

Abstract
Background
Systemic lupus erythematosus (SLE) is an autoimmune inflammatory rheumatic disease. SLE susceptibility is affected by multiple genetic elements, environmental factors, and their interactions. We aimed in this study to statistically and functionally characterize a gene-gene interaction (epistasis) recently documented to affect SLE risk.
Methods
Two single-nucleotide polymorphisms, rs2230926 in TNFAIP3 (A20) gene and rs131654 in UBE2L3 (UBCH7) gene, were genotyped in all 3525 Korean participants, and their SLE risk association and epistasis were statistically analyzed by calculating odds ratio (OR), 95% confidence interval (CI), and P values in genotype comparisons between 1318 SLE patients and 2207 healthy controls. Furthermore, their effects on gene functions were assessed by comparatively examining separate and combined effects of TNFAIP3 and UBE2L3 knockdowns on NF-κB transcription factor activity in human cells.
Results
SLE susceptibility is associated with TNFAIP3 rs2230926 (OR = 1.9, 95% CI 1.6–2.4, P = 8.6 × 10−11) and UBE2L3 rs131654 (OR = 1.2, 95% CI 1.1–1.4, P = 1.1 × 10−4) in a Korean population of this study. Their risk-associated alleles synergistically elevate SLE susceptibility in both multivariate logistic regression analysis (ORinteraction = 1.6, P = 0.0028) and genotype-stratified analysis (ORinteraction = 2.4), confirming the synergistic TNFAIP3-UBE2L3 interaction in SLE risk. Additionally, the SLE-susceptible alleles confer decreased TNFAIP3 expression (P = 1.1 × 10−6, n = 610) and increased UBE2L3 expression (P = 9.5 × 10−11, n = 475), respectively, in B cell analysis of the International HapMap Project individuals with adjustment for ethnicity. Furthermore, when compared with TNFAIP3 non-knockdown and UBE2L3 knockdown in human HeLa cells, TNFAIP3 knockdown and UBE2L3 non-knockdown synergistically increase three cytokines, CCL2, CXCL8 (IL8), and IL6, all regulated by NF-κB in the human TNFR signaling pathway.
Conclusions
A synergistic interaction between TNFAIP3 and UBE2L3 genes is observed in SLE risk, as being evident in comparison of genotype distributions between SLE patients and controls. Additionally, the synergistic gene-gene interaction is functionally validated, as TNFAIP3 reduction and UBE2L3 augment exert synergism in activation of NF-κB and subsequent induction of inflammatory cytokines. Accordingly, SLE inflammation and risk could be synergistically alleviated by TNFAIP3 upregulation and UBE2L3 downregulation.
Keywords: Autoimmune disease, Cytokine, Epistasis, Gene-gene interaction, Gene polymorphism, Inflammatory disease, Rheumatic disease, Synergistic interaction, Systemic lupus erythematosus, TNFR pathway
Background
Systemic lupus erythematosus (SLE) [MIM 152700], the most common type of lupus, is an autoimmune inflammatory rheumatic disease. SLE is a complex disease, as its susceptibility is affected by multiple genetic and nongenetic factors. Its genetic heritability is 44–66% [1, 2], but all the SLE-associated genetic elements discovered so far can explain roughly only 24% [3]. The missing genetic heritability can be partly attributed to non-linear effects arising from gene-gene interactions, among others [4].
The genetic interaction where the phenotypic effect of a gene allele depends on the presence of one or more modifier gene alleles is referred to as gene-gene interaction, or epistasis [5]. This phenotypic dependence could result from an underlying functional interaction of the genes. Some epistases have been identified for SLE, but no functional validation has been achieved to explain how the interacting genetic variants can influence SLE development.
Recently, an interaction between two NF-κB modulator genes in SLE risk has been found in Han Chinese [6] and European populations [7]. The interaction was synergistic between TNFAIP3 gene encoding TNF alpha-induced protein 3 (A20) and UBE2L3 gene encoding ubiquitin-conjugating enzyme E2 L3 (UBCH7). However, the examined single-nucleotide polymorphisms (SNPs) were different in the two case-control studies, reporting very different P values for the SLE risk interaction, P = 3 × 10−14 versus 0.04 [6, 7]. In this study, we reexamined their gene-gene interaction in Korean SLE patients and controls, and explored their functional consequences in human cells.
SNPs of the two genes are associated with multiple autoimmune diseases including SLE [8], and their proteins are involved in diverse regulations of NF-κB transcription factor activity [9–17]. TNFAIP3 plays a potent suppressor of NF-κB pathways for immune homeostasis using its diverse ubiquitin-related activities. First, TNFAIP3 is an E3 ubiquitin ligase that catalytically modifies substrate proteins with a Lys-48-linked ubiquitin chain using a C-terminal domain (K48ub writer) [9, 10]. Second, TNFAIP3 is a deubiquitinating enzyme that catalytically removes a Lys-63-linked ubiquitin chain from modified proteins using the N-terminal ovarian tumor domain (K63ub eraser) [9]. Third, TNFAIP3 is a ubiquitin receptor that non-catalytically binds a Met-1-linked ubiquitin chain of partner proteins using a zinc finger domain (M1ub reader) [11–13].
In the human TNFR signaling pathway (Fig. 1), TNFAIP3 inhibits NF-κB activation in multiple ways. First, TNFAIP3 mediates degradation of RIPK1. As a K48ub writer, TNFAIP3 attaches K48ub to RIPK1 so that RIPK1 is degraded by proteasome [9]. Second, TNFAIP3 inactivates RIPK1, which regulates IκB kinase (IKK) [14]. As a K63ub eraser, TNFAIP3 detaches K63ub from RIPK1 so that RIPK1 cannot interact with other modulators [9]. Third, TNFAIP3 inhibits IKK activation. As an M1ub reader, TNFAIP3 binds M1ub of NEMO (IKKγ), which forms IKK complex with IKKα and IKKβ, and inhibits the phosphorylation of IKKβ by TAK1 [11–13]. All these activities of TNFAIP3 disable IKK activation in the cytoplasm to reduce NF-κB activity in the nucleus.
Fig. 1.
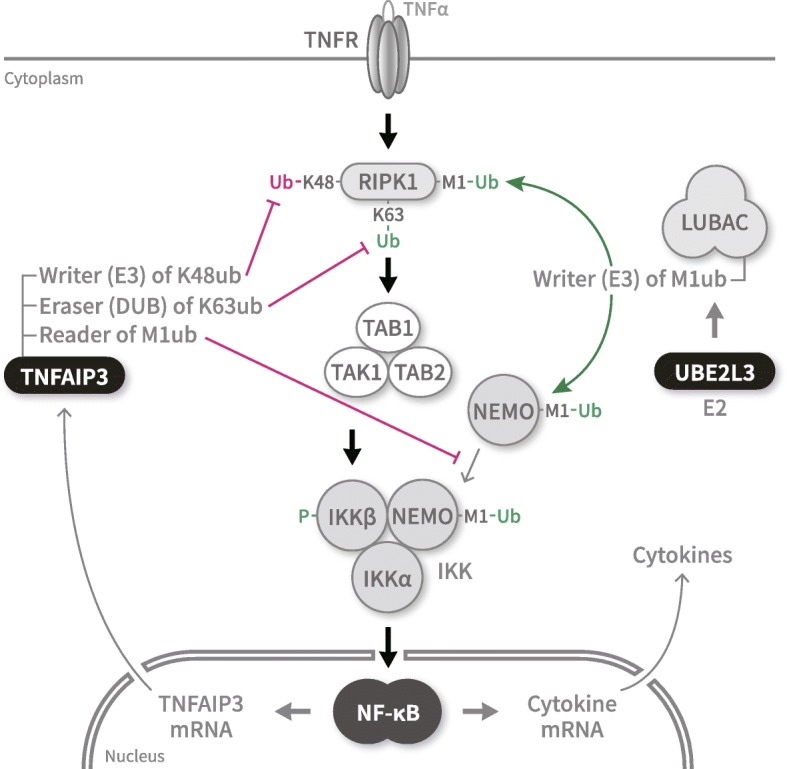
Participation of TNFAIP3 and UBE2L3 in regulation of NF-κB activity under TNFα signaling. The human TNFR pathway transduces a signal of extracellular TNFα through cytoplasmic IKK to latent transcription factor NF-κB for cytokine induction in the nucleus, among others. TNFAIP3 negatively regulates NF-κB (marked by red arrows) (1) by mediating RIPK1 degradation via K48 ubiquitylation (E3), (2) by inactivating RIPK1 via K63 deubiquitylation (DUB), and (3) by inhibiting IKK via binding ubiquitylated M1 of NEMO. In contrast, UBE2L3 (E2) positively regulates NF-κB (marked by green arrows) by helping LUBAC (E3) to activate RIPK1 and MEMO via M1 ubiquitylation
On the other hand, UBE2L3 is an E2 ubiquitin-conjugating enzyme that pairs up with an E3 ubiquitin ligase of HECT (homologous to the E6AP carboxyl terminus) type or RBR (ring between ring) family, but not of ring type, for catalytic ubiquitylation [15]. In the human TNFR pathway (Fig. 1), UBE2L3 E2 enzyme provides activated ubiquitin to LUBAC E3 enzyme (RBR family) for M1 ubiquitylation of RIPK1 and NEMO [16, 17]. All these activities of UBE2L3 enable the cytoplasmic IKK activation to enhance the nuclear NF-κB activity.
Thus, TNFAIP3 and UBE2L3 participate in the same pathway but exert opposite effects on sequential activation of IKK and NF-κB and subsequent induction of inflammatory cytokines (Fig. 1). In principle, two genes functionally participating in one pathway can interact with each other in affecting disease susceptibility even without their direct protein-protein interaction [18]. Accordingly, we aimed in this study to validate whether TNFAIP3 reduction and UBE2L3 augment influence each other’s effect on NF-κB transcription factor activation for cytokine induction.
Materials and methods
Human subjects and SNP genotyping
Korean participants were recruited at six university-affiliated hospitals in Seoul, Daejeon, and Daegu, Republic of Korea, in a study approved by the Institutional Review Board of Hanyang University Medical Center, and all provided written informed consent [19, 20]. Upon approval from the Institutional Review Board of Korea Advanced Institute of Science and Technology, peripheral blood samples from all subjects were genotyped for SNPs using the MassARRAY® system from Sequenom [20, 21].
Statistical analyses
Logistic regression analysis of the PLINK v1.07 program [22] was performed to calculate odds ratio (OR), 95% confidence interval (CI), and P values for SLE risk association and epistasis with adjustment for age and gender of participants by using the -logistic and -covar options [20, 23–25]. SNP association was corrected for multiple testing.
For testing multiplicativity in gene-gene interaction, we added an interaction term in logistic regression: logit{P(disease = 1)|SNP1 = A, SNP2 = B, interaction SNP1 × SNP2 = AB, age = C, and gender = D} = β0 + β1A + β2B + β3AB + β4C + β5D. When OR of the risk-associated genotype in one SNP is OR1 = eβ1, that in the other SNP is OR2 = eβ2, and that in both SNPs is OR1&2 = eβ3, a multiplicative interaction between the two genes is represented by ORint = OR1&2/(OR1 × OR2) [18, 26].
We used CRAN R package “epiR” to calculate an attributable proportion (AP) due to interaction, 95% confidence interval (CI), and P value in Rothman’s additive model [27]. The combined relative risk of the two SNPs (RR11) was estimated from their individual relative risks (RR10 and RR01) by RR11 = RR10 + RR01 – 1, as the presence of risk allele is denoted by subscript 1 and its absence by subscript 0. AP is a derivative measure of the relative excess risk caused by additive interaction: AP = (RR11 – RR10 + 1)/RR11 [26, 28].
Gene knockdowns
Human HeLa cells purchased from the American Type Culture Collection were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum by incubation at 37 °C in 5% CO2 atmosphere. The cells were seeded in 6-well plates at 1 × 105 cells/well, and 24 h later transfected with one or two small interfering RNAs (siRNAs) or a control of 15 nM each. At 72 h after transfection, the cells were treated with 10 ng/ml of human recombinant TNFα, 210-TA from R&D System, for 0, 2, 4, 8, and 12 h.
Three siRNAs, HS S110861 and HS S110862 from Thermo Fisher Scientific and SI00086989 from Qiagen, were individually used for TNFAIP3 knockdown and are denoted here by siA20-1, siA20-2, and siA20-3, respectively. An anti-UBE2L3 siRNA SI05191242 from Qiagen, denoted here by siUBE, was used to knockdown UBE2L3 [17]. AccuTarget Control siRNA SN-1002 from Bioneer, denoted here by siCon, served as a negative control.
Protein and RNA quantifications
The whole-cell lysates were obtained using the passive lysis buffer from Promega. The protein extracts (50 μg) were subjected to western blotting using anti-TNFAIP3 antibody 59A426 from Merck Millipore, anti-UBE2L3 antibody 3848S from Cell Signaling Technology, and anti-β-actin antibody SC-1616-R from Santa Cruz Biotechnology.
Total RNA was isolated from cells using the RNAspin kit from iNtRON Biotechnology and used for cDNA library construction using oligo-dT primer and ImProm-II reverse transcriptase from Promega. Quantitative polymerase chain reaction (qPCR) was performed with SYBR green fluorescent dye using CFX96™ real-time qPCR detection system from Bio-Rad Lab and gene-specific primers such as GTCTCCTCTGACTTCAACAGCG (forward) and ACCACCCTGTTGCTGTAGCCAA (reverse) for GAPDH [29], GAAAGTCTCTGCCGCCCTT and ATTGATTGCATCTGGCTGAGCG for CCL2 [7], AGCTCTGTCTGGACCCCAAG and GAATTCTCAGCCCTCTTCAAAAAC for CXCL8 [30], and CCCCCAGGAGAAGATTCCAA and GCTGCTTTCACACATGTTACTCTTG for IL6 [31]. Relative mRNA levels were estimated using the comparative Ct method, ΔΔCt method [32].
Results
TNFAIP3-UBE2L3 interaction in SLE risk
In this study, a total of 3525 Korean participants were all genotyped for TNFAIP3 rs22230926 and UBE2L3 rs131654 SNPs, which had shown the lowest P value among the SNPs of each respective gene locus in three previous genome-wide association studies on Asian SLE susceptibility [19, 33, 34]. For example, their P values were 1 × 10−17 and 3 × 10−16, respectively, in a Han Chinese population [33]. The SNP genotype distributions among the control subjects were in Hardy-Weinberg equilibria.
The SNP genotypes were statistically compared between 1318 SLE patients (34.4 ± 12.4 years; 93% female) and 2207 healthy controls (40.7 ± 15.4 years old; 77% female) using logistic regression analysis (Table 1). Parts of these Korean samples previously demonstrated no genetic stratification in principal component analyses [19, 35], but the case and control groups of this study were different in age distribution and gender ratio, so the logistic regression was adjusted for age and gender of the participants. SLE susceptibility is associated with both TNFAIP3 rs2230926 (OR = 1.9, 95% CI 1.6–2.4, P = 8.8 × 10−11) and UBE2L3 rs131654 (OR = 1.2, 95% CI 1.1–1.4, P = 1.1 × 10−4) SNPs, as their P values are much lower than a significance threshold of Bonferroni correction for 2-SNP testing, α = 0.05/2 = 0.025.
Table 1.
Genotype distributions and SLE risk associations of TNFAIP3 and UBE2L3 SNPs
| Controls | Cases | SLE risk associationb | ||
|---|---|---|---|---|
| SNP major>minora | nn/nr/rra | nn/nr/rra | OR (95% CI) | P |
| TNFAIP3 rs2230926 T>G | 1989/213/5 | 1069/240/9 | 1.9 (1.6–2.4) | 8.8 × 10−11 |
| UBE2L3 rs131654 T>G | 572/1116/519 | 274/669/375 | 1.2 (1.1–1.4) | 1.1 × 10−4 |
SLE systemic lupus erythematosus, SNP single-nucleotide polymorphism, OR odds ratio, CI confidence interval
aMajor alleles are more frequent than minor alleles in controls. Shown in bold are the risk-associated alleles (r), which are more frequent in SLE cases than healthy controls. The nonrisk-associated alleles (n) are more frequent in controls than cases
bUsing logistic regression with adjustment for age and gender of the participants
Additionally, the gene-gene interaction was examined using this SNP pair. A multivariate logistic regression analysis with their interaction term according to a multiplicative model [26] showed a synergistic interaction with the two SNPs (ORint = 1.6, P = 0.0028), indicating a synergistic interaction between TNFAIP3 and UBE2L3 genes in conferring SLE risk. Furthermore, this SNP pair revealed a similar interaction (AP of interaction = 0.42; 95% CI 0.18–0.66, P = 0.049) additionally in Rothman’s additive model based on additivity of the individual relative risks [27].
This TNFAIP3-UBE2L3 interaction was further tested by using genotype-stratified analysis. According to the genotypes of the two SNPs, all study subjects are stratified into six rather than nine subgroups in Table 2, as heterozygotes in the TNFAIP3 SNP are grouped together with homozygotes of the risk-associated minor allele G, which are too few to form a subgroup alone. Thus, the TNFAIP3 SNP genotypes are classified into risk-associated allele carriage (R) and non-carriage (N), while the UBE2L3 SNP genotypes are denoted by nn, nr, and rr, comprising of the risk-associated major allele T (r) and the nonrisk-associated minor allele G (n).
Table 2.
Genotype-stratified analysis of the TNFAIP3-UBE2L3 interaction in SLE risk
| Subgroup | N/nn | N/nr | N/rr | R/nn | R/nr | R/rr |
|---|---|---|---|---|---|---|
| Genotypea | TT/GG | TT/TG | TT/TT | (TG+GG)/GG | (TG+GG)/TG | (TG+GG)/TT |
| Controls | 509 | 999 | 481 | 63 | 117 | 38 |
| Cases | 232 | 540 | 297 | 42 | 129 | 78 |
| Pb | – | 0.13 | 0.0090 | 0.21 | 1.6 × 10−7 | 8.4 × 10−11 |
| ORb | 1.0 | 1.2 | 1.3 | 1.3 | 2.3 | 4.1 |
| ORint | – | – | – | – | 1.5 | 2.4 |
SLE systemic lupus erythematosus, OR odds ratio, CI confidence interval
aIn the genotypes of TNFAIP3 rs2230926/UBE2L3 rs131654, shown in bold are their risk-associated alleles, which are more frequent in patient cases than healthy controls
bUsing logistic regression with adjustment for age and gender of the participants
ORs of the R/rr, R/nn, and N/rr subgroups are 4.1-, 1.3-, and 1.3-fold greater than the N/nn subgroup, yielding ORint = 4.1/(1.3 × 1.3) = 2.4, a synergistic interaction. That is, the combined effect of R and rr is 2.4-fold greater than the sum of their separate effects versus N and nn. Additionally, a comparison among the ORs for R/nr, R/nn, and N/nr subgroups versus N/nn subgroup yields a 1.5-fold increased combined effect, ORint = 2.3/(1.3 × 1.2) = 1.5.
A similar gene-gene interaction was recently found in Han Chinese susceptibility to SLE with TNFAIP3 rs2230926 and UBE2L3 rs463426 SNPs (P = 3.1 × 10−14) in generalized multifactor dimensionality reduction analysis [6]. Another similar interaction was found in European susceptibility to SLE with TNFAIP3 rs80126770 and UBE2L3 rs140490 SNPs (ORint = 1.3), but the P value was not very low (P = 0.039) [7]. Although the documented SNP pairs were all different, the three independent epistasis studies appear to support for the TNFAIP3-UBE2L3 interaction in multiple ethnic populations.
Expression quantitative trait loci (eQTL) of TNFAIP3 and UBE2L3
It was then examined whether the SLE risk-associated alleles confer increase or decrease in their respective gene expression. First, we used gene expression and genotype data of Epstein-Barr virus (EBV)-transformed B cell lines derived from the International HapMap Project participants [36] to calculate eQTL P values with an additive linear function with adjustment for ethnic background using R MatrixEQTL package [37, 38]. The SLE risk-associated allele G of rs2230926 is associated with decreased TNFAIP3 mRNA (P = 1.1 × 10−6) in 610 individuals (Fig. 2, left). In contrast, increased UBE2L3 mRNA is associated with the risk-associated allele T of rs131654 (P = 9.5 × 10−11) in 475 individuals (Fig. 2, right). Thus, the SLE risk-associated variants are associated with decreased TNFAIP3 and increased UBE2L3, both elevating NF-κB transcription factor activity.
Fig. 2.
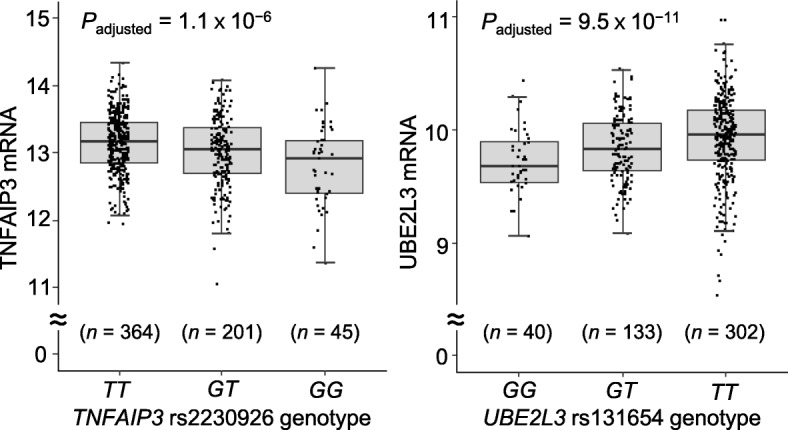
Association of TNFAIP3 and UBE2L3 gene expressions with two SNPs. The genotype and mRNA data of 726 EBV-transformed B cell samples of the HapMap Project were obtained from the Array Express Database (series accession number E-MTAB-264). TNFAIP3 rs2230926 genotypes of 610 individuals (left graph) and UBE2L3 rs131654 genotypes of 475 individuals (right graph) were obtained from the Ensemble genome browser. Correlations between genotypes and gene expressions were analyzed using linear regression with adjustment for ethnic backgrounds of the samples
Second, we analyzed the Genotype-Tissue Expression Project data (V8 release) [39]. TNFAIP3 mRNA is not associated with rs2230926 in any of the 47 analyzed tissues, although it is associated with some other SNPs in subcutaneous adipose and brain hippocampus tissues. UBE2L3 mRNA is marginally associated with rs131654 in skeletal muscle (P = 0.0008), whole blood (P = 0.0023), subcutaneous adipose (P = 0.012), tibial artery (P = 0.019), and esophagus muscularis (P = 0.045) tissues, as their P values are less than 0.05 but higher than a gene-wise significance threshold for each tissue.
Functional assay of TNFAIP3-UBE2L3 interaction
TNFAIP3 and UBE2L3 enzymes participate together in the human TNFR [9–13, 40, 41], IL1R [41, 42], NOD2 [43, 44], RIG1 [45, 46], and TLR4 [42, 43] immune pathways, among others. Their cellular and molecular functions have been well characterized in the TNFR pathway, which was chosen in this study for examining their concurrent effects on NF-κB activity for cytokine induction. Transcript mRNAs produced from three NF-κB-dependent cytokine genes, CCL2, CXCL8 (IL8), and IL6, were quantified in TNFα-treated HeLa cells.
In order to compare the effects of relatively higher and lower levels of TNFAIP3 and UBE2L3 on NF-κB activity, four different HeLa cell derivatives were constructed using combinations of TNFAIP3 and UBE2L3 knockdowns. The first cell line without TNFAIP3 knockdown but with UBE2L3 knockdown has relatively high TNFAIP3 and low UBE2L3, and serves as a reference. The second cell line with both knockdowns has low TNFAIP3 and UBE2L3, and provides the TNFAIP3 reduction effect compared with the reference. The third without any knockdown has high TNFAIP3 and UBE2L3, and shows the UBE2L3 augment effect. The fourth with TNFAIP3 knockdown alone has low TNFAIP3 and high UBE2L3, and reveals the combined effect of TNFAIP3 reduction and UBE2L3 augment.
TNFAIP3 is effectively knocked down by siA20-1, siA20-2, or siA20-3 separately, although they target different sequences in TNFAIP3 mRNA (Fig. 3). The UBE2L3 knockdown is effective with siUBE, as its efficiency and specificity have been demonstrated under the same conditions as this study, i.e., in HeLa cells under TNFR activation [17]. Furthermore, siCon is an adequate negative control as it does not affect CCL2, CXCL8, or IL6 expression.
Fig. 3.

Knockdowns of TNFAIP3 and UBE2L3 for functional assays. After HeLa cells were transfected with various combinations of siRNAs (siCon, siUBE, and siA20-1 or siA20-2), protein levels of TNFAIP3 (A20), UBE2L3, and β-actin were measured using western blotting at the indicated time points. NS means nonspecific band
Synergistic activation of NF-κB
From the four cell lines constructed using different combinations of siA20-1, siUBE, and siCon, the three cytokine mRNAs were quantified in a time-course manner at 0, 4, 8, and 12 h after TNFα treatment (Fig. 4). Either TNFAIP3 reduction or UBE2L3 augment alone increases all three cytokine mRNAs. The combined effect of TNFAIP3 reduction and UBE2L3 augment on each cytokine (fold change of mRNA) is greater than the sum of their individual effects at any of the three time points, indicating their consistent synergistic effects.
Fig. 4.

Functional assays for synergistic induction of NF-κB-mediated cytokine expressions. Four HeLa cell-line derivatives with relatively high or low TNFAIP3 and high or low UBE2L3 levels were treated with TNFα for varying periods. Cellular levels of CCL2, CXCL8 (IL8), and IL6 mRNAs were separately measured using qPCR as normalized against GAPDH mRNA. The mRNA levels were measured in triplicate samples and each sample by triplicate qPCR experiments. The nine measurements were averaged per cytokine at each time point. P values for comparisons of cytokine expressions between the reference cell line, transfected with siUBE and siCon, and each of the other three derivatives were calculated in unpaired t tests, * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001. The synergy (ŝ) is calculated as ŝ = combined effect ÷ (TNFAIP3 reduction effect × UBE2L3 augment effect)
When the synergy (ŝ) is calculated as ŝ = combined effect ÷ (TNFAIP3 reduction effect × UBE2L3 augment effect), the synergy on CCL2 or CXCL8 expression is higher at 12 h (1.3- or 1.7-fold, respectively) than at 4 or 8 h after TNFα treatment. By contrast, the synergy on IL6 expression is maximal at the earliest time point, 4 h (1.4-fold). Thus, the synergy peaks with IL6 expression sooner than CCL2 or CXCL8 expression.
This set of experiments was repeated using two other anti-TNFAIP3 siRNAs, siA20-2 and siA20-3, separately in place of siA20-1. Although the interaction extents vary with the siRNAs, their effects on CCL2, CXCL8, and IL6 expressions are reproduced (Fig. 5), excluding possible off-target effects of anti-TNFAIP3 siRNAs, and confirming the synergistic effects of TNFAIP3 reduction and UBE2L3 augment on their activation of NF-κB transcription factor and subsequent elevation of inflammatory cytokines.
Fig. 5.
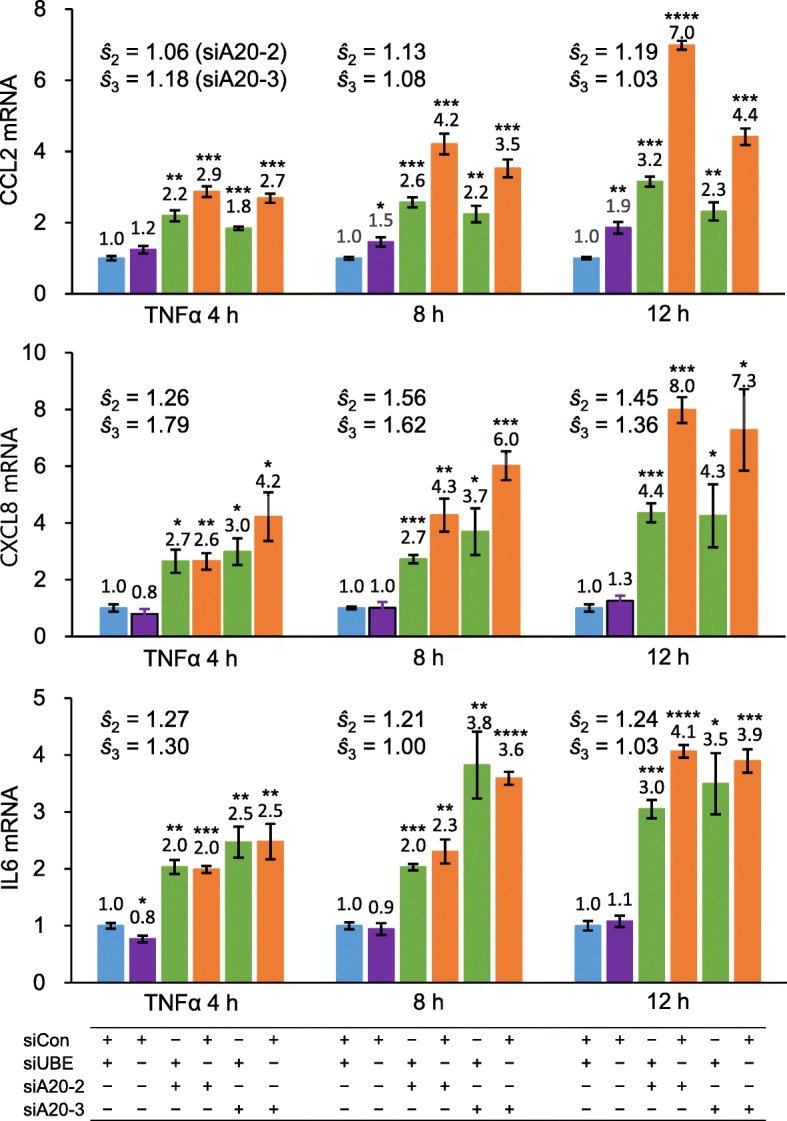
Reproduced functional assays for synergistic cytokine induction. In place of siA20-1, two other anti-TNFAIP3 siRNAs with different target sequences, siA20-2 and siA20-3, were used in HeLa cells to demonstrate the absence of off-target effects on synergistic induction of NF-κB-mediated cytokine expressions. After treatment with TNFα for varying periods, CCL2, CXCL8, and IL6 mRNAs were measured against GAPDH mRNA using qPCR. See the legend of Fig. 4
Discussion
Here, we report the first functional validation of gene-gene interaction in conferring susceptibility to SLE, an autoimmune inflammatory rheumatic disease. First, a synergistic interaction between TNFAIP3 and UBE2L3 in SLE risk is evident with their respective SNPs, rs2230926 and rs131654 (Table 2). Second, their risk-associated alleles are associated with decreased TNFAIP3 mRNA and increased UBE2L3 mRNA, respectively (Fig. 2). Third, while TNFAIP3 reduction and UBE2L3 augment separately elevate NF-κB transcription factor activity, they together additionally show a synergism in NF-κB activation for cytokine induction (Figs. 4 and 5).
The synergistic activation of NF-κB in the nucleus can be ascribed to a mechanistic connection between TNFAIP3 and UBE2L3 in the cytoplasm. In the human TNFR pathway (Fig. 1), an extracellular activation signal is transduced to latent transcription factor NF-κB through a series of sequential events in the cytoplasm: activation of IKK, inactivation of IκB, and activation and nuclear import of NF-κB [11–13]. There is another positive route for NF-κB activation: UBE2L3 E2 enzyme supplies activated ubiquitin to LUBAC E3 enzyme for linear ubiquitylation of NEMO and subsequent activation of IKK [17, 47]. Then, activated NF-κB induces TNFAIP3 expression to exert a negative feedback on NF-κB activation, among others. Accordingly, with higher UBE2L3, NF-κB could be less inhibited by TNFAIP3.
The synergistic effect on NF-κB is evident when UBE2L3 stimulation overrides TNFAIP3 inhibition (Figs. 4 and 5). UBE2L3 augment increases the three cytokines 1.1–2.7-fold at 4, 8, and 12 h time points in comparisons of the two cell lines without TNFAIP3 knockdown. The same cytokines increase 1.3–3.7-fold in comparisons of the two cell lines with TNFAIP3 knockdown. Thus, the UBE2L3 augment effect tends to be greater with low TNFAIP3 than with high TNFAIP3, although the ranges overlap.
The functional consequences of this gene-gene interaction in NF-κB signaling need to be replicated in human immune cells instead of cervical HeLa cells used in this study. As a key modulator of diverse immune and inflammatory responses [48], NF-κB induces inflammatory cytokines and chemokines in innate immune cells, modulates inflammatory T cell functions [49, 50], and regulates inflammasome activation [51], among others. Thus, deregulated NF-κB activation is associated with multiple chronic inflammatory diseases including SLE.
For example, NF-κB promotes proliferation and survival of B cells [52], and is constitutively activated in peripheral B cells from SLE patients [53]. Elevation of GLK expression in SLE patients activates NF-κB signaling under T cell receptor, and is correlated with disease severity [54]. FcγRIIB-deficient mice with SLE-like symptoms have elevated NF-κB function and immunogenicity in their splenic dendritic cells [55].
While NF-κB is ubiquitously involved in diverse immune cell types, both TNFAIP3 and UBE2L3 participate in B cell differentiation, and SLE risk-associated alleles of their genes affect NF-κB activation in B cells. B cell-specific ablation of Tnfaip3 in mice exaggerates NF-κB response to CD40-induced signals, leading to SLE-like features, including elevation of germinal center B cells and plasma cells, production of autoreactive immunoglobulins, and immune complex deposits [56–58]. UBE2L3 augment increases basal and CD40-stimulated NF-κB activation in CD19+ B cells. An SLE risk-associated allele of UBE2L3 is associated with increased plasmablasts and plasma cells in SLE patients [7]. Accordingly, B cells could be one of the immune cells where the said synergism is connected with SLE phenotypes.
TNFAIP3 rs2230926 SNP affects TNFAIP3 gene expression (Fig. 2, left). This SNP is perfectly correlated (r2 = 1.0) with another SLE risk-associated dinucleotide polymorphism, rs148314165 and rs200820567 (TT>A) located in a TNFAIP3-downstream enhancer, according to the Asian population data of the 1000 Genomes Project. The risk-associated minor allele A renders the enhancer to lack NF-κB binding and reduce TNFAIP3 expression in EBV-transformed B cells and HEK293T cells [59–61].
UBE2L3 rs131654 SNP also affects UBE2L3 gene expression (Fig. 2, right). This SNP is highly correlated with two other SLE risk-associated SNPs, rs140490 in the promoter (r2 = 0.76) and rs7444 in the 3′ untranslated region (r2 = 0.74) according to the Asian data of the 1000 Genomes Project. Their risk-associated alleles have been associated with increased UBE2L3 mRNA or protein in EBV-transformed B cells, CD19+ B cells, CD14+ monocytes [7, 62].
Conclusions
TNFAIP3 reduction and UBE2L3 augment together confer synergistic activation of NF-κB transcription factor and subsequent synergistic induction of inflammatory cytokines. This functional demonstration supports for the TNFAIP3-UBE2L3 gene-gene interaction that is statistically observed in synergistic elevation of SLE risk. Accordingly, inflammation in SLE could be synergistically reduced by concurrent upregulation of TNFAIP3 and downregulation of UBE2L3, and an inclusion of this synergistic genetic effect in weighted genetic risk score would improve SLE risk prediction.
Acknowledgements
We thank Prof. Kwangwoo Kim at Kyung Hee University, Prof. Suk-Jo Kang at Korea Advanced Institute of Science and Technology, and Prof. Gang Min Hur at Chungnam National University for their discussions and advices.
Authors’ contributions
C.K. and T.K. conceived and designed the study. T.K. and S.C.B. acquire the data. C.K. supervised the biology part, and S.C.B. supervised the clinical part. All analyzed and interpreted the data, were involved in drafting the article, and approved the final version to be published.
Funding
This work was supported by the National Research Foundation of Korea (2015052849 to CK and 2017M3A9B4050335 to SCB).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the Institutional Review Boards of Hanyang University Medical Center and Korea Advanced Institute of Science and Technology. Every participant provided written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sang-Cheol Bae, Email: scbae@hanyang.ac.kr.
Changwon Kang, Email: ckang@kaist.ac.kr.
References
- 1.Lawrence JS, Martins CL, Drake GL. A family survey of lupus erythematosus. 1. Heritability. J Rheumatol. 1987;14:913–921. [PubMed] [Google Scholar]
- 2.Wang J, Yang S, Chen JJ, Zhou SM, He SM, Liang YH, et al. Systemic lupus erythematosus: a genetic epidemiology study of 695 patients from China. Arch Dermatol Res. 2007;298:485–491. doi: 10.1007/s00403-006-0719-4. [DOI] [PubMed] [Google Scholar]
- 3.Sun C, Molineros JE, Looger LL, Zhou XJ, Kim K, Okada Y, et al. High-density genotyping of immune-related loci identifies new SLE risk variants in individuals with Asian ancestry. Nat Genet. 2016;48:323–330. doi: 10.1038/ng.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cordell HJ. Detecting gene-gene interactions that underlie human diseases. Nat Rev Genet. 2009;10:392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snitkin ES, Segre D. Epistatic interaction maps relative to multiple metabolic phenotypes. PLoS Genet. 2011;7:e1001294. doi: 10.1371/journal.pgen.1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zuo XB, Sheng YJ, Hu SJ, Gao JP, Li Y, Tang HY, et al. Variants in TNFSF4, TNFAIP3, TNIP1, BLK, SLC15A4 and UBE2L3 interact to confer risk of systemic lupus erythematosus in Chinese population. Rheumatol Int. 2014;34:459–464. doi: 10.1007/s00296-013-2864-3. [DOI] [PubMed] [Google Scholar]
- 7.Lewis MJ, Vyse S, Shields AM, Boeltz S, Gordon PA, Spector TD, et al. UBE2L3 polymorphism amplifies NF-kappaB activation and promotes plasma cell development, linking linear ubiquitination to multiple autoimmune diseases. Am J Hum Genet. 2015;96:221–234. doi: 10.1016/j.ajhg.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parkes M, Cortes A, van Heel DA, Brown MA. Genetic insights into common pathways and complex relationships among immune-mediated diseases. Nat Rev Genet. 2013;14:661–673. doi: 10.1038/nrg3502. [DOI] [PubMed] [Google Scholar]
- 9.Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430:694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 10.Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, et al. Ubiquitin binding to A20 ZnF4 is required for modulation of NF-kappaB signaling. Mol Cell. 2010;40:548–557. doi: 10.1016/j.molcel.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell. 2011;44:559–571. doi: 10.1016/j.molcel.2011.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tokunaga F, Nishimasu H, Ishitani R, Goto E, Noguchi T, Mio K, et al. Specific recognition of linear polyubiquitin by A20 zinc finger 7 is involved in NF-kappaB regulation. EMBO J. 2012;31:3856–3870. doi: 10.1038/emboj.2012.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verhelst K, Carpentier I, Kreike M, Meloni L, Verstrepen L, Kensche T, et al. A20 inhibits LUBAC-mediated NF-kappaB activation by binding linear polyubiquitin chains via its zinc finger 7. EMBO J. 2012;31:3845–3855. doi: 10.1038/emboj.2012.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Israel A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb Perspect Biol. 2010;2:a000158. doi: 10.1101/cshperspect.a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature. 2011;474:105–108. doi: 10.1038/nature09966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 17.Fu B, Li S, Wang L, Berman MA, Dorf ME. The ubiquitin conjugating enzyme UBE2L3 regulates TNFalpha-induced linear ubiquitination. Cell Res. 2014;24:376–379. doi: 10.1038/cr.2013.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J, Yum S, Kang C, Kang SJ. Gene-gene interactions in gastrointestinal cancer susceptibility. Oncotarget. 2016;7:67612–67625. doi: 10.18632/oncotarget.11701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee HS, Kim T, Bang SY, Na YJ, Kim I, Kim K, et al. Ethnic specificity of lupus-associated loci identified in a genome-wide association study in Korean women. Ann Rheum Dis. 2014;73:1240–1245. doi: 10.1136/annrheumdis-2012-202675. [DOI] [PubMed] [Google Scholar]
- 20.Kim K, Cho SK, Han TU, Kim JH, Kang SJ, Kang C, et al. A redundant epistatic interaction between IRF5 and STAT4 of the type I interferon pathway in susceptibility to lupus and rheumatoid arthritis. Lupus. 2013;22:1336–1340. doi: 10.1177/0961203313504479. [DOI] [PubMed] [Google Scholar]
- 21.Han TU, Lee HS, Kang C, Bae SC. Association of joint erosion with SLC22A4 gene polymorphisms inconsistently associated with rheumatoid arthritis susceptibility. Autoimmunity. 2015;48:313–317. doi: 10.3109/08916934.2015.1016219. [DOI] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim K, Brown EE, Choi CB, Alarcon-Riquelme ME, Biolupus, Kelly JA, et al. Variation in the ICAM1-ICAM4-ICAM5 locus is associated with systemic lupus erythematosus susceptibility in multiple ancestries. Ann Rheum Dis. 2012;71:1809–1814. doi: 10.1136/annrheumdis-2011-201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim K, Bang SY, Joo YB, Kim T, Lee HS, Kang C, et al. Response to intravenous cyclophosphamide treatment for lupus nephritis associated with polymorphisms in the FCGR2B-FCRLA locus. J Rheumatol. 2016;43:1045–1049. doi: 10.3899/jrheum.150665. [DOI] [PubMed] [Google Scholar]
- 25.Han TU, Cho SK, Kim T, Joo YB, Bae SC, Kang C. Association of an activity-enhancing variant of IRAK1 and an MECP2-IRAK1 haplotype with increased susceptibility to rheumatoid arthritis. Arthritis Rheum. 2013;65:590–598. doi: 10.1002/art.37804. [DOI] [PubMed] [Google Scholar]
- 26.VanderWeele TJ, Knol MJ. A tutorial on interaction. Epidemiol Methods. 2014;3:33–72. [Google Scholar]
- 27.Rothman KJ. Epidemiology: an introduction. 2nd ed. New York: Oxford University Press; 2012.
- 28.Kim K. Massive false-positive gene-gene interactions by Rothman’s additive model. Ann Rheum Dis. 2019;78:437–439. doi: 10.1136/annrheumdis-2018-214297. [DOI] [PubMed] [Google Scholar]
- 29.Nabokina SM, Inoue K, Subramanian VS, Valle JE, Yuasa H, Said HM. Molecular identification and functional characterization of the human colonic thiamine pyrophosphate transporter. J Biol Chem. 2014;289:4405–4416. doi: 10.1074/jbc.M113.528257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ea CK, Baltimore D. Regulation of NF-kappaB activity through lysine monomethylation of p65. Proc Natl Acad Sci U S A. 2009;106:18972–18977. doi: 10.1073/pnas.0910439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi KC, Lee YH, Jung MG, Kwon SH, Kim MJ, Jun WJ, et al. Gallic acid suppresses lipopolysaccharide-induced nuclear factor-kappaB signaling by preventing RelA acetylation in A549 lung cancer cells. Mol Cancer Res. 2009;7:2011–2021. doi: 10.1158/1541-7786.MCR-09-0239. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. 2009;41:1234–1237. doi: 10.1038/ng.472. [DOI] [PubMed] [Google Scholar]
- 34.Yang W, Shen N, Ye DQ, Liu Q, Zhang Y, Qian XX, et al. Genome-wide association study in Asian populations identifies variants in ETS1 and WDFY4 associated with systemic lupus erythematosus. PLoS Genet. 2010;6:e1000841. doi: 10.1371/journal.pgen.1000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freudenberg J, Lee HS, Han BG, Shin HD, Kang YM, Sung YK, et al. Genome-wide association study of rheumatoid arthritis in Koreans: population-specific loci as well as overlap with European susceptibility loci. Arthritis Rheum. 2011;63:884–893. doi: 10.1002/art.30235. [DOI] [PubMed] [Google Scholar]
- 36.Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 2012;8:e1002639. doi: 10.1371/journal.pgen.1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–1358. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fagny M, Paulson JN, Kuijjer ML, Sonawane AR, Chen CY, Lopes-Ramos CM, et al. Exploring regulation in tissues with eQTL networks. Proc Natl Acad Sci U S A. 2017;114:E7841–E7E50. doi: 10.1073/pnas.1707375114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Consortium GT. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 41.Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature. 2011;471:633–636. doi: 10.1038/nature09815. [DOI] [PubMed] [Google Scholar]
- 42.Shembade N, Ma A, Harhaj EW. Inhibition of NF-kappaB signaling by A20 through disruption of ubiquitin enzyme complexes. Science. 2010;327:1135–1139. doi: 10.1126/science.1182364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hitotsumatsu O, Ahmad RC, Tavares R, Wang M, Philpott D, Turer EE, et al. The ubiquitin-editing enzyme A20 restricts nucleotide-binding oligomerization domain containing 2-triggered signals. Immunity. 2008;28:381–390. doi: 10.1016/j.immuni.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Damgaard RB, Nachbur U, Yabal M, Wong WW, Fiil BK, Kastirr M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell. 2012;46:746–758. doi: 10.1016/j.molcel.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Parvatiyar K, Barber GN, Harhaj EW. TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem. 2010;285:14999–15009. doi: 10.1074/jbc.M110.109819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. 2011;41:354–365. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Witt A, Vucic D. Diverse ubiquitin linkages regulate RIP kinases-mediated inflammatory and cell death signaling. Cell Death Differ. 2017;24:1160–1171. doi: 10.1038/cdd.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu T, Zhang L, Joo D, Sun SC. NF-kappaB signaling in inflammation. Signal Transduct Target Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107:7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1:a001651. doi: 10.1101/cshperspect.a001651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci. 2014;1319:82–95. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, et al. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W, Shi Q, Xu X, Chen H, Lin W, Zhang F, et al. Aberrant CD40-induced NF-kappaB activation in human lupus B lymphocytes. PLoS One. 2012;7:e41644. doi: 10.1371/journal.pone.0041644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chuang HC, Lan JL, Chen DY, Yang CY, Chen YM, Li JP, et al. The kinase GLK controls autoimmunity and NF-kappaB signaling by activating the kinase PKC-theta in T cells. Nat Immunol. 2011;12:1113–1118. doi: 10.1038/ni.2121. [DOI] [PubMed] [Google Scholar]
- 55.Kalergis AM, Iruretagoyena MI, Barrientos MJ, Gonzalez PA, Herrada AA, Leiva ED, et al. Modulation of nuclear factor-kappaB activity can influence the susceptibility to systemic lupus erythematosus. Immunology. 2009;128:e306–e314. doi: 10.1111/j.1365-2567.2008.02964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu Y, Vahl JC, Kumar D, Heger K, Bertossi A, Wojtowicz E, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011;117:2227–2236. doi: 10.1182/blood-2010-09-306019. [DOI] [PubMed] [Google Scholar]
- 57.Hovelmeyer N, Reissig S, Xuan NT, Adams-Quack P, Lukas D, Nikolaev A, et al. A20 deficiency in B cells enhances B-cell proliferation and results in the development of autoantibodies. Eur J Immunol. 2011;41:595–601. doi: 10.1002/eji.201041313. [DOI] [PubMed] [Google Scholar]
- 58.Tavares RM, Turer EE, Liu CL, Advincula R, Scapini P, Rhee L, et al. The ubiquitin modifying enzyme A20 restricts B cell survival and prevents autoimmunity. Immunity. 2010;33:181–91.56. doi: 10.1016/j.immuni.2010.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adrianto I, Wen F, Templeton A, Wiley G, King JB, Lessard CJ, et al. Association of a functional variant downstream of TNFAIP3 with systemic lupus erythematosus. Nat Genet. 2011;43:253–258. doi: 10.1038/ng.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang S, Wen F, Wiley GB, Kinter MT, Gaffney PM. An enhancer element harboring variants associated with systemic lupus erythematosus engages the TNFAIP3 promoter to influence A20 expression. PLoS Genet. 2013;9:e1003750. doi: 10.1371/journal.pgen.1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang S, Wen F, Tessneer KL, Gaffney PM. TALEN-mediated enhancer knockout influences TNFAIP3 gene expression and mimics a molecular phenotype associated with systemic lupus erythematosus. Genes Immun. 2016;17(3):165–170. doi: 10.1038/gene.2016.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S, Adrianto I, Wiley GB, Lessard CJ, Kelly JA, Adler AJ, et al. A functional haplotype of UBE2L3 confers risk for systemic lupus erythematosus. Genes Immun. 2012;13:380–387. doi: 10.1038/gene.2012.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.