHg, where it would be needed most. Additionally, based upon the symmetry of Hb nitrite reductase activity at the P50, RBCs traversing vascular beds with PO2 at 25 or 75 would generate equal NO-based activity91, where different blood flow demands are required.
Vasoregulation by RBC-derived Adenosine Triphosphate (ATP)
ATP has long been known to act as an endothelium dependent vasodilator in humans45, binding to P2Y purinergic receptors to induce local and conducted vasodilation via stimulation of vasoactive signals including endothelial NO, prostaglandins, and endothelial-derived hyperpolarization factors (EDHFs). More recently, RBCs have been identified as sources of vascular ATP45,96, with release stimulated by conditions associated with diminished O2 supply relative to demand,, hypoxia, hypercapnia, and low pH45,97. O2 offloading from membrane associated Hb is thought to initiate RBC ATP release96, stimulating heterotrimeric G protein98, as a result of membrane deformation. This leads to activation of adenylyl cyclase and an increase in cAMP99, which activates protein kinase A (PKA)99. PKA stimulates cystic fibrosis transmembrane conductance regulator (CFTR)100, which activates release of ATP from the RBC via pannexin 1101. Release of ATP via this pathway requires an increase in intracellular cAMP, which is controlled by the relative activities of adenylyl cyclase and phosphodiesterase 3 (PDE3B)50.
Despite potential as a HVD mediator, RBC derived ATP falls short on two fronts. Firstly, HVD is unaltered by both endothelial denudation and eNOS deletion48, however ATP vasoactivity is endothelial dependent. Secondly, blood levels of ATP rise and fall over a period of minutes, which is not commensurate with the HVD response that occurs in the course of A-V transit over a couple of seconds. Despite its shortcomings in terms of acting as a primary mediator of HVD, it is likely that Hb and ATP serve complementary vasoactive roles, in acute local and prolonged systemic hypoxia respectively48.
RBC Energetics and Consequences of Antioxidant System Failure
RBCs produce ATP by glycolysis only, with two branches:102 the Embden Meyerhof Pathway (EMP) and the Hexose Monophosphate Pathway (HMP).103 Importantly, the HMP is the sole means for recycling NADPH,104 which powers the thiol-based antioxidant system.104 HMP flux is gated by protein complex assembly upon the cytoplasmic domain of the Band 3 membrane protein (cdB3 ‘metabolon’).105–112 HMP flux oscillates with pO2, as a function of Hb conformation and cdB3 phosphorylation (Figure 6 A–B).113–119 Of note, RBC antioxidant systems fail when HMP flux is blunted by altered cdB3 protein assembly/phosphorylation caused by aberrant Hbs or hypoxia.120,121 Strikingly similar perturbations to cdB3 are reported in sepsis,122,123 possibly arising from caspase 3 activation124–126 and/or direct endotoxin or complement membrane binding127–134 (altering metabolon assembly, glycolysis and ROS clearance, Figure 2c).135–137 As such, it appears that that sepsis (particularly, in the setting of hypoxic and/or uremic/oxidative138–145 environments) disturbs cdB3-based metabolic control (Figure 6c), leading to: 1) EMP activation, 2) limited glucose-6-phosphate availability, 3) HMP flux constraint, 4) depowered NADPH/GSH recycling, 4) antioxidant system failure, and 5) injury to proteins/lipids that are key to O2 delivery homeostasis (SiRD). This full pattern has been reported in other settings impacting protein assembly at cdB3;120,121 further, such HMP constraint has functional similarity to G6PD deficiency,120 which amplifies vulnerability to sepsis.146–148 Moreover, hypoxia critically limits RBC energetics and depowers RBC antioxidant systems149,150. In health, O2• abundance is tightly regulated by the superoxide dismutase (SOD) family;151 however, overwhelming O2• genesis152 is implicated in sepsis-associated injury cascades153,154 Of note, sepsis-associated O2• excess injures RBCs, impairing O2-delivery by altering: control of O2 affinity,9–12 NO processing,155–157 rheology,20,23,130,131,158–160 and adhesion.161,162 O2• excess also disrupts vasoregulation via NO consumption and catecholamine inactivation in plasma.163–169 Specifically, ROS sourced directly to RBCs170–173 injure vessels.174,175 Such reciprocal injuries mutually escalate and as such, the dysoxia characteristic of septic shock (ischemia despite adequate blood O2 content and cardiac output),176–179 may arise from SiRD ~ vascular interactions.27,28 Notably, ROS excess is also a common consequence of uremia/kidney injury138–145, particularly during sepsis180–186. As such, the combination of lung injury (hypoxia) and kidney injury (uremia) simultaneously constrain RBC energetics and antioxidant systems and present substantive oxidant loading conditions, meaningfully increasing RBC injury risk.
Figure 6.
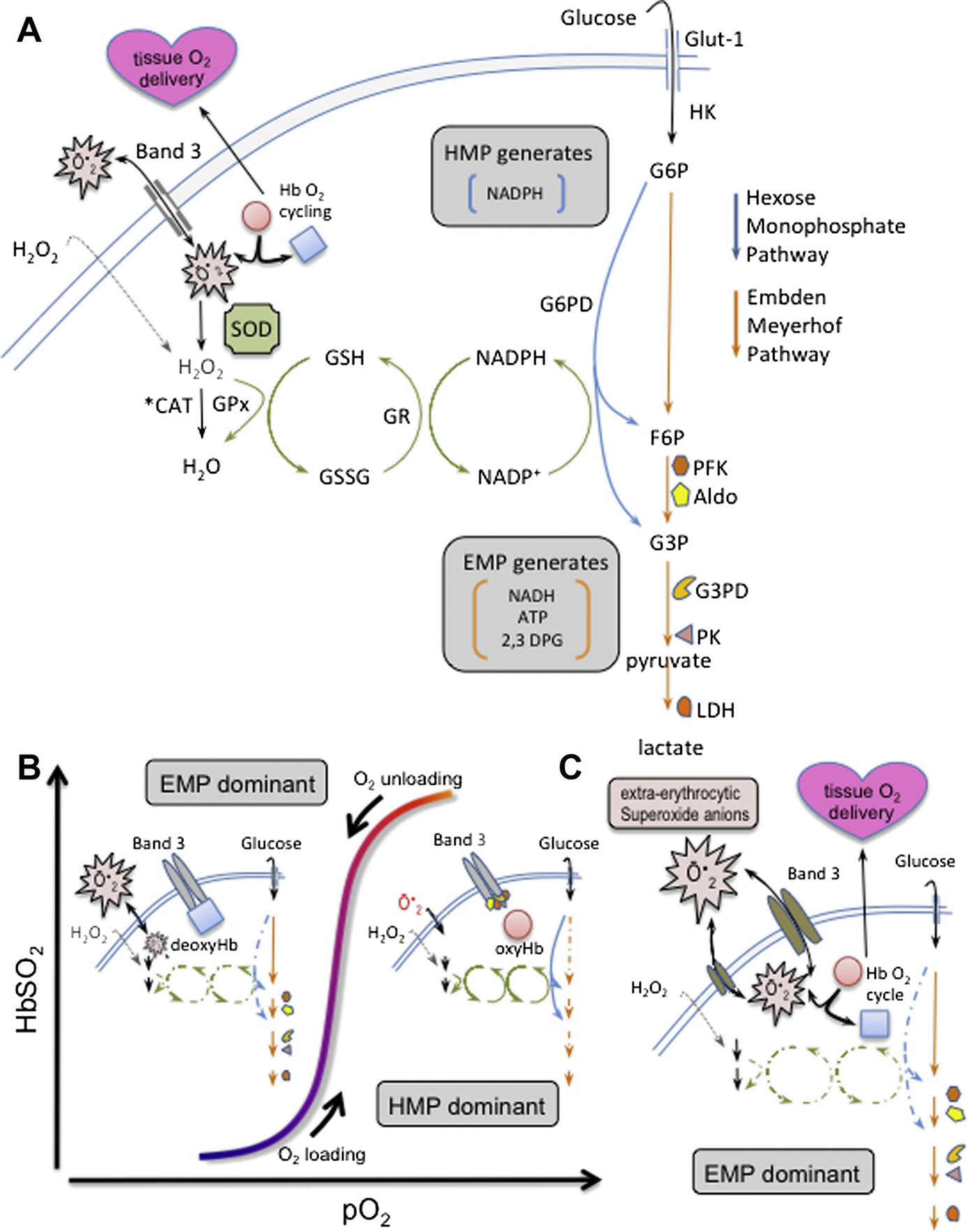
Simplified scheme of cdB3-based control of RBC metabolism and proposed causal path for sepsis induced red cell dysfunction: (A) Energy metabolism in RBCs proceeds through either the Embden-Meyerhof pathway (EMP, orange arrows), or the hexose monophosphate pathway (HMP, blue arrows, AKA ‘pentose shunt’). Both share glucose-6 phosphate (G6P) as initial substrate. The HMP is the sole source of NADPH in RBCs and generates fructose-6-phosphate (F6P) or glyceraldehyde-3-phosphate (G3P), which rejoin the EMP prior to glyceraldehyde-3-phosphate dehydrogenase (G3PD/GAPDH), a key regulatory point. The EMP generates NADH (utilized by metHb reductase), as well as ATP (to drive ion pumps) and 2,3-DPG (to modulate hemoglobin P50). Hydrogen peroxide (H2O2) and superoxide anion (O2−) are the principal endogenous reactive O2 species (ROS) that are generated / encountered by RBCs. Both ROS are generated internally in the course of HbO2 cycling.230–232 Notably, only H2O2 can cross the membrane directly. O2− enters/departs RBCs via the Band 3 channel (anion exchange protein 1, or AE-1). O2− and H2O2 are ultimately reduced to water by catalase (CAT) or glutathione peroxidase (GPx). (B) O2 content modulates EMP/HMP balance via reciprocal binding for cdB3 between deoxyHb and key EMP enzymes (PFK, Aldo, G3PD, PK, and LDH). In oxygenated RBCs (right half of stylized O2 dissociation plot), EMP enzyme sequestration to cdB3 inactivates this pathway, resulting in HMP dominance and maximal NADPH (and thus GSH) recycling capacity. In deoxygenated RBCs (left half of O2 dissociation plot), deoxyHb binding to cdB3 disperses bound EMP enzymes, activating the EMP, creating G6P substrate competition, constraining HMP flux, limiting NADPH and GSH recycling capacity and weakening resilience to ROS, such as O2−. (C) In sepsis, data suggest cdB3-complex assembly may be prevented (particularly, with coincident hypoxia, see text). As in settings similarly impacting the cdB3 complex, it appears that this disturbs normal EMP/HMP balance (disfavoring HMP), depowering antioxidant systems and rendering RBCs vulnerable to oxidant attack. GSH, glutathione; GR, glutathione reductase; NADPH, nicotinamide adenine dinucleotide phosphate; PFK, phosphofructokinase; Aldo, aldolase; PK, pyruvate kinase; LDH, lactate dehydrogenase
Acquired RBC Injury, Eryptosis and Clearance
After maturation to an anucleated cell furnished with the metabolic systems described above, the estimated normal life span of a mature RBC is 110–120 days187. To date, clearance of normal senescent RBC has not been clearly understood. Two mechanisms have been proposed, clustering of the band 3 (B3) membrane protein188–191 and externalization of membrane phosphatidyl serine (PS)192–195, both of these processes may be accelerated in the setting of critical illness, impairing oxygen transport capacity. Oxidatively modified hemoglobin (Hb) forms hemichrome aggregates, which associate with the cytoplasmic domain of the abundant membrane protein B3. Subsequent clustering of B3 exofascial domains increases affinity of naturally occurring anti-B3 autoantibodies, which activate the complement system leading to RBC uptake and destruction by macrophages196. Normally, PS is asymmetrically distributed in the plasma membrane (a process regulated by flippases). Disruption of this pattern is a well-documented mark of RBC senescence192–195, signaling RBC removal by the reticulo-endothelial system195. Alternatively, RBCs may proceed through a form of ‘stimulated suicide’ similar to apoptosis (termed eryptosis), which is characterized by cell shrinkage and cell membrane scrambling, that is stimulated by Ca2+ entry through Ca2+−permeable, PGE2-activated cation channels, by ceramide, caspases, calpain, complement, hyperosmotic shock, energy depletion, oxidative stress, and deranged activity of several kinases (e.g. AMPK, GK, PAK2, CK1α, JAK3, PKC, p38-MAPK). Eryptosis has been described in the setting of ethanol intoxication, malignancy, hepatic failure, diabetes, chronic renal insufficiency, hemolytic uremic syndrome, dehydration, phosphate depletion, fever, sepsis, mycoplasma infection, malaria, iron deficiency, sickle cell anemia, thalassemia, G6PD deficiency, and Wilson’s disease195,197,198.
Influence of RBCs on hemostasis
The principle impact of RBCs in clot formation in vivo is rheological, since RBC laminar shearing promotes platelet margination199, as well as RBC aggregation and deformability of RBCs, which also support clot assembly/retraction200. In addition, RBCs interact directly and indirectly with endothelial cells and platelets during thrombosis201. Both the stiffness of RBCs and the extent to which they form a procoagulant surface to generate thrombin through exposure of phosphatidylserine appear to play an important role, both in clot initiation and completion202,203. Moreover, RBC-derived MPs transfused with stored RBCs or formed in various pathological conditions associated with hemolysis have strong procoagulant potential along with prothrombotic effects of the extracellular hemoglobin and heme204. Additionally, RBCs directly interact with fibrin(ogen) and affect the structure, mechanical properties, and lytic resistance of clots and thrombi205. Finally, tessellated polyhedral RBCs (polyhedrocytes) are recognized to be a significant structural component of contracted clots, enabling the impermeable barrier important for hemostasis and wound healing206.
Summary: RBC Dysfunction disrupts of O2 delivery during critical illness
Evidence is mounting in support of a causal relationship between acquired RBC dysfunction and a host of perfusion-related morbidities that complicate critical illness82,171,207–221. Recently, it has been observed that levels of SNO-Hb are altered in several disease states characterized by disordered tissue oxygenation82,83,155,156,222–227. In addition, where examined, RBCs from such patients exhibit impaired vasodilatory capacity78,82,83,224,226–228. These data suggest that altered RBC-derived NO bioactivity may contribute to human pathophysiology. Specifically, alterations in thiol-based RBC NO metabolism have been reported in congestive heart failure82, diabetes83,223, pulmonary hypertension81,222 and sickle cell disease224,229, all of which are conditions characterized by inflammation, oxidative stress and dysfunctional vascular control. Moreover, known cross-talk between SNO signaling and cellular communication via carbon monoxide, serotonin, prostanoids, catecholamines and endothelin may permit broad dispersal of signals generated by dysfunctional RBCs. Precise understanding of the roles of dysregulated RBC-based NO transport in the spread of vasomotor dysfunction from stressed vascular beds may open novel therapeutic approaches to a range of pathologies.
Synopsis.
Oxygen (O2) delivery, the maintenance of which is fundamental to supporting those with critical illness, is a function of blood O2 content and flow. Here, we review red blood cell (RBC) physiology and dysfunction relevant to disordered O2 delivery in the critically ill. Flow (rather than content) is the focus of O2 delivery regulation: O2 content is relatively fixed, whereas flow fluctuates by several orders of magnitude. Thus, blood flow volume and distribution vary to maintain coupling between O2 delivery and demand. The trapping, processing and delivery of vasoactive effectors (NO and ATP) by RBCs has emerged as a conserved mechanism through which regional blood flow is linked to biochemical cues of perfusion sufficiency. We will review conventional RBC physiology influencing O2 delivery (O2 affinity & rheology) and introduce a new paradigm for O2 delivery homeostasis based on coordinated gas transport and vascular signaling by RBCs. By coordinating vascular signaling in a fashion that links O2 and NO flux, RBCs couple vessel caliber (and thus blood flow) to O2 need in tissue. Malfunction of this signaling system is implicated in a wide array of pathophysiologies and may be explanatory for the dysoxia frequently encountered in the critical care setting.
Key Points.
Together, all red blood cells (RBC) at each stage of development, may be considered an organ (termed the erythron) now appreciated to participate in active regulation of regional blood flow distribution as well as O2 and CO2 transport.
RBCs are subject to intense biochemical, biomechanical and physiologic stress during repeated circulatory transit and as such, possess unique properties and robust energetic and antioxidant systems to maintain functionality for a 3–4-month lifetime.
RBCs actively regulate blood flow volume and distribution to maintain coupling between O2 delivery and demand. The trapping, processing and delivery of nitric oxide (NO) by RBCs has emerged as a conserved mechanism through which regional blood flow is linked to biochemical cues of perfusion sufficiency.
A new paradigm for O2 delivery homeostasis has emerged, based on coordinated gas transport and vascular signaling by RBCs. By coordinating vascular signaling in a fashion that links O2 and nitric oxide (NO) flux, RBCs couple vessel caliber (and thus blood flow) to O2 need in tissue. Malfunction of this signaling system is implicated in a wide array of pathophysiologies and may be in part explanatory for the dysoxia frequently encountered in the critical care setting.
Funding:
NIH R01GM113838, R42HL135965, U01AI126610 and Department of Defense W81XWH-17-1-0668
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest:
AD has received research funding and/or consulting fees from: Viasys Inc., Entegrion Inc., Terumo BCT, Fresenius Kabi, Galleon Pharmaceuticals, Nitrox LLD, Nitric BioTherapeutics, Galera Inc., and Novartis. AD holds intellectual property related to biosynthetic artificial RBCs and holds equity in and is CSO of KaloCyte, Inc – which is developing this technology. SR has no conflicts to declare.
References
- 1.Kaushansky K Williams Hematology. 8e ed: McGraw-Hill; 2010. [Google Scholar]
- 2.Doctor A, Stamler JS. Nitric oxide transport in blood: a third gas in the respiratory cycle. Compr Physiol. 2011;1(1):541–568. [DOI] [PubMed] [Google Scholar]
- 3.Hsia CC. Respiratory function of hemoglobin. N Engl J Med. 1998;338(4):239–247. [DOI] [PubMed] [Google Scholar]
- 4.Edsall JT. Understanding blood and hemoglobin: an example of international relations in science. Perspectives in biology and medicine. 1986;29(3 Pt 2):S107–123. [DOI] [PubMed] [Google Scholar]
- 5.Edsall JT. Hemoglobin and the origins of the concept of allosterism. Federation proceedings. 1980;39(2):226–235. [PubMed] [Google Scholar]
- 6.Winslow RM. The role of hemoglobin oxygen affinity in oxygen transport at high altitude. Respir Physiol Neurobiol. 2007;158(2–3):121–127. [DOI] [PubMed] [Google Scholar]
- 7.Bohr CHK, Krogh A. Ueber einen in biologischer Beziehung wichtigen Einfluss, den die Kohlensäurespannung des Blutes auf dessen Sauerstoffbindung übt. Skand Arch Physiol. 1904;16:402–412. [Google Scholar]
- 8.Margaria RGA. The first dissociation constant, pK 1, of carbonic acid in hemoglobin solutions and its relation to the existence of a combination of hemoglobin with carbon dioxide. J Biol Chem. 1933;102:611–634. [Google Scholar]
- 9.Leon K, Pichavant-Rafini K, Quemener E, et al. Oxygen blood transport during experimental sepsis: effect of hypothermia*. Crit Care Med. 2012;40(3):912–918. [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim Eel D, McLellan SA, Walsh TS. Red blood cell 2,3-diphosphoglycerate concentration and in vivo P50 during early critical illness. Crit Care Med. 2005;33(10):2247–2252. [DOI] [PubMed] [Google Scholar]
- 11.Tuynman HA, Thijs LG, Straub JP, Koopman PA, Bezemer PD, Bronsveld W. Effects of glucose-insulin-potassium (GIK) on the position of the oxyhemoglobin dissociation curve, 2.3-diphosphoglycerate, and oxygen consumption in canine endotoxin shock. The Journal of surgical research. 1983;34(3):246–253. [DOI] [PubMed] [Google Scholar]
- 12.Johnson G Jr., McDevitt NB, Proctor HJ. Erythrocyte 2,3-diphosphoglycerate in endotoxic shock in the subhuman primate: response to fluid and-or methylprednisolone succinate. Ann Surg. 1974;180(5):783–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunn HF, Jandl JH. Control of hemoglobin function within the red cell. N Engl J Med. 1970;282(25):1414–1421. [DOI] [PubMed] [Google Scholar]
- 14.Severinghaus JW. Oxyhemoglobin dissociation curve correction for temperature and pH variation in human blood. J Appl Physiol. 1958;12(3):485–486. [DOI] [PubMed] [Google Scholar]
- 15.Robin Fahraeus TL. The viscosity of the blood in narrow capillary tubes. American Journal of Physiology. 1931;96:562–568. [Google Scholar]
- 16.Copley AL. Fluid mechanics and biorheology. Thrombosis research. 1990;57(3):315–331. [DOI] [PubMed] [Google Scholar]
- 17.McHedlishvili G, Varazashvili M, Gobejishvili L. Local RBC aggregation disturbing blood fluidity and causing stasis in microvessels. Clin Hemorheol Microcirc. 2002;26(2):99–106. [PubMed] [Google Scholar]
- 18.Baskurt OK, Meiselman HJ. Blood rheology and hemodynamics. Seminars in thrombosis and hemostasis. 2003;29(5):435–450. [DOI] [PubMed] [Google Scholar]
- 19.Baskurt OK, Temiz A, Meiselman HJ. Effect of superoxide anions on red blood cell rheologic properties. Free Radic Biol Med. 1998;24(1):102–110. [DOI] [PubMed] [Google Scholar]
- 20.Piagnerelli M, Boudjeltia KZ, Vanhaeverbeek M, Vincent JL. Red blood cell rheology in sepsis. Intensive Care Med. 2003;29(7):1052–1061. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki Y, Nakajima T, Shiga T, Maeda N. Influence of 2,3-diphosphoglycerate on the deformability of human erythrocytes. Biochim Biophys Acta. 1990;1029(1):85–90. [DOI] [PubMed] [Google Scholar]
- 22.Todd JC 3rd, Mollitt DL. Effect of sepsis on erythrocyte intracellular calcium homeostasis. Crit Care Med. 1995;23(3):459–465. [DOI] [PubMed] [Google Scholar]
- 23.Piagnerelli M, Boudjeltia KZ, Brohee D, et al. Alterations of red blood cell shape and sialic acid membrane content in septic patients. Crit Care Med. 2003;31(8):2156–2162. [DOI] [PubMed] [Google Scholar]
- 24.Machiedo GW, Powell RJ, Rush BF Jr., Swislocki NI, Dikdan G. The incidence of decreased red blood cell deformability in sepsis and the association with oxygen free radical damage and multiple-system organ failure. Arch Surg. 1989;124(12):1386–1389. [DOI] [PubMed] [Google Scholar]
- 25.Somer T, Meiselman HJ. Disorders of blood viscosity. Annals of medicine. 1993;25(1):31–39. [DOI] [PubMed] [Google Scholar]
- 26.Baskurt OK, Meiselman HJ. Activated polymorphonuclear leukocytes affect red blood cell aggregability. J Leukoc Biol. 1998;63(1):89–93. [DOI] [PubMed] [Google Scholar]
- 27.Aird WC. The hematologic system as a marker of organ dysfunction in sepsis. Mayo Clinic proceedings Mayo Clinic. 2003;78(7):869–881. [DOI] [PubMed] [Google Scholar]
- 28.Goyette RE, Key NS, Ely EW. Hematologic changes in sepsis and their therapeutic implications. Seminars in respiratory and critical care medicine. 2004;25(6):645–659. [DOI] [PubMed] [Google Scholar]
- 29.Anniss AM, Sparrow RL. Variable adhesion of different red blood cell products to activated vascular endothelium under flow conditions. American journal of hematology. 2007;82(6):439–445. [DOI] [PubMed] [Google Scholar]
- 30.Tissot Van Patot MC, MacKenzie S, Tucker A, Voelkel NF. Endotoxin-induced adhesion of red blood cells to pulmonary artery endothelial cells. Am J Physiol. 1996;270(1 Pt 1):L28–36. [DOI] [PubMed] [Google Scholar]
- 31.Zoukourian C, Wautier MP, Chappey O, et al. Endothelial cell dysfunction secondary to the adhesion of diabetic erythrocytes. Modulation by iloprost. International angiology : a journal of the International Union of Angiology. 1996;15(3):195–200. [PubMed] [Google Scholar]
- 32.Sirois E, Charara J, Ruel J, Dussault JC, Gagnon P, Doillon CJ. Endothelial cells exposed to erythrocytes under shear stress: an in vitro study. Biomaterials. 1998;19(21):1925–1934. [DOI] [PubMed] [Google Scholar]
- 33.Munn LL, Melder RJ, Jain RK. Role of erythrocytes in leukocyte-endothelial interactions: mathematical model and experimental validation. Biophys J. 1996;71(1):466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Migliorini C, Qian Y, Chen H, Brown EB, Jain RK, Munn LL. Red blood cells augment leukocyte rolling in a virtual blood vessel. Biophys J. 2002;83(4):1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zennadi R, Chien A, Xu K, Batchvarova M, Telen MJ. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood. 2008;112(8):3474–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohandas N, Shohet SB. The role of membrane-associated enzymes in regulation of erythrocyte shape and deformability. Clin Haematol. 1981;10(1):223–237. [PubMed] [Google Scholar]
- 37.Mohandas N The red blood cell membrane In: Hoffman RBE, Shattil SJ, Furie B, Cohen HJ ed. Hematology: basis, principles and practice. New York: Churchill-Livingstone; 1991:264–269. [Google Scholar]
- 38.Rendell M, Luu T, Quinlan E, et al. Red cell filterability determined using the cell transit time analyzer (CTTA): effects of ATP depletion and changes in calcium concentration. Biochimica et biophysica acta. 1992;1133(3):293–300. [DOI] [PubMed] [Google Scholar]
- 39.Kayar E, Mat F, Meiselman HJ, Baskurt OK. Red blood cell rheological alterations in a rat model of ischemia-reperfusion injury. Biorheology. 2001;38(5–6):405–414. [PubMed] [Google Scholar]
- 40.Baskurt O Activated granulocyte induced alterations in red blood cells and protection by antioxidant enzymes. Clinical hemorheology and microcirculation. 1996;16:49–56. [Google Scholar]
- 41.Ross JM, Fairchild HM, Weldy J, Guyton AC. Autoregulation of blood flow by oxygen lack. Am J Physiol. 1962;202:21–24. [DOI] [PubMed] [Google Scholar]
- 42.Roy CS, Brown JG. The Blood-Pressure and its Variations in the Arterioles, Capillaries and Smaller Veins. J Physiol. 1880;2(5–6):323–446 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stein JC, Ellsworth ML. Capillary oxygen transport during severe hypoxia: role of hemoglobin oxygen affinity. Journal of applied physiology. 1985;75(4):1601–1607. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez-Alonso J, Richardson RS, Saltin B. Exercising skeletal muscle blood flow in humans responds to reduction in arterial oxyhaemoglobin, but not to altered free oxygen. J Physiol. 2001;530(Pt 2):331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. The erythrocyte as a regulator of vascular tone. Am J Physiol. 1995;269(6 Pt 2):H2155–2161. [DOI] [PubMed] [Google Scholar]
- 46.Jia L, Bonaventura C, Bonaventura J, Stamler JS. S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature. 1996;380(6571):221–226. [DOI] [PubMed] [Google Scholar]
- 47.Stamler JS, Jia L, Eu JP, et al. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276(5321):2034–2037. [DOI] [PubMed] [Google Scholar]
- 48.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. [DOI] [PubMed] [Google Scholar]
- 49.Gladwin MT, Shelhamer JH, Schechter AN, et al. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of regional blood flow in humans. Proc Natl Acad Sci U S A. 2000;97(21):11482–11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda). 2009;24:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373–376. [DOI] [PubMed] [Google Scholar]
- 52.Furchgott RF. Endothelium-derived relaxing factor: discovery, early studies, and identification as nitric oxide. Bioscience reports. 1999;19(4):235–251. [DOI] [PubMed] [Google Scholar]
- 53.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84(24):9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524–526. [DOI] [PubMed] [Google Scholar]
- 55.Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate cyclase and increases guanosine 3’:5’-cyclic monophosphate levels in various tissue preparations. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(8):3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gonzalez-Alonso J, Mortensen SP, Dawson EA, Secher NH, Damsgaard R. Erythrocytes and the regulation of human skeletal muscle blood flow and oxygen delivery: role of erythrocyte count and oxygenation state of haemoglobin. The Journal of physiology. 2006;572(Pt 1):295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kantrow SP, Huang YC, Whorton AR, et al. Hypoxia inhibits nitric oxide synthesis in isolated rabbit lung. Am J Physiol. 1997;272(6 Pt 1):L1167–1173. [DOI] [PubMed] [Google Scholar]
- 58.Rengasamy A, Johns RA. Determination of Km for oxygen of nitric oxide synthase isoforms. The Journal of pharmacology and experimental therapeutics. 1996;276(1):30–33. [PubMed] [Google Scholar]
- 59.Mamone G, Sannolo N, Malorni A, Ferranti P. In vitro formation of S-nitrosohemoglobin in red cells by inducible nitric oxide synthase. FEBS Lett. 1999;462(3):241–245. [DOI] [PubMed] [Google Scholar]
- 60.Angelo M, Singel DJ, Stamler JS. An S-nitrosothiol (SNO) synthase function of hemoglobin that utilizes nitrite as a substrate. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(22):8366–8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schechter AN, Gladwin MT. Hemoglobin and the paracrine and endocrine functions of nitric oxide. The New England journal of medicine. 2003;348(15):1483–1485. [DOI] [PubMed] [Google Scholar]
- 62.Hobbs AJ. Soluble guanylate cyclase: the forgotten sibling. Trends in pharmacological sciences. 1997;18(12):484–491. [DOI] [PubMed] [Google Scholar]
- 63.Gow AJ, Ischiropoulos H. Nitric oxide chemistry and cellular signaling. J Cell Physiol. 2001;187(3):277–282. [DOI] [PubMed] [Google Scholar]
- 64.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR Jr. Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem. 1998;273(30):18709–18713. [DOI] [PubMed] [Google Scholar]
- 65.Huang KT, Han TH, Hyduke DR, et al. Modulation of nitric oxide bioavailability by erythrocytes. Proc Natl Acad Sci U S A. 2001;98(20):11771–11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liao JC, Hein TW, Vaughn MW, Huang KT, Kuo L. Intravascular flow decreases erythrocyte consumption of nitric oxide. Proc Natl Acad Sci U S A. 1999;96(15):8757–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gow AJ, Farkouh CR, Munson DA, Posencheg MA, Ischiropoulos H. Biological significance of nitric oxide-mediated protein modifications. American journal of physiology Lung cellular and molecular physiology. 2004;287(2):L262–268. [DOI] [PubMed] [Google Scholar]
- 68.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nature reviews. 2005;6(2):150–166. [DOI] [PubMed] [Google Scholar]
- 69.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258(5090):1898–1902. [DOI] [PubMed] [Google Scholar]
- 70.Stamler JS, Jaraki O, Osborne J, et al. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(16):7674–7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stamler JS, Simon DI, Osborne JA, et al. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(1):444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnelle DR, Stamler JS. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318(2):279–285. [DOI] [PubMed] [Google Scholar]
- 73.Ferranti P, Malorni A, Mamone G, Sannolo N, Marino G. Characterisation of S-nitrosohaemoglobin by mass spectrometry. FEBS Lett. 1997;400(1):19–24. [DOI] [PubMed] [Google Scholar]
- 74.Chan NL, Rogers PH, Arnone A. Crystal structure of the S-nitroso form of liganded human hemoglobin. Biochemistry. 1998;37(47):16459–16464. [DOI] [PubMed] [Google Scholar]
- 75.Luchsinger BP, Rich EN, Gow AJ, Williams EM, Stamler JS, Singel DJ. Routes to S-nitroso-hemoglobin formation with heme redox and preferential reactivity in the beta subunits. Proc Natl Acad Sci U S A. 2003;100(2):461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gow AJ, Stamler JS. Reactions between nitric oxide and haemoglobin under physiological conditions. Nature. 1998;391(6663):169–173. [DOI] [PubMed] [Google Scholar]
- 77.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409(6820):622–626. [DOI] [PubMed] [Google Scholar]
- 78.Doctor A, Platt R, Sheram ML, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A. 2005;102(16):5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Palmer LA, Doctor A, Chhabra P, et al. S-nitrosothiols signal hypoxia-mimetic vascular pathology. J Clin Invest. 2007;117(9):2592–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Diesen DL, Hess DT, Stamler JS. Hypoxic vasodilation by red blood cells: evidence for an s-nitrosothiol-based signal. Circ Res. 2008;103(5):545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McMahon TJ, Moon RE, Luschinger BP, et al. Nitric oxide in the human respiratory cycle. Nat Med. 2002;8(7):711–717. [DOI] [PubMed] [Google Scholar]
- 82.Datta B, Tufnell-Barrett T, Bleasdale RA, et al. Red blood cell nitric oxide as an endocrine vasoregulator: a potential role in congestive heart failure. Circulation. 2004;109(11):1339–1342. [DOI] [PubMed] [Google Scholar]
- 83.James PE, Lang D, Tufnell-Barret T, Milsom AB, Frenneaux MP. Vasorelaxation by red blood cells and impairment in diabetes: reduced nitric oxide and oxygen delivery by glycated hemoglobin. Circ Res. 2004;94(7):976–983. [DOI] [PubMed] [Google Scholar]
- 84.Saltin B, Radegran G, Koskolou MD, Roach RC. Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand. 1998;162(3):421–436. [DOI] [PubMed] [Google Scholar]
- 85.Fox-Robichaud A, Payne D, Kubes P. Inhaled NO reaches distal vasculatures to inhibit endothelium- but not leukocyte-dependent cell adhesion. Am J Physiol. 1999;277(6 Pt 1):L1224–1231. [DOI] [PubMed] [Google Scholar]
- 86.Fox-Robichaud A, Payne D, Hasan SU, et al. Inhaled NO as a viable antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. J Clin Invest. 1998;101(11):2497–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kubes P, Payne D, Grisham MB, Jourd-Heuil D, Fox-Robichaud A. Inhaled NO impacts vascular but not extravascular compartments in postischemic peripheral organs. Am J Physiol. 1999;277(2 Pt 2):H676–682. [DOI] [PubMed] [Google Scholar]
- 88.Cannon RO 3rd, Schechter AN, Panza JA, et al. Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. J Clin Invest. 2001;108(2):279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McMahon TJ, Ahearn GS, Moya MP, et al. A nitric oxide processing defect of red blood cells created by hypoxia: deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(41):14801–14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reutov VP, Sorokina EG. NO-synthase and nitrite-reductase components of nitric oxide cycle. Biochemistry (Mosc). 1998;63(7):874–884. [PubMed] [Google Scholar]
- 91.Gladwin MT. Evidence mounts that nitrite contributes to hypoxic vasodilation in the human circulation. Circulation. 2008;117(5):594–597. [DOI] [PubMed] [Google Scholar]
- 92.Luchsinger BP, Rich EN, Yan Y, Williams EM, Stamler JS, Singel DJ. Assessments of the chemistry and vasodilatory activity of nitrite with hemoglobin under physiologically relevant conditions. Journal of inorganic biochemistry. 2005;99(4):912–921. [DOI] [PubMed] [Google Scholar]
- 93.Huang Z, Shiva S, Kim-Shapiro DB, et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115(8):2099–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rifkind JM, Ramasamy S, Manoharan PT, Nagababu E, Mohanty JG. Redox reactions of hemoglobin. Antioxidants & redox signaling. 2004;6(3):657–666. [DOI] [PubMed] [Google Scholar]
- 95.Crawford JH, Isbell TS, Huang Z, et al. Hypoxia, red blood cells, and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2006;107(2):566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jagger JE, Bateman RM, Ellsworth ML, Ellis CG. Role of erythrocyte in regulating local O2 delivery mediated by hemoglobin oxygenation. Am J Physiol Heart Circ Physiol. 2001;280(6):H2833–2839. [DOI] [PubMed] [Google Scholar]
- 97.Bergfeld GR, Forrester T. Release of ATP from human erythrocytes in response to a brief period of hypoxia and hypercapnia. Cardiovasc Res. 1992;26(1):40–47. [DOI] [PubMed] [Google Scholar]
- 98.Sprague RS, Bowles EA, Olearczyk JJ, Stephenson AH, Lonigro AJ. The role of G protein beta subunits in the release of ATP from human erythrocytes. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society. 2002;53(4 Pt 1):667–674. [PubMed] [Google Scholar]
- 99.Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal-transduction pathway relating erythrocyte deformation to ATP release. American journal of physiology Cell physiology. 2001;281(4):C1158–1164. [DOI] [PubMed] [Google Scholar]
- 100.Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires CFTR activity. The American journal of physiology. 1998;275(5 Pt 2):H1726–1732. [DOI] [PubMed] [Google Scholar]
- 101.Sridharan M, Adderley SP, Bowles EA, et al. Pannexin 1 is the conduit for low oxygen tension-induced ATP release from human erythrocytes. Am J Physiol Heart Circ Physiol. 2010;299(4):H1146–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rogers SC, Said A, Corcuera D, McLaughlin D, Kell P, Doctor A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. Faseb J. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bossi D, Giardina B. Red cell physiology. Mol Aspects Med. 1996;17(2):117–128. [DOI] [PubMed] [Google Scholar]
- 104.Siems WG, Sommerburg O, Grune T. Erythrocyte free radical and energy metabolism. Clinical nephrology. 2000;53(1 Suppl):S9–17. [PubMed] [Google Scholar]
- 105.Sterling D, Reithmeier RAF, Casey JR. A Transport Metabolon. Journal of Biological Chemistry. 2001;276(51):47886–47894. [DOI] [PubMed] [Google Scholar]
- 106.Bruce LJ, Beckmann R, Ribeiro ML, et al. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101(10):4180–4188. [DOI] [PubMed] [Google Scholar]
- 107.Messana I, Orlando M, Cassiano L, et al. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett. 1996;390(1):25–28. [DOI] [PubMed] [Google Scholar]
- 108.Barvitenko NN, Adragna NC, Weber RE. Erythrocyte signal transduction pathways, their oxygenation dependence and functional significance. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2005;15(1–4):1–18. [DOI] [PubMed] [Google Scholar]
- 109.Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci U S A. 2005;102(7):2402–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Low PS, Rathinavelu P, Harrison ML. Regulation of glycolysis via reversible enzyme binding to the membrane protein, band 3. J Biol Chem. 1993;268(20):14627–14631. [PubMed] [Google Scholar]
- 111.Chu H, Low PS. Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochemical Journal. 2006;400(1):143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chu H, Breite A, Ciraolo P, Franco RS, Low PS. Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: implications for O2 regulation of erythrocyte properties. Blood. 2008;111(2):932–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hald B, Madsen MF, Dano S, Quistorff B, Sorensen PG. Quantitative evaluation of respiration induced metabolic oscillations in erythrocytes. Biophys Chem. 2009;141(1):41–48. [DOI] [PubMed] [Google Scholar]
- 114.Barvitenko NN, Adragna NC, Weber RE. Erythrocyte Signal Transduction Pathways, their Oxygenation Dependence and Functional Significance. Cellular Physiology and Biochemistry. 2005;15(1–4):1–18. [DOI] [PubMed] [Google Scholar]
- 115.Messana I, Orlando M, Cassiano L, et al. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Letters. 1996;390(1):25–28. [DOI] [PubMed] [Google Scholar]
- 116.Kinoshita A, Tsukada K, Soga T, et al. Roles of Hemoglobin Allostery in Hypoxia-induced Metabolic Alterations in Erythrocytes: simulation and its verification by metabolome analysis. Journal of Biological Chemistry. 2007;282(14):10731–10741. [DOI] [PubMed] [Google Scholar]
- 117.Barbul A, Zipser Y, Nachles A, Korenstein R. Deoxygenation and elevation of intracellular magnesium induce tyrosine phosphorylation of band 3 in human erythrocytes. FEBS Lett. 1999;455(1–2):87–91. [DOI] [PubMed] [Google Scholar]
- 118.Bordin L, Ion-Popa F, Brunati AM, Clari G, Low PS. Effector-induced Syk-mediated phosphorylation in human erythrocytes. Biochim Biophys Acta. 2005;1745(1):20–28. [DOI] [PubMed] [Google Scholar]
- 119.Harrison ML, Rathinavelu P, Arese P, Geahlen RL, Low PS. Role of band 3 tyrosine phosphorylation in the regulation of erythrocyte glycolysis. J Biol Chem. 1991;266(7):4106–4111. [PubMed] [Google Scholar]
- 120.Rogers SC, Said A, Corcuera D, McLaughlin D, Kell P, Doctor A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. The FASEB Journal. 2009;23(9):3159–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rogers SC, Ross JG, d’Avignon A, et al. Sickle hemoglobin disturbs normal coupling between erythrocyte O2 content, glycolysis and antioxidant capacity. Blood. 2013;121(9):1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Condon MR, Feketova E, Machiedo GW, Deitch EA, Spolarics Z. Augmented erythrocyte band-3 phosphorylation in septic mice. Biochim Biophys Acta. 2007;1772(5):580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Serroukh Y, Djebara S, Lelubre C, Zouaoui Boudjeltia K, Biston P, Piagnerelli M. Alterations of the Erythrocyte Membrane during Sepsis. Critical care research and practice. 2012;2012:702956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Suzuki Y, Ohkubo N, Aoto M, et al. Participation of caspase-3-like protease in oxidation-induced impairment of erythrocyte membrane properties. Biorheology. 2007;44(3):179–190. [PubMed] [Google Scholar]
- 125.Foller M, Huber SM, Lang F. Erythrocyte programmed cell death. IUBMB Life. 2008;60(10):661–668. [DOI] [PubMed] [Google Scholar]
- 126.Mandal D, Baudin-Creuza V, Bhattacharyya A, et al. Caspase 3-mediated proteolysis of the N-terminal cytoplasmic domain of the human erythroid anion exchanger 1 (band 3). J Biol Chem. 2003;278(52):52551–52558. [DOI] [PubMed] [Google Scholar]
- 127.Birmingham DJ. Erythrocyte complement receptors. Critical reviews in immunology. 1995;15(2):133–154. [DOI] [PubMed] [Google Scholar]
- 128.Pascual M, Schifferli JA. The binding of immune complexes by the erythrocyte complement receptor 1 (CR1). Immunopharmacology. 1992;24(2):101–106. [DOI] [PubMed] [Google Scholar]
- 129.Pascual M, Schifferli JA. Another function of erythrocytes: transport of circulating immune complexes. Infusionstherapie und Transfusionsmedizin. 1995;22(5):310–315. [DOI] [PubMed] [Google Scholar]
- 130.Poschl JM, Leray C, Ruef P, Cazenave JP, Linderkamp O. Endotoxin binding to erythrocyte membrane and erythrocyte deformability in human sepsis and in vitro. Crit Care Med. 2003;31(3):924–928. [DOI] [PubMed] [Google Scholar]
- 131.Todd JC 3rd, Poulos ND, Mollitt DL. The effect of endotoxin on the neonatal erythrocyte. J Pediatr Surg. 1993;28(3):334–336; discussion 336–337. [DOI] [PubMed] [Google Scholar]
- 132.Todd JC 3rd, Poulos ND, Davidson LW, Mollitt DL. Role of the leukocyte in endotoxin-induced alterations of the red cell membrane. Second place winner of the Conrad Jobst Award in the Gold Medal paper competition. The American surgeon. 1993;59(1):9–12. [PubMed] [Google Scholar]
- 133.Todd JC 3rd, Poulos ND, Mollitt DL. The effect of endotoxin on neonatal erythrocyte intracellular calcium concentration. J Pediatr Surg. 1994;29(6):805–807. [DOI] [PubMed] [Google Scholar]
- 134.Godin DV, Tuchek JM, Garnett ME. Studies on the interaction of Escherichia coli endotoxin with erythrocyte membranes. Canadian journal of physiology and pharmacology. 1982;60(7):977–985. [DOI] [PubMed] [Google Scholar]
- 135.Lewis IA, Campanella ME, Markley JL, Low PS. Role of band 3 in regulating metabolic flux of red blood cells. Proc Natl Acad Sci U S A. 2009;106(44):18515–18520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Low PS. Structure and function of the cytoplasmic domain of band 3: center of erythrocyte membrane--peripheral protein interactions. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1986;864(2):145–167. [DOI] [PubMed] [Google Scholar]
- 137.Low PS, Rathinavelu P, Harrison ML. Regulation of glycolysis via reversible enzyme binding to the membrane protein, band 3. Journal of Biological Chemistry. 1993;268(20):14627–14631. [PubMed] [Google Scholar]
- 138.Buranakarl C, Trisiriroj M, Pondeenana S, Tungjitpeanpong T, Jarutakanon P, Penchome R. Relationships between oxidative stress markers and red blood cell characteristics in renal azotemic dogs. Res Vet Sci. 2009;86(2):309–313. [DOI] [PubMed] [Google Scholar]
- 139.Vaziri ND. Oxidative stress in uremia: nature, mechanisms, and potential consequences. Seminars in nephrology. 2004;24(5):469–473. [DOI] [PubMed] [Google Scholar]
- 140.Zingraff J, Kamoun P, Lebreton P, Drueke T, Man NK, Jungers P. Plasma inhibitors of the erythrocyte hexose monophosphate shunt in uraemia. Proc Eur Dial Transplant Assoc. 1979;16:475–480. [PubMed] [Google Scholar]
- 141.Yawata Y, Jacob HS. Abnormal red cell metabolism in patients with chronic uremia: Nature of the defect and its persistence despite adequate hemodialysis. Blood. 1975;45(2):231–239. [PubMed] [Google Scholar]
- 142.Poulianiti KP, Kaltsatou A, Mitrou GI, et al. Systemic Redox Imbalance in Chronic Kidney Disease: A Systematic Review. Oxid Med Cell Longev. 2016;2016:8598253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Eggert W, Scigalla P, Noack C, Devaux S, Wolf S. [Effect of uremic plasma fractions of different molecular sizes on the filterability of red blood cells]. Z Urol Nephrol. 1981;74(5):391–399. [PubMed] [Google Scholar]
- 144.Eggert W, Schmidt G, Devaux S. [Uremic plasma as cause of metabolic changes in red blood cells]. Z Urol Nephrol. 1981;74(2):141–147. [PubMed] [Google Scholar]
- 145.Lucchi L, Bergamini S, Iannone A, et al. Erythrocyte susceptibility to oxidative stress in chronic renal failure patients under different substitutive treatments. Artificial organs. 2005;29(1):67–72. [DOI] [PubMed] [Google Scholar]
- 146.Spolarics Z, Condon MR, Siddiqi M, Machiedo GW, Deitch EA. Red blood cell dysfunction in septic glucose-6-phosphate dehydrogenase-deficient mice. AJP - Heart and Circulatory Physiology. 2004;286(6):H2118–H2126. [DOI] [PubMed] [Google Scholar]
- 147.van Wijk R, van Solinge WW. The energy-less red blood cell is lost: erythrocyte enzyme abnormalities of glycolysis. Blood. 2005;106(13):4034–4042. [DOI] [PubMed] [Google Scholar]
- 148.Lang F, Abed M, Lang E, Foller M. Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal. 2013. [DOI] [PubMed] [Google Scholar]
- 149.Rogers SC, Ross JG, d’Avignon A, et al. Sickle hemoglobin disturbs normal coupling among erythrocyte O2 content, glycolysis, and antioxidant capacity. Blood. 2013;121(9):1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Rogers SC, Said A, Corcuera D, McLaughlin D, Kell P, Doctor A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2009;23(9):3159–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Droge W Free radicals in the physiological control of cell function. Physiological Reviews. 2001;82:47–95. [DOI] [PubMed] [Google Scholar]
- 152.Fantone JC, Ward PA. Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. The American journal of pathology. 1982;107(3):395–418. [PMC free article] [PubMed] [Google Scholar]
- 153.Andrades ME, Morina A, Spasic S, Spasojevic I. Bench-to-bedside review: sepsis - from the redox point of view. Crit Care. 2011;15(5):230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Salvemini D, Cuzzocrea S. Oxidative stress in septic shock and disseminated intravascular coagulation. Free Radic Biol Med. 2002;33(9):1173–1185. [DOI] [PubMed] [Google Scholar]
- 155.Crawford JH, Chacko BK, Pruitt HM, Piknova B, Hogg N, Patel RP. Transduction of NO-bioactivity by the red blood cell in sepsis: novel mechanisms of vasodilation during acute inflammatory disease. Blood. 2004;104(5):1375–1382. [DOI] [PubMed] [Google Scholar]
- 156.Liu L, Yan Y, Zeng M, et al. Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell. 2004;116(4):617–628. [DOI] [PubMed] [Google Scholar]
- 157.Doctor A, Platt R, Sheram ML, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proceedings of the National Academy of Sciences. 2005;102(16):5709–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bellary SS, Anderson KW, Arden WA, Butterfield DA. Effect of lipopolysaccharide on the physical conformation of the erythrocyte cytoskeletal proteins. Life sciences. 1995;56(2):91–98. [DOI] [PubMed] [Google Scholar]
- 159.Piagnerelli M, Cotton F, Van Nuffelen M, Vincent JL, Gulbis B. Modifications in erythrocyte membrane protein content are not responsible for the alterations in rheology seen in sepsis. Shock. 2012;37(1):17–21. [DOI] [PubMed] [Google Scholar]
- 160.Condon MR, Kim JE, Deitch EA, Machiedo GW, Spolarics Z. Appearance of an erythrocyte population with decreased deformability and hemoglobin content following sepsis. Am J Physiol Heart Circ Physiol. 2003;284(6):H2177–2184. [DOI] [PubMed] [Google Scholar]
- 161.Eichelbronner O, Sielenkamper A, Cepinskas G, Sibbald WJ, Chin-Yee IH. Endotoxin promotes adhesion of human erythrocytes to human vascular endothelial cells under conditions of flow. Crit Care Med. 2000;28(6):1865–1870. [DOI] [PubMed] [Google Scholar]
- 162.Eichelbronner O, Sibbald WJ, Chin-Yee IH. Intermittent flow increases endotoxin-induced adhesion of human erythrocytes to vascular endothelial cells. Intensive Care Med. 2003;29(5):709–714. [DOI] [PubMed] [Google Scholar]
- 163.Behonick GS, Novak MJ, Nealley EW, Baskin SI. Toxicology update: the cardiotoxicity of the oxidative stress metabolites of catecholamines (aminochromes). Journal of applied toxicology : JAT. 2001;21 Suppl 1:S15–22. [DOI] [PubMed] [Google Scholar]
- 164.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320(6061):454–456. [DOI] [PubMed] [Google Scholar]
- 165.Jonsson LM, Rees DD, Edlund T, Marklund SL. Nitric oxide and blood pressure in mice lacking extracellular-superoxide dismutase. Free Radic Res. 2002;36(7):755–758. [DOI] [PubMed] [Google Scholar]
- 166.Macarthur H, Westfall TC, Riley DP, Misko TP, Salvemini D. Inactivation of catecholamines by superoxide gives new insights on the pathogenesis of septic shock. Proc Natl Acad Sci U S A. 2000;97(17):9753–9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Misra HP, Fridovich I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 1972;247(10):3170–3175. [PubMed] [Google Scholar]
- 168.Thomas DD, Ridnour LA, Espey MG, et al. Superoxide fluxes limit nitric oxide-induced signaling. J Biol Chem. 2006;281(36):25984–25993. [DOI] [PubMed] [Google Scholar]
- 169.Wink DA, Cook JA, Kim SY, et al. Superoxide modulates the oxidation and nitrosation of thiols by nitric oxide-derived reactive intermediates. Journal of Biological Chemistry. 1997;272:11147–11151. [DOI] [PubMed] [Google Scholar]
- 170.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood. 2008;111(10):5205–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Huertas A, Das SR, Emin M, et al. Erythrocytes induce proinflammatory endothelial activation in hypoxia. American journal of respiratory cell and molecular biology. 2013;48(1):78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rattan V, Shen Y, Sultana C, Kumar D, Kalra VK. Diabetic RBC-induced oxidant stress leads to transendothelial migration of monocyte-like HL-60 cells. Am J Physiol. 1997;273(2 Pt 1):E369–375. [DOI] [PubMed] [Google Scholar]
- 173.Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood. 1998;92(10):3924–3935. [PubMed] [Google Scholar]
- 174.Darley-Usmar V, Halliwell B. Blood radicals: reactive nitrogen species, reactive oxygen species, transition metal ions, and the vascular system. Pharmaceutical research. 1996;13(5):649–662. [DOI] [PubMed] [Google Scholar]
- 175.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. AJP - Regulatory, Integrative and Comparative Physiology. 2004;287(5):R1014–R1030. [DOI] [PubMed] [Google Scholar]
- 176.Abraham E, Singer M. Mechanisms of sepsis-induced organ dysfunction. Crit Care Med. 2007;35(10):2408–2416. [DOI] [PubMed] [Google Scholar]
- 177.Sibbald WJ, Messmer K, Fink MP. Roundtable conference on tissue oxygenation in acute medicine, Brussels, Belgium, 14–16 March 1998. Intensive Care Med. 2000;26(6):780–791. [DOI] [PubMed] [Google Scholar]
- 178.Elbers PW, Ince C. Mechanisms of critical illness--classifying microcirculatory flow abnormalities in distributive shock. Crit Care. 2006;10(4):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ince C Mechanism of impaired oxygen extraction in sepsis. Shunt and the pO2 gap. Minerva anestesiologica. 1999;65(6):337–339. [PubMed] [Google Scholar]
- 180.Martensson J, Bellomo R. Sepsis-Induced Acute Kidney Injury. Crit Care Clin. 2015;31(4):649–660. [DOI] [PubMed] [Google Scholar]
- 181.McCullough PA, Shaw AD, Haase M, et al. Diagnosis of acute kidney injury using functional and injury biomarkers: workgroup statements from the tenth Acute Dialysis Quality Initiative Consensus Conference. Contrib Nephrol. 2013;182:13–29. [DOI] [PubMed] [Google Scholar]
- 182.Nguyen MT, Devarajan P. Biomarkers for the early detection of acute kidney injury. Pediatric nephrology. 2008;23(12):2151–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.O’Neal JB, Shaw AD, Billings FTt. Acute kidney injury following cardiac surgery: current understanding and future directions. Crit Care. 2016;20(1):187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Okusa MD, Jaber BL, Doran P, et al. Physiological biomarkers of acute kidney injury: a conceptual approach to improving outcomes. Contrib Nephrol. 2013;182:65–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Prowle JR, Bellomo R. Sepsis-associated acute kidney injury: macrohemodynamic and microhemodynamic alterations in the renal circulation. Seminars in nephrology. 2015;35(1):64–74. [DOI] [PubMed] [Google Scholar]
- 186.Zuk A, Bonventre JV. Acute Kidney Injury. Annual review of medicine. 2016;67:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Landaw SA. Factors that accelerate or retard red blood cell senescence. Blood cells. 1988;14(1):47–67. [PubMed] [Google Scholar]
- 188.Kay MM. Localization of senescent cell antigen on band 3. Proc Natl Acad Sci U S A. 1984;81(18):5753–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kay MM, Bosman GJ, Johnson GJ, Beth AH. Band-3 polymers and aggregates, and hemoglobin precipitates in red cell aging. Blood cells. 1988;14(1):275–295. [PubMed] [Google Scholar]
- 190.Low PS, Waugh SM, Zinke K, Drenckhahn D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science. 1985;227(4686):531–533. [DOI] [PubMed] [Google Scholar]
- 191.Lutz HU, Bussolino F, Flepp R, et al. Naturally occurring anti-band-3 antibodies and complement together mediate phagocytosis of oxidatively stressed human erythrocytes. Proc Natl Acad Sci U S A. 1987;84(21):7368–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Boas FE, Forman L, Beutler E. Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc Natl Acad Sci U S A. 1998;95(6):3077–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bratosin D, Mazurier J, Tissier JP, et al. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie. 1998;80(2):173–195. [DOI] [PubMed] [Google Scholar]
- 194.Kiefer CR, Snyder LM. Oxidation and erythrocyte senescence. Curr Opin Hematol. 2000;7(2):113–116. [DOI] [PubMed] [Google Scholar]
- 195.Kuypers FA, de Jong K. The role of phosphatidylserine in recognition and removal of erythrocytes. Cellular and molecular biology. 2004;50(2):147–158. [PubMed] [Google Scholar]
- 196.Lutz HU. Naturally occurring anti-band 3 antibodies in clearance of senescent and oxidatively stressed human red blood cells. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie. 2012;39(5):321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lang F, Abed M, Lang E, Foller M. Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal. 2014;21(1):138–153. [DOI] [PubMed] [Google Scholar]
- 198.Lang KS, Lang PA, Bauer C, et al. Mechanisms of suicidal erythrocyte death. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2005;15(5):195–202. [DOI] [PubMed] [Google Scholar]
- 199.Aarts PA, van den Broek SA, Prins GW, Kuiken GD, Sixma JJ, Heethaar RM. Blood platelets are concentrated near the wall and red blood cells, in the center in flowing blood. Arteriosclerosis. 1988;8(6):819–824. [DOI] [PubMed] [Google Scholar]
- 200.Hellem AJ, Borchgrevink CF, Ames SB. The role of red cells in haemostasis: the relation between haematocrit, bleeding time and platelet adhesiveness. Br J Haematol. 1961;7:42–50. [DOI] [PubMed] [Google Scholar]
- 201.Andrews DA, Low PS. Role of red blood cells in thrombosis. Curr Opin Hematol. 1999;6(2):76–82. [DOI] [PubMed] [Google Scholar]
- 202.Turitto VT, Weiss HJ. Red blood cells: their dual role in thrombus formation. Science. 1980;207(4430):541–543. [DOI] [PubMed] [Google Scholar]
- 203.Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood. 2012;120(18):3837–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Du VX, Huskens D, Maas C, Al Dieri R, de Groot PG, de Laat B. New insights into the role of erythrocytes in thrombus formation. Seminars in thrombosis and hemostasis. 2014;40(1):72–80. [DOI] [PubMed] [Google Scholar]
- 205.van Gelder JM, Nair CH, Dhall DP. Erythrocyte aggregation and erythrocyte deformability modify the permeability of erythrocyte enriched fibrin network. Thrombosis research. 1996;82(1):33–42. [DOI] [PubMed] [Google Scholar]
- 206.Gersh KC, Nagaswami C, Weisel JW. Fibrin network structure and clot mechanical properties are altered by incorporation of erythrocytes. Thromb Haemost. 2009;102(6):1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339(9):584–590. [DOI] [PubMed] [Google Scholar]
- 208.Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001;345(17):1230–1236. [DOI] [PubMed] [Google Scholar]
- 209.Hart RG, Kanter MC. Hematologic disorders and ischemic stroke. A selective review. Stroke. 1990;21(8):1111–1121. [DOI] [PubMed] [Google Scholar]
- 210.Cirillo M, Laurenzi M, Trevisan M, Stamler J. Hematocrit, blood pressure, and hypertension. The Gubbio Population Study. Hypertension. 1992;20(3):319–326. [DOI] [PubMed] [Google Scholar]
- 211.Stephansson O, Dickman PW, Johansson A, Cnattingius S. Maternal hemoglobin concentration during pregnancy and risk of stillbirth. JAMA. 2000;284:2611–2617. [DOI] [PubMed] [Google Scholar]
- 212.Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion Requirements in Critical Care Investigators, Canadian Critical Care Trials Group. N Engl J Med. 1999;340(6):409–417. [DOI] [PubMed] [Google Scholar]
- 213.Vincent JL, Baron JF, Reinhart K, et al. Anemia and blood transfusion in critically ill patients. JAMA. 2002;288(12):1499–1507. [DOI] [PubMed] [Google Scholar]
- 214.Crawford JH, Chacko BK, Kevil CG, Patel RP. The Red Blood Cell and Vascular Function in Health and Disease. Antioxidants & Redox Signaling. 2004;6(6):992–999. [DOI] [PubMed] [Google Scholar]
- 215.Rao SV, Jollis JG, Harrington RA, et al. Relationship of Blood Transfusion and Clinical Outcomes in Patients With Acute Coronary Syndromes. JAMA. 2004;292(13):1555–1562. [DOI] [PubMed] [Google Scholar]
- 216.Qing DY, Conegliano D, Shashaty MG, et al. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am J Respir Crit Care Med. 2014;190(11):1243–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Mohanty JG, Nagababu E, Rifkind JM. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Frontiers in physiology. 2014;5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kiefmann R, Rifkind JM, Nagababu E, Bhattacharya J. Red blood cells induce hypoxic lung inflammation. Blood. 2008;111(10):5205–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Purtle SW, Moromizato T, McKane CK, Gibbons FK, Christopher KB. The association of red cell distribution width at hospital discharge and out-of-hospital mortality following critical illness*. Crit Care Med. 2014;42(4):918–929. [DOI] [PubMed] [Google Scholar]
- 220.Bazick HS, Chang D, Mahadevappa K, Gibbons FK, Christopher KB. Red cell distribution width and all-cause mortality in critically ill patients. Crit Care Med. 2011;39(8):1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Reggiori G, Occhipinti G, De Gasperi A, Vincent JL, Piagnerelli M. Early alterations of red blood cell rheology in critically ill patients. Crit Care Med. 2009;37(12):3041–3046. [DOI] [PubMed] [Google Scholar]
- 222.McMahon TJ, Ahearn GS, Moya MP, et al. A nitric oxide processing defect of red blood cells created by hypoxia: Deficiency of S-nitrosohemoglobin in pulmonary hypertension. Proceedings of the National Academy of Sciences. 2005;102(41):14801–14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Milsom AB, Jones CJ, Goodfellow J, Frenneaux MP, Peters JR, James PE. Abnormal metabolic fate of nitric oxide in Type I diabetes mellitus. Diabetologia. 2002;45(11):1515–1522. [DOI] [PubMed] [Google Scholar]
- 224.Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proceedings of the National Academy of Sciences. 2005;102(7):2531–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sonveaux P, Kaz AM, Snyder SA, et al. Oxygen Regulation of Tumor Perfusion by S-Nitrosohemoglobin Reveals a Pressor Activity of Nitric Oxide. Circulation Research. 2005;96(10):1119–1126. [DOI] [PubMed] [Google Scholar]
- 226.Reynolds JD, Ahearn GS, Angelo M, Zhang J, Cobb F, Stamler JS. S-nitrosohemoglobin deficiency: A mechanism for loss of physiological activity in banked blood. Proceedings of the National Academy of Sciences. 2007;104(43):17058–17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Bennett-Guerrero E, Veldman TH, Doctor A, et al. Evolution of adverse changes in stored RBCs. Proceedings of the National Academy of Sciences. 2007;104(43):17063–17068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.James PE, Tufnell-Barret T, Milsom AB, Frenneaux MP, Lang D. Red Blood Cell-Mediated Hypoxic Vasodilatation: A Balanced Physiological Viewpoint. Circulation Research. 2004;95(2):e8–e9. [PubMed] [Google Scholar]
- 229.Hammerman SI, Klings ES, Hendra KP, et al. Endothelial cell nitric oxide production in acute chest syndrome. AJP - Heart and Circulatory Physiology. 1999;277(4):H1579–H1592. [DOI] [PubMed] [Google Scholar]
- 230.Balagopalakrishna C, Manoharan PT, Abugo OO, Rifkind JM. Production of superoxide from hemoglobin-bound oxygen under hypoxic conditions. Biochemistry. 1996;35(20):6393–6398. [DOI] [PubMed] [Google Scholar]
- 231.Misra HP, Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. J Biol Chem. 1972;247(21):6960–6962. [PubMed] [Google Scholar]
- 232.Cimen MYB. Free radical metabolism in human erythrocytes. Clinica Chimica Acta. 2008;390(1–2):1–11. [DOI] [PubMed] [Google Scholar]