

Abstract
Excess of Aβ42 peptide is considered a hallmark of the disease. Here we express the human Aβ42 peptide to assay the neuroprotective effects of PI3K in adult Drosophila melanogaster. The neuronal expression of the human peptide elicits progressive toxicity in the adult fly. The pathological traits include reduced axonal transport, synapse loss, defective climbing ability and olfactory perception, as well as lifespan reduction. The Aβ42-dependent synapse decay does not involve transcriptional changes in the core synaptic protein encoding genes bruchpilot, liprin and synaptobrevin. All toxicity features, however, are suppressed by the coexpression of PI3K. Moreover, PI3K activation induces a significant increase of 6E10 and thioflavin-positive amyloid deposits. Mechanistically, we suggest that Aβ42-Ser26 could be a candidate residue for direct or indirect phosphorylation by PI3K. Along with these in vivo experiments, we further analyze Aβ42 toxicity and its suppression by PI3K activation in in vitro assays with SH-SY5Y human neuroblastoma cell cultures, where Aβ42 aggregation into large insoluble deposits is reproduced. Finally, we show that the Aβ42 toxicity syndrome includes the transcriptional shut down of PI3K expression. Taken together, these results uncover a potential novel pharmacological strategy against this disease through the restoration of PI3K activity.
INTRODUCTION
Aβ peptide is a diagnostic biomarker of Alzheimer’s disease (AD). The peptide originates from the sequential cleavage of the amyloid precursor protein (APP) by β and γ secretases in the amyloidogenic pathway, which is opposed to the nonamyloidogenic alternative (De Strooper et al., 2010; Corbett and Hooper, 2018). Through this cleavage, two predominant forms of 40 or 42 amino acid–long polypeptides (Aβ40 and Aβ42) are produced (Kummer and Heneka, 2014). Only when the equilibrium between APP processing and its degradation by neprilysin, insulin-degrading enzyme or endothelin-converting enzyme (Turner et al., 2004) is imbalanced, the amyloid cascade is initiated and Aβ42 becomes synaptotoxic (Hardy and Higgins, 1992; Kummer and Heneka, 2014). The relative increase in Aβ42/Aβ40 due to excessive accumulation of β-amyloid yields protein aggregation, which involves misfolding of Aβ into soluble and insoluble assemblies. Such aggregation process is thought to be critical for AD progression, as the different Aβ assemblies differ in their toxicity (Goure et al., 2014). Whereas monomers are innocuous, when they self-associate into oligomers and prefibrillar aggregates they become toxic. Insoluble Aβ plaques are considered rather benign species that could serve as a sink for oligomers, although they are also a potential source of toxic aggregates (Caughey and Lansbury, 2003; Stefani, 2010; DaRocha-Souto et al., 2011; Moreth et al. 2013a). The amyloid cascade hypothesizes that plaques can sequester Aβ oligomers until reaching a physical limit after which the oligomers diffuse to the surrounding membranes and hydrophobic cell surfaces (Esparza et al., 2013).
Extracellular Aβ peptide can be detected in the cerebrospinal fluid (CSF) of AD patients (Kollhoff et al., 2018; Verberk et al., 2018). External Aβ deposits originate from APP amyloidogenic proteolysis by β-secretase, which occurs in the extracellular domain. The subsequent cleavage of the intramembranous domain by γ-secretase releases the Aβ peptide into the extracellular space or vesicle lumen (Shoji et al., 1992; Haass et al., 2012). On the other hand, intracellular Aβ accumulation has also been found inside synaptic terminals of AD patients by high-resolution electron microscopy (LaFerla et al., 2007; Gouras et al., 2010; Zhao et al., 2015). The nature of intracellular Aβ deposits has not yet been fully clarified, as they could be a result of endosomal membrane APP-derived Aβ, be in their way to secretion, targeted for lysosomal degradation, or be a result from uptake of previously secreted Aβ oligomers. Whether the first source of toxic oligomers is the extracellular or the intracellular β-amyloid remains still an open question. Nevertheless, it is well documented that Aβ oligomers produce early synaptic alterations that eventually cause synapse loss (Li et al., 2018). However, the mechanisms by which β-amyloid peptides elicit synaptotoxicity are still largely unknown.
Drosophila is an experimental system suitable to analyze cellular and molecular mechanisms relevant to AD. Different forms of human Aβ peptides can be genetically expressed in selected neurons in a time-controlled manner using the Gal4/UAS expression system (Brand and Perrimon, 1993). Fly models of AD follow different strategies. Some studies are based on the expression of both human APP and human BACE (β-secretase) describing the consequent Aβ plaque formation and the age-dependent neurodegeneration (Greeve et al., 2004). These events include decreased presynaptic connections, altered mitochondrial localization, and reduced postsynaptic protein levels (Mhatre et al., 2014). Other models are based on the expression of human β-amyloid peptides including Aβ40 (Iijima et al., 2004), which does not produce plaque formation but causes age-dependent learning defects; Aβ42 that elicits synaptic alterations and locomotor, survival, and learning impairments (Iijima et al., 2008; Martin-Peña et al., 2017, 2018), and other Aβ aggregation-prone models containing human AD mutations like Aβ42-Arc (Crowther et al., 2005; Iijima et al., 2008). In addition, the UAS-Aβ42(2X) construct, consisting in two tandem copies of the human Aβ42 fused to a secretion signal, yields stronger neurotoxic phenotypes than other single-copy models. This model shows extensive neuronal death and induces unconventional splicing of the transcription factor XBP1 (Casas-Tinto et al., 2011). Besides, Aβ42-induced neurodegeneration is rescued by XBP1 expression (Casas-Tinto et al., 2011). Thus, we have employed this construct throughout this study to generate Aβ42-dependent synaptotoxicity.
Class I phosphoinositide 3 kinase (PI3K) is involved in a signaling pathway that leads to synapse formation in Drosophila (Martín-Peña et al., 2006) and vertebrates (Cuesto et al., 2011). This synaptogenic pathway includes the type II BMP receptor Wit (wishfull thinking) and several MAPKs, but not mTOR or S6K, which are characteristic of other PI3K classical signaling pathways (Jordán-Álvarez et al., 2017). The noncanonical PI3K-mediated pathway for synaptogenesis is counterbalanced by a Mad/Medea-regulated anti-synaptogenesis pathway (Jordán-Álvarez et al., 2017). In turn, inhibitors of the PI3K pathway, as GSK3-β, reduce the number of synapses (Cuesto et al., 2015). Actually, genetic manipulations of GSK3-β attenuate or suppress several AD traits in Drosophila (Sofola et al., 2010). Several studies have described GSK3-β activation upon Aβ expression, which can produce APP transport alterations by affecting kinesin-1 and dynein (Weaver et al., 2013). Consistently, GSK3-β inhibition can restore some of the Aβ toxic effects, similar to lithium or Congo red treatments (Crowther et al., 2004; Sofola et al., 2010; Sofola-Adesakin et al., 2014). By contrast, other reports found no restoration when GSK3-β was down-regulated in Aβ-expressing neurons, claiming no Wnt-Aβ interaction in Drosophila AD models (Lüchtenborg and Katanaev, 2014). PI3K inhibition by down-regulation of the p65 regulatory subunit has also been used in Aβ experiments, showing prevention of Aβ-induced neuronal electrophysiological defects (Chiang et al., 2010).
Synapse-promoting actions of PI3K are age-independent, as their activation at different times along adulthood generates new, supernumerary and fully functional synapses (Martín-Peña et al., 2006; Acebes et al., 2011, 2012). Recently, we reported the protective activity of PI3K over the Aβ42 toxicity caused in nonneuronal cells (Arnés et al., 2017). Unraveling the mechanisms that underlie these protective effects of PI3K would be of potential therapeutic interest, in particular if the early onset of AD could be diagnosed. Thus, considering that the phosphorylation of Aβ peptide has consequences in AD pathology (Kumar et al., 2011, 2012), we set out to investigate the potential involvement of PI3K in the mechanisms of neuroprotection against Aβ42 synaptotoxicity.
RESULTS
PI3K prevents Aβ42-induced synapse loss in an age-independent manner
To determine whether PI3K can rescue the loss of synapses induced by Aβ42, we overexpressed a constitutively active form of PI3K and the human Aβ42 peptide with the pan-neural elav-Gal4 driver. To discard an effect during development, we used the temperature-sensitive Gal80TS construct to activate the expression of the Gal4 system only in adult stages by shifting the rearing temperature to 29°C. To estimate the number of synapses, we aged adult flies for 7, 15, and 25 d, and dissected adult abdomens to visualize the ventral longitudinal muscle (VLM) and their neuromuscular junctions (NMJs) in the third abdominal segment. Synapses were revealed by the monoclonal antibody nc82 anti-Bruch pilot (brp), whereas anti-HRP antibody was used to identify the motor neuron membrane.
At day 7 post-Gal4 system activation, all genotypes had similar extents of synaptic areas, indicating that no neurodegeneration was evident when measured at the first time point in the experimental design (Figure 1A). However, at day 15, Aβ42-expressing flies showed significant decrease in the total synaptic area. In addition, PI3K and Aβ42 coexpression showed also a statistically significant increase in synaptic surface compared with Aβ42-expressing flies (Figure 1B). This recovery in the synaptic area indicates that the synaptogenic effect of PI3K could prevent the synapse loss induced by Aβ42 in adult motor neurons after 15 d of expression. Immune-labeled puncta in Aβ42-expressing flies showed disorganized, nonspherical positive dots of BRP signal (Figure 1A). This deposit-like morphology was absent in all the other genotypes. Owing to this observation, the quantification of synapses was done in terms of total surface of BRP-positive signal for all the experiments, instead of the usual counting of puncta. At 25 d postexpression PI3K flies showed increased total synaptic surface, in concordance with previously published data (Martín-Peña et al., 2006). On another note, most Aβ42-expressing flies were dead and could not be analyzed, but Aβ42/PI3K flies were still alive and showed synapse areas similar to controls (Figure 1B). Thus, PI3K expression prevents Aβ42 synaptotoxic effects also at later time points in adulthood.
FIGURE 1:

Human Aβ42 causes progressive reduction of synapses in adult motor neurons and PI3K activation suppresses this effect with no transcriptional changes in core synaptic genes. (A) Representative confocal images of neuromuscular junctions (NMJs) of the ventral longitudinal muscle in the third abdominal segment of adult female at 15 d postexpression. Active zones monitored as nc82 immune-positive area. (B) Time course of synaptic effects. At 7, 15, and 25 d posttriggering genetic expression, the Aβ42 flies show a drastic reduction of synaptic area, while at 25 d most synapses have disappeared. Note that PI3K flies increase the synaptic signal along the three time points and the suppression effects on Aβ42 are still effective at 25 d. One-way ANOVA test with ***, p < 0.001. (C) RT-qPCR analysis of three genes encoding synapse proteins, bruchpilot, liprin, and synaptobrevin, carried out in 15-d-old adult female heads. Results of mRNA levels (Y axis) are represented as fold induction in triplicate experiments normalized with respect to the control genotype (see below). Histogram differences are not statistically significant (Student’s t test). Genotypes: Control (UAS-LacZ/elavC155-Gal4; +/+; Tub-Gal80TS/+), PI3K (UAS-PI3KCAAX/elavC155-Gal4; +/+; Tub-Gal80TS/+), Aβ42 (elavC155-Gal4/+; UAS-Aβ42(2x)/+; Tub-Gal80TS/+), PI3K/Aβ42 (UAS-PI3KCAAX/elavC155-Gal4; UAS-Aβ42(2x)/+; Tub-Gal80TS/+). Data represent mean ± SD. Data in B are processed by IMARIS Bitplane. Scale bar is 20 μm.
To investigate a possible molecular mechanism by which PI3K rescues Aβ42-induced synapse loss, we analyzed the transcriptional status of several synaptic genes, such as bruchpilot, liprin, and synaptobrevin, in 15-d-old adult fly heads of the four genotypes studied here. We found no significant differences in mRNA expression in any of these genes (Figure 1C), suggesting that PI3K does not restore synapses through transcriptional modulation of synaptic genes.
Taking the results together, we conclude that human Aβ42 expression causes progressive morphological alterations and synapse loss in adult fly motor neurons, and that these deleterious effects can be prevented by PI3K activation in an age-independent manner without modifying gene transcription.
PI3K prevents Aβ42-induced microtubule dynamics defects
Because Aβ42 can alter the neuronal cytoskeleton leading to defective neurite outgrowth (Mokhtar et al., 2013), and this could lead to deficits in intracellular transport, we tested whether PI3K activation could prevent these alterations as a mechanism to protect synapses. To this end, we analyzed microtubule dynamics by live time-lapse imaging in third instar whole-mount larvae expressing the UAS-EB1-GFP construct. EB1 is a plus-end microtubule binding protein that reports microtubule dynamics, as it binds only to growing microtubules, but it detaches from shrinking microtubules resulting in dynamic GFP signals known as “comets” (Morrison et al., 1998).
We recorded EB1-GFP comets of third instar larval ddA (dendritic arborization dorsal cluster) sensory neurons (Figure 2, A–D). Track speed in PI3K and PI3K/Aβ42 genotypes showed similar values, being both significantly different compared with controls (Figure 2, E and F). In turn, track length and duration remained constant in all genotypes (Figure 2, G and H) indicating that the growing process of the microtubule, once started, is unaffected by Aβ42, PI3K, or PI3K/Aβ42 expression. By contrast, track density was significantly decreased in Aβ42-expressing neurons, but not in PI3K and PI3K/Aβ42-expressing neurons (Figure 2I), indicating that PI3K prevents the reduction in comet density caused by Aβ42 expression.
FIGURE 2:

PI3K prevents Aβ42-induced microtubule dynamics deficits. (A–D) Still images of live recordings of EB1-GFP in ddA sensory neurons of third instar larvae. (E) Cartoon depicting the EB1 position in the plus-end of microtubules. (F–I) Quantification of GFP-tagged comet’s speed (F), length (G), duration (H), and density (I). Note the significant reduction of comet density by Aβ42 and its suppression by PI3K. Data represent mean ± SD. One-way ANOVA significances are indicated with **, p < 0.01 and *, p < 0.05 for comparisons with the control group. Scale bar is 50 µm.
We concluded that Aβ42 alters microtubule dynamics by reducing the number of growing events per surface unit, which is direct evidence of intracellular transport deficits. Interestingly, PI3K/Aβ42 neurons had the same number of growing microtubules per surface unit as control neurons, indicating that PI3K can prevent microtubule dynamics defects induced by Aβ42, thus allowing a fully functional microtubule-associated intracellular transport.
PI3K attenuates Aβ42-induced functional deficits in locomotion, olfaction, and lifespan
To test the functionality of PI3K-protected synapses under Aβ42 overexpression, we analyzed locomotion performance and olfaction in adult flies. In locomotion assays, three different groups of flies were annotated: 1) flies that reached the 4-cm line threshold within a given time (gray histograms), 2) flies that did not reach this line but climbed along the tube (orange histograms), and 3) flies that stayed at the bottom of the tube (blue histograms; Figure 3A). Control and PI3K flies showed normal locomotion until 30 d of age, as expected for healthy flies grown at 29°C (Figure 3, B and C). However, Aβ42-expressing flies exhibited reduced locomotor activity as soon as 11 d of age, with a significant group of flies that stayed at the bottom of the tube during the climbing assay (black histograms; Figure 3D). The total percentage of flies that did not climb at all increased progressively as the flies aged, and this proportion of flies was higher than 50% at 18 d of age. When combined with Aβ42, PI3K overexpression delayed the appearance of the nonclimbing phenotype to 15 d, which reached 50% of the total population at 25 d of age, 7 d later than the Aβ42-expressing flies (Figure 3E).
FIGURE 3:

Aβ42-induced functional effects are restored by PI3K. (A) Cartoon representing the three color-coded fly populations considered in the negative geotaxis assay. (B–E) Histograms show normalized values of flies climbing to the top of the tube (gray), not reaching the 4-cm line (orange), and not climbing (purple). Numerical data are shown in Supplemental Table S1.
To analyze potential changes in olfactory perception, we evaluated the odorant choice index in a 10−3–10−1 (vol/vol) concentration range for two different volatiles: ethyl butyrate (EB) and isoamyl acetate (IAA) in a GH298-Gal4 fly line (Figure 4 and Supplemental Figure S1). This line expresses the Gal4 driver mainly in 30–32 inhibitory local interneurons of each antennal lobe, the first olfactory neuropil in the insect brain (Ng et al., 2002; Acebes et al., 2011). In these experiments we used an inducible UAS-PI3K92D construct driven by GH298-Gal4, instead of using the constitutively active form UAS-PI3KCAAX, to test whether the protective effects by PI3K would be different depending on the inducible versus the constitutively active forms of this kinase. First, we addressed the effects of Aβ42 introgression in the GH298-Gal4 domain by using two genotypes: UAS-Aβ42 and UAS-Aβ42/UAS-GFPnls. Flies overexpressing Aβ42 in either combination showed more negative olfactory indexes (Figure 4A). However, the simultaneous coexpression of the inducible PI3K restored normal olfactory responses. In turn, flies expressing PI3K exhibited comparable olfactory responses with respect to control. Equivalent results were obtained in olfaction assays using IAA as odorant stimulus (Supplemental Figure S1).
FIGURE 4:

Olfactory perception assays. (A) Adult flies (5–7 d old) subjected to ethyl butyrate (EB) along three concentrations (vol/vol). Data represent mean ± SEM with 350–400 individuals per group and concentration. When two different Aβ42 constructs were expressed in the GH298-Gal4 domain (genotypes: GH298-Gal4/UAS-Aβ42 and GH298-Gal4/UAS-Aβ42/UAS-GFPnls), olfactory responses to 10−3 and 10−2 concentrations were consistently repulsive (full and dotted gray lines, respectively) compared with GH298-Gal4 controls (black line) and GH298-Gal4/UAS-PI3K flies (blue line). However, when PI3K (inducible form, UAS-PI3K92D) and Aβ42 were coexpressed in GH298 neurons (genotype: GH298-Gal4/UAS-PI3K/UAS-Aβ42(2X)), the olfactory index returned to the normal profile (red line). Black and gray asterisks: comparison between GH298-Gal4/UAS-Aβ42 and GH298-Gal4/UAS-Aβ42/UAS-GFPnls flies with respect to control. ζ = **, p < 0.001; comparison between GH298-Gal4/UAS-Aβ42 and GH298-Gal4/UAS-PI3K/UAS-Aβ42 flies. This return to normal olfactory perception when PI3K and Aβ42 are simultaneously addressed to GH298 neurons is also reproduced with IAA odorant (Supplemental Figure S1). (B) Equivalent assay in the krasavietz-Gal4 domain. The MB-Gal80 represses the Gal4 driver in the mushroom body neurons. Olfactory responses from krasavietz-Gal4/MBGal80/UAS-Aβ42 flies were altered (full gray line) compared with krasavietz-Gal4/MBGal80 control individuals (full black line). The simultaneous coexpression of PI3K (constitutively active form, UAS-PI3KCAAX) and Aβ42 restores the normal response to [10−3] and [10−1]. Note the olfactory response of theses flies at [10−2] above control values (red asterisk = *, p < 0.05). Black asterisks: comparisons between krasavietz-Gal4/MBGal80/UAS-Aβ42 vs. krasavietz-Gal4/MB-Gal80 control flies. ζ = ***, p < 0.0001;, comparison between krasavietz-Gal4/MBGal80/UAS-Aβ42 and krasavietz-Gal4/MB-Gal80/UAS-PI3K/UAS-Aβ42. *, p < 0.05; **, p < 0.001; ***, p < 0.0001 (Student’s t test with the Welch’s correction).
Gal4 expression domains often include cell subsets beyond those aimed in behavioral experiments, inhibitory local interneurons in the case of GH298, or are transiently expressed during development in other tissues (Casas-Tintó et al., 2017). To confirm the PI3K protective effect in other subsets of olfactory neurons, we carried out a similar experiment using the constitutively active PI3K construct, UAS-PI3KCAAX, under the krasavietz-Gal4 driver, which targets a group of five to eight excitatory local interneurons mainly. Owing to the fact that the krasavietz domain also extends to extrinsic mushroom body neurons, we employed a MB-Gal80 construct to silence the krasavietz-Gal4 in these cells. The effects on EB perception were akin to those observed with the inhibitory local interneurons of GH298 (Figure 4B). Thus, we can conclude that the protective effects of PI3K upon the Aβ42-dependent toxicity are reproduced in olfactory neurons, both inhibitory and excitatory, and the protection by PI3K can result, from either the constitutively active or the inducible forms.
Aβ42 is also known to severely affect the organism lifespan when expressed throughout the nervous system, using a pan-neural elav-Gal4 driver (Iijima et al., 2004). Therefore, here we aimed to study the overexpression of PI3K with the same adult-onset design. As expected, Aβ42 flies decreased their lifespan, whereas PI3K increased it (Supplemental Figure S2A). The joint coexpression, PI3K/Aβ42, significantly ameliorated the negative effect of Aβ42, albeit partially (Supplemental Figure S2A).
Taken together, these data indicate that PI3K overexpression rescues Aβ42-dependent locomotor deficits and olfactory perception changes and ameliorates organism longevity defects. Thus, the PI3K-protected synapses seem functional throughout all neurons targeted here to the extent of improving the fly lifespan.
PI3K extends cell survival of human neurons exposed to Aβ42 oligomers
To validate whether the results of PI3K in fly neurons affected by Aβ42 can be reproduced in human cells, we examined the effects of PI3K activation in human neuroblastoma SH-SY5Y cells treated with Aβ42 oligomers. Cells were cultured and differentiated with retinoic acid and 1% serum for 48 h before Aβ42 oligomers treatments. Aβ42 oligomers were added to the cell culture 72 h after cell seeding (48 h after differentiation), at three different concentrations (Kayed and Glabe, 2006). To stimulate PI3K activity we used the peptide PTD4-PI3KAc, previously proved to trigger PI3K activation both in vitro and in vivo (Cuesto et al., 2011, 2015; Enriquez-Barreto et al., 2014). PTD4-PI3KAc was administered at three different time points: before Aβ42 (pretreatment), simultaneously with Aβ42 (cotreatment), or after Aβ42 addition (posttreatment; Figure 5). As expected, Aβ42 oligomers did not affect cell viability at low concentrations (18 μg/ml). However, the percentage of cells alive after the treatment was significantly decreased at 36 and 72 μg/ml concentrations. These results confirmed the effectiveness of the oligomer preparation protocol and were used as positive internal controls in the experiment (Figure 5).
FIGURE 5:
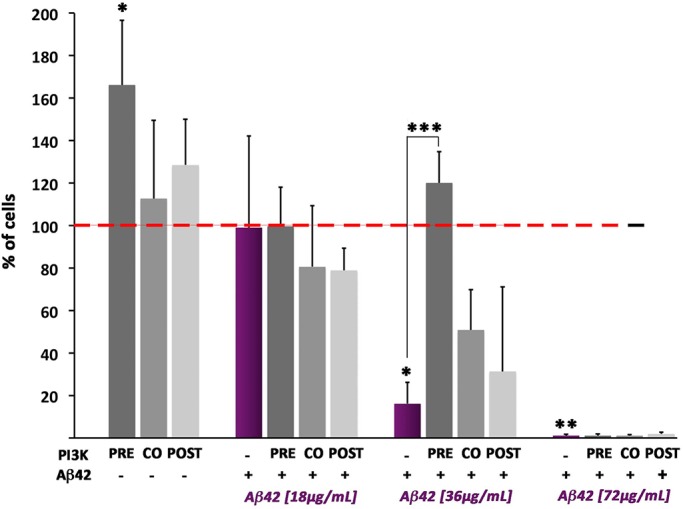
PI3K neuroprotection against Aβ42 effects is reproduced in human cells. Differentiated neuroblastoma SH-SY5Y human cells were treated with increasing concentrations of Aβ42 oligomers (18, 36, and 72 µg/ml) and PTD-PI3KAc peptide (50 µg/ml) administered 24 h prior (pre), at the same time (co) or 24 h after (post) the Aβ42 treatment. Data represent mean ± SD from CellTiter-Glo luminescence is shown as percentage of viability compared with control values of cells exposed neither to Aβ42 nor to PTD-PI3KAc (red-dotted line). At 36 µg/ml Aβ42 treatment, the deleterious effects of the oligomers are evident and the PI3K protection is most effective if cultures are pre- or cotreated. At 72 µg/ml Aβ42 treatment, the effects are so deleterious that no protection seems possible. One-way ANOVA significances are indicated with ***, p < 0,001; **, p < 0.01; and *, p < 0.05.
PTD4-PI3KAc peptide showed no cell viability effects in non–Aβ42-treated cells at 48 or 72 h, but it significantly increased cell viability when administered 24 h after differentiation. This effect can be attributed to the brief period of starvation and differentiation that the cells had at this time (24 h) that could allow cells to respond to mitogen stimuli, like PI3K pathway effectors. More interestingly, PI3K increases cell viability only when PTD4-PI3KAc peptide is added 24 h before or simultaneously with Aβ42, at an oligomer concentration of 36 μg/ml (Figure 5). No protection was found at higher Aβ42 concentrations (72 μg/ml).
These data prove that PTD4-PI3KAc protects human neurons from Aβ42 oligomers toxic effects, validating the in vivo results obtained in flies. Presumably, the protective effect of this peptide will be elicited through the activation of the cell endogenous PI3K because the peptide has no kinase activity per se (see Materials and Methods).
Protection from synaptic effects takes place through noncanonical PI3K signaling
Being PI3K is a shared element in several signaling pathways (Acebes and Morales, 2012), we explored two possibilities: 1) the insulin receptor canonical PI3K/AKT pathway, which is known to elicit prosurvival and cell growth responses (Basu et al., 2012; Dyer et al., 2016), and 2) the recently revealed prosynaptogenesis pathway (Jordán-Álvarez et al., 2017). To assay the effect of the classical pathway effector, we overexpressed in flies the mammalian target of rapamycin (mTOR), a downstream factor of PI3K, and we analyzed the synaptic signal in abdominal motor neurons of 15-d-old adults. The quantification of the synaptic BRP signal indicates that mTOR does not protect neurons from Aβ42 toxicity (Figure 6, A, B, D, E, and G). This result is consistent with previous reports in Drosophila and mammals where mTOR does not change synapse number (Martín-Peña et al., 2006; Cuesto et al., 2011). On the other hand, Medea, a member of the prosynaptogenesis pathway acting downstream from PI3K (Jordán-Álvarez et al., 2017), fully prevents the deleterious effects of Aβ42 on synapses when down-regulated (Figure 6, C, F, and G).
FIGURE 6:

Suppression of Aβ42 effects is mediated through PI3K synaptogenic pathway. (A–F) Representative confocal images of 15-d adult females NMJ corresponding to the ventral muscle in the third abdominal segment. Active zones are revealed by nc82 antibody. (G) Quantification of synaptic area at 15 d. (H–M) Climb assay with color-coded performance following the same protocol and criteria as in Figure 2. Note the severe enhancement of Aβ42 defects by mTOR, not a member of the synaptogenesis PI3K signaling, while MedeaRNAi, a member of that pathway, does suppress them fully. Data represent mean ± SD. One-way ANOVA significances are indicated with ***, p < 0.001. Note the neuroprotection elicited by MedeaRNAi but not by mTOR or S6K. Numerical data are shown in Supplemental Table S1. (N) Adult eyes coexpressing Aβ42 and members of PI3K signaling pathways from genotypes ♀ GMR-Gal4, UAS-Aβ42(2X)/+; UAS-X/+ raised at 25°C. Symbols ↑ and ↓ indicate overexpression of the normal factor or its corresponding RNAi, respectively.
To test the behavioral outcome of the synapse loss prevention by Medea down-expression, we performed climbing experiments using the same genotypes tested for synapse quantification. mTOR/Aβ42 flies showed the same reduction in climbing activity as either of both factors separately (Figure 6, H–K). Nonclimbing flies began to be detected at day 9 (Aβ42) or day 15 (mTOR), while, in controls, this class became evident from day 29 onward. By contrast, the down-regulation of Medea rescued the climbing phenotype of Aβ42 to a large extent (Figure 6, L and M).
To explore additional PI3K-related signals and to validate the protection effect on different types of neurons, we studied its effects in eye photoreceptors (Figure 6N). In these neurons, PI3K up-regulation rescued to a large extent the Aβ42 damage. Down-regulating Medea also had a noticeable protection, whereas its up-regulation had no effect. Up-regulating S6K showed very little protection, if at all, and either up- or down-regulation of mTOR failed to modify the Aβ42 defects in the eye.
Knowing that PI3K ameliorates lifespan in Aβ42-expressing flies, we wondered whether mTOR overexpression or Medea down-expression could also affect longevity. Lifespan studies showed a significant decrease in mTOR/Aβ42 flies, as well as in mTOR overexpression alone (Supplemental Figure S2B). Actually, mTOR overexpression aggravated the lifespan reduction caused by Aβ42 alone, while down-regulating Medea led to a normal lifespan in Aβ42-expressing flies. Thus, PI3K overexpression and Medea attenuation, two manipulations previously shown to up-regulate synapse number, effectively decrease Aβ42 synaptotoxicity, allowing a better locomotor performance and increasing fly lifespan. Nevertheless, protection by PI3K was largely more effective than that elicited by down-regulating Medea (see below). Taken together, these data indicate that the protective effects of PI3K over Aβ42 toxicity rely on the synaptogenic pathway signaling, rather than on the canonical insulin receptor pathway.
PI3K increases Aβ42 insoluble aggregates in Drosophila and human neurons
To understand the causative effects of PI3K over Aβ42 accumulation, we analyzed total Aβ42 accumulation in 15-d-old fly brains by immunohistochemistry. Aβ42-overexpressing adult brains showed a noticeable 6E10 positive signal, which was absent in control or PI3K-overexpressing brains. Surprisingly, this positive signal was increased in intensity and extent in PI3K/Aβ42 brains (Figure 7, A–D). To confirm this result, same aged adult head homogenates were analyzed by Western blot (Figure 7E). Total Aβ42 protein levels were 2.5-fold higher in PI3K/Aβ42 compared with Aβ42 alone. To rule out a putative effect on transcription, reverse transcription quantitative PCR (RT-qPCR) was performed with same aged adult head homogenates and the results showed no changes in total Aβ42 mRNA, discarding this possibility (Figure 7F). Thus, mechanistically, PI3K protects from Aβ42 toxicity and elicits an increase of the amyloid protein deposits but does not affect the transcription of the Aβ42 construct.
FIGURE 7:

PI3K activation increases Aβ42 protein deposits without affecting transcription. (A–D) Representative confocal images of 15-d-old adult heads stained with 6E10 antibody of the indicated genotypes. Note the large increase of amyloid deposits (white) revealed by the 6E10 antibody. Cell nuclei are marked by DAPI (magenta). (E) Western blot of adult heads of the same genotypes and age. Data correspond to three independent Western blots normalized for tubulin content as internal control. Note the significant increase of the 6E10 signal elicited by PI3K activation. One-way ANOVA significance is indicated with **, p < 0.01. (F) RT-qPCR data from the same genotypes and ages. Data represent mean ± SD. Statistical significance was analyzed by Student’s t test. Scale bar is 50 µm.
Aiming to clarify whether the PI3K protection and the mild one observed by down-regulating Medea would operate through the same mechanism, we performed 6E10 staining in adult heads coexpressing Aβ42 and (Supplemental Figure S3). The data clearly show that the down-regulation of Medea does not increase the number and size of Aβ deposits. This suggests that the mild protection elicited by Medea is mediated by a different mechanism than that of PI3K, and justifies our focus on PI3K in this study.
Because the various amyloid aggregates of Aβ42 can evolve different levels of toxicity, we tested the nature of the Aβ42 deposits by the criterion of thioflavin-S staining. This dye binds specifically to proteins with β-sheet conformation and labels Aβ42 fibers and plaques, constituents of the insoluble fraction of β-amyloid aggregates. Quantification of the thioflavin-S signal in 7- and 15-d-old brains demonstrated a significant increase in the insoluble fraction of Aβ42 deposits at 15 d of age in the PI3K/Aβ42 brains compared with age-matched Aβ42 alone (Figure 8, A and B). This result was confirmed by a fourfold increase of the Aβ42 fraction of insoluble protein extracts from 15-d-old adult heads (Figure 8C). These data show that PI3K triggers a change in Aβ42 aggregation into more insoluble species.
FIGURE 8:

PI3K increases amyloid Aβ42 insoluble fraction. (A) Representative confocal images of 7- and 15-d-old adult brains stained with thioflavin. Scale bar is 50 µm. (B) Quantification of thioflavin signal. Note the progressive signal increment with age and, in particular, with PI3K coexpression. (C) Immunoblot showing insoluble Aβ levels in control, PI3K, Aβ42, and Aβ42/PI3K adult heads of 15-d-old flies. Histograms represent mean Aβ levels visualized by 6E10 antibody and normalized to tubulin on three independent blots. One-way ANOVA significances are indicated with ***, p < 0.001 and **, p < 0.01.
Beyond the effects in fly neurons, we tested whether human neurons could also exhibit the PI3K-induced increase of Aβ42 aggregation deposits. SH-SY5Y cell-line human neuroblastoma cells were treated with Aβ42 oligomers (36 μg/ml) and PTD4-PI3KAc peptide (50 μg/ml) following differentiation. As expected, no 6E10-positive deposits were found in cells treated either with vehicles or with PTD4-PI3K alone (Figure 9). For practical reasons, only Aβ42 and PTD4-PI3KAc/Aβ42 experiments were compared. Treatment with PTD4-PI3KAc in combination with Aβ42 oligomers produced a significant decrease in the total number of deposits compared with those found in Aβ42 treatment (Figure 9). The deposits, however, were larger in cell cultures cotreated with Aβ42 oligomers and PI3K-activating peptide (Figure 9E).
FIGURE 9:

PI3K increases amyloid Aβ42 insoluble fraction in human cells. (A) Cartoon representing the experimental design in SHT-SY5Y cells differentiated and treated with Aβ42 oligomers (36 μg/ml) and PTD4-PI3KAc peptide (50 μg/ml) for 48 h. (B–E) Histograms representing total deposits (B), total area (C), deposits to area ratio (D), and deposit size distribution (E) in the cultures. (F) Representative confocal images of cell cultures showing Aβ (6E10; green) and DAPI (magenta) signals. Note the larger amyloid deposits in PI3K/Aβ42 with respect to Aβ42 (E, F). Data represent mean ± SD analyzed by Student’s t test. Scale bar is 10 µm. One-way ANOVA significance is indicated with *, p < 0.05.
To further analyze the aggregation effect of Aβ42 oligomers, we questioned whether these aggregates could occur in the extracellular space, where the majority of deposits are usually found. Four different experiments were designed: 1) an internal control to test Aβ42 oligomers aggregation, 2) cells treated with Aβ42 oligomers previously treated with PTD4-PI3KAc (pretreatment), 3) cells treated with Aβ42 oligomers and PTD4-PI3KAc at the same time (cotreatment), and 4) cells treated with Aβ42 oligomers later treated with PTD4-PI3KAc (posttreatment). Supernatants were recovered and deposits were stained with 6E10 antibody (Supplemental Figure S4).
The internal control of Aβ42 oligomers showed no 6E10-positive deposits, in the given time of 48 h. We found no significant differences when comparing either the total number of deposits in the PTD4-PI3K pretreatment, cotreatment, and posttreatment experiments or in the total deposit area (Supplemental Figure S4, C, and D). However, the three treatments showed significant differences when the individual size of each deposit was analyzed (Supplemental Figure S4B). PTD4-PI3K cotreatment had up to a 10-fold increase in deposit size, when compared with PTD4-PI3KAc pretreatment and posttreatment. PTD4-PI3KAc posttreatment showed the smallest deposits among the three experiments of the study (Supplemental Figure S4E).
These data indicate that the effects in Aβ42 aggregation elicited by PI3K in adult flies are reproduced in human SH-SY5Y differentiated neurons, which can occur in the extracellular space.
PI3K increases phosphorylation of Aβ42
Building on previous findings that point to phosphorylation as a factor affecting Aβ42 seeding in the initial phase of the plaque formation (Kumar et al., 2011, 2016; Kumar and Walter, 2011), we addressed whether the PI3K kinase activity could play a role in the mechanism of Aβ42 aggregation. To explore this hypothesis, we tested NetPhos 2.0 server prediction software (www.cbs.dtu.dk) and identified three putative residues: pSer8 (score: 0.963), pTyr10 (score: 0.870), and pSer26 (score: 0.787) as candidates (Figure 10A).
FIGURE 10:

Aβ42 residues are potentially targeted by PI3K. (A) Serine and tyrosine residues in Aβ42 sequence subject to phosphorylation. (B) Images of p-Ser (green) and DAPI (magenta) stainings in 15-d-old adult brains of the corresponding genotypes (see Figure 1 legend). Equivalent images of brains stained for p-Tyr (green) are shown in the right panels. Note the increment of p-Ser, but not p-Tyr, immunosignal in PI3K/Aβ42 brains. (C) Images of p-Ser26 (green), p-Ser8 (red), and DAPI (magenta) immunostaining in 15-d-old adult brains of the corresponding genotypes. (D) Quantification of pixel intensities in 10 images from three brains as those shown in panels B and C. One-way ANOVA significances are indicated with ***, p < 0.001. Scale bar is 10 µm (B) and 50 µm (C).
Specific antibodies against phospho-Tyr and phospho-Ser were applied in 15-d-old brains in the four genotypes under study. The phospho-Ser signal was only incremented in PI3K/Aβ42 brains and had a deposit-like morphology (Figure 10, B and D). By contrast, the phospho-Tyr signal was comparable in all genotypes and no deposit-like morphology was found (Figure 10, B and D). Colocalization of 6E10 and both phospho-Ser and phospho-Tyr was not feasible due to the requirement of formic acid for 6E10 immunostaining, which strongly alters phosphopeptides and membrane stability.
Aiming to discriminate between the two serine residues, Ser-8 and Ser-26, as candidates for phosphorylation, we tested p-Ser-8-Aβ42 (1E4E11) and p-Ser-26-Aβ42 (7H3D6) antibodies (Kumar et al., 2013, 2016). No changes were found in pSer8-Aβ42 immunostainings in Aβ42 or PI3K/Aβ42 genotypes (Figure 10, C and D). Phospho-Ser26-Aβ42 immunostainings showed deposits with positive signal in both Aβ42 and PI3K/Aβ42. Unfortunately, the p-Ser-26-Aβ42 antibody seems to detect both phosphorylated and nonphosphorylated forms of Aβ42.
The data prompted us to conclude that PI3K overexpression in an Aβ42 background drives the accumulation of phosphoserine-positive deposits.
The Aβ42-induced toxicity syndrome includes the drastic reduction of PI3K expression
So far, this study has revealed that PI3K protects neurons from Aβ42 toxicity. However, this protection could result from a novel cell activity, or, alternatively, from the restoration of a lost property in the affected cell. To place the PI3K protective effect within the context of the Aβ42-induced toxicity, we questioned whether it would be part of the repertoire of abnormal features elicited by the amyloid peptide accumulation. Quantitative PCR assays were carried out in adult heads after 7 d of Aβ42 expression monitoring PI3K and AKT (Figure 11). The data show a drastic reduction in the transcriptional expression of these two genes as compared with sibling controls. This result demonstrates that PI3K, a recognized cell survival factor, is abolished by Aβ42 accumulation. In turn, this feature of the toxicity syndrome explains the protection observed when PI3K expression is genetically restored in the Aβ42 background.
FIGURE 11:
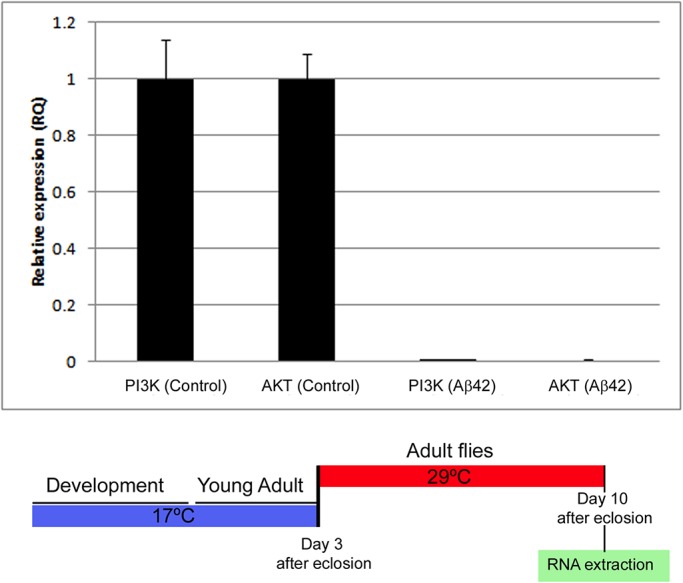
Aβ42 reduces the expression of PI3K and AKT. RT-qPCR assays in triplicate from male adult heads in which Aβ42 expression has been maintained for 7 d. Gal4 driver expression was triggered at day 3 of adulthood and flies were processed at day 10 of adulthood of the corresponding genotypes: ♂ elav-Gal4; CyO/+; Gal80TS/+ and elav-Gal4; UAS-Aβ42(2X)/+; Gal80TS/+. Values are normalized to controls.
DISCUSSION
Synapse loss is one of the first steps in the neurodegenerative process in AD. Previous in vitro and in vivo studies have demonstrated the age-independent synaptogenic actions of PI3K (Martín-Peña et al., 2006; Acebes et al., 2011; Cuesto et al., 2011). Thus, we set out to study the potential effects of PI3K in ameliorating the AD features in an adult-onset Drosophila model. The focus on PI3K is justified because, as we show in this study, the toxicity of Aβ42 includes a drastic reduction of PI3K gene expression. Benefitting from the temporal control of the Gal4/UAS binary system, we have driven the expression of human Aβ42 peptide in the adult central nervous system and followed the progression of the synaptic toxicity. Although another study has used a similar strategy based in the GeneSwitch system by feeding mifepristone (RU486; Sofola et al., 2010), most AD models in Drosophila and other organisms, follow constitutive expression of the Aβ peptide, APP, PS1, and tau, in their mutated or wild-type forms (Ashe and Zahs, 2010; Iijima-Ando and Iijima, 2010; Wisniewski and Sigurdsson, 2010; De Felice and Munoz, 2016).
PI3K mechanisms in Aβ42-induced synapse toxicity
Protein kinases (PKs) in general, can operate inside or outside the cell, where they can phosphorylate cell-surface proteins or extracellular substrates (Shaltiel et al., 1993; Walter et al., 1994; Redegeld et al., 1999). These Ecto- and Exo-PKs can use extracellular ATP, which is present in the brain at nanomolar concentrations, and can also increase locally upon certain stimuli (el-Moatassim et al., 1992; Melani et al., 2005; Gourine et al., 2007). The extracellular Aβ42 deposits identified in this study could, thus, be originated directly outside of the cell or within the presynaptic side, where they are genetically coexpressed, followed by their extrusion.
In turn, PTEN is a lipid phosphatase that inhibits PI3K activation (Maehama and Dixon, 1998), and it mediates Aβ42 synaptic and cognitive impairments (Knafo et al., 2016). PTEN can regulate LTD (Jurado et al., 2010) and it can be targeted by Aβ42 in synaptic spines. Furthermore, pharmacological administration of a PTEN membrane-permeable peptide, unable to interact with Aβ42, rescues synapse deterioration (Knafo et al., 2016). Other PI3K regulators, like the signaling inhibitor GSK3-β, are known to elicit anti-synaptogenic actions in Drosophila and mice (Martín-Peña et al., 2006; Cuesto et al., 2015), with concomitant effects in LTP and long-term memory (Hooper et al., 2008). In addition, Jun kinase/AP-1 and Wnt signaling are also members of the synaptogenic pathway, as they are regulated by GSK3-β (Franciscovich et al., 2008). In AD, GSK3-β is known to phosphorylate tau and regulate microtubule dynamics (reviewed in Llorens-Martín et al., 2014). PIP3, synthetized by PI3K, regulates PSD-95 clustering and synaptic AMPA receptors in spines, affecting synaptic strength (Arendt et al., 2010). All these features compose a scenario in which PI3K is situated in a strategic position to influence opposite pro- and anti-synaptogenic signaling, whose equilibrium ultimately determines the number of synapses that a neuron establishes (Jordán-Álvarez et al., 2017).
Transcriptional changes do not seem to be part of the mechanism elicited by PI3K synaptogenic effect. Our data suggest that neither Aβ42 nor PI3K promotes mRNA changes in three major synaptic active zone (AZ) components (bruchpilot, liprin, and synaptobrevin genes). Nonetheless, other transcriptional alterations, or even posttranscriptional regulations triggered by PI3K, could modify synaptic proteins availability, turnover, degradation, and/or trafficking, leading to the synaptic changes described here. Noticeably, Aβ42 accumulation does seem to affect transcription of other genes; PI3K and AKT, at least.
Microtubule dynamics showed that Aβ42 reduces EB1 comets density, and that this effect was also suppressed by PI3K activation. Defects in microtubule dynamics alter protein trafficking along the axon (Guedes-Dias et al., 2019). BRP, as well as other components of the AZ and the postsynaptic density (PSD), need to be transported via microtubules, and decreased microtubule stability can hamper not only the formation of new AZ or PSD structures, but also synaptic protein turnover. Aβ42 effects on microtubule dynamics might explain the reduction in the synaptic area in adult NMJs that did not depend on transcriptional changes. How PI3K suppresses the Aβ42-dependent impairment of microtubule dynamics has not been addressed in this study. However, among the wide diversity of PI3K-dependent signaling pathways, some are linked to actin cytoskeleton via Rac activation and are AKT-independent (Lien et al., 2017), while others connect microtubules with insulin receptors via GSK3-β (Chiu et al., 2008). If equivalent pathways would exist in Drosophila, they could support the observed suppression of defective microtubule dynamics by PI3K.
Aiming to discriminate among PI3K effector pathways, we assayed here mTOR, S6K, and Medea as potential mediators of Aβ42. Only the latter proved to be effective to some extent, although the neuroprotection mechanisms by PI3K and Medea are different because only the first elicits amyloid deposit increase. In addition, Medea down-regulation is known to attenuate BMP signaling (Wisotzkey et al., 1998), which is altered in AD (Crews et al., 2010; Kang et al., 2014), whereas PI3K does not. Concerning mTOR, it appears to enhance the neurotoxic effects of Aβ42 in lifespan. This result is in line with others, where mTOR is found to participate in memory formation and consolidation, and seems targeted by Aβ42 (Lin et al., 2014; Uddin et al., 2018; Mueed et al., 2019). Thus, mTOR inhibition could become yet another experimental strategy, independent from PI3K activation, to be considered in future AD studies. Conceivably, a combination of strategies could yield even better results.
PI3K mechanisms in Aβ42 dynamics
Aberrant protein phosphorylation has been described in the brain of AD patients (Chung, 2009). Here, we identified a new role of PI3K in Aβ42 accumulation with in vivo and in vitro results that show that PI3K activation induces aggregation of Aβ42, leading to insoluble, less toxic aggregates. Previous studies consider insoluble deposits of Aβ42 as rather benign species (DaRocha-Souto et al., 2011; Moreth et al., 2013b), in contrast to soluble Aβ42 oligomers. Although insoluble deposits would seem refractory to degradation, at least three enzymes are known to degrade Aβ42, neprilysin, insulin-degrading enzyme (IDE), and endothelium-converting enzyme (ECE; Turner et al., 2004; Grimm et al., 2013).
Posttranslationally modified Aβ species have been identified (Kuo et al., 2001; Kummer and Heneka, 2014), including truncation (Härtig et al., 2010), racemization (Murakami et al., 2008), isomerization (Shimizu et al., 2000), pyroglutamination (Kuo et al., 1997), metal-induced oxidation (Dong and Bai, 2003), and phosphorylation (Milton, 2001; Kumar et al., 2011). Particularly, phosphorylation can be potentially achieved at three different Aβ42 residues: Ser-8, Ser-26, and Tyr-10. In addition, Aβ is reported to undergo phosphorylation by protein kinase A (PKA) and by cyclin-dependent kinase 2 (CDC-2) in vitro (Milton, 2001). Phosphorylation at Ser-8 by PKA was observed in free extracellular Aβ rather than in its precursors (APP or β-CTF) and promoted the formation of toxic aggregates. Also, phosphorylated deposits from APP transgenic mice were found concentrated in the center of the plaque (Kumar et al., 2011). Phosphorylation in Ser-8 has been documented to induce Aβ resistance to degradation by IDE (Kumar et al., 2012).
One study described Aβ phosphorylation at Ser-26 by CDC-2 in vitro (Milton, 2001), whereas another showed that pSer26-Aβ42 is abundant in intraneuronal deposits at very early stages of AD (Kumar et al., 2016). However, potential changes in Aβ42 aggregation were not investigated. Nevertheless, cell treatment with olomoucine prevents cytotoxicity and Aβ phosphorylation, suggesting that aggregative forms favored by Ser-26 phosphorylation enhance Aβ42 progressive toxicity. In contrast, our data suggest that PI3K-induced phosphorylation in Ser-26 ameliorates Aβ42 effects and promotes insoluble Aβ42 assembly formation. A recent in vitro analysis of Aβ40 phosphorylated at Ser-26 describes that this modification impairs fibrillization while stabilizing monomers and nontoxic soluble aggregates. Computational studies predicted that a phosphate group at Ser-26 could rigidify the region and interfere with a fibril-specific salt bridge (Rezaei-Ghaleh et al., 2014).
Structural considerations and perspectives
A specific region in the Aβ sequence comprising residues 25–29 generates a bend-like structure that juxtaposes the hydrophobic faces of the two cross-β units. This bend is important for Aβ pathogenic aggregation (Grant et al., 2007; Fawzi et al., 2008; Murray et al., 2009). Other studies have further emphasized the importance of the Gly-25–Ser-26 dipeptide in Aβ42 monomer structure by finding changes in aggregative properties upon biochemical modifications in these two residues (Roychaudhuri et al., 2014). The potential use of drugs that could alter the interactions of this dipeptide with neighboring side-chain atoms, or with the core sequence of the peptide, has been proposed.
Moreover, specific mutations in APP, such as Arctic (E22G), Italian (E22K), Dutch (E22Q), Osaka (E22Δ), and Iowa (D23N), are known to induce aggregation. These variants can cause early-onset familial AD (Benilova et al., 2012). Thus, given the biochemical and biophysical properties of Aβ peptide, and the evidence reported here, new strategies based on specific residue modifications like phosphorylation or dephosphorylation could be explored.
The suppression mechanism of neurotoxicity elicited by PI3K activation is very different from others, such as lithium treatment, where the inhibition of translation seems to be the targeted step (Sofola-Adesakin et al., 2014). The seemingly counterintuitive observation that increasing Aβ deposits is neuroprotective must be evaluated in the context of the equilibrium between soluble, albeit more toxic, oligomers and the relatively innocuous insoluble aggregates. PI3K seems to displace the equilibrium toward the insoluble aggregate forms. Given the conserved nature of PI3K pathways (Ruggero and Sonenberg, 2005), including synaptogenesis signaling (Cuesto et al., 2011; Enriquez-Barreto et al., 2014; Jordán-Álvarez et al., 2017), the data reported here invite one to consider PI3K activation as a suitable candidate to prevent AD progression. It is worth emphasizing the fact that the PI3K activation is, actually, a repair of an early deficit elicited by Aβ42 toxicity.
MATERIALS AND METHODS
Fly strains
The following strains were obtained from the Bloomington Stock Center (National Institutes of Health [NIH P40OD018537; http://flystocks.bio.indiana.edu/): elavc155-Gal4, BL-458, BL-8765, BL-8760 (Lin and Goodman, 1994; Luo et al., 1994); D42-Gal4, BL-8816 (Chan, 2002); Tubulin-Gal80TS, BL-7019 (McGuire et al., 2003); UAS-LacZ, BL-1776 (Brand and Perrimon, 1993); UAS-GFPnls, BL-4776 ; UAS-PI3K92D (Leevers et al., 1996), and UAS-PI3KCAAX, BL-8294 (Parrish et al., 2009). Driver GH298-Gal4 was a gift from Rehinard. F. Stocker (University of Fribourg; Wong et al., 2002), whereas krasavietz-Gal4 was provided by Gero Miessenbock (Oxford University, UK; Dubnau et al., 2003; Shang et al., 2007). Line UAS-Aβ42(2x) was a gift from Pedro Fernández-Fúnez (University of Florida; Casas-Tinto et al., 2011), and UAS-EB1-GFP was a gift from Melisa Rolls (Penn State University; Rolls et al., 2007). The UAS-Aβ42(2x) construct contains two copies of the gene encoding the human Aβ42 peptide, which has proven effective to cause β-amyloid deposits immune-positive for 6E10 antibody (Covance). Other UAS lines include UAS-mTOR, BL-7013 (Hennig and Neufeld, 2002); UAS-bsk, BL-9310 (Boutros et al., 1998), and UAS-MedeaRNAi, BL-31028 (Ni et al., 2009).
Immunostaining
Adults were dissected and fixed with 4% formaldehyde in phosphate-buffered saline (PBS-1X) for 20 min, washed three times with PBS-1 × 0.1% Triton X-100, and mounted in Vectashield medium with DAPI (4′,6-diamidino-2-phenylindole), or incubated with primary and secondary antibodies. The following antibodies and dilutions were used: mouse anti-Bruchpilot (nc82) 1:20 (DSHB); rabbit anti-HRP 1:200 (DSHB); mouse anti-Aβ42 (6E10) 1:1500 (Covance); mouse anti-phospho-Ser 1:200 (Abcam); rabbit anti-phospho-Tyr 1:200 (Abcam); mouse anti-phosphoSer8-Aβ42 1:1000, and rat anti-phosphoSer26-Aβ42 1:1000 (W. Jochen, Universitätklinikum Bonn, Germany); mouse anti-β-tubulin 1:10,000 (Abcam). As secondary antibodies, we used anti-mouse Alexa 488 and anti-rabbit Alexa 568. Preparations were imaged in a Leica SP5 confocal microscope and images were processed by ImageJ. Fluorescence quantification was performed with Imaris (Bitplane) software.
Adult NMJ synapse analysis
Adult females of 7, 15, and 25 (±0–3) d were dissected in order to isolate the abdominal muscles and their motor neurons. Flies were anesthetized with CO2 and immobilized with dissection pins in Silgar polymer plates with their dorsal side up. Flies were then submerged in a drop PBS-1X containing formaldehyde (4%), allowing the fixing process to start. A dorso-longitudinal cut was done along the abdomen and dissection pins were used to open the abdomen in a wide-open-book manner. Fat and not required tissues were removed. After a total fixation process of 20 min, the abdomens were washed with PBS-1X containing 1% Triton X-100 and transferred to four-well plates for immunostaining.
Synapses were revealed by the primary mouse monoclonal antibody nc82, which recognizes the CAST homologue Bruchpilot protein, a constituent of the AZ of the synapse. Anti-HRP was used to label and identify the motor neuron membrane. Samples were mounted in Vectashield medium after incubation with secondary antibodies. To quantify the AZ area, we measured the total nc82-positive signal in ventral muscle packs of motor neurons in the third abdominal segment with Imaris (Bitplane) software. This estimation procedure was required as some of the experimental conditions render the nc82 signal diffuse over the motor neuron membrane as opposed to the regular appearance as single puncta (see Results and Figure 1B).
PI3K-activating peptides
Activation of PI3K was achieved by using the peptide PTD4-PI3KAc (Cuesto et al., 2011). The peptide includes a PTD4-transduction domain that allows membrane permeabilization (Tyr-Ala-Arg-Ala-Ala-Ala-Arg-Gln-Ala-Arg-Ala; Ho et al., 2001), fused to a phosphopeptide containing the intracellular phosphorylated SH2 domain of the PDGF receptor (Gly-Ser-Asp-Gly-Gly-p-Tyr-Met-Asp-Met-Ser; Derossi et al., 1998). This peptide has been shown to induce class I PI3K activation, independently of tyrosine kinase dimerization, both in vitro and in vivo (Cuesto et al., 2011). PTD4-PI3KAc peptide was a gift from Miguel Morales (Biophysics Institute, CSIC-UPV/EHU, Spain).
Cell culture and cell viability assay
SH-SY5Y human neuroblastoma cells were purchased from the American Type Culture Collection (ref. CRL-2266). Cells were seeded at 5 × 104 cells/cm2 and used when cultures reached a 70–80% confluence. Culture media contained RPMI (Sigma-Aldrich) supplemented with 0.5 mM glutamine (Sigma-Aldrich), penicillin (50 mg/ml), streptomycin (50 U/ml) from Sigma-Aldrich, and 10% fetal bovine serum (FBS; Sigma-Aldrich). Cells were serum starved and differentiated with retinoic acid (10 μM) and RPMI containing 1% FBS for 24 h before treatment. For cell viability assays, PTD4-PI3KAc peptide was added 24 h after setting cell cultures to the wells for pretreatment studies. Aβ42 oligomers were added to the media 24 h after the first treatment of PTD4-PI3KAc. For cotreatment experiments, PTD4-PI3KAc peptide was added at the same time as Aβ42 oligomers. Finally, for posttreatment studies, PTD4-PI3KAc peptide was added to the media 24 h after oligomer addition. PTD4-PI3KAc was added to a 50 μg/ml concentration in all cases. The concentration of Aβ42 oligomers was 18, 36, or 72 μg/ml, as indicated.
Aβ42 intra- and extracellular aggregation assays
Aβ42 oligomers (36 μg/ml) and PTD4-PI3KAc (50 μg/ml) were added to the cell culture media. Cells were incubated with the stimuli for another 48-h period and then the coverslips were subject to the standard immunostaining protocol. The 6E10 immune-positive signal was analyzed in the total surface of the coverslip and quantified with Imaris (Bitplane) software. For extracellular aggregation assays, cells were seeded and differentiated as described above. In this assay, four different approaches were designed: 1) Cells supernatant was transferred to a new well, with a coverslip, after 48 h of differentiation. Aβ42 oligomers were added to the supernatant and incubated for 48 h before immunostaining. This experiment was an internal control for Aβ42 aggregation. 2) Differentiated cells were treated with PTD4-PI3KAc and incubated for 48 h; the supernatant was then transferred to a new well, with a coverslip, where Aβ42 oligomers were added and incubated for another 48 h before immunostaining. This experiment was named PI3K pretreatment. 3) The supernatant of differentiated cells was transferred to a new well, with a coverslip, after 48 h of differentiation. In the new well, the supernatant was treated with PTD4-PI3KAc and Aβ42 oligomers at the same time, and incubated for 48 h before immunostaining. This experiment was named PI3K cotreatment. 4) Differentiated cells were treated with Aβ42 oligomers for 48 h before transferring the supernatant to a new well, with a coverslip. The supernatant was then treated with PTD4-PI3KAc and incubated for another 48 h before immunostaining. This experiment was named PI3K posttreatment. The 6E10-positive dots in the total surface of the coverslip were analyzed and counted with Imaris (Bitplane) software.
Preparation of Aβ42 oligomers
Soluble oligomers were prepared by dissolving 0.3 mg Aβ42, previously resolubilized in formic acid, in 200 μl of hexafluoroisopropanol (HFIP) for 20 min, at room temperature. This Aβ42 solution (200 μl) was added to 1 ml double-distilled H2O in a siliconized Eppendorf tube. The samples were then stirred at 500 rpm using a Teflon-coated micro stir bar for 8 h at 22°C for evaporation of the HFIP and progressive formation of oligomers. Before using Aβ42 oligomers for cytotoxicity assays, samples were sonicated to break and prevent incipient fibers (Kayed and Glabe, 2006).
Protein extraction and Western blot analysis
Ten fly heads per genotype were homogenized in 20 μl of lysis buffer containing PBS-1X, 0.05% Tween-20, 150 mM NaCl, and Complete protease inhibitors (Roche) and centrifuged at 1300 rpm for 5 min. A monomerization step (see below) was done before tricine-SDS and 5% β-mercaptoethanol loading buffer were added to each sample. Samples were then boiled at 95°C for 5 min. Protein extracts were fractionated by SDS–PAGE in a 15% Bis–Tris gel under reducing conditions and electroblotted into a 0.22-μm nitrocellulose membrane. Membranes were then blocked in PBS containing 5% nonfat milk, and probed against Aβ42 (6E10) and β-tubulin antibodies. To improve reactivity of 6E10 antibody, membranes were boiled for 5 min in PBS-1X before blocking. Immunoreactive bands were visualized by enhanced chemiluminescence (ECL; Amersham). Quantification of relative expression was done from three independent experiments using a loading control for normalization. The signal intensity was quantified using ImageJ (NIH) software.
Insoluble vs soluble protein separation
Ten fly heads were hand homogenized in 240 μl of lysis buffer and centrifuged for 1 min at 1300 rpm. The supernatant was stored as the soluble fraction. The remaining pellet was resuspended in 30 μl of PBS-1X with 1% Triton X-100 and 30 μl 70% formic acid. After sonication, vortex shaking, and 800 rpm centrifugation for 1 min, the supernatant was dried with SpeedVac for 1 h. The dried pellet was finally resuspended in 240 μl of PBS-1X with 1% Triton X-100, sonicated, and centrifuged again at 1300 rpm. The resulting supernatant was stored and named insoluble fraction.
Monomerization
Monomerization buffer (20 μl; 9 M urea, 1% SDS, 25 mM Tris-HCl, 1 mM EDTA) was added to total, insoluble, or soluble protein fractions. The samples were then sonicated and incubated for 1 h at 55°C, followed by centrifugation at 14,000 rpm for 2 min. The supernatants were then mixed with loading buffer and boiled at 95°C for 5 min.
Thioflavin-S histochemistry
Female brains (7 and 15 d [±0–3 d] old) were fixed in 4% formaldehyde and permeabilized in PBS containing 0.4% Triton X-100. The brains were incubated in 50% ethanol containing 0.1% thioflavin-S (Sigma-Aldrich) for 10 min and later washed in 50% ethanol and PBS-1X.
Functional assays
For EB1-GFP live imaging of microtubule dynamics, third instar larvae were washed in PBS-1X, dried, and placed in a drop of halocarbon oil 700 and 10% chloroform mixture. Dendritic arborization of dorsal cluster sensory neurons (ddAs) live imaging was performed in the dorsal side of the extended larva in a spinning-disk microscope. Laser intensity was maintained to 15% in all experiments. The time-lapse acquisition was done for a total time of 3 min with 2-s intervals. For negative geotaxis and lifespan assays, a total number of 30–48 female flies per genotype grown at 17°C were shifted to 29°C after hatching, allowing the inactivation of the Gal80TS repressor and, consequently, Gal4/UAS system to be activated. Flies were separated and placed in plastic vials containing 10–15 of them, and kept at 29°C, 70% humidity, under a 12/12-h light/dark cycle. The counting was done at 25°C every 2–3 d in a plastic vial that was gently tapped to the bottom. The number of flies that reached the 4-cm-high threshold line was counted after 10 s of climbing; at the same time, the number of flies that reached below the 4-cm criterion line and those that stayed at the bottom of the tube, were also annotated. Each counting was repeated eight times. Lifespan assays were carried out with a similar protocol and food vials were changed every 2–3 d, and the dead flies were counted at that time. Experiments were carried out in triplicate.
The olfactory response index, OI, was obtained in a T-maze test as described previously (Acebes and Ferrús, 2001; Acebes et al., 2011). OI score represents the number of flies counted in the odorant compartment subtracted by the number of flies trapped in the control compartment, divided by the total number of flies. OI values range from 1 (total attraction) to −1 (total repulsion) in a range of 10−3–10−1 concentrations; 0 value means indifference. Each data point represents the response of 350–400 individuals aged 5–7 d old at 25°C (GH-298-Gal4) or 22–23 d old at 17°C (krasavietz-Gal4) distributed in 14–16 replicates of 25 flies that were subjected only once to one odorant concentration choice. Experiments in which more than one-third of the flies did not make any choice were discarded. All experiments were conducted in the dark at room temperature randomizing control and experimental fly groups. No significant locomotion effects were found in any of the genotypes analyzed. The two odorants employed, EB and IAA were of the highest purity (Fluka, purity >95%).
Statistical analysis
Data are shown as mean ± SD of the mean (SD), or mean ± SE of the mean (SEM), as indicated. Statistical significance was calculated using one-way analysis of variance (ANOVA) or unpaired Student’s two-tailed t test as indicated in the corresponding figure legends. Olfactory experiments were subjected to an unpaired t test with Welch’s correction, with significant differences between compared groups noted by *, p ≤ 0.05; **, p ≤ 0.01; and ***, p ≤ 0.001.
Supplementary Material
Acknowledgments
We appreciate the Bloomington Stock Center and the VDRC repository for fly strains. We thank David Van Vactor (Harvard University) for hosting M.A. during a working stay to perform the live-imaging experiments on microtubule dynamics. We also appreciate the help of Cristina Martin-Higueras for paper editing. Research was funded by Grant no. BFU2015-65685-P and Grant no. PGC2018-094630-B-100 from the Spanish Ministry of Economy.
Abbreviations used:
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- AZ
active zone
- Aβ42
amyloid beta 42 peptide
- BMP
bone morphogenetic protein
- DSHB
Drosophila Hybridome Bank
- EB1
end-binding protein 1
- GFP
green fluorescent protein
- GSK3β
gluthation S kinase 3 beta
- HRP
horseradish peroxidase
- IAA
isoamyl acetate
- LTD
long-term depression
- LTP
long-term potentiation
- MAPK
mitogen-associated kinase
- Medea
maternal-effect dominant embryonic arrest
- mTOR
mammalian target of rapamycin
- PDGF
platelet derived growth factor
- PI3K
phosphoinositide 3 kinase
- PK
protein kinase
- PS1
presenilin 1
- PSD95
postsynaptic density protein of 95 kDa
- PTEN
phosphatase and tensin homologue.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-05-0303) on December 26, 2019.
REFERENCES
- Acebes A, Devaud J-M, Arnés M, Ferrús A. (2012). Central adaptation to odorants depends on PI3K levels in local interneurons of the antennal lobe. J Neurosci , 417–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebes A, Ferrús A. (2001). Increasing the number of synapses modifies olfactory perception in Drosophila. J Neurosci , 6264–6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebes A, Martín-Peña A, Chevalier V, Ferrús A. (2011). Synapse loss in olfactory local interneurons modifies perception. J Neurosci , 2734–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acebes A, Morales M. (2012). At a PI3K crossroads: lessons from flies and rodents. Rev Neurosci , 29–37. [DOI] [PubMed] [Google Scholar]
- Arendt KL, Royo M, Fernández-Monreal M, Knafo S, Petrok CN, Martens JR, Esteban JA. (2010). PIP3 controls synaptic function by maintaining AMPA receptor clustering at the postsynaptic membrane. Nat Neurosci , 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnés M, Casas-Tintó S, Malmendal A, Ferrús A. (2017). Amyloid β42 peptide is toxic to non-neural cells in Drosophila yielding a characteristic metabolite profile and the effect can be suppressed by PI3K. Biol Open , 1664–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashe KH, Zahs KR. (2010). Probing the biology of Alzheimer’s disease in mice. Neuron , 631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Rajakaruna S, Menko AS. (2012). Insulin-like growth factor receptor-1 and nuclear factor κB are crucial survival signals that regulate caspase-3-mediated lens epithelial cell differentiation initiation. J Biol Chem , 8384–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I, Karran E, De Strooper B. (2012). The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci , 349–357. [DOI] [PubMed] [Google Scholar]
- Boutros M, Paricio N, Strutt DI, Mlodzik M. (1998). Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell , 109–118. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development (Cambridge, England) , 401–415. [DOI] [PubMed] [Google Scholar]
- Casas-Tintó S, Arnés M, Ferrús A. (2017). Drosophila enhancer-Gal4 lines show ectopic expression during development. R Soc Open Sci , 170039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas-Tinto S, Zhang Y, Sanchez-Garcia J, Gomez-Velazquez M, Rincon-Limas DE, Fernandez-Funez P. (2011). The ER stress factor XBP1s prevents amyloid-β neurotoxicity. Hum Mol Genet , 2144–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. (2003). Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci , 267–298. [DOI] [PubMed] [Google Scholar]
- Chan HYE. (2002). Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet , 2895–2904. [DOI] [PubMed] [Google Scholar]
- Chiang H-C, Wang L, Xie Z, Yau A, Zhong Y. (2010). PI3 kinase signaling is involved in Aβ-induced memory loss in Drosophila. Proc Natl Acad Sci USA , 7060–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S-L, Chen C-M, Cline HT. (2008). Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron , 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S-H. (2009). Aberrant phosphorylation in the pathogenesis of Alzheimer’s disease. BMB Rep , 467–474. [DOI] [PubMed] [Google Scholar]
- Corbett NJ, Hooper NM. (2018). Soluble amyloid precursor protein α: friend or foe? Adv Exp Med Biol , 177–183. [DOI] [PubMed] [Google Scholar]
- Crews L, Adame A, Patrick C, Delaney A, Pham E, Rockenstein E, Hansen L, Masliah E. (2010). Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J Neurosci , 12252–12262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Miranda E, Page R, Curry JA, Duthie FAI, Gubb DC, Lomas DA. (2005). Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience , 123–135. [DOI] [PubMed] [Google Scholar]
- Crowther DC, Kinghorn KJ, Page R, Lomas DA. (2004). Therapeutic targets from a Drosophila model of Alzheimer’s disease. Curr Opin Pharmacol , 513–516. [DOI] [PubMed] [Google Scholar]
- Cuesto G, Enriquez-Barreto L, Caramés C, Cantarero M, Gasull X, Sandi C, Ferrús A, Acebes Á, Morales M. (2011). Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J Neurosci , 2721–2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesto G, Jordán-Álvarez S, Enriquez-Barreto L, Ferrús A, Morales M, Acebes Á. (2015). GSK3β inhibition promotes synaptogenesis in Drosophila and mammalian neurons. PLoS One , e0118475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DaRocha-Souto B, Scotton TC, Coma M, Serrano-Pozo A, Hashimoto T, Serenó L, Rodríguez M, Sánchez B, Hyman BT, Gómez-Isla T. (2011). Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol , 360–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice FG, Munoz DP. (2016). Opportunities and challenges in developing relevant animal models for Alzheimer’s disease. Ageing Res Rev , 112–114. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Vassar R, Golde T. (2010). The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol , 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derossi D, Williams EJ, Green PJ, Dunican DJ, Doherty P. (1998). Stimulation of mitogenesis by a cell-permeable PI 3-kinase binding peptide. Biochem Biophys Res Commun , 148–152. [DOI] [PubMed] [Google Scholar]
- Dong H-T, Bai Y. (2003). [Clinical research of the influence of cognizing potential on AD patients by electroacupuncture treatment]. Zhongguo Ying Yong Sheng Li Xue Za Zhi , 94–96. [PubMed] [Google Scholar]
- Dubnau J, Chiang A-S, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, et al (2003). The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol , 286–296. [DOI] [PubMed] [Google Scholar]
- Dyer AH, Vahdatpour C, Sanfeliu A, Tropea D. (2016). The role of insulin-like growth factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience , 89–99. [DOI] [PubMed] [Google Scholar]
- el-Moatassim C, Dornand J, Mani JC. (1992). Extracellular ATP and cell signalling. Biochim Biophys Acta , 31–45. [DOI] [PubMed] [Google Scholar]
- Enriquez-Barreto L, Cuesto G, Dominguez-Iturza N, Gavilán E, Ruano D, Sandi C, Fernández-Ruiz A, Martín-Vázquez G, Herreras O, Morales M. (2014). Learning improvement after PI3K activation correlates with de novo formation of functional small spines. Front Mol Neurosci , 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL. (2013). Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol , 104–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawzi NL, Phillips AH, Ruscio JZ, Doucleff M, Wemmer DE, Head-Gordon T. (2008). Structure and dynamics of the Aβ(21–30) peptide from the interplay of NMR experiments and molecular simulations. J Am Chem Soc , 6145–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franciscovich AL, Mortimer ADV, Freeman AA, Gu J, Sanyal S. (2008). Overexpression screen in Drosophila identifies neuronal roles of GSK-3 β/shaggy as a regulator of AP-1-dependent developmental plasticity. Genetics , 2057–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. (2010). Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol , 523–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goure WF, Krafft GA, Jerecic J, Hefti F. (2014). Targeting the proper amyloid-β neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther , 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourine AV, Dale N, Llaudet E, Poputnikov DM, Spyer KM, Gourine VN. (2007). Release of ATP in the central nervous system during systemic inflammation: real-time measurement in the hypothalamus of conscious rabbits. J Physiol (Pt 1), 305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, Yamin G, Rigby AC, Teplow DB. (2007). Familial Alzheimer’s disease mutations alter the stability of the amyloid β-protein monomer folding nucleus. Proc Natl Acad Sci USA , 16522–16527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve I, Kretzschmar D, Tschäpe J-A, Beyn A, Brellinger C, Schweizer M, Nitsch RM, Reifegerste R. (2004). Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci , 3899–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm MOW, Mett J, Stahlmann CP, Haupenthal VJ, Zimmer VC, Hartmann T. (2013). Neprilysin and Aβ clearance: impact of the APP intracellular domain in NEP regulation and implications in Alzheimer’s disease. Front Aging Neurosci , 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedes-Dias P, Nirschl JJ, Abreu N, Tokito MK, Janke C, Magiera MM, Holzbaur ELF. (2019). Kinesin-3 responds to local microtubule dynamics to target synaptic cargo delivery to the presynapse. Curr Biol , 268–282.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Kaether C, Thinakaran G, Sisodia S. (2012). Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med , a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. (1992). Alzheimer’s disease: the amyloid cascade hypothesis. Science , 184–185. [DOI] [PubMed] [Google Scholar]
- Härtig W, Goldhammer S, Bauer U, Wegner F, Wirths O, Bayer TA, Grosche J. (2010). Concomitant detection of β-amyloid peptides with N-terminal truncation and different C-terminal endings in cortical plaques from cases with Alzheimer’s disease, senile monkeys and triple transgenic mice. J Chem Neuroanat , 82–92. [DOI] [PubMed] [Google Scholar]
- Hennig KM, Neufeld TP. (2002). Inhibition of cellular growth and proliferation by dTOR overexpression in Drosophila. Genesis , 107–110. [DOI] [PubMed] [Google Scholar]
- Ho A, Schwarze SR, Mermelstein SJ, Waksman G, Dowdy SF. (2001). Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Res , 474–477. [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. (2008). The GSK3 hypothesis of Alzheimer’s disease. J Neurochem , 1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Chiang H-C, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. (2008). Aβ42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS One , e1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima K, Liu H-P, Chiang A-S, Hearn SA, Konsolaki M, Zhong Y. (2004). Dissecting the pathological effects of human Aβ40 and Aβ42 in Drosophila: a potential model for Alzheimer’s disease. Proc Natl Acad Sci USA , 6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Iijima K. (2010). Transgenic Drosophila models of Alzheimer’s disease and tauopathies. Brain Struct Funct , 245–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordán-Álvarez S, Santana E, Casas-Tintó S, Acebes Á, Ferrús A. (2017). The equilibrium between antagonistic signaling pathways determines the number of synapses in Drosophila. PLoS One , e0184238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Benoist M, Lario A, Knafo S, Petrok CN, Esteban JA. (2010). PTEN is recruited to the postsynaptic terminal for NMDA receptor-dependent long-term depression. EMBO J , 2827–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang MJ, Hansen TJ, Mickiewicz M, Kaczynski TJ, Fye S, Gunawardena S. (2014). Disruption of axonal transport perturbs bone morphogenetic protein (BMP)–signaling and contributes to synaptic abnormalities in two neurodegenerative diseases. PLoS One , e104617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Glabe CG. (2006). Conformation-dependent anti-amyloid oligomer antibodies. Methods Enzymol , 326–344. [DOI] [PubMed] [Google Scholar]
- Knafo S, Sánchez-Puelles C, Palomer E, Delgado I, Draffin JE, Mingo J, Mingo J, Wahle T, Kaleka K, Mou L. (2016). PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat Neurosci , 443–453. [DOI] [PubMed] [Google Scholar]
- Kollhoff AL, Howell JC, Hu WT. (2018). Automation vs. experience: measuring Alzheimer’s β-amyloid 1–42 peptide in the CSF. Front Aging Neurosci , 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Rezaei-Ghaleh N, Terwel D, Thal DR, Richard M, Hoch M, Mc Donald JM, Wüllner U, Glebov K, Heneka MT, et al (2011). Extracellular phosphorylation of the amyloid β-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J , 2255–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Singh S, Hinze D, Josten M, Sahl H-G, Siepmann M, Walter J. (2012). Phosphorylation of amyloid-β peptide at serine 8 attenuates its clearance via insulin-degrading and angiotensin-converting enzymes. J Biol Chem , 8641–8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Walter J. (2011). Phosphorylation of amyloid beta (Aβ) peptides—a trigger for formation of toxic aggregates in Alzheimer’s disease. Aging , 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wirths O, Stüber K, Wunderlich P, Koch P, Theil S, Rezaei-Ghaleh N, Zweckstetter M, Bayer TA, Brüstle O, et al (2016). Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol , 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Wirths O, Theil S, Gerth J, Bayer TA, Walter J. (2013). Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease. Acta Neuropathol , 699–709. [DOI] [PubMed] [Google Scholar]
- Kummer MP, Heneka MT. (2014). Truncated and modified amyloid-beta species. Alzheimers Res Ther , 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Beach TG, Sue LI, Scott S, Layne KJ, Kokjohn TA, Kalback WM, Luehrs DC, Vishnivetskaya TA, Abramowski D, et al (2001). The evolution of Aβ peptide burden in the APP23 transgenic mice: implications for Aβ deposition in Alzheimer disease. Mol Med , 609–618. [PMC free article] [PubMed] [Google Scholar]
- Kuo YM, Emmerling MR, Woods AS, Cotter RJ, Roher AE. (1997). Isolation, chemical characterization, and quantitation of Aβ 3-pyroglutamyl peptide from neuritic plaques and vascular amyloid deposits. Biochem Biophys Res Commun , 188–191. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. (2007). Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci , 499–509. [DOI] [PubMed] [Google Scholar]
- Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. (1996). The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J , 6584–6594. [PMC free article] [PubMed] [Google Scholar]
- Li K, Wei Q, Liu F-F, Hu F, Xie A-J, Zhu L-Q, Liu D. (2018). Synaptic dysfunction in Alzheimer’s disease: Aβ, tau, and epigenetic alterations. Mol Neurobiol , 3021–3032. [DOI] [PubMed] [Google Scholar]
- Lien EC, Dibble CC, Toker A. (2017). PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol , 62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. (1994). Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron , 507–523. [DOI] [PubMed] [Google Scholar]
- Lin J-Y, Wang W-A, Zhang X, Liu H-Y, Zhao X-L, Huang F-D. (2014). Intraneuronal accumulation of Aβ42 induces age-dependent slowing of neuronal transmission in Drosophila. Neurosci Bull , 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Martín M, Jurado J, Hernández F, Avila J. (2014). GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci , 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüchtenborg A-M, Katanaev VL. (2014). Lack of evidence of the interaction of the Aβ peptide with the Wnt signaling cascade in Drosophila models of Alzheimer’s disease. Mol Brain , 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Liao YJ, Jan LY, Jan YN. (1994). Distinct morphogenetic functions of similar small GTPases: Drosophila Drac1 is involved in axonal outgrowth and myoblast fusion. Genes Dev , 1787–1802. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem , 13375–13378. [DOI] [PubMed] [Google Scholar]
- Martín-Peña A, Acebes A, Rodríguez J-R, Sorribes A, de Polavieja GG, Fernández-Fúnez P, Ferrús A. (2006). Age-independent synaptogenesis by phosphoinositide 3 kinase. J Neurosci , 10199–10208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Peña A, Rincon-Limas DE, Fernandez-Funez P. (2017). Anti-Aβ single-chain variable fragment antibodies restore memory acquisition in a Drosophila model of Alzheimer’s disease. Sci Rep , 11268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Peña A, Rincón-Limas DE, Fernandez-Fúnez P. (2018). Engineered Hsp70 chaperones prevent Aβ42-induced memory impairments in a Drosophila model of Alzheimer’s disease. Sci Rep , 9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL. (2003). Spatiotemporal rescue of memory dysfunction in Drosophila. Science , 1765–1768. [DOI] [PubMed] [Google Scholar]
- Melani A, Turchi D, Vannucchi MG, Cipriani S, Gianfriddo M, Pedata F. (2005). ATP extracellular concentrations are increased in the rat striatum during in vivo ischemia. Neurochem Int , 442–448. [DOI] [PubMed] [Google Scholar]
- Mhatre SD, Satyasi V, Killen M, Paddock BE, Moir RD, Saunders AJ, Marenda DR. (2014). Synaptic abnormalities in a Drosophila model of Alzheimer’s disease. Dis Model Mech , 373–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton NG. (2001). Phosphorylation of amyloid-beta at the serine 26 residue by human cdc2 kinase. Neuroreport , 3839–3844. [DOI] [PubMed] [Google Scholar]
- Mokhtar SH, Bakhuraysah MM, Cram DS, Petratos S. (2013). The beta-amyloid protein of Alzheimer’s disease: communication breakdown by modifying the neuronal cytoskeleton. Int J Alzheimers Dis , 910502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreth J, Kroker KS, Schwanzar D, Schnack C, von Arnim CAF, Hengerer B, Rosenbrock H, Kussmaul L. (2013). Globular and protofibrillar aβ aggregates impair neurotransmission by different mechanisms. Biochemistry , 1466–1476. [DOI] [PubMed] [Google Scholar]
- Moreth J, Mavoungou C, Schindowski K. (2013). Passive anti-amyloid immunotherapy in Alzheimer’s disease: what are the most promising targets? Immun Ageing , 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison EE, Wardleworth BN, Askham JM, Markham AF, Meredith DM. (1998). EB1, a protein which interacts with the APC tumour suppressor, is associated with the microtubule cytoskeleton throughout the cell cycle. Oncogene , 3471–3477. [DOI] [PubMed] [Google Scholar]
- Mueed Z, Tandon P, Maurya SK, Deval R, Kamal MA, Poddar NK. (2019). Tau and mTOR: the hotspots for multifarious diseases in Alzheimer’s development. Front Neurosci , 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami K, Uno M, Masuda Y, Shimizu T, Shirasawa T, Irie K. (2008). Isomerization and/or racemization at Asp23 of Aβ42 do not increase its aggregative ability, neurotoxicity, and radical productivity in vitro. Biochem Biophys Res Commun , 745–751. [DOI] [PubMed] [Google Scholar]
- Murray MM, Krone MG, Bernstein SL, Baumketner A, Condron MM, Lazo ND, Teplow DB, Wyttenbach T, Shea JE, Bowers MT. (2009). Amyloid β-protein: experiment and theory on the 21–30 fragment. J Phys Chem B , 6041–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng M, Roorda RD, Lima SQ, Zemelman BV, Morcillo P, Miesenböck G. (2002). Transmission of olfactory information between three populations of neurons in the antennal lobe of the fly. Neuron , 463–474. [DOI] [PubMed] [Google Scholar]
- Ni J-Q, Liu L-P, Binari R, Hardy R, Shim H-S, Cavallaro A, Booker M, Pfeiffer BD, Markstein M, Wang H, et al (2009). A Drosophila resource of transgenic RNAi lines for neurogenetics. Genetics , 1089–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish JZ, Xu P, Kim CC, Jan LY, Jan YN. (2009). The microRNA bantam functions in epithelial cells to regulate scaling growth of dendrite arbors in Drosophila sensory neurons. Neuron , 788–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redegeld FA, Caldwell CC, Sitkovsky MV. (1999). Ecto-protein kinases: ecto-domain phosphorylation as a novel target for pharmacological manipulation? Trends Pharmacol Sci , 453–459. [DOI] [PubMed] [Google Scholar]
- Rezaei-Ghaleh N, Amininasab M, Giller K, Kumar S, Stündl A, Schneider A, Becker S, Walter J, Zweckstetter M. (2014). Turn plasticity distinguishes different modes of amyloid-β aggregation. J Am Chem Soc , 4913–4919. [DOI] [PubMed] [Google Scholar]
- Rolls MM, Satoh D, Clyne PJ, Henner AL, Uemura T, Doe CQ. (2007). Polarity and intracellular compartmentalization of Drosophila neurons. Neural Dev , 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychaudhuri R, Lomakin A, Bernstein S, Zheng X, Condron MM, Benedek GB, Bowers M, Teplow DB. (2014). Gly25-Ser26 amyloid β-protein structural isomorphs produce distinct Aβ42 conformational dynamics and assembly characteristics. J Mol Biol , 2422–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Sonenberg N. (2005). The Akt of translational control. Oncogene , 7426–7434. [DOI] [PubMed] [Google Scholar]
- Shaltiel S, Schvartz I, Korc-Grodzicki B, Kreizman T. (1993). Evidence for an extra-cellular function for protein kinase A. Mol Cell Biochem , 283–291. [DOI] [PubMed] [Google Scholar]
- Shang Y, Claridge-Chang A, Sjulson L, Pypaert M, Miesenböck G. (2007). Excitatory local circuits and their implications for olfactory processing in the fly antennal lobe. Cell , 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Watanabe A, Ogawara M, Mori H, Shirasawa T. (2000). Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch Biochem Biophys , 225–234. [DOI] [PubMed] [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, et al (1992). Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science , 126–129. [DOI] [PubMed] [Google Scholar]
- Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C, Hardy J, Lovestone S, Partridge L. (2010). Inhibition of GSK-3 ameliorates Aβ pathology in an adult-onset Drosophila model of Alzheimer’s disease. PLoS Genet , e1001087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola-Adesakin O, Castillo-Quan JI, Rallis C, Tain LS, Bjedov I, Rogers I, Li L, Martinez P, Khericha M, Cabecinha M, et al (2014). Lithium suppresses Aβ pathology by inhibiting translation in an adult Drosophila model of Alzheimer’s disease. Front Aging Neurosci , 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M. (2010). Biochemical and biophysical features of both oligomer/fibril and cell membrane in amyloid cytotoxicity. FEBS J , 4602–4613. [DOI] [PubMed] [Google Scholar]
- Turner AJ, Fisk L, Nalivaeva NN. (2004). Targeting amyloid-degrading enzymes as therapeutic strategies in neurodegeneration. Ann NY Acad Sci , 1–20. [DOI] [PubMed] [Google Scholar]
- Uddin MS, Stachowiak A, Mamun AAl, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, Stankiewicz AM. (2018). Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci , 04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verberk IMW, Slot RE, Verfaillie SCJ, Heijst H, Prins ND, van Berckel BNM, Scheltens P, Teunissen CE, van der Flier WM. (2018). Plasma amyloid as prescreener for the earliest Alzheimer pathological changes. Ann Neurol , 648–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Kinzel V, Kübler D. (1994). Evidence for CKI and CKII at the cell surface. Cell Mol Biol Res , 473–480. [PubMed] [Google Scholar]
- Weaver C, Leidel C, Szpankowski L, Farley NM, Shubeita GT, Goldstein LSB. (2013). Endogenous GSK-3/shaggy regulates bidirectional axonal transport of the amyloid precursor protein. Traffic , 295–308. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Sigurdsson EM. (2010). Murine models of Alzheimer’s disease and their use in developing immunotherapies. Biochim Biophys Acta , 847–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisotzkey RG, Mehra A, Sutherland DJ, Dobens LL, Liu X, Dohrmann C, Attisano L, Raftery LA. (1998). Medea is a Drosophila Smad4 homolog that is differentially required to potentiate DPP responses. Development , 1433–1445. [DOI] [PubMed] [Google Scholar]
- Wong AM, Wang JW, Axel R. (2002). Spatial representation of the glomerular map in the Drosophila protocerebrum. Cell , 229–241. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, et al (2015). Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci , 978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.