

Abstract
Eisosomes are membrane furrows at the cell surface of yeast that have been shown to function in two seemingly distinct pathways, membrane stress response and regulation of nutrient transporters. We found that many stress conditions affect both of these pathways by changing plasma membrane tension and thus the morphology and composition of eisosomes. For example, alkaline stress causes swelling of the cell and an endocytic response, which together increase membrane tension, thereby flattening the eisosomes. The flattened eisosomes affect membrane stress pathways and release nutrient transporters, which aids in their down-regulation. In contrast, glucose starvation or hyperosmotic shock causes cell shrinking, which results in membrane slack and the deepening of eisosomes. Deepened eisosomes are able to trap nutrient transporters and protect them from rapid endocytosis. Therefore, eisosomes seem to coordinate the regulation of both membrane tension and nutrient transporter stability.
INTRODUCTION
Budding yeast Saccharomyces cerevisiae developed efficient stress response systems that allow adaptation of this unicellular organism to rapidly changing environmental conditions. Recent studies identified eisosomes as integral parts of major stress response pathways. Eisosomes are furrows in the plasma membrane of yeast and other fungi that represent stable membrane domains with unique lipid and protein compositions. The membrane of eisosomes is thicker than the surrounding membrane (Bharat et al., 2018) and it is enriched in sphingolipids and ergosterol (Grossmann et al., 2007; Stradalova et al., 2009). Therefore, eisosomes have the characteristics of lipid rafts and other related lipid domains such as caveolae. Several transmembrane and membrane-associated proteins have been identified that play key roles in the formation and function of eisosomes. Among them are Pil1 and Lsp1, two homologous BAR domain proteins that form a half-pipe-shaped polymer associated with the curved base of the eisosome, and tetraspan proteins including Nce102 that might cluster lipids and transmembrane proteins to form the membrane domain of the eisosome (for a review, see Douglas and Konopka, 2014).
Two functions have been attributed to eisosomes: regulation of amino acid-polyamine-organocation (APC) transporters (Bianchi et al., 2018; Gournas et al., 2018; Moharir et al., 2018) and sensing of membrane stress (Athanasopoulos et al., 2015; Kabeche et al., 2015). APC transporters are evolutionary conserved nutrient transporters that import small molecules such as amino acids and nucleobases. Based on detailed studies of a few of these transporters, it has been predicted that the 26 yeast APC transporters use the proton gradient across the plasma membrane to drive import of the nutrients. Activity of APC transporters and thus the import of the nutrients they pump is mainly regulated by endocytosis and degradation of these proteins. The rate-determining and key regulatory step of this down-regulation is the ubiquitination of the transporters by the ubiquitin ligase Rsp5, which tags the APC transporters for endocytosis and lysosomal degradation via the multivesicular body pathway. Ubiquitination efficiency is affected by both the activity of transporter (pumping transporters expose degron-like sequences; Keener and Babst, 2013) and the regulation of Rsp5 adaptor proteins, called ARTs (Lin et al., 2008; Nikko et al., 2008). In addition, eisosomes function as storage compartments of APC transporters that protect inactive transporters from ubiquitination (Grossmann et al., 2008; Spira et al., 2012; Gournas et al., 2018; Moharir et al., 2018). This is accomplished possibly by the special lipid environment of the eisosomes that is predicted to stabilize the ground state of the transporters and/or by blocking access of the Rsp5 ubiquitin ligase to the eisosome-localized transporter.
The membrane stress sensing function of eisosomes is linked to the two homologue proteins Slm1 and Slm2 that localize to eisosomes (Kamble et al., 2011; Olivera-Couto et al., 2011). These proteins contain BAR domains and PIP2 (phosphoinositide-4,5 bisphosphate)-binding PH domains. Membrane stress or tension causes the release of Slm1/2 from eisosomes and the association of these proteins with the Tor complex 2 (TORC2) signaling complex, which activates TORC2 kinase and triggers a membrane stress response (Berchtold et al., 2012; Niles et al., 2012). One consequence of this stress response is the phosphorylation and inactivation of Orm1 and Orm2, two ER proteins that inhibit sphingolipid synthesis. Orm1/2 are phosphorylated by Ypk1/2, the major effector kinases that operate directly downstream of TORC2 (Roelants et al., 2011). TORC2–Ypk1/2-mediated inactivation of Orm1/2 causes up-regulation of sphingolipid synthesis, which ultimately helps to relieve membrane tension. Furthermore, it has been suggested that during acute membrane tension eisosomes flatten, thereby providing additional membrane to the cell surface (Kabeche et al., 2015). However, this model was based on data obtained from osmotically stressed spheroplasts (yeast without cell wall), resulting in nonphysiological swelling of the cells.
In our study we revisited the response of eisosomes to different stress conditions and found a link between membrane tension and APC transporter regulation, explaining why these two types of regulation localize to the same structure. Furthermore, we found an unexpected delay in the derepression of sphingolipid synthesis on membrane stress, suggesting other sources contribute membrane to the cell surface during acute cell expansion.
RESULTS
The addition of 50 mM Tris, pH 8, buffer to the growth medium of a yeast culture increases the extracellular pH from ∼4 to ∼7.5, thereby disrupting the plasma membrane proton gradient. This high-pH stress causes rapid endocytosis and down-regulation of APC transporters, nutrient transporters that depend on the proton gradient for import activity (Moharir et al., 2018; see also Figure 4A later in this article). In addition to APC transporter endocytosis, loss of the proton gradient also results in restructuring of eisosomes; the membrane compartments that harbor APC transporters. Specifically we observed on pH shift the redistribution of the eisosome components Nce102 and Slm1, whereas Pil1 remained associated with eisosomes (Moharir et al., 2018). The move of Nce102 out of eisosomes was of particular interest since mutation of this eisosome component has been shown to result in the formation of shallow eisosomes or eisosome remnants (Loibl et al., 2010). This observation suggested that Tris treatment might mimic the loss of Nce102 function and thus cause eisosomes to flatten.
FIGURE 4:

Effect of different stresses on Fur4 localization and on cell surface area. (A) Fluoresence microscopy of wild-type, end3-1, and rsp5-1 cells expressing Fur4-GFP (SEY6210 pJK19, BWY1346 pJK19, MYY880 pJK19). The pictures show a single optical section through the center of the cells. Cells were grown in SDcomp-ura or in the case of the hypoosmotic shock (-sorbitol) in SDcomp-ura +1 M sorbitol. These cells were shifted into medium either containing 50 mM Tris, pH 8, containing 1 M sorbitol (+sorbitol), lacking glucose (-glucose), or lacking sorbitol (-sorbitol). (B) Quantification of the cell surface area change of 50 cells after treatment in the microfluidics system (15 min treatment if not indicated otherwise). The black line indicates the median of each data set.
To quantify the redistribution of Nce102, we analyzed by fluorescence microscopy cells expressing Nce102-mCherry growing in a microfluidics system, which allowed us to observe the same cells before and after the addition of Tris buffer to the growth medium. We quantified the effect of the Tris treatment on Nce102 localization using software that is part of the microscope analysis tools called 2D Polygon Analysis. The algorithm identifies objects of a certain size and with a brightness above a certain threshold relative to the surrounding area (see Materials and Methods). In wild type, the addition of 50 mM Tris, pH 8, caused a large portion of Nce102 to leave eisosomes. This redistribution reduced the eisosome signal of Nce102 and increased the signal of the surrounding membrane (see example in Figure 1C). As a consequence, the polygon analysis no longer recognized many of these objects, resulting in an ∼90% drop in the number of Nce102-labeled eisosomes (15 min after the addition of 50 mM Tris, pH 8; #2 in Figure 1, A and B). In contrast, the polygon analysis on mock-treated samples showed no reduction in Nce102 structures (#1 in Figure 1, A and B). It should be noted that the algorithm recognized only objects that are clearly brighter than the surrounding area (set by a threshold parameter) and thus the polygon analysis did not count all eisosomes present (see example in Figure 1C). However, the algorithm used the same parameters to analyze the microscopy pictures of the same cells before and after treatment and therefore provided a reliable and unbiased analysis of the change in Nce102 localization.
FIGURE 1:

Eisosomes respond differently to various stress conditions. (A) Wild type expressing Nce102-mCherry (AMY4) was monitored by fluorescence microscopy for changes in localization of Nce102. The pictures show the projection of 10 optical sections representing the bottom half of the cell. Cells were maintained in a microfluidics system under optimal growth conditions (SDcomp medium at 30°C) (1) and then shifted to different stress conditions for 15 min. To disrupt the proton gradient, 50 mM Tris buffer, pH 8, was added to conditioned growth medium (medium obtained by centrifugation of the starting culture, which is identical to the medium of the control cells), which changed the pH from 4 to 7.5 (2). For glucose and leucine starvation, SDcomp medium buffered to pH 4 was used lacking either glucose or leucine (3 and 4). For the hypoosmotic shock (1 M > 0 M sorbitol) (5), cells were grown in SDcomp + 1 M sorbitol and shifted to conditioned SDcomp lacking sorbitol (this medium was obtained from a parallel culture that grew to the same cell density in absence of sorbitol). (B) Quantification of eisosomes using a 2D-polygon analysis (see Materials and Methods). The error bars indicate the SD of four to five analyzed microscopy pictures containing on average ∼40 cells each. (C) Example of the 2D polygon analysis used to quantify changes in Nce102 localization (B). The microscopy pictures are identical to those shown in A2. Red outlines mark the by the algorithm identified objects. The line graph shows the intensity profile along the line indicated in the micrographs. The intensity profiles were standardized to the last peak (set to 1.0). (D) Wild-type cells expressing Nce102-mCherry (AMY4) were grown in SDcomp + 1 M sorbitol, shifted for 3 min to conditioned medium lacking sorbitol, and then shifted back for 3 min to conditioned medium containing 1 M sorbitol. (E) Wild-type, end3-1, and rsp5-1 cells expressing Nce102-mCherry (AMY4, DAY20, DAY21) grown in SDcomp were treated with 50 mM Tris buffer, pH 8, in conditioned medium either in the presence or in the absence of 1 M sorbitol.
The reduced eisosome signal of Nce102 after Tris treatment might suggest a reduction in the depth of the membrane furrows under these stress conditions. The depth of wild-type eisosomes is in the range of 50 nm (Stradalova et al., 2009), too small for standard fluorescence microscopy to observe. Therefore, we attempted to determine a potential Tris-induced change in eisosome morphology using transmission electron microscopy (TEM). In a first set of experiments, we prepared control samples for TEM either by plunge freezing followed by chemical fixation or by direct chemical fixation, and in both cases, only few and very small invaginations of the plasma membrane were observed (see control in Figure 10 later in this article), suggesting that these methods were not able to preserve eisosomes. After testing additional sample preparations for TEM, we identified one method that was able to preserve eisosome-like structures at the plasma membrane (examples in Figure 2A; see Materials and Methods). We used this method to analyze cells that were chemically fixed either before or 15 min after the addition of 50 mM Tris, pH 8. The resulting micrographs showed eisosome-like structures (30–80 nm deep invaginations) both in the treated and in the untreated samples (Figure 2A). In fact, the Tris-treated sample showed a slight increase in the number of invaginations per plasma membrane surface area (from ∼0.9 to ∼1.3/ µm2; Figure 2A). These TEM results suggested that, in contrast to our predictions, the redistribution of Nce102 observed during alkaline stress did not cause a loss of eisosome structures; rather, it increased the presence of these membrane invaginations. However, we noticed in the micrographs that the plasma membranes of the Tris-treated cells were detached from the cell wall and the average size of these cells was reduced compared with untreated samples (Figure 2A). In contrast, fluorescence microscopy of live cells indicated that Tris treatment resulted in swelling of the cells (Figure 4B, discussed later), suggesting the TEM data on Tris-treated cells are questionable. Chemical fixation causes loss of plasma membrane integrity and as a consequence loss of membrane tension (cells lose their turgor pressure). If the morphology of eisosomes is affected by the tension of the plasma membrane, chemical fixation is likely to result in artifacts.
FIGURE 10:

Glucose starvation causes shrinking of the cells. Wild-type cells (BY4741) were glucose starved for 3 or 24 h and analyzed by TEM. Deep invaginations of the plasma membrane are marked by arrowheads.
FIGURE 2:

Plasma membrane morphology observed by TEM. (A) The micrographs show representative cells either before or 15 min after Tris treatment. Black arrowheads indicate invaginations at the plasma membrane. The calculation of the number of invaginations per µm2 was based on the assumption that the invaginations represent membrane furrows (eisosomes) with a length of 300 nm (the number of invaginations found in the 60 nm-thin sections was corrected by dividing by 5). The numbers represent the mean ± SD. (B) The changes in cell size after chemical fixation was determined using fluorescence microscopy of cells expressing Fur4-GFP. Cells were imaged in a microfluidics system and the cell diameter of the same cells before and after 15 min of fixation was measured.
To test the effect of chemical fixation on cell size (and thus on membrane tension) we performed the fixation procedure used for the EM sample preparations in the microfluidics system and measured by fluorescence microscopy the diameter of 50 Tris-treated cells before and after addition of the fixation solution. The data indicated that chemical fixation caused on average a 9% decrease in cell diameter, resulting in a 17% lower surface area (Figure 2B). This observation supported the notion that the EM sample preparation we used for the experiments in Figure 2A resulted in the loss of turgor pressure and thus caused an artificial shrinking of the cell.
Because EM-based analysis of eisosomes seemed to be prone to artifact, we analyzed potential changes in eisosome depth by superresolution microscopy. This technique commonly uses photoactivatable or blinking dyes, which allows the detection of single fluorescent molecules and the calculation of the center of the point spread function. GFP and mCherry are not considered blinking dyes and therefore are not commonly used in superresolution microscopy. In fact, techniques have been developed using GFP-targeted antibody-dye conjugates to overcome this problem (Platonova et al., 2015). Surprisingly, we were able to obtain superresolution images of live cells expressing Pil1-mCherry (BAR domain protein localizing to the curved part of the furrow) and Fur4-GFP (APC transporter that imports uracil) that showed clear separation of the two eisosome-localized proteins (Figure 3A). GFP and GFP derivatives have been shown to blink for short periods of time (Dickson et al., 1997) and together with the high concentration of tagged proteins at the eisosomes, it seems that enough blinking events were identified by the microscope software to allow for high precision localization of Pil1 and Fur4 (∼25 nm resolution in X/Y and ∼50 nm in Z). The results in Figure 3A show the cross-section of yeast cells growing in a microfluidics chamber under optimal conditions (30°C, SDcomp-ura medium). In these micrographs, Pil1 localized to small structures that most likely represent eisosomes. Half of these structures were separated by 50–80 nm from the Fur4-labeled structures (from center to center of labeled structures; Figure 3C), suggesting that in these structures Fur4 localized closer to the surface of the eisosome, whereas Pil1 was concentrated at the curved bottom of the membrane furrow (see model in Figure 11 later in this article). A similar difference in distribution has been observed by electron microscopy between Pil1 and the eisosome component Sur7 (Stradalova et al., 2009). In contrast, conventional fluorescence microscopy is not able to resolve the difference in Fur4 versus Pil1 localization (Figure 3B).
FIGURE 3:

Loss of proton gradient causes flattening of eisosomes. (A) Superresolution microscopy of live yeast cells in a microfluidics system. The pictures show a single optical section of 50 nm through the center of the cells. Wild-type or end3-1 cells expressing Pil1-mCherry and Fur4-GFP (AMY6 pJK19, DAY27 pJK19) were grown in SDcomp-ura and shifted to conditioned medium containing 50 mM Tris, pH 8. To determine the effect of glucose starvation, cells expressing a stabilized, N-terminally deleted Fur4-GFP, Fur4(∆N)-GFP (AMY6 pJK30; the N-terminal deletion prevents ubiquitination and thus impairs down-regulation of Fur4 [ Keener and Babst, 2013]) were shifted to SDcomp-ura medium adjusted to pH 4 and lacking glucose. Because of the rapid endocytic response, after 15 min glucose starvation, wild-type Fur4-GFP was no longer present at the plasma membrane. (B) Fluorescence microscopy of wild type expressing Pil1-mCherry and Fur4-GFP (AMY6 pJK19). The picture shows a single optical section comparable with the superresolution pictures shown in A. (C) Quantification of Fur4-Pil1 colocalization of over 400 eisosomes (numbers of eisosomes in the control cells indicated; 30–40 cells). Colocalization was defined by <25 nm distance between the centers of the Fur4 and Pil1 signals.
FIGURE 11:

Model of the response of eisosomes to high membrane tension. Under optimal growth conditions, eisosomes harbor inactive APC-type nutrient transporters that are in an equilibrium with active transporters outside of eisosomes. The tetraspan protein Nce102 and the membrane stress sensor Slm1 both localize to eisosomes. Loss of the proton gradient causes swelling of the cell and a rapid endocytic response. The resulting increase in membrane tension flattens the eisosomes. As a consequence, Nce102, APC transporters and Slm1 move out of eisosomes. Outside of eisosomes the APC transporters can be targeted for ubiquitination, endocytosis, and degradation. Similar to high membrane tension caused by high extracellular pH, heat-shock conditions also cause eisosome flattening and trigger degradation of APC transporters. Therefore, eisosomes seem to respond to increased fluidity of the membrane.
Eisosomes with overlapping Pil1 and Fur4 signal were often found on one side of the cell, whereas the opposite side showed clear separation between these proteins. This distribution was likely a result of optical sections that were not precisely at the equator of the yeast cells, placing the two signals in different positions on the z-axis. In comparison to x/y-axes, the z-axis has a much lower optical resolution (∼50–60 nm), resulting in overlapping Pil1/Fur4 signals. Therefore, it is likely that the actual percentage of eisosomes with signal separation is much higher than the observed ∼50%.
Although Fur4 is concentrated adjacent to the Pil1-marked eisosomes (reflecting the fact that Fur4 is concentrated in eisosomes), it is also more broadly distributed along the plasma membrane. However, the superresolution pictures showed clear gaps in the plasma membrane distribution of Fur4 that were not observed in the conventional microscopy analysis (compare Figure 3, A and B). These gaps were at least in part caused by the filtering algorithms of the superresolution microscopy software, which eliminated signals from molecules that either moved during the acquisition time (50 ms) or that did not produce enough photons and thus were considered of low quality (low resolution). This statement was based on the observation that lowering the filtering stringency closed many of the gaps. It is important to note that Figure 3A is not a camera picture but a software generated map of high-resolution events (25 nm in X/Y or better) presented as spheres of 25 nm diameter.
The addition 50 mM Tris, pH 8, to the growth medium of the microfluidics chamber reduced the distance between Pil1 and Fur4 (Figure 3A). After 15 min in the high-pH medium almost all Pil1 structures overlapped with the Fur4 signal. A manual survey of more than 400 eisosomes indicated that the number of Pil1 structures/clusters spatially separated from Fur4 dropped from 47% to 4% (Figure 3C). An automated cluster analysis of the same data set suggested a drop in separated Fur4-Pil1 structures from 52% to 21% (the different results are likely caused by differences in defining the borders of the clusters). The high pH treatment also caused a 22% drop in the total number of Pil1 structures (Figure 3C). Together, these data suggested that the addition of Tris buffer caused not only rapid endocytosis of APC transporters but also flattening of the eisosomes and the disassembly of some of the eisosomes. Furthermore, the move of Nce102 out of eisosomes we observed after Tris treatment (#2 in Figure 1A) seemed to indicate flattening of the eisosomes, consistent with the published observation that deletion of Nce102 causes the presence of shallow eisosome structures (Loibl et al., 2010).
The eisosome flattening we observed after Tris treatment could be the result of rapid APC transporters endocytosis in these stressed cells. Yeast encodes 26 different APC transporters that together represent a large pool of the plasma membrane proteins. The rapid endocytosis of these transporters could cause increased membrane tension that might trigger the eisosome flattening. To test this idea, we analyzed the Nce102 localization before and after the addition of 50 mM Tris, pH 8, to the growth medium of end3-1, a yeast strain mutated for a key endocytic factor (expresses a truncated, loss of function mutant of End3; Whitworth et al., 2014). As expected, the end3-1 strain exhibited no Tris-induced endocytosis of Fur4-GFP (Figure 4A). Furthermore, after Tris treatment we observed no obvious change in the Nce102-localization to eisosomes (#9 in Figure 1E), suggesting that the block in endocytosis might have prevented eisosome flattening.
The polygon analysis indicated that Tris treatment of end3-1 cells caused only an ∼20% drop in Nce102 marked eisosomes (#9 in Figure 1B). Consistent with these data, we observed by superresolution microscopy only an ∼20% increase in collapsed eisosomes after Tris treatment of end3-1 (compared with over 90% collapsed eisosomes in wild type; Figure 2, A and C). Also, the total number of Pil1 marked eisosomes did not change significantly in Tris-treated end3-1 cells (Figure 3C). Similarly, mutating Rsp5 (hypomorphic allele rsp5-1; Wang et al., 1999), the E3 ligase responsible for the ubiquitination of cell surface proteins, not only blocked endocytosis of Fur4 after Tris treatment (Figure 4A) but also impaired the redistribution of Nce102 out of eisosomes (#10 in Figure 1, B and E). Together, the data suggested the endocytic response caused by the loss of the proton gradient was an important factor for the flattening of eisosomes and the concomitant redistribution of Nce102.
To test whether any stress-induced endocytic response is sufficient to trigger eisosome flattening, we determined the localization of Nce102 after glucose or leucine starvation. These starvation conditions have been shown to result in endocytic responses similar to those observed after Tris treatment (Jones et al., 2012; Lang et al., 2014; see glucose starvation in Figure 4A). Leucine starvation showed no effect on Nce102 localization to eisosomes, suggesting that the rapid endocytosis of APC transporters alone might not be sufficient to trigger eisosome flattening (#4 in Figure 1, A and B). Glucose starvation, on the other hand, resulted in a partial loss of Nce102-marked eisosomes (#3 in Figure 1, A and B). This response varied from cell to cell, with some cells showing no Nce102 redistribution, whereas other cells exhibited an obvious loss of Nce102 localization to eisosomes (#3 in Figure 1A). The partial Nce102 redistribution was reflected in the flattening of a portion of the eisosomes (observed by superresolution microscopy and quantified in Figure 3C; Figure 3A shows an example of a cell that did not exhibit a change in eisosome depth). However, unlike Tris treatment, glucose starvation did not cause a change in eisosome number (judged by Pil1 localization), suggesting that the basic structure of the eisosomes remained intact (Figure 3C). Glucose starvation results in a broad endocytic response that affects not only APC transporters but also many other cell surface proteins (Lang et al., 2014), suggesting this stress response is likely to cause membrane loss that is greater than that triggered by leucine starvation. This difference in endocytic response might explain the difference in flattening of eisosomes: the larger the membrane loss the larger the portion of the eisosomes that flatten and even disassemble.
In summary, our data indicated that many common methods for EM sample preparation are not suited for a detailed analysis of eisosome morphology. Some methods cause swelling of the cell and thus flatten the eisosomes (see EM micrographs of control cells in Figure 10 later in this article), whereas other methods result in cell shrinking and eisosome formation (Figure 2). Therefore, superresolution fluorescence microscopy of live cells might be a better method to determine changes to eisosome structures. However, our conclusion that eisosomes flatten after Tris treatment is based on the model that even during stress conditions Pil1 marks the curved bottom of the membrane furrow (see Figure 11 later in this article), an assumption we are currently not able to test.
Changes in cell size
The endocytic response alone was not able to explain why the addition of Tris buffer affected eisosomes much stronger than glucose starvation. One important difference we observed in the response to these two stress conditions was an increase in cell size after Tris treatment but not after initiating glucose starvation. The addition of Tris buffer caused yeast to swell by almost half a micron, resulting in an increase of surface area of the cells by ∼12% (based on changes in diameters and assuming spherical cells; #2 in Figure 4B). In contrast, the 15 min mock-treated cells showed no change in size (0.1% increase; #1 in Figure 4B), whereas glucose starvation caused a small reduction in cell size (–2.4%; #3 in Figure 4B). These changes in surface area together with the endocytic response collectively represented the actual stress on the plasma membrane and presumably added up to explain the effect of the different treatments on eisosomes. Tris treatment causes swelling and endocytosis, whereas glucose starvation triggers a very large endocytic response but at the same time reduces the cell size. Therefore, membrane tension seems to be larger in Tris-treated cells than after glucose depletion, explaining the efficient eisosome flattening in the latter case. Leucine starvation did not change cell size (#4 in Figure 4B) and likely triggered a more moderate endocytic response compared with that of glucose starvation.
Why are Tris treatment and glucose starvation causing opposite effects on cell size? The addition of Tris buffer causes loss of the proton gradient and thus is expected to impair the activity of the proton-driven sodium/potassium pump Nha1. In addition, we have shown that Tris treatment results in a drop in ATP levels for ∼2 min (Moharir et al., 2018) thereby reducing the activity of the ATP-driven sodium/potassium pump Ena1. Therefore, we propose that on Tris treatment the salt concentration in the cytoplasm rises, resulting in an increased influx of water and the subsequent swelling of the cells.
Using uracil-import assays we found that in contrast to Tris treatment, 10 min of glucose starvation does not cause loss of the proton gradient (uracil import is proton driven; Figure 5A). This result was unexpected, since maintaining the proton gradient is energy intensive. However, yeast stores glucose in the form of glycogen that can be rapidly mobilized when an extracellular concentration of glucose drops (reviewed in Wilson et al., 2010), possibly explaining how the cells can maintain the proton gradient 10 min into glucose starvation.
FIGURE 5:
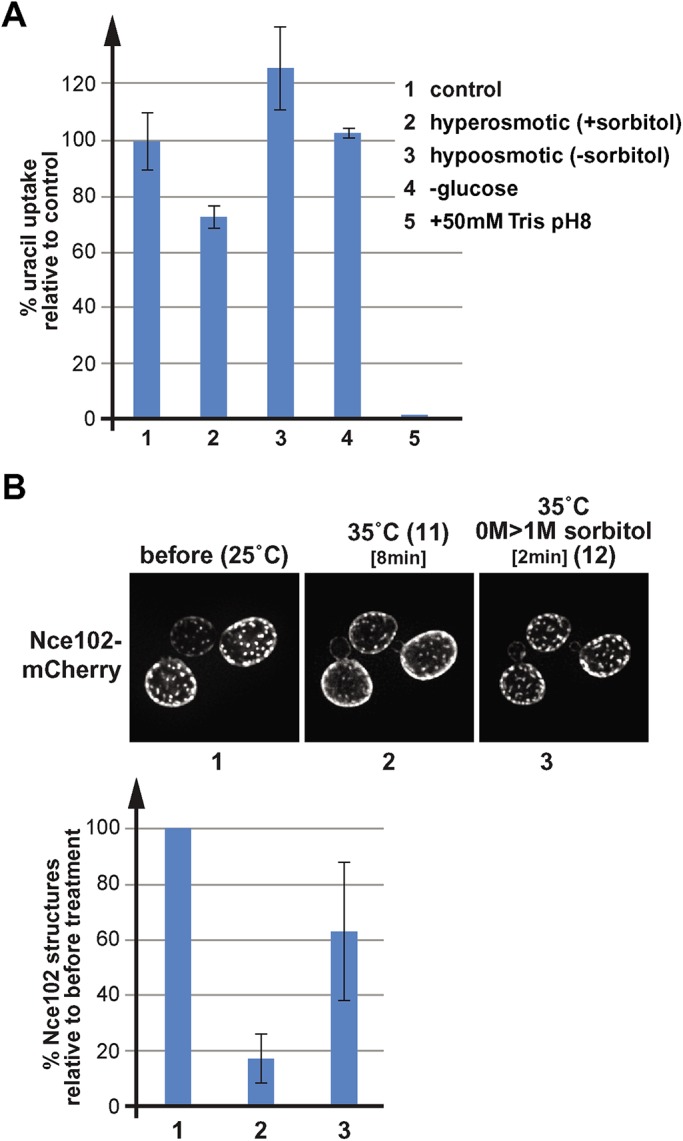
Fur4 uracil import activity under different stress conditions and the effect of heat-shock on Nce102 localization. (A) Uracil import assays of wild type expressing Fur4(∆N)-GFP (N-terminal deletion that lacks the ubiquitination sites; SEY6210 pJK88). Cells were grown in SDcomp-ura or SDcomp-ura + 1 M sorbitol (for the hypoosmotic shock) and treated for 10 min as indicated. Uracil (20 μg/ml) was added for 10 min, and the cells were harvested and analyzed for the uracil content using normal-phase chromatography. The bar graph shows the average and standard deviations of three measurements. The data were standardized relative to the control samples (= 100%). (B) Fluorescence microscopy of wild-type cells expressing Nce102-mCherry (AMY4). The cells were grown at 25°C and imaged in the microfluidics system at 25°C (control). The temperature of the microfluidics chamber was rapidly increased to 35°C by switching to a heated objective. After imaging the heat-shocked cells (8 min), the growth medium was switched to a medium containing 1 M sorbitol. The bar graph illustrates the quantification of Nce102 objects performed by 2D polygon analysis and standardized to starting conditions (100% = 1854 objects).
If our model about eisosome flattening was correct, we would expect that the combination of Tris treatment with hyperosmotic shock should preserve the eisosomes. Therefore, we exposed yeast in the microfluidics system to medium containing both 50 mM Tris, pH 8, and 1 M sorbitol. Fluorescence microscopy indicated that the cells reduced in size (Figure 4B8) and Nce102-mCherry did not redistribute, suggesting that indeed the presence of 1 M sorbitol suppressed Tris-induced membrane stress and thus inhibited eisosome flattening (#8 in Figure 1, B and E).
We have shown in Figure 1D that inhibiting endocytosis of APC transporters, either by mutating the endocytic factor End3 or by impairing ubiquitination (rsp5-1 mutant), blocked the redistribution of Nce102 and eisosome flattening on Tris treatment. Cell size measurements indicated that this effect is the consequence not only of impaired endocytosis but also of a reduced swelling of the cells. Both mutants reduced expansion of the cell surface area after high pH stress compared with wild type (#9 and #10 in Figure 4B). The reason for this reduced expansion in cell size can most likely be attributed to trafficking problems of the chitin synthase Chs3. Although chitin is a minor component of the cell wall, it plays a key role in the stability and stiffness of this polymeric structure. Both end3-1 and rsp5-1 mutant strains are known to stabilize Chs3 on the cell surface, thereby increasing the deposition of chitin in the cell wall. As a consequence, both mutant strains are known to be sensitive for the chitin-binding drug calcofluor white (Tang et al., 2000; Kaminska et al., 2005). This increased stability of the cell wall found in end3-1 and rsp5-1 could explain the reduced swelling of the cells after Tris treatment.
In summary, our data suggested that eisosomes respond to rapid increase of membrane tension by flattening and redistribution of eisosome-associated proteins such as Nce102 and APC transporters. The combination of endocytic response and changes in cell size determines the effect of stress conditions on eisosome structure.
Hypoosmotic shock causes eisosome flattening
To test whether eisosome flattening requires an endocytic response or can be triggered just by cell expansion, we grew yeast for 24 h in medium containing 1 M sorbitol to adjust the cells to high osmolarity and then switched these cells in the microfluidics system to medium lacking sorbitol. This hypoosmotic shock did not trigger an endocytic response (based on localization of Fur4-GFP; Figure 4A) but caused the cells to swell and increase its surface area by ∼16% (15 min treatment; #5 in Figure 4B). As a consequence, the number of Nce102 structures (eisosomes) recognized by the polygon analysis dropped to 48% of starting conditions (#5 in Figure 1, A and B), suggesting that the hypoosmotic shock flattened a large number of eisosomes. However, we noticed that in contrast to Tris treatment, the response to hypoosmolarity was faster and much more pronounced at earlier time points (see 3 min treatment; #6 in Figure 1, B and D). After 15 min hypoosmotic shock, Nce102 had already moved partially back into eisosomes, resulting in an increase in Nce102 structures from 16% at 3 min to the 48% at 15 min. Furthermore, we found that Nce102 redistribution was rapidly reversible. Switching the hypoosmotically shocked cells after 3 min back to medium containing 1 M sorbitol restored most of the eisosome localization of Nce102 within 3 min (from 16 to 63%; #6 and #7 in Figure 1, B and D). Together the data suggested that yeast is able to recover and reform the eisosomes much faster after exposure to hypoosmotic conditions compared with high pH conditions. Hypoosmotic shock does not affect the proton gradient (see uracil import in Figure 5A) or cause an endocytic response (Figure 4A), possibly explaining why cells alleviate membrane stress much faster than after Tris treatment.
Heat shock causes eisosome flattening
Both hypoosmotic conditions and Tris treatment increase cell surface tension by increasing cell size and in the latter case by triggering a strong endocytic response. We propose that this increase in membrane tension is causing the flattening of eisosomes. With regard to membrane properties, an increase in tension mimics an increase in temperature. Both parameter changes are expected to cause an increase in lipid fluidity, thinning of the membrane, and disruption of membrane structures such as eisosomes or lipid rafts. To test this prediction, we caused a rapid increase in temperature of the microfluidics system and monitored changes in Nce02-mCherry localization. For this experiment, yeast was grown at 25°C, flushed into the microfluidics chamber at the same temperature. After taking fluorescence microscopy pictures at 25°C (room temperature), we switched to an objective that was heated to 35°C and monitored the change in Nce102 localization (the oil-emersion objective will rapidly increase the temperature of the contacted microfluidics chamber). After 8 min we observed an obvious loss of eisosome-localized Nce102, resulting in a >80% drop in eisosomes identified by the 2D polygon analysis (Figure 5B). The addition of sorbitol to the heat-shocked cells caused within ∼2 min the recovery of many of the Nce102-labeled eisosomes (2D polygon analysis indicated an increase from ∼20% to ∼60% eisosomes; Figure 5B). These observations are consistent with the prediction that tension and temperature affect membranes in a similar way. Therefore, eisosomes flattening is not caused by a mechanical pull on the furrow structure, but is the result of increased lipid fluidity and mixing, which disrupt the structure of the eisosome membrane domain.
Slm1 relocalizes after eisosome flattening
A previous study found that plasma membrane stress (hypoosmotic conditions or the addition of sphingolipid synthesis inhibitor myriocin) resulted in the redistribution of the eisosome component Slm1 to the TORC2 complex (Berchtold et al., 2012). Consistently, we observed, after a hypoosmotic shock (1 M > 0 M sorbitol), a dramatic decrease in the number of Slm1 structures localized to eisosomes (from ∼85% to ∼30%; Figure 6B). TORC2 localizes to the plasma membrane and its kinase activity is regulated by stressors that impact cell wall and the plasma membrane integrity (reviewed in Gaubitz et al., 2016). Slm1 localization to TORC2 is thought to activate the kinase complex. In turn, TORC2 activates Ypk1/2, which then derepresses the serine-palmitoyl-transferase (SPT, encoded by Lcb1/2) complex by phosphorylating Orm1/2 at the ER (Roelants et al., 2011). The result of this phosphorylation cascade is an increase in sphingolipid biosynthesis. This regulatory feedback seems to maintain plasma membrane tension in a normal range, thereby ensuring plasma membrane integrity.
FIGURE 6:

Loss of proton gradient causes relocalization of Slm1. (A) Wild type expressing Slm1-GFP and Pil1-mCherry (LGY31) was analyzed by fluorescence microscopy using a microfluidics device. The pictures show projections of 10 optical sections representing the bottom half of the cells. The cells were grown in SDcomp and shifted to conditioned SDcomp +50 mM Tris, pH 8. (B, D) Quantification of Slm1 localization relative to start conditions. Wild-type and end3-1 mutant cells (LGY31, DAY28) were treated either with 50 mM Tris, pH 8, or by moving the culture from growth in the presence of 1 M sorbitol to conditioned medium without sorbitol. The number above the bar graph indicates the total number of Slm1-GFP structures counted in the control cells (= 100%; ∼50 cells analyzed). (C) Quantification of the Slm1-GFP distribution (cell surface/total) in LGY31 cells before and 15 min after the addition of 50 mM Tris, pH 8 (same data set as used for quantification in B). (E) Fluorescence microscopy of wild-type cells expressing Slm1-GFP and Avo3-mCherry (DAY53). The pictures show a projection of 10 optical sections (250 nm each) representing the bottom half of the cells. The bar graph represents the quantification of 30 cells, determining the number Slm1-GFP only, Avo3-mCherry only, and colocalizing structures per half-cell. The quantification was performed manually. Small structures >4 pixels were omitted.
Slm1 is a soluble protein that dynamically localizes to the cytoplasm and puncta at the plasma membrane, a majority of which are eisosomes. In a previous publication, we showed that Slm1 leaves eisosomes after Tris treatment (Moharir et al., 2018), suggesting that eisosome flattening triggers the release of this protein from eisosomes (Figure 6A). We quantified this effect and found that after 15 min of Tris treatment, the number of plasma membrane–localized Slm1 puncta decreased by 34% (Figure 6B). Furthermore, after Tris treatment plasma membrane Slm1 puncta redistributed from 99 to 68% eisosome localization (Figure 6B). However, the ratio of plasma membrane to total Slm1-GFP signal did not change after Tris treatment, indicating that Slm1 remained associated with the plasma membrane and did not move to the cytoplasmic pool (Figure 6C). These results suggested that loss of the proton gradient caused Slm1 to move from eisosomes to other surface-localized puncta.
As a control, we analyzed the localization of Slm1-GFP in end3-1 cells before and after treatment either with 50 mM Tris, pH 8, or with hypoosmotic shock. These mutant cells exhibit an endocytosis defect and show a diminished increase in cell size after Tris treatment (#9 in Figures 1E and 4B). Accordingly, stress-induced relocalization of Slm1 was dramatically reduced (Figure 6B), which was consistent with the model that membrane tension is required for both Slm1 and Nce102 to redistribute.
Hypoosmotic conditions, which cause increased membrane tension, have been shown to localize Slm1 to TORC2 (Berchtold et al., 2012), which might explain the noneisosomal puncta we observed after Tris treatment. Therefore, we analyzed the localization of Slm1-GFP in cells expressing the tagged TORC2 subunit Avo3-mCherry. Surprisingly, we observed negligible colocalization of the two tagged proteins both before and after Tris treatment (Figure 6E), suggesting that the peripheral, noneisosomal Slm1 structures do not correspond to TORC2. At this point we do not have an explanation why our localization data contradict the previous published Slm1-TORC2 colocalization (Berchtold et al., 2012).
Orm1 and Orm2 are ER-localized proteins and components of the SPOTS complex (SPT, Orm1/2, Tsc3, and Sac1) that inhibit the synthesis of sphingolipids. Orm1/2 are phosphorylated by the TORC2 effector kinases Ypk1/2, which release the inhibitory effect of these proteins and thus increase sphingolipid synthesis (Roelants et al., 2011). Hypoosmotic conditions cause redistribution of Slm1 from eisosomes to TORC2, which activates TORC2 and as a consequence releases the inhibition on sphingolipid synthesis by phosphorylating Orm1/2 (Berchtold et al., 2012). Using Phos-tag gels we analyzed the changes of Orm2 phosphorylation caused by Tris treatment. We observed an initial drop in Orm2 phosphorylation after the addition of Tris (see two examples in Figure 7, A, lanes 1–4, and B, lanes 1–4). Approximately 40 min after the addition of Tris buffer, the levels of phospho-Orm2 started increasing and after 1 h reached levels equal to or higher than observed before treatment. To control these Phos-tag experiments, we repeated the Tris treatment using a strain expressing a form of Orm2 that is mutated in the Ypk1/2 phosphorylation sites (S46,47,48A). As expected, we did not observe phosphorylation of the mutant Orm2 protein (Figure 7A, lanes 5–8). As an additional control, we treated cells after 1 h Tris treatment with the sphingolipid-synthesis inhibitor drug myriocin, which resulted in a dramatic increase of the phosphorylated form of wild-type Orm2 (Figure 7A, lane 9, as expected from previous publications; Roelants et al., 2011). Finally, to test whether the observed Tris-induced changes in Orm2 phosphorylation were caused by membrane tension, we treated the cells simultaneously with Tris and sorbitol. This combined treatment is expected to disrupt the proton gradient and to cause shrinking of the cell, thereby reducing membrane tension (see #8 in Figures 1E and 4B). The experiment showed that addition of sorbitol did not affect the initial drop in phospho-Orm2 but suppressed the increase in Orm2 phosphorylation at later time points (40-60 min; Figure 7B, lanes 5–8). These fluctuations in Orm2 phsophorylation did not correlate with the Slm1 localization data, since quantifications indicated no change in localization of Slm1 between 15 min and 1 h of Tris treatment (Figure 6D).
FIGURE 7:
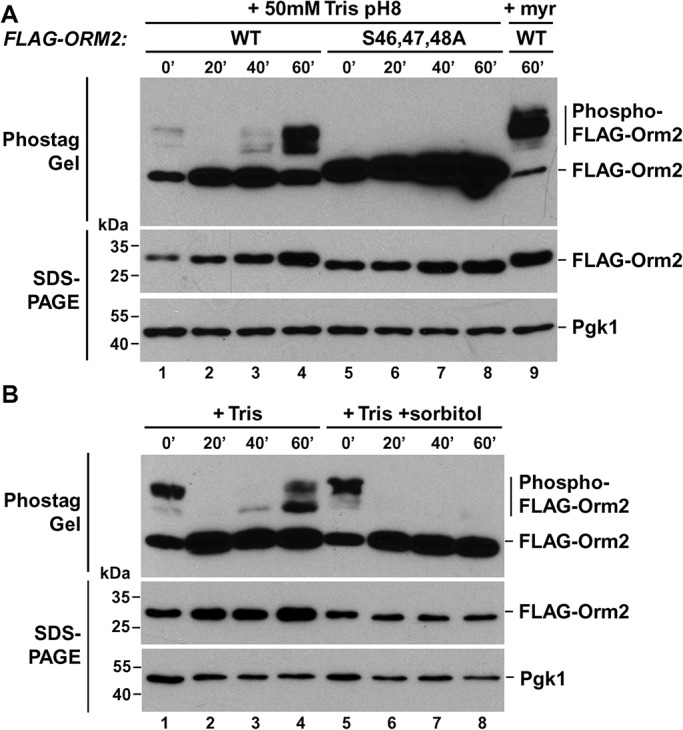
Membrane stress-induced changes in Orm2 phosphorylation. (A, B) Cells expressing wild-type or the S46,47,48A mutant FLAG-Orm2 in an orm2∆ background (YWY005 pOS129, YWY005 pYW001) were grown in SD medium adjusted to pH 4 and were treated with 50 mM Tris, pH 8 (+ Tris), in the presence or absence of 1 M sorbitol (+ sorbitol), and samples were taken at different time points. As a control, a sample was prepared from the 1 h + Tris cells that were treated for an additional 1 h with 2.5 µM myriocin (+ myr). The cell lysates were separated by Phostag Gel or standard SDS–PAGE, and proteins were detected by Western blot (Pgk1 served as a loading control).
In summary, the increased membrane tension observed after the loss of the proton gradient did not result in localization of Slm1 to TORC2 or an increased phosphorylation of the downstream effector of the TORC2 pathway, Orm2. In other words, the observed phosphorylation status of Orm2 did not correlate with the flattening of the eisosomes and the concurrent redistribution of Slm1. After the initial Orm2 dephosphorylation, Orm2 phosphorylation increased ∼1 h after Tris treatment and this increase was dependent on membrane tension (the addition of sorbitol suppressed both the Tris-induced flattening of eisosomes and Orm2 phosphorylation; Figures 1E #8 and 7B). The data suggested that sphingolipid synthesis is suppressed during acute alkaline stress and resumed only after cells adapted to the new conditions (cells will continue to grow after Tris treatment, although slowly; unpublished data). As a consequence, it is unlikely that new sphingolipid synthesis is involved in the membrane stress response after loss of the proton gradient. Consistent with this idea, the presence of myriocin did not change the cell expansion during hypoosmotic stress (#12 in Figure 4B), again supporting the idea that TORC2-induced sphingolipid synthesis is not necessary to accommodate the rapid increase in cell surface area.
Eisosomes are not necessary for cell expansion
Studies on yeast spheroplasts reported that eisosomes might function as membrane reservoirs that prevent lysis during acute cell expansion (Kabeche et al., 2015). This proposed function would be consistent with our observed eisosome flattening during membrane stress. To further test this idea, we treated a NCE102 deletion strain (exhibiting shallow eisosomes; Loibl et al., 2010) with Tris and measured the change in cell size. We observed the same increase in surface area of nce102∆ as with the wild-type strain (∼12%; #11 in Figure 4B), suggesting that the eisosome membranes were not required to accommodate the large Tris-induced increase in cell size. We also determined the amount of cell lysis that might occur as a consequence of a hypoosmotic shock. Wild-type, nce102∆, and pil1∆ cells exhibited minor increases in the number of lysed cells after shifting the cells from medium with 1 M sorbitol to medium lacking sorbitol (flow cytometry of propidium iodide [PI]-stained cells; Figure 8A), indicating that eisosome mutants are not lysing during a hypoosmotic shock. In contrast, 16% of growing end3-1 mutant cells were positive for PI, a number that increased to 26% after the hypoosmotic shock (Figure 8A). END3 mutants are defective in the proper delivery of chitin synthase to the growing bud, which might explain why these cells are sensitive to osmotic stress.
FIGURE 8:
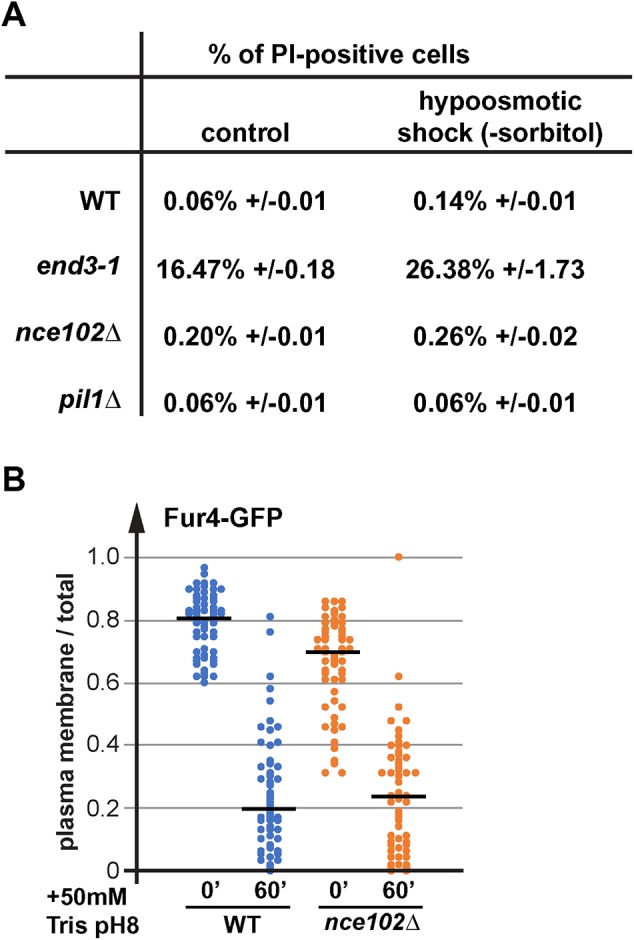
Eisosome mutations do not impair endocytosis or cell integrity. (A) Wild-type and mutant cells were grown in SDcomp + 1 M sorbitol. The cells of the midlog yeast cultures were moved into SDcomp pH 4 medium and after 10 min the cells were stained with PI and analyzed by flow cytometry. (B) Microscopy pictures of Fur4-GFP expressing cells (WT, SEY6210 pJK19; nce102∆, AMY41 pJK19) in the presence or absence of 50 mM Tris, pH 8, were quantified by determining the signal at the plasma membrane of Fur4 relative to the total signal. The graph shows the result of ∼50 cells for each strain and condition. The black line indicates the median of the data set.
The rapid endocytic response after Tris treatment requires sufficient plasma membrane, which might not be available in cells lacking eisosomes. Therefore, we tested whether the eisosome mutant nce102∆ is impaired in Tris-induced endocytosis. We determined, by microscopy, the ratio of cell surface-localized Fur4-GFP relative to total Fur4-GFP signal in wild-type and nce102∆ cells before and after 1 h addition of 50 mM Tris, pH 8. Consistent with previously published results (Moharir et al., 2018), in nce102∆ prior to treatment we observed a reduction of the cell surface pool of Fur4 compared with wild-type cells (Figure 8B). However, after Tris treatment both strains showed a similar endocytic response, resulting in the removal of most of Fur4 from the plasma membrane.
In summary, our phenotypic analysis did not indicate any major role of eisosomes in the acute response to osmotic stress or in stress-induced endocytosis. A simple calculation also questions the role of eisosomes in plasma membrane expansion. On the basis of the estimated number of eisosomes (∼50 per cell) and the approximate size (∼50 nm deep and ∼300 nm long) (Stradalova et al., 2009), we calculate that eisosomal membrane constitutes only ∼2% of the total surface area of a 5µm diameter yeast cell (0.05 µm × 0.3 µm × 2 × 50 = 1.5 µm2; surface area of a sphere with 5 µm diameter = 79 µm2). Therefore, we were not able to support the idea that eisosomes function as membrane reservoirs during severe membrane stress conditions. However, eisosomes might provide membrane for the fine tuning of the plasma membrane tension.
Hyperosmotic shock causes deepening of eisosomes
Since high membrane tension seemed to cause flattening of eisosomes, we tested whether the opposite, membrane slack, would result in eisosome deepening. We treated cells with 1 M sorbitol, which causes a hyperosmotic shock and subsequent shrinking of the cells (Miermont et al., 2013). This stress condition did not affect the proton gradient (see uracil import in Figure 5A) and did not cause a strong endocytic response (Figure 4A). Using a standard fluorescence microscope, we observed a separation of the Pil1-mCherry signal from the Fur4-GFP signal after just 5 min, suggesting a distance of at least 250 nm between the two proteins (Figure 9A). In contrast, the control cells (before treatment) exhibited almost complete colocalization of Fur4 with Pil1 (Figure 9A), consistent with the estimated ∼50 nm depth of eisosomes (Stradalova et al., 2009). The separation between Pil1-mCherry and Fur4-GFP signals was also observed after the combined treatment of cells with 50 mM Tris, pH 8, and 1 M sorbitol (Figure 9A). Similar to Tris treatment alone, most of Fur4-GFP was found internalized after 1 h treatment with Tris/sorbitol. However, the presence of sorbitol caused a pool of Fur4 to remain in eisosomes (Figure 9A), suggesting the deepened eisosomal structures prevent efficient Fur4 endocytosis. In comparison, the small amount of Fur4 remaining at the cell surface after 1 h Tris treatment localized to membrane areas adjacent to eisosomes (Figure 9A), consistent with the previously observed loss of eisosome-localized Fur4 in Tris-treated cells (Moharir et al., 2018).
FIGURE 9:
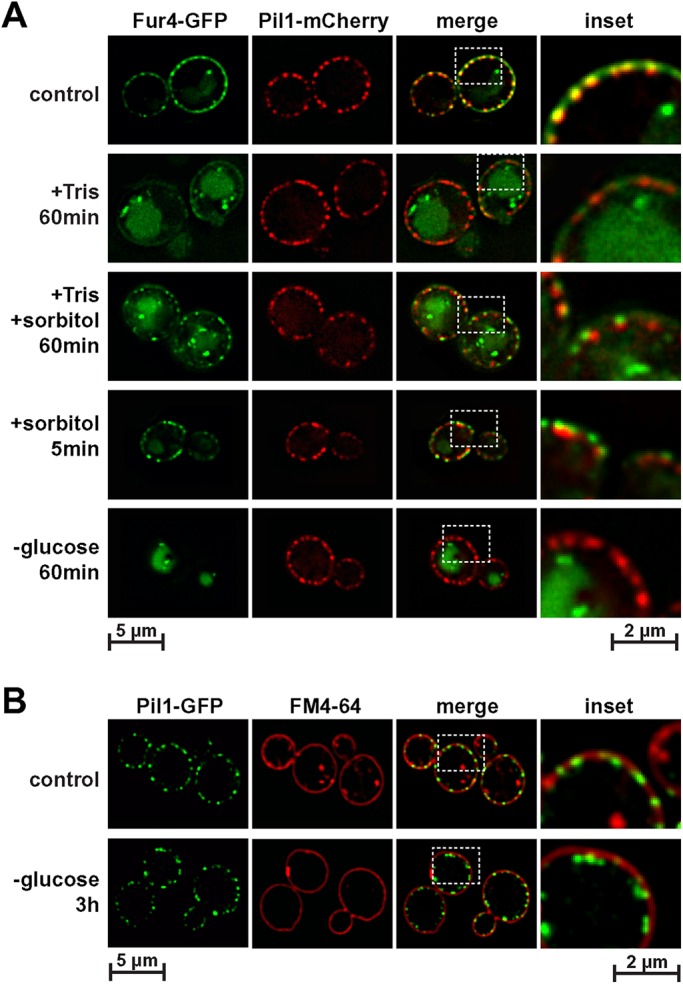
Eisosomes deepen after a hyperosmotic shock or glucose starvation. (A) Wild-type cells expressing Pil1-mCherry and Fur4-GFP (AMY6 pJK19) were treated with 50 mM Tris, pH 8 (+ Tris), with 1 M sorbitol (+ sorbitol) or combined with Tris and sorbitol. For the glucose starvation (- glucose) cells were transferred from the growth medium (SDcomp-ura) to growth medium lacking glucose. (B) Wild-type cells expressing Pil1-GFP (AMY6) were analyzed by fluorescence microscopy before (control) and after 3 h of glucose starvation. Before microscopy the cells were stained with FM4-64 (2 µg/ml) for 5 min at room temperature.
The experiments in Figures 1 and 2 indicated that 15 min of glucose starvation caused modest shrinking of the cells and partial effects on eisosome structure. We observe that prolonged glucose starvation (24 h) further reduced the cell size, even causing the detachment of the plasma membrane from the cell wall (see electron microscopy in Figure 10). At earlier time points of glucose starvation (3 h), electron microscopy indicated the presence of deep invaginations that might represent enlarged eisosomes (Figure 10). The size of these structures was consistent with the observed 250–300 nm separation of Fur4-GFP from Pil1-mCherry after sorbitol addition (Figure 9A). Since glucose starvation causes efficient removal of Fur4-GFP from the cell surface (Figures 4A and 9A), we used the lipophilic dye FM4-64 as a stain for the plasma membrane and found in glucose-starved cells a clear separation between the FM4-64 and the Pil1-GFP signal (Figure 9B), suggesting at least 250 nm distance between the plasma membrane and the curved bottom of the membrane furrow. Together, the data suggested that prolonged glucose starvation caused cells to shrink, resulting in membrane slack. To maintain proper membrane tension, the cell seemed to respond by deepening eisosomes.
DISCUSSION
Previous studies indicated that eisosomes have seemingly two discrete functions: regulating APC transporter turnover and maintaining plasma membrane integrity. We found that under certain stress conditions, these two functions overlap and become part of the same stress response pathway. For example, loss of the proton gradient (here caused by the addition of Tris buffer) triggered the rapid endocytosis of APC transporters and caused a membrane stress response. Membrane stress seemed to be the result of the strong endocytic response (possibly removing all 26 types of APC transporters from the cell surface) and swelling of the cell, which might be caused by sodium influx (the proton gradient is used to excrete sodium ions). Superresolution fluorescence microscopy suggested that the resulting membrane tension causes flattening of eisosomes and the partial disassembly of these membrane domains. The tetraspan protein Nce102, together with the APC transporters, moved out of the flattened eisosomes, which is predicted to aid in the ubiquitination and subsequent endocytosis of the transporters (eisosomes inhibit APC transporter turnover; Gournas et al., 2018; Moharir et al., 2018). Furthermore, Slm1 dissociated from flattened eisosomes and localized to other structures on the plasma membrane. Therefore, eisosome flattening is a stress response that affects both the membrane integrity and the nutrient transporter regulation.
The eisosome flattening we observed with Tris-treated live cells using superresolution fluorescence microscopy could not be confirmed by electron microscopy. Eisosomes seem to respond very fast to changes in membrane fluidity caused by changes in either tension (as observed by hypoosmotic shocks in Figure 1) or temperature (Figure 5B). Therefore, the rather slow chemical fixation procedure is likely not able to preserve the actual eisosome morphology present at the time of sample preparation. Chemical fixation disrupts plasma membrane integrity causing loss in turgor pressure and membrane tension, which can be observed as cell shrinking. As a result, flattened eisosomes might reform during the fixation procedure. High-pressure freezing of yeast samples should be able to preserve the morphology. However, even this method is problematic since preparing yeast cultures for the freezing process requires centrifugation or filtration, methods that are known to disrupt normal metabolism (lack of nutrients and oxygen). A drop in ATP levels is expected to cause loss of the proton gradient and thus expansion of the cell (as shown for Tris treatment). Therefore, we argue that superresolution fluorescence microscopy of live cells is currently the preferred method of studying changes in eisosome morphology as a consequence of high membrane tension.
APC transporters are among the largest consumers of the plasma membrane proton gradient. It is therefore not surprising that yeast responds to the loss of the proton gradient by removing these transporters from the cell surface, which aids in the recovery of the proton gradient. Furthermore, in a previous study, we found that without proton gradient APC transporters act as permeases that secrete nutrients from the cell (Moharir et al., 2018), highlighting the importance of transporter endocytosis during stress conditions that weaken the proton gradient. Since yeast can grow prototrophically, the nutrients APC transporters import (amino acids and nucleobases, among others) are not essential for growth and thus their loss can be compensated by using part of the imported glucose anabolically as a carbon source (glucose transporters are not proton driven and thus are not affected by alkaline stress).
The effect of eisosome flattening on the regulation of the membrane integrity pathway does not fit to published models, which would have predicted that relocalization of Slm1 on Tris-induced membrane tension should activate the TORC2-Ypk1/2 kinase system. One consequence of this activation is the phosphorylation of the Orm1/2 proteins, which releases the inhibition of these proteins on sphingolipid synthesis (Roelants et al., 2011). The resulting increase in lipid synthesis is proposed to alleviate the high membrane tension. Surprisingly, we found that loss of the proton gradient caused the opposite response, a rapid dephosphorylation of Orm2 followed by an increase in Orm2 phosphorylation after 1 h of the stress conditions. This result suggested that the acute membrane tension caused by Tris treatment was not relieved by rapid up-regulation of sphingolipid synthesis; rather, the cells seemed to have responded by a transient down-regulation of lipid synthesis. The combined treatment of cells with Tris and sorbitol, which causes shrinking of the cells, did not suppress the initial dephosphorylation of Orm2. However, this combined treatment blocked the Orm2 phosphorylation usually observed after 1 h Tris treatment. Together the data suggested that loss of the proton gradient initially suppressed sphingolipid synthesis, independent of the presence or absence of membrane stress. At a later time point (∼1 h), sphingolipid synthesis was up-regulated and this part of the stress response was dependent on increased membrane tension. We did not observe colocalization of Slm1 with TORC2 before or after Tris treatment and therefore it is not clear how the Tris-induced regulation of Orm2 is accomplished. Together, our observations questioned the role of the Orm1/2 pathway and sphingolipid synthesis in the release of dramatic membrane tension.
In contrast to Tris treatment, a hypoosmotic shock by itself (shift cells from medium containing 1 M sorbitol to no sorbitol) resulted in a much faster recovery of membrane tension. Even though the cell swelling and the initial effects on Nce102 localization to eisosomes were more dramatic than the response we observed with Tris treatment, the cells seemed to recover from this membrane stress much faster. Within 5–10 min after the hypoosmotic shock, Nce102 started to move back into eisosomal structures. This observation suggested that within minutes, the flattened membrane domains were reforming into membrane furrows, indicating a rapid recovery of normal membrane tension. This fast eisosome recovery was consistent with published kinetics of Ypk1 phosphorylation after hypoosmotic shock; activation of Ypk1 peaked within 30 s after the osmotic shock and reached pretreatment levels 5 min later (Riggi et al., 2018). However, it is difficult to comprehend how, in such a short time, the cell would be able to synthesize enough lipids and deliver them to the plasma membrane to relive the membrane tension caused by >15% increase in surface area. Furthermore, a block of sphingolipid synthesis (addition of myriocin) did not impair the rapid increase in cell size caused by hypoosmotic conditions. Therefore, it is unlikely that up-regulation of sphingolipid synthesis is responsible for relieving the increased membrane tension after shift to a hypoosmotic environment, which implies that other sources provide the membrane necessary to prevent loss of cell integrity during acute membrane stress.
Studies on spheroplasted yeast suggested that eisosomes are important to provide membrane for the rapid and dramatic expansion during hypoosmotic shock. Furthermore, the study found that under these membrane stress conditions, spheroplasts disassemble the cell surface Pil1 structures, suggesting a complete loss of eisosomal structures. In contrast, the presence of a cell wall dramatically reduced the cell swelling and prevented the loss of Pil1 structures (Kabeche et al., 2015). We found that yeast with an intact cell wall expanded during Tris treatment to the same extent in the presence or absence of eisosomes. We observed only an ∼20% loss of Pil1-labeled eisosomes after Tris treatment nonspheroplasted yeast and the lack of eisosomes did not increase the number of lysed cells after a hypoosmotic shock. Finally, on the basis of size and numbers of eisosomes we calculated that these membrane furrows are only able to provide ∼2% of additional membrane to the cell surface, which is not sufficient membrane during Tris treatment or hypoosmotic shock, stress conditions that increase the surface area of the cell by 12–16%. Together, our observations suggest a model in which eisosomes mainly function in sensing rather than relieving of membrane tension. However, eisosomal membrane could relieve some membrane tension under less dramatic stress conditions.
Hyperosmotic shock causes rapid shrinking of yeast, resulting in plasma membrane slack. We observed by fluorescence microscopy that under these conditions eisosomes seemed to deepen, indicated by an ∼250 nm separation of the Fur4-GFP signal from the Pil1-mCherry signal. Glucose starvation also caused shrinking of the cell, which after 24 h of starvation became so dramatic that many cells showed detachment of the plasma membrane from the cell wall. After 3 h of glucose starvation, cells exhibited ∼300 nm deep invaginations of the plasma membrane that were reminiscent of eisosomes during hyperosmotic conditions. Together, these results suggested that eisosomes not only responded to an increase in membrane tension (eisosome flattening) but also to a lack of membrane tension (eisosome deepening). Therefore, eisosomes might function to fine-tune membrane tension, a parameter that has important consequences for membrane fluidity and organization (reviewed in Pontes et al., 2017).
Increased eisosome depth seems to affect the movement of Fur4 out of these membrane domains. Whereas flattened eisosomes cause redistribution of Fur4 into the surrounding membrane, the deep eisosomes that form after a hyperosmotic shock trap a pool of Fur4 that is protected from stress-induced endocytosis (remains in eisosomes after Tris treatment; Figure 9). A previous study suggested that the eisosomal pool of APC transporters is inactive and that transporters move out of eisosomes for efficient nutrient import (Moharir et al., 2018). Consistent with this idea, we observed that hypoosmotic conditions, which flatten eisosomes and redistribute Fur4, increased import activity. However, a hyperosmotic shock that trapped Fur4 in eisosomes decreased the uracil import by Fur4 (Figure 5A). Therefore, changes in membrane tension impact both activity and turnover of APC transporters.
It is important to note that an increase in temperature or membrane tension has similar effects on membrane properties. In both cases, the fluidity of lipids in membranes increases and membrane domains/rafts are being disassembled (Portet et al., 2012; Chen and Santore, 2014). Consistent with this idea, we observed eisosome disassembly during both hypoosmotic conditions and heat shock. Therefore, eisosome flattening is a consequence of increased membrane fluidity, which dissolves the raft-like membrane domain of the eisosome. As we have observed with Tris treatment, heat-shock conditions also trigger the rapid endocytosis of nutrient transporters, which in the case of Fur4 has been shown to be dependent on the eisosome-localized proteins Slm1/2 (Bultynck et al., 2006).
In summary, membrane tension, and thus eisosomes, are not only affected by osmotic changes in the environment but can also be changed by lack of nutrients or increase in extracellular pH. Furthermore, temperature affects eisosomes in a similar way as membrane tension. Therefore, eisosomes might function as general sensors for environmental changes that relay this information via Slm1/2 proteins and via APC transporter regulation to the cell’s metabolism (Figure 11). This system is particularly sensible considering the stresses experienced in the natural environment of yeast. The dramatic change from growing in fruit juice to being washed away by a rain storm triggers all the stressors mentioned above simultaneously: nutrient starvation, hypoosmotic shock and increase in pH (affecting the proton gradient), and possibly a change in temperature. It is therefore not surprising that all these stress response pathways intersect at eisosomes.
MATERIALS AND METHODS
Strains, media, and plasmids
The yeast strains and plasmids used in this study are listed in Table 1. Yeast was grown in synthetic dextrose medium (SD; yeast nitrogen base, 2% glucose), either in the presence of all amino acids and essential supplements (SDcomp) or in the case of plasmid selection lacking certain amino acids/supplements (e.g., SDcomp-ura: SDcomp lacking uracil). To induce expression from CUP1 promoter-driven constructs we added 0.1 mM CuSO4 to the medium. Chromosomal gene tagging or deletion was performed by homologues recombination (Longtine et al., 1998). The modifications were confirmed by PCR analysis.
TABLE 1:
Strains and plasmids used in this study
| Descriptive name | Genotype or description | Reference or source | |
|---|---|---|---|
| Strains | |||
| SEY6210 | WT | MATα leu2-3,112 ura3-52 his3-∆200 trp1-∆901 lys2-801 suc2-∆9 GAL | Robinson et al., 1988 |
| SEY6210.1 | WT | MATa leu2-3,112 ura3-52 his3-∆200 trp1-∆901 lys2-801 suc2-∆9 GAL | Robinson et al., 1988 |
| BY4741 | WT | MATa leu2-3112, ura3-52, his3-∆200 | Winston et al., 1995 |
| AMY6 | PIL1-mCherry | SEY6210, PIL1-mCherry, KANMX6 | Moharir et al., 2018 |
| AMY4 | NCE102-mCherry | SEY6210, NCE102-mCherry, KANMX6 | Moharir et al., 2018 |
| AMY41 | nce102∆ | SEY6210, NCE102::KANMX6 | Moharir et al., 2018 |
| AMY42 | pil1∆ | SEY6210, PIL1::KANMX6 | Moharir et al., 2018 |
| LGY31 | SLM1-GFP, PIL1-mCherry | SEY6210, SLM1-GFP TRP1, PIL1-mCherry KANMX6 | This study |
| BWY1346 | end3-1 | MATαtrp 1 leu2 ura3 lys2 end3-1 | Whitworth et al., 2014 |
| MYY808 | rsp5-1 | MATa, MDM1, smm1, his3, leu2, ura3 | Fisk and Yaffe, 1999 |
| DAY20 | NCE102-mCherry, end3-1 | BWY1346, NCE102-mCherry KANMX6 | This study |
| DAY21 | NCE102-mCherry, rsp5-1 | MYY808, NCE102-mCherry KANMX6 | This study |
| DAY27 | PIL1-mCherry, end3-1 | BWY1346, PIL1-mCherry KANMX6 | This study |
| DAY28 | SLM1-GFP, PIL1-mCherry, end3-1 | BWY1346, PIL1-mCherry KANMX6, SLM1-GFP TRP1 | This study |
| YWY005 | orm2∆ | SEY6210.1, ORM2::TRP1 | This study |
| DAY53 | SLM1-GFP, AVO3-mCherry | SEY6210, SLM1-GFP KANMX6, AVO3-mCherry URA3 | This study |
| Plasmids | |||
| pJK19 | P(CUP1)-FUR4-GFP | URA3 (pRS416) P(CUP1)-FUR4-GFP | Keener and Babst, 2013 |
| pJK30 | P(CUP1)-fur4(ΔN)-GFP | URA3 (pRS416) P(CUP1)-fur4(Δ2-60)-GFP | Keener and Babst, 2013 |
| PJK88 | P(SNF7)-fur4(ΔN)-GFP | URA3 (pRS416) P(SNF7)-fur4(Δ2-60)-GFP | Moharir et al., 2018 |
| pOS129 | FLAG-ORM2 | URA3 (pRS416) P(ORM2)-3xFLAG-ORM2 | This study |
| pYW001 | FLAG-orm2(S47,48,49A) | URA3 (pRS416) P(ORM2)-3xFLAG-orm2- S47,48,49A | This study |
| pRS416 | vector | URA3(pRS416) | Christianson et al., 1992 |
Fluorescence microscopy
For fluorescence microscopy, cells were grown to midlog phase (OD600 ∼0.7) and imaged using a deconvolution microscope (DeltaVision; GE, Fairfield, CT). In Figures 1 and 4, the microscopy was performed using a microfluidics system (CellASIC ONIX; Millipore Sigma) at 30°C and with constant flow of medium (4 psi). It was important that the medium was identical to that of the cell culture. It was obtained by centrifugation of the culture used for analysis (fresh medium is different in pH and nutrient content and therefore affects the experiments).
For the 2D-Polygon Analysis, Z-stacks were deconvolved and projected after image collection. To assess eisosome localization, the Softworx image software package, and 2D polygon Analysis, were used to select polygons based on a set of given parameters. These parameters were set manually. The “Maximum Perimeter” parameter was set at 50 pixels to exclude diffuse signal from the cell perimeter. The “Fluorescent Threshold” was set as low as possible without obvious inclusion of aberrant polygons or polygons that outside of the cell being selected. Fluorescent threshold levels were set for the control and kept the same for the corresponding experimental image. Using these defined parameters, the 2D Polygon Analysis tool assessed the number of eisosomes present.
The cell surface analyses shown in Figure 4B are approximations based on measured cell diameters and the assumption that the yeast cells are spherical. For the diameter measurements, the optical sections of cells observed in a microfluidics system were projected and the diameter was measured at the same position before and after treatment. These measurements were used to determine the percentage of change in surface area for each cell based on the diameter of the control cell (assuming a spherical cell).
Superresolution microscopy
Yeast cells expressing Fur4-GFP and Pil1-mCherry were deposited into a microfluidic chamber (see above for details). Images of cells were recorded with a Vutara SR 200 microscope (Bruker) based on the single-molecule localization biplane technology with a 60× Olympus water immersion objective (1.2 NA). Before treatments, yeast cells from several fields (20 × 20 µm) were imaged for 55 s (mCherry: 1000 frames, 30 ms/frame; GFP: 500 frames, 50 ms/frame). The maximum powers used for the readout lasers for GFP and mCherry were 7.5 and 3.75 mW/cm2, respectively. Fifteen minutes after treatments, the same yeast cells were imaged with the same settings.
The centers of GFP and mCherry clusters were localized in the recorded frames using a 3D localization algorithm of the Vutara SRX software (Juette et al., 2008). Briefly, pixels were sorted according to brightness for each frame. Nonoverlapping square boxes (10 pixels wide) were created to center the brightest pixels, and the images within were cut out. The centers of the “cutouts” were localized and their 3D coordinates were recorded. Each GFP or mCherry cluster may include several cutouts for a given frame. Because the intensities within the cluster fluctuated, different cutout centers were localized between frames. All the centers of GFP and mCherry were displayed in the SRX software as 3D balls of 25-nm radius. In short, the centroids of GFP and mCherry cutouts defined the clusters and allowed us to visualize eisosomes below the diffraction limits. Distances between GFP and mCherry clusters in the same yeast cells are measured before and after treatments.
Electron microscopy
For electron microscopy shown in Figure 10, cells were fixed with 4% glutaraldehyde and cell walls were permeabilized using 1% sodium metaperiodate as described previously (Wright, 2000). The cell pellets were postfixed with 1% osmium tetroxide/1.25% potassium ferrocyanide, stained en bloc with 2% aqueous uranyl acetate, dehydrated in ethanol and propylene oxide, and embedded in Spurr’s resin. Ultrathin sections (70 nm) were mounted on copper grids followed by staining with uranyl acetate and lead citrate. Grids were observed and photographed using a LEO EM910 transmission electron microscope (Carl Zeiss NTS, LLC, Peabody, MA) equipped with a Gatan Orius SC1000 digital camera and Digital Micrograph 3.11.0 software (Gatan, Pleasanton, CA).
For electron microscopy shown in Figure 2, yeast cultures grown to midlogarithmic phase were rapidly mixed with an equal volume of fixative and gently mixed for 20–30 min. We used 4% formaldehyde (EMS, 15710) and 0.4% glutaraldehyde (EMS, 16200) in PHEM buffer (EMS, 11163) as the standard fixative throughout the experiment (Griffith et al., 2008). After the initial fixation, cells were pelleted at 1800 RCF (relative centrifugal force) for 10 min, and then half of the supernatant volume was replaced with fresh fixative. Once resuspended in fresh fixative, cells were mixed gently for 3 h. After the secondary fixation, cells were washed 3× with PHEM buffer to remove the fixative. To infiltrate cells, cell pellet was resuspended in 12% gelatin in PHEM buffer at 37°C and incubated for 10 min in a 37°C water bath. Cells were concentrated at 17k RCF to increase cell density and placed on ice to solidify gelatin. Gelatin-embedded cells were cut into cubes of approximately one cubic mm in size. These blocks were infiltrated with 2.3 M sucrose overnight at 4°C. Each infiltrated block was mounted on a cryopin and covered with a fresh drop of 2.3 M sucrose. These pins were placed to vitrify inside a FC7 cryochamber that was precooled to –150°C. Frozen pins were transferred to liquid nitrogen for storage. For both trimming and sectioning, we used Leica UC7 ultramicrotome equipped with FC7 cryochamber and a single micromanipulator-grid holder combination. In preparation for cryosectioning, the top surface of the cell-cube mounted on cryopins was trimmed with 45° cryotrim tool (diatome) at –150°C. Ribbons of cryosections were collected at –150°C with a 35° cryoimmuno knife (diatome) with cutting speed set to 1 mm/s and knife advance to 60 nm. Sections were directly collected on 200 mesh hex grids with formvar-carbon film. Ribbons were adhered to the grid using crion ionizer (Leica). Grids containing sections were picked up using a metal loop and thawed to room temperature on a droplet of pickup solution containing phosphate buffer with 2% methylcellulose (Sigma, M-6385) and 0.4% UA (Liou et al., 1996). On immediate removal from the cryochamber, grids were floated, section-side down, on a cold 50-μl droplet of pickup solution. Grids were incubated on this droplet for 5 min on ice for simultaneous embedding and staining. Grids were picked up with a loop of diameter slightly larger than that of the grids. Excess solution was wicked off using a filter paper, leaving a thin layer of solution coating the section-containing side. These were air-dried overnight at room temperature and transferred to grid storage boxes. Sections were imaged using a TF20 electron microscope held at 200 kV. Images were collected with K2 direct electron detector (Gatan). A 3 × 3 montage centered at each cell profile was collected at 0.7 nm pixel size using serialEM (Mastronarde, 2005). Montages were brought into register using Etomo and postprocessed using Fiji (Schindelin et al., 2012). Images of cell profiles were annotated and quantified using TrakEM2. We used Microsoft Excel and GraphPad Prism for statistical data analysis. Two groups were compared using nonparametric t test with Mann–Whitney correction.
Flow cytometry
Cells grown to midlog phase were analyzed using the CytoFlex S flow cytometer (Beckman Coulter, Brea, CA). The data of ≥150,000 events were collected for each sample. Events with a forward scatter light between 100,000 and 1,400,000 were used for analysis. Data were collected and analyzed using the CytExpert software package. Cells were incubated for 2 min at room temperature in 1 μg/ml PI and then immediately analyzed, and all experiments were completed in triplicate. Cells were considered PI-positive if they had ≥10,000 fluorescent units in PerCP-Cy5-5 channel.
Uracil import assay
Wild-type cells expressing fur4(∆N)-GFP (pJK88) were used for uracil import assays. The Fur4(∆N) mutant lacks the N-terminal 60 amino acids, which includes the ubiquitination sites. Therefore, this protein is not regulated by substrate and remains stable at the cell surface. The cells were grown in SDcomp-ura to midlog phase, 20 µg/ml uracil was added and the cells were incubated at 30°C for 10 min. One-milliliter samples were taken and placed on ice. After 3 min the cells were collected by centrifugation at 4°C, washed 3× with ice-cold water and extracted with 50 µl methanol at 55°C for 5 min. The cells were removed by centrifugation and 5 µl of the supernatant and was used for the analysis by normal phase chromatography (acetonitrile/water gradient from 100 to 80% in 10 min; Luna-NH2 column; Phenomenex). Uracil was detected by absorption spectroscopy at 260 nm. The measured amount of uracil was standardized by the optical density of the cell culture.
SDS–PAGE, Phos-tag SDS–PAGE, and Western blot
Cells (4OD600nm) were harvested by centrifugation and lysed by a modified postalkaline lysis protocol (Kushnirov, 2000) including phosphatase inhibitors (10 mM NaF; 10 mM β-glycerophosphate; 0.1× PhosStop mix (Roche) in all steps. Protein extracts were dissolved in 200 µl Lämmli sample buffer and 5–10 µl was separated on 12.5% acrylamide SDS–PAGE gels (Bio-Rad Mini Protean) and transferred to PVDF membranes by semidry electroblotting.
Twelve and a half percent acrylamide Phos-tag gels (50 µM Phos-tag acrylamide [Wako]; 100 µm MnCl2) were prepared according to the manufacturer’s recommendations. Gels were run in a standard Lämmli electrophoresis buffer at 200 V, 40 mA for 1.5 h, afterward rinsed in Western blot transfer buffer with 10 mM EDTA for 20 min, and then equilibrated in transfer buffer without EDTA, followed by standard semidry electroblotting to PVDF membranes.
Antibodies used in this study are mouse monoclonal anti-FLAG M2 (Sigma; Cat. # F3165); mouse monoclonal anti-PGK1 22C5D8 (Invitrogen; Cat. # 459250); goat anti-mouse immunoglobulin G–peroxidase (Sigma; Cat. # A4416).
Acknowledgments
We thank Victoria Madden and the Microscopy Services Laboratory, Department of Pathology and Laboratory Medicine, University of North Carolina at Chapel Hill, for help with the electron microscopy. The research was supported by grants from the National Institutes of Health (NIGMS R01 GM123147 to M.B., NIGMS R01 GM092741 to M.C.D. and NINDS NS034307 to E.M.J.). E.M.J. is an investigator of the Howard Hughes Medical Institute. E.M.J.’s licensed intellectual property U-4587 “Automated, 3-Dimensional, Multi-Channel, Biplane PALM Microscope with TIRF” was used in this study.
Abbreviations used:
- APC
amino acid-polyamine-organocation
- PI
propidium iodide
- SD
synthetic dextrose
- SPT
serine-palmitoyl-transferase
- TEM
transmission electron microscopy
- TORC2
Tor complex 2.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-04-0218) on December 18, 2019.
REFERENCES
- Athanasopoulos A, Gournas C, Amillis S, Sophianopoulou V. (2015). Characterization of AnNce102 and its role in eisosome stability and sphingolipid biosynthesis. Sci Rep , 15200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold D, Piccolis M, Chiaruttini N, Riezman I, Riezman H, Roux A, Walther TC, Loewith R. (2012). Plasma membrane stress induces relocalization of Slm proteins and activation of TORC2 to promote sphingolipid synthesis. Nat Cell Biol , 542–547. [DOI] [PubMed] [Google Scholar]
- Bharat TAM, Hoffmann PC, Kukulski W. (2018). Correlative microscopy of vitreous sections provides insights into BAR-domain organization in situ. Structure , 879–886.e873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi F, Syga L, Moiset G, Spakman D, Schavemaker PE, Punter CM, Seinen AB, van Oijen AM, Robinson A, Poolman B. (2018). Steric exclusion and protein conformation determine the localization of plasma membrane transporters. Nat Commun , 501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultynck G, Heath VL, Majeed AP, Galan JM, Haguenauer-Tsapis R, Cyert MS. (2006). Slm1 and slm2 are novel substrates of the calcineurin phosphatase required for heat stress-induced endocytosis of the yeast uracil permease. Mol Cell Biol , 4729–4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Santore MM. (2014). Large effect of membrane tension on the fluid-solid phase transitions of two-component phosphatidylcholine vesicles. Proc Natl Acad Sci USA , 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. (1992). Multifunctional yeast high-copy-number shuttle vectors. Gene , 119–122. [DOI] [PubMed] [Google Scholar]
- Dickson RM, Cubitt AB, Tsien RY, Moerner WE. (1997). On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature , 355–358. [DOI] [PubMed] [Google Scholar]
- Douglas LM, Konopka JB. (2014). Fungal membrane organization: the eisosome concept. Annu Rev Microbiol , 377–393. [DOI] [PubMed] [Google Scholar]
- Fisk HA, Yaffe MP. (1999). A role for ubiquitination in mitochondrial inheritance in Saccharomyces cerevisiae. J Cell Biol , 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaubitz C, Prouteau M, Kusmider B, Loewith R. (2016). TORC2 structure and function. Trends Biochem Sci , 532–545. [DOI] [PubMed] [Google Scholar]
- Gournas C, Gkionis S, Carquin M, Twyffels L, Tyteca D, Andre B. (2018). Conformation-dependent partitioning of yeast nutrient transporters into starvation-protective membrane domains. Proc Natl Acad Sci USA , E3145–E3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith J, Mari M, De Maziere A, Reggiori F. (2008). A cryosectioning procedure for the ultrastructural analysis and the immunogold labelling of yeast Saccharomyces cerevisiae. Traffic , 1060–1072. [DOI] [PubMed] [Google Scholar]
- Grossmann G, Malinsky J, Stahlschmidt W, Loibl M, Weig-Meckl I, Frommer WB, Opekarova M, Tanner W. (2008). Plasma membrane microdomains regulate turnover of transport proteins in yeast. J Cell Biol , 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann G, Opekarova M, Malinsky J, Weig-Meckl I, Tanner W. (2007). Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J , 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CB, Ott EM, Keener JM, Curtiss M, Sandrin V, Babst M. (2012). Regulation of membrane protein degradation by starvation-response pathways. Traffic , 468–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, Bewersdorf J. (2008). Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat Methods , 527–529. [DOI] [PubMed] [Google Scholar]
- Kabeche R, Howard L, Moseley JB. (2015). Eisosomes provide membrane reservoirs for rapid expansion of the yeast plasma membrane. J Cell Sci , 4057–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamble C, Jain S, Murphy E, Kim K. (2011). Requirements of Slm proteins for proper eisosome organization, endocytic trafficking and recycling in the yeast Saccharomyces cerevisiae. J Biosci , 79–96. [DOI] [PubMed] [Google Scholar]
- Kaminska J, Kwapisz M, Grabinska K, Orlowski J, Boguta M, Palamarczyk G, Zoladek T. (2005). Rsp5 ubiquitin ligase affects isoprenoid pathway and cell wall organization in S. cerevisiae. Acta Biochim Pol , 207–220. [PubMed] [Google Scholar]
- Keener JM, Babst M. (2013). Quality control and substrate-dependent downregulation of the nutrient transporter fur4. Traffic , 412–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. (2000). Rapid and reliable protein extraction from yeast. Yeast , 857–860. [DOI] [PubMed] [Google Scholar]
- Lang MJ, Martinez-Marquez JY, Prosser DC, Ganser LR, Buelto D, Wendland B, Duncan MC. (2014). Glucose starvation inhibits autophagy via vacuolar hydrolysis and induces plasma membrane internalization by down-regulating recycling. J Biol Chem , 16736–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, MacGurn JA, Chu T, Stefan CJ, Emr SD. (2008). Arrestin-related ubiquitin-ligase adaptors regulate endocytosis and protein turnover at the cell surface. Cell , 714–725. [DOI] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot JW. (1996). Improving structural integrity of cryosections for immunogold labeling. Histochem Cell Biol , 41–58. [DOI] [PubMed] [Google Scholar]
- Loibl M, Grossmann G, Stradalova V, Klingl A, Rachel R, Tanner W, Malinsky J, Opekarova M. (2010). C terminus of Nce102 determines the structure and function of microdomains in the Saccharomyces cerevisiae plasma membrane. Eukaryot Cell , 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast , 953–961. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. (2005). Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol , 36–51. [DOI] [PubMed] [Google Scholar]
- Miermont A, Waharte F, Hu S, McClean MN, Bottani S, Leon S, Hersen P. (2013). Severe osmotic compression triggers a slowdown of intracellular signaling, which can be explained by molecular crowding. Proc Natl Acad Sci USA , 5725–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moharir A, Gay L, Appadurai D, Keener J, Babst M. (2018). Eisosomes are metabolically regulated storage compartments for APC-type nutrient transporters. Mol Biol Cell , 2113–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikko E, Sullivan JA, Pelham HR. (2008). Arrestin-like proteins mediate ubiquitination and endocytosis of the yeast metal transporter Smf1. EMBO Rep , 1216–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niles BJ, Mogri H, Hill A, Vlahakis A, Powers T. (2012). Plasma membrane recruitment and activation of the AGC kinase Ypk1 is mediated by target of rapamycin complex 2 (TORC2) and its effector proteins Slm1 and Slm2. Proc Natl Acad Sci USA , 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivera-Couto A, Grana M, Harispe L, Aguilar PS. (2011). The eisosome core is composed of BAR domain proteins. Mol Biol Cell , 2360–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platonova E, Winterflood CM, Ewers H. (2015). A simple method for GFP- and RFP-based dual color single-molecule localization microscopy. ACS Chem Biol , 1411–1416. [DOI] [PubMed] [Google Scholar]
- Pontes B, Monzo P, Gauthier NC. (2017). Membrane tension: a challenging but universal physical parameter in cell biology. Semin Cell Dev Biol , 30–41. [DOI] [PubMed] [Google Scholar]
- Portet T, Gordon SE, Keller SL. (2012). Increasing membrane tension decreases miscibility temperatures; an experimental demonstration via micropipette aspiration. Biophys J , L35–L37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggi M, Niewola-Staszkowska K, Chiaruttini N, Colom A, Kusmider B, Mercier V, Soleimanpour S, Stahl M, Matile S, Roux A, et al. (2018). Decrease in plasma membrane tension triggers PtdIns(4,5)P2 phase separation to inactivate TORC2. Nat Cell Biol , 1043–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JS, Klionsky DJ, Banta LM, Emr SD. (1988). Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol Cell Biol , 4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelants FM, Breslow DK, Muir A, Weissman JS, Thorner J. (2011). Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc Natl Acad Sci USA , 19222–19227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods , 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira F, Mueller NS, Beck G, von Olshausen P, Beig J, Wedlich-Soldner R. (2012). Patchwork organization of the yeast plasma membrane into numerous coexisting domains. Nat Cell Biol , 640–648. [DOI] [PubMed] [Google Scholar]
- Stradalova V, Stahlschmidt W, Grossmann G, Blazikova M, Rachel R, Tanner W, Malinsky J. (2009). Furrow-like invaginations of the yeast plasma membrane correspond to membrane compartment of Can1. J Cell Sci , 2887–2894. [DOI] [PubMed] [Google Scholar]
- Tang HY, Xu J, Cai M. (2000). Pan1p, End3p, and S1a1p, three yeast proteins required for normal cortical actin cytoskeleton organization, associate with each other and play essential roles in cell wall morphogenesis. Mol Cell Biol , 12–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang J, Huibregtse JM. (1999). Functional domains of the Rsp5 ubiquitin-protein ligase. Mol Cell Biol , 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitworth K, Bradford MK, Camara N, Wendland B. (2014). Targeted disruption of an EH-domain protein endocytic complex, Pan1-End3. Traffic , 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Roach PJ, Montero M, Baroja-Fernandez E, Munoz FJ, Eydallin G, Viale AM, Pozueta-Romero J. (2010). Regulation of glycogen metabolism in yeast and bacteria. FEMS Microbiol Rev , 952–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston F, Dollard C, Ricupero-Hovasse SL. (1995). Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast , 53–55. [DOI] [PubMed] [Google Scholar]
- Wright R. (2000). Transmission electron microscopy of yeast. Microsc Res Tech , 496–510. [DOI] [PubMed] [Google Scholar]