

Abstract
Alzheimer’s disease is characterized by a compromised blood-brain barrier and disrupted intracellular calcium homeostasis in the brain. Therefore, rectifying the blood-brain barrier (BBB) integrity and restoring calcium homeostasis could provide an effective strategy to treat Alzheimer’s disease. Recently, we developed a high throughput-screening assay to screen for compounds that enhance a cell-based BBB model integrity, which identified multiple hits among which is granisetron, a Food and Drug Administration approved drug. Here, we evaluated the therapeutic potential of granisetron against Alzheimer’s disease. Granisetron was tested in C57Bl/6J young and aged wild-type mice, and in a transgenic mouse model of Alzheimer’s disease namely TgSwDI for its effect on BBB intactness and Aβ-related pathology. Our study findings showed that granisetron enhanced BBB integrity in both aged and TgSwDI mice. This effect was associated with an overall reduction in amyloid-β load and neuroinflammation in TgSwDI mice brains. In addition, and supported by proteomics analysis, granisetron significantly reduced amyloid-β induced calcium influx in vitro, and rectified calcium dyshomeostasis in TgSwDI mice brains by restoring calmodulin-dependent protein kinase II/cAMP-response element binding protein pathway, which was associated with cognitive improvement. These results support granisetron repurposing as a potential drug to hold, slow and/or treat Alzheimer’s disease.
Keywords: Alzheimer’s disease, Granisetron, Repurposing, Calcium homeostasis, CamKII/CREB pathway
Background
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and form of dementia, which accounts for more than 60–80% of all cases worldwide [1]. According to the 2018 world Alzheimer’s report, someone develops dementia every 3 seconds with about 50 million people living with AD, a number that is expected to increase to 132 million by 2050 [2]. AD alone kills more than breast cancer and prostate cancer combined [3].
Several neuropathological hallmarks of AD have been recognized since Alois Alzheimer’s description of the disease over a century ago. While the major cause remains unknown, AD is described clinically by the gradual deterioration in cognitive function and memory loss, leading to slow and progressive behavioral changes [4]. The pathological characteristics of the disease are the presence of extracellular amyloid-β (Aβ) deposition in brain parenchyma as senile plaques and in vessels as cerebral amyloid angiopathy (CAA), intracellular neurofibrillary tangles (NFTs) composed of hyper-phosphorylated tau protein deposits, and the shrinkage of the cerebral cortex due to extensive neuronal loss [5]. In addition, AD is characterized by a chronic brain inflammation, elevated intracellular calcium (Ca+2), and a disrupted blood-brain barrier (BBB) [6–9].
To date, drug discovery and development in AD has been largely unsuccessful in finding any disease modifying treatment. Currently approved medications for AD only address its symptoms once they have reached the stage of clinical dementia. Two classes of medications are available, the cholinesterase inhibitors and N-methyl-D-aspartate (NMDA) antagonists. Besides, numerous AD clinical trials have failed for several reasons including the lack of extent in molecular targets of new treatments, which have predominantly focused on amyloid to provoke disease modification [4]. Thus, there is an urgent need to find disease-modifying therapies that will prevent, delay the onset and/or slow the progression of AD.
One important therapeutic target is the BBB. Current evidence suggests that aging, cerebrovascular damage, and/or Aβ or tau accumulation can induce BBB dysfunction by affecting different components of the neurovascular unit [5,10–14]. BBB disruption not only causes neuronal damage, but also compromises Aβ clearance, which could result in a vicious cycle between Aβ accumulation and BBB dysfunction during AD progression. Therefore, to maintain a healthy and/or restore the BBB function could provide a novel approach to prevent or treat AD. Recently, we have developed a cell-based BBB model with CAA characteristics to high-throughput screen (HTS) thousands of compounds to identify drugs that enhance the BBB model integrity and restore its function against Aβ toxicity [15]. Among the hit compounds, granisetron emerged as a potential drug to protect the BBB model against Aβ toxicity. Granisetron, a 5-hydroxytryptamine-3 (5-HT3) receptor antagonist, is widely used to treat chemotherapy-induced nausea and vomiting [16]. 5-HT3 receptor is a ligand-gated ion channel that increases intracellular cations such as calcium (Ca+2), sodium and potassium. Stimulation of 5-HT3 receptor induces rapid and transient depolarization and excitation of neurons, and 5-HT3 receptor antagonists, such as granisetron, directly decrease depolarization and elevation of intracellular Ca+2 by blocking the receptor [17].
The overall objective of this work is to evaluate the therapeutic potential of granisetron against AD. To investigate this objective, in vitro and in vivo studies in young and aged wild-type mice as well as in AD mouse model were performed. Proteomics analysis of young and aged mice brains was also performed to examine the proteomic response to granisetron.
Materials and methods
Materials
Granisetron HCl was purchased from TCI America (Portland, OR). Texas red-Dextran 10 KDa and FITC-Dextran 40 KDa, bovine serum albumin (BSA) and Thioflavin-S (Thio-S) were purchased from Sigma-Aldrich (St. Louis, MO). Fetal bovine serum (FBS) was purchased from Atlanta Biological (Flowery Branch, GA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Grand Island, NY), Fluo-4 Direct™ Calcium Assay Kit was purchased form ThermoFisher Scientific Inc. (Waltham, MA). Total protein measurement reagents with the bicinchoninic acid (BCA) method were obtained from Pierce (Rockford, IL). Choline/Acetylcholine assay kit was purchased from Abcam (Cambridge, MA). All other chemicals and reagents were of analytical grade and were readily available from commercial sources.
Antibodies
Antibodies against light chain LRP1 and BACE1 were obtained from Abcam, P-glycoprotein (P-gp) monoclonal antibody (C219) and Alexa-fluor 488-labeled 6E10 human-specific anti-Aβ antibody were purchased from BioLegend (San Diego, CA). Zona-occludin-1 (ZO-1), claudin-5, occludin and BDNF were purchased from ThermoFisher. Junctional adhesion molecule-1 (JAM-1), actin, tubulin, glial fibrillary acidic protein (GFAP), insulin degrading enzyme (IDE), MMP-9, RAGE, neprilysin (NEP) and Fluorescein-conjugated donkey anti-IgG were purchased from Santa Cruz Biotechnology Inc (Santa Cruz, CA). HRP-labeled secondary anti-rabbit and anti-mouse antibodies were purchased from Invitrogen (Carlsbad, CA), HRP-labeled secondary anti-goat antibody was purchased from R&D systems (Minneapolis, MN). Nicastrin, cAMP-dependent protein kinase (PKAc), IP3 receptor, phospho-IP3 receptor (Ser1756), CREB, phospho-CREB (Ser133), caspase-3, Bcl-2, αCaMKII, phospho-CaMKII (Thr286) and calbindin D28K were purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-collagen-IV antibody was purchased from Millipore (Burlington, MA). Specific antibodies against soluble APPα (sAPPα) and soluble APPβ (sAPPβ) were purchased from Immuno-Biological Laboratories (Minneapolis, MN). Synaptic markers, PSD-95 and SNAP-25 antibodies were purchased form GeneTex, Inc. (Irvin, CA) and Synapsin-1 antibody was purchase from Cell Signaling Technology, Inc. (Danvers, MA). Secondary antibodies used for collagen-IV were either CFL594-conjugated donkey anti-rabbit IgG (Santa Cruz Biotechnology) or goat anti-rabbit IgG (H+L) F(ab′)2 fragment, CF™350 from Sigma-Aldrich.
Cell culture
The mouse brain endothelial cells (bEnd3; ATCC, Manassas, VA), passage 27–33, were cultured in DMEM growth medium supplemented with 10% FBS, and the antibiotics penicillin G (100 units/ml) and streptomycin (100 g/ml). Cells were grown to confluence in 75 cm2 cell culture flasks for one to two days in a humidified atmosphere (5%CO2/95% air) at 37° C. Neuronal cultures were prepared from SH-SY5Y cell line transfected with APP695 (hereafter SH-SY5Y-APP). Cells were maintained in DMEM media supplemented with 10% FBS containing geneticin (Gibco) added at 400 μg/ml to SH-SY5Y-APP cells. Cultures were maintained in a humidified atmosphere (5% CO2/95% air) at 37°C and media were changed every other day.
Calcium flux measurement
Cells were plated at 100,000 cells/cm2 in 96-well black-walled clear bottom microplates (Costar® 3603; ThermoFisher). Twenty-four hours later, cells were treated with 50 μl media (control) or 50 μl media containing granisetron in a final concentration of 5 and 10 μM for 24 h. To determine effect on Ca+2 flux, cells were loaded with the fluorescent Ca+2 sensor Fluo-4 Direct for 30 min at 37 °C, 5% CO2, and then equilibrated to room temperature for another 30 min. Fluorescence intensity was measured at excitation and emission wavelengths of 490 and 520 nm, respectively, for baseline (10 reads at 32-s intervals) followed by another 10 reads after Aβ42 addition (final concentration of 2 μM) as described preciously [18]. Cytation™ 5 cell imaging multi-mode reader (BioTek Instruments, Inc., Winooski, VT) was used. Ca+2 flux values (ΔF/F0) were expressed as the difference between mean baseline and immediately following Aβ application.
Animals
All animal experiments and procedures were approved by The Institutional Animal Care and Use Committee of the University of Louisiana at Monroe and according to the National Institutes of Health guidelines Principles of laboratory animal care. Wild type C57Bl/6 male mice at age of 4 months, representing the young group, and 18 months, representing the aged group, and TgSwDI transgenic mice at age of 4 (a CAA/AD mouse model) were purchased from Jackson Laboratories (Bar Harbor, ME). TgSwDI mice express human APP under control of Thy 1.2 neuronal promoter harboring double Swedish mutations and the Dutch and Iowa vasculotropic Aβ mutations leading to early and aggressive Aβ accumulation associated with inflammatory astrocytes activation and cognitive decline. In the brain of TgSwDI mice, Aβ begins to accumulate at age 2 to 3 months and deposits extensively at age of 12 months [19]. All mice were housed in plastic cages under standard conditions, 12-h light/dark cycle, 22°C, 35% relative humidity, and ad libitum access to water and food.
Animals’ treatment
To evaluate the effect of granisetron on the BBB integrity, young (4 months) and aged (18 months) wild type mice were divided into 2 groups each, control and treatment groups (n=7 mice/group). Treatment groups of young and aged mice received 3 mg/kg/day granisetron and the control groups received sterile distilled water as vehicle for 28 days using ALZET® (DURECT Corporation, Cupertino, CA) osmotic minipumps (model 1004). Granisetron dose was selected based on previous reports used granisetron in the range of 0.1–10 mg/kg/day to test its effect against several diseases/conditions other than nauseas and vomiting [20–24]. Based on these reports and for a proof-of-concept, in the current study granisetron at 3 mg/kg/day dose was used to evaluate its effect against AD. Pumps were implanted subcutaneously by making a small cut in the midscapulary region to insert the pump, and closing the wound with clips as previously described [25]. Pumps delivered 100 μl of solution at a rate of 0.11 μl/h for 28 days. For TgSwDI mice experiments, mice (4 months old, male) were divided into two groups (n=5 mice/group), control group received sterile water as a vehicle administered intraperitonially (IP), and treatment group that received 3 mg/kg/day granisetron administered IP for 28 days. During the treatment period, animals’ body weights were monitored every week, the average mouse weight was 27±3 g and 28±2 for the wild type aged control and granisetron treated groups, respectively. For young wild type mice, the average weight was 24±2 g and 23±3 g for control and granisetron treated groups, respectively. In addition, the average body weights of TgSwDI mice during the treatment period were 24±2g for control group compared to 25±2 g for granisetron group. Health status and normal behavior were checked daily with no observed side effects.
Florescent tagged-Dextran to monitor BBB permeability in wild type young and aged mice
At the end of treatment period, vehicle and granisetron treated mice were intravenously (IV) injected with 100 μl solution of two different molecular size florescent tagged dextran; 10 kDa Texas-red dextran and 40 kDa FITC-dextran (20 mg/ml of each dextran dissolved in 0.9% saline) through the mice tail vein as previously reported [26]. Four hours after the injection, mice were deeply anesthetized with ketamine (100 mg/kg)/xylazine (12.5 mg/kg) cocktail IP. Mice were transcardially perfused with heparinized-PBS using a peristaltic pump (Harvard apparatus, Holliston, MA) for 4–5 min until the PBS starts to run clear from the right atrium cut as described previously [27]. Brains were then collected for immunochemical and biochemical analysis.
Brain microvessels extraction
Brain microvessels were isolated from the brains of young and aged wild type mice, and TgSwDI mice as described previously [28]. In brief, freshly isolated brain hemispheres were immediately homogenized in ice-cold DPBS (2.7 mM KCl, 1.46 mM KH2PO4, 136.9 mM NaCl, 8.1 mM Na2HPO4, 0.9 mM CaCl2, and 0.5 mM MgCl2 supplemented with 5 mM D-glucose, 1 mM sodium pyruvate, pH 7.4). Brain homogenate samples were added to 30% ficoll in 1:1 volume, mixed thoroughly and centrifuged at 5800×g for 15 min at 4°C. Supernatant was discarded and pellet-containing microvessels was gently re-suspended in ice-cold DPBS containing 1% BSA and collected by separation over glass beads containing column. Microvessels adhered to glass beads were collected by gentle shaking followed by centrifugation. Pelleted microvessels were lysed in radioimmunoprecipitation assay (RIPA) buffer containing 1% protease inhibitor and were used for Western blot analysis.
Western Blot analysis
Twenty-five micrograms of protein samples were loaded and resolved using 10% SDS-polyacrylamide gel at 140V for 1 h and transferred electrophoretically to PVDF membrane (Millipore) at 300 mA for 3 h at 4°C. Nonspecific binding was blocked by pre-incubation of the PVDF membrane in PBS solution containing 1% skimmed milk with rocking for 1 h at room temperature followed by overnight incubation at 4°C with primary antibodies. Primary antibodies used to immunoblot bEnd3 cells lysate and isolated microvessels proteins are P-gp (C-219), LRP1 (light chain), ZO-1, claudin-5, occludin, JAM-1 and β-actin. For brain homogenate samples, primary antibodies used are sAPPα, sAPPβ, PSD-95, SNAP-25, Synapsin-1, Iba-1, GFAP, IDE, neprilysin, MMP-9, RAGE, BACE-1, IP3 receptor-1, phospho-IP3 receptor (Ser1756), CREB, phospho-CREB (Ser133), PKA-c, Nicastrin, caspase-3, Bcl-2, αCaMKII, phospho-CaMKII (Thr286), calbindin D28K, BDNF, β-actin, and β-Tubulin. Secondary antibodies used were: HRP-conjugated anti-mouse IgG secondary antibodies (1:1000 dilutions) for P-gp, PSD-95, sAPPα, claudin-5, β-actin, IDE, Bcl-2 and β-tubulin; HRP-conjugated anti-rabbit IgG secondary antibodies (1:1000 dilutions) for LRP1, SNAP-25, GFAP, neprilysin, IP3 receptor, phospho-IP3 receptor (Ser1756), CREB, phospho-CREB (Ser133), caspase-3, αCaMKII, phospho-CaMKII (Thr286), calbindin D28K, BDNF, sAPPβ, ZO-1, occludin and JAM-1. Proteins’ blots were developed using a chemiluminescence detection kit (SuperSignal West Femto substrate; ThermoFisher). Bands were visualized using ChemiDoc™ MP Imaging System (Bio-Rad Hercules, CA, USA) and quantified by densitometric analysis using Image Lab™ Software V.6.0 (Bio-Rad).
Immunohistochemical analysis
Brain sections of 16 μm-thick were prepared using Leica CM3050S Research Cryostat (Buffalo Grove, IL, USA); mice hemispheres were snap-frozen in dry-ice cold liquid 2-methylbutane (Sigma-Aldrich) and stored at −80°C. Subsequently, sections were fixed by incubation in methanol or 10 min at −20°C. Sections were washed 5 times in PBS and blocked in PBS containing 10% donkey serum for 1 h at room temperature. Immunostaining was performed for brains hippocampi of the wild type young and aged mice and TgSwDI mice. The entire hippocampus region was included in the analysis spanning the dentate gyrus and CA1-CA3 regions. To assess IgG extravasation from brain microvessels, sections were probed by dual immunohistochemical staining for collagen IV and mouse IgG using rabbit anti-collagen-IV and fluorescein-conjugated donkey anti-IgG (both at 1:200 dilution), respectively. The secondary antibody used for collagen-IV was CFL594-conjugated donkey anti-rabbit IgG. For dextran extravasation, brain sections from young and aged wild type mice were fixed and blocked, as described above, incubated with the rabbit anti-collagen-IV at 1:200 dilution, then probed with CF™ 350 goat anti-rabbit IgG (H+L) secondary antibody for collagen-IV. For total Aβ detection, TgSwDI brain sections were immunostained with Alexa-fluor 488 labeled 6E10 human-specific anti-Aβ antibody at 1:200 dilution. For detection of Aβ-plaque load in hippocampus, the brain tissue sections were stained with a freshly prepared and filtered 0.02% Thio-S solution in 70 % ethanol for 30 min followed by incubation in 70% ethanol, as described previously [29]. For reactive astrocytes, TgSwDI sections were probed with rabbit anti-GFAP polyclonal IgG at 1:100 dilution followed by anti-rabbit IgG-CFL594 secondary antibody. For each treatment, image acquisition was performed in 10 tissue sections spanning the hippocampus, each separated by 150 μm (total of 40 sections per mouse). Total Aβ load and Thio-S were captured and quantified at a total magnification of 4X, GFAP, IgG and dextran extravasation images were capture at a total magnification of 20X. Quantification of fluorescence intensity was performed using ImageJ® version 1.6.0 software (Research Services Branch, National Institute of Mental Health/National Institutes of Health, Bethesda, MD) after adjusting for threshold. All images were visualized using Nikon Eclipse Ti-S inverted fluorescence microscope (Melville, NY).
Human Aβ40 and Aβ42 determination by ELISA
To quantitatively extract soluble Aβ from TgSwDI mice brain, 150 mg of brain tissue was polytron homogenized in diethylamine (DEA) buffer (50 mM NaCl and 0.2% DEA), with complete mammalian protease inhibitor (Sigma-Aldrich) and centrifuged at 21,000g for 45 min at 4°C. Supernatants were collected and analyzed for Aβ40 and Aβ42 brain levels using commercially available ELISA kits according to the manufacturer instructions (ThermoFisher). All samples were run at least in triplicates and corrected to the total protein amount in each sample using BCA assay.
Analysis of brain acetylcholine levels
Acetylcholinesterase (AChE) activity in TgSwDI mice brains’ homogenates was determined indirectly by measuring acetylcholine (ACh) levels. For this, we used choline/acetylcholine assay kit from Abcam. Samples from brain homogenates were processed according to the protocol provided by the kit and detection of choline was performed fluorometrically at excitation/emission of 535/590 nm. This assay measures free choline and total choline (free choline and ACh). Thus, ACh level can be determined as follows: ACh = total choline – free choline.
Behavioral testing
The Morris water maze (MWM) test was performed for TgSwDI mice to assess learning and memory performance at the end of the treatment using protocols similar to those described previously [30]. All mice underwent training 3 times a day for 4 consecutive days. The platform was kept at the same quadrant during the entire course of the experiment. Mice were required to find the hidden platform utilizing the distal spatial cues available in the room. Conditions were maintained the same during all the experiments. An overhead camera connected to a computerized tracking system (SMART 3.0 Platform, Panlab Harvard apparatus (Holliston, MA)) was used to record the movements of the mice. The results including latency and swimming distance were collected and used for statistical analysis.
Sample preparation for tandem mass tagging labeling and proteomic analysis
Three of each granisetron-treated mice and control mice were used for proteomic experiment in both age groups. Proteins in tissue lysate were reduced, alkylated, and digested using filter-aided sample preparation (FASP) based on our previous method with a minor modification [31]. Tissues were lysed and solubilized in lysis buffer containing 2% SDS, 100mM Tris-HCl pH 7.6, 0.1M DTT. Protein extracts were alkylated using FASP in 50mM iodoacetamide in 8M urea, 100 mM Tris-HCl pH 8.5. Proteins were then digested with sequencing grade porcine trypsin (Promega, Madison, WI). Digested peptides were cleaned by 50 mg Sep-Pak SPE (Waters Milford, MA). Resuspended tryptic peptides were then labeled using a tandem mass tag 6-plex isobaric label reagent set (Thermo, Rockford, IL) following the manufacturer’s instructions. Aged and young mice were grouped into two different TMT experiments. The combined samples of each TMT experiment were cleaned by 50 mg Sep-Pak SPE immediately before offline high pH fractionation.
For the offline high pH fractionation, labeled peptides were separated into 13 fractions on a 100×1.0 mm Acquity BEH C18 column (Waters, Milford, MA) using an UltiMate 3000 UHPLC system (Thermo) with a 40 min gradient from 99:1 to 60:40 buffer A:B ratio under basic pH conditions, and then consolidated into 13 super-fractions. Buffer A = 0.1% formic acid, 0.5% acetonitrile; buffer B = 0.1% formic acid, 99.9% acetonitrile. Both buffers adjusted to pH 10 with ammonium hydroxide for offline separation. Each super-fraction was then further separated by reverse phase Jupiter Proteo resin (Phenomenex, Torrance, CA) on an in-line 200 × 0.075 mm column using a nanoAcquity UPLC system (Waters Corporation, Milford, MA). Peptides were eluted using a 60 min gradient from 97:3 to 67:33 buffer A:B ratio described above. Eluted peptides were ionized by electrospray (2.15 kV) followed by mass spectrometric analysis on an Orbitrap Fusion Tribrid mass spectrometer (Thermo) using multi-notch MS3 parameters. MS data were acquired using the FTMS analyzer in top-speed profile mode at a resolution of 240,000 over a range of 375 to 1500 m/z. Following CID activation with normalized collision energy of 35.0, MS/MS data were acquired using the ion trap analyzer in centroid mode over a range of 400–2000 m/z. Using synchronous precursor selection, up to 10 MS/MS precursors were selected for HCD activation with normalized collision energy of 65.0, followed by acquisition of MS3 reporter ion data using the FTMS analyzer in profile mode at a resolution of 30,000 over a range of 100–500 m/z.
Protein identification and quantification
Proteins were identified and reporter ions were quantified using MaxQuant (Max Planck Institute of Biochemistry, Martinsried, Germany; version 1.5.8.3) with a parent ion tolerance of 3 ppm, a fragment ion tolerance of 0.5 Da, and a reporter ion tolerance of 0.01 Da. Scaffold Q+S (Proteome Software Inc., Portland, OR; version 4.8.5) was used to verify MS/MS based peptide and protein identifications as well as to quantitate TMT Label Based Quantitation and data analysis. Peptide identifications were accepted if they could be established at greater than 8.0% probability to achieve an FDR less than 1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 51.0% probability to achieve an FDR less than 1.0% and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm 2 and to perform reporter ion-based statistical analysis.
Ingenuity pathway analysis and bioinformatics analysis
Normalization was performed iteratively (across samples and spectra) on intensities by Scaffold Q+S, as described in Statistical Analysis of Relative Labeled Mass Spectrometry Data from Complex Samples Using ANOVA and medians were used for averaging 3. Spectra data were log-transformed, pruned of those matched to multiple proteins, and weighted by an adaptive intensity-weighting algorithm. Of 102146 spectra in the experiment at the given thresholds, 77419 (76%) were included in quantitation. Differentially expressed proteins were determined by applying Mann-Whitney Test with significance level P<0.05 corrected by Benjamini-Hochberg. To further understand the biological events in relation to the treatment, the entire complement of proteins that were statistically significantly different (FDR<0.05) between treatment and control groups containing 2 fold up- or down-regulated proteins were analyzed by Ingenuity Pathway Analysis version 42012434 (Ingenuity ® Systems, www.ingenuity.com, Mountain View, CA). Right-tailed Fisher’s exact test was used to calculate the P value in the canonical pathway. Calcium pathways-related protein networks analysis with differently expressed proteins were evaluated.
Statistical analysis
Data are presented as box-and-whisker plots representing median and interquartile range (IQR), with minimum and maximum values of n=7 mice/group for wild type mice and n=5 mice/group for TgSwDI mice. The experimental results were statistically analyzed for significant difference using Student’s t-test for two groups and one-way ANOVA for more than two-group analysis. For behavior studies, two-way ANOVA with Bonferroni post-hoc analysis was used. Values of P < 0.05 were considered statistically significant. Data analyses were performed using GraphPad Prism, version 6.0. In the figures, the fold change for control groups was calculated by dividing each control value on the average of all control mice values for a specific protein. Treatment values were then normalized to control (1.0).
Results
Effect of granisetron on endogenous IgG and exogenous dextran extravasations across the BBB and on the expression of BBB tight junction proteins
Effect of granisetron on BBB leakiness was evaluated by immunostaining. As shown in Fig. 1, the extravasation of 10 KDa Texas-red dextran and 40 KDa FITC-dextran was significantly increased with aging (Fig. 1B). In young mice, however, dextran extravasation was very low for the low molecular weight dextran (10 KDa) and was not observed with 40 KDa dextran (Fig. 1A,F). In aged mice, compared to vehicle treatment, granisetron significantly reduced extravasation of the florescence-tagged 10 and 40 KDa dextran by 40 and 60%, respectively (Fig. 1B, G and K). On the other hand, granisetron has no significant effect on dextran extravasation in young mice (Fig. 1A, F and K). To evaluate granisetron effect on an endogenous and higher molecular weight BBB permeability marker, IgG extravasation was tested (Fig. 1C–E, H–J, L). Similarly, IgG extravasation significantly increased with aging (Fig. 1D), which was significantly reduced by granisetron (Fig. 1I and L), and such effect was not observed in the young mice (Fig. 1C, H and L). Next, the effect of granisetron on BBB integrity was evaluated in TgSwDI mouse model for AD, and consistent with the wild-type aged mice results, granisetron significantly reduced IgG extravasation by 80% (Fig. 1 E, J and L). Compared to young mice, aged and TgSwDI mice demonstrated significantly higher IgG extravasation approximately by 3- and 1.9 fold, respectively.
Figure 1. Granisetron enhanced BBB tightness in mice brains.
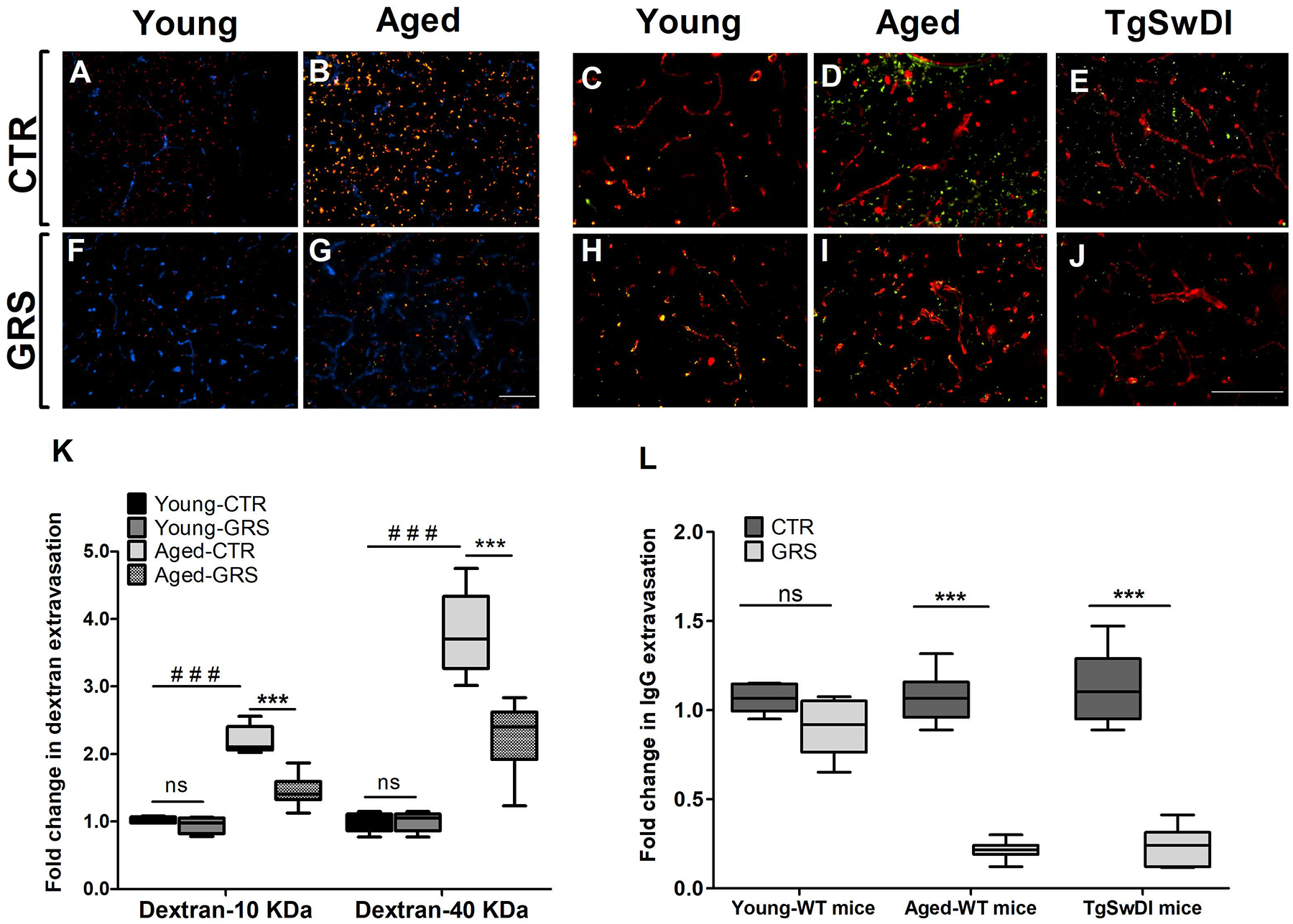
Mice were treated with granisetron (3 mg/day) for 28 days. (A,B,F,G) Representative brain sections measured using two different molecular weight fluorescents tagged dextran 10KD (red) 40 KD (green) as exogenous markers for BBB permeability and anti-Collagen IV antibody (Blue) to detect microvessels where (A) young control group, (F) young granisetron group, (B) aged control group, and (G) aged granisetron group. (K) Optical density semi-quantification of dextran 10 KD and 40 KD extravasation. (C-E, H-J) Representative brain sections stained with anti-mouse IgG antibody to detect IgG extravasation (green) and anti-collagen antibody (red) in wild type and TgSWDI mice brains and IgG levels quantification where (C) young control group, (H) young granisetron group, (D) aged control group, (I) aged granisetron group, (E) control TgSwDI group, (J) granisetron TgSwDI group, (L) optical density semi-quantification of IgG extravasation. All values in K were normalized to Young-CTR (1.0). GRS treatment values in L were normalized for each group corresponding CTR (1.0). Data are presented as box-and-whisker plots representing median and interquartile range (IQR), with minimum and maximum values. CTR is for control group i.e. vehicle-treated mice, GRS for granisetron, ns = not significant, ***P < 0.001, ###P<0.001 (Students t-test, n=5–7 mice). Scale bar, 200 μm.
This increase in BBB tightness was associated with increased expression of tight junction proteins as determined by Western blot. Granisetron significantly increased the tight junctions in the isolated microvessels from both young and aged mice, and TgSwDI mice. In young mice, granisetron increased ZO-1, occludin, claudin-5, and JAM-1 by 2.0-, 1.4-, 1.7- and 1.3-fold (Fig. 2A), and in aged mice by 2.5-, 1.4-, 2.1- and 1.6-fold, respectively (Fig. 2B). Similarly, granisetron treatment increased ZO-1, occludin, claudin-5 and JAM-1 by 2.8-, 3.1, 2.9- and 1.5-fold, respectively in TgSwDI mice (Fig. 2C).
Figure 2. Granisetron (GRS) increased tight junction proteins expression in isolated microvessels from mice brains compared to vehicle-treated mice (control, CTR).
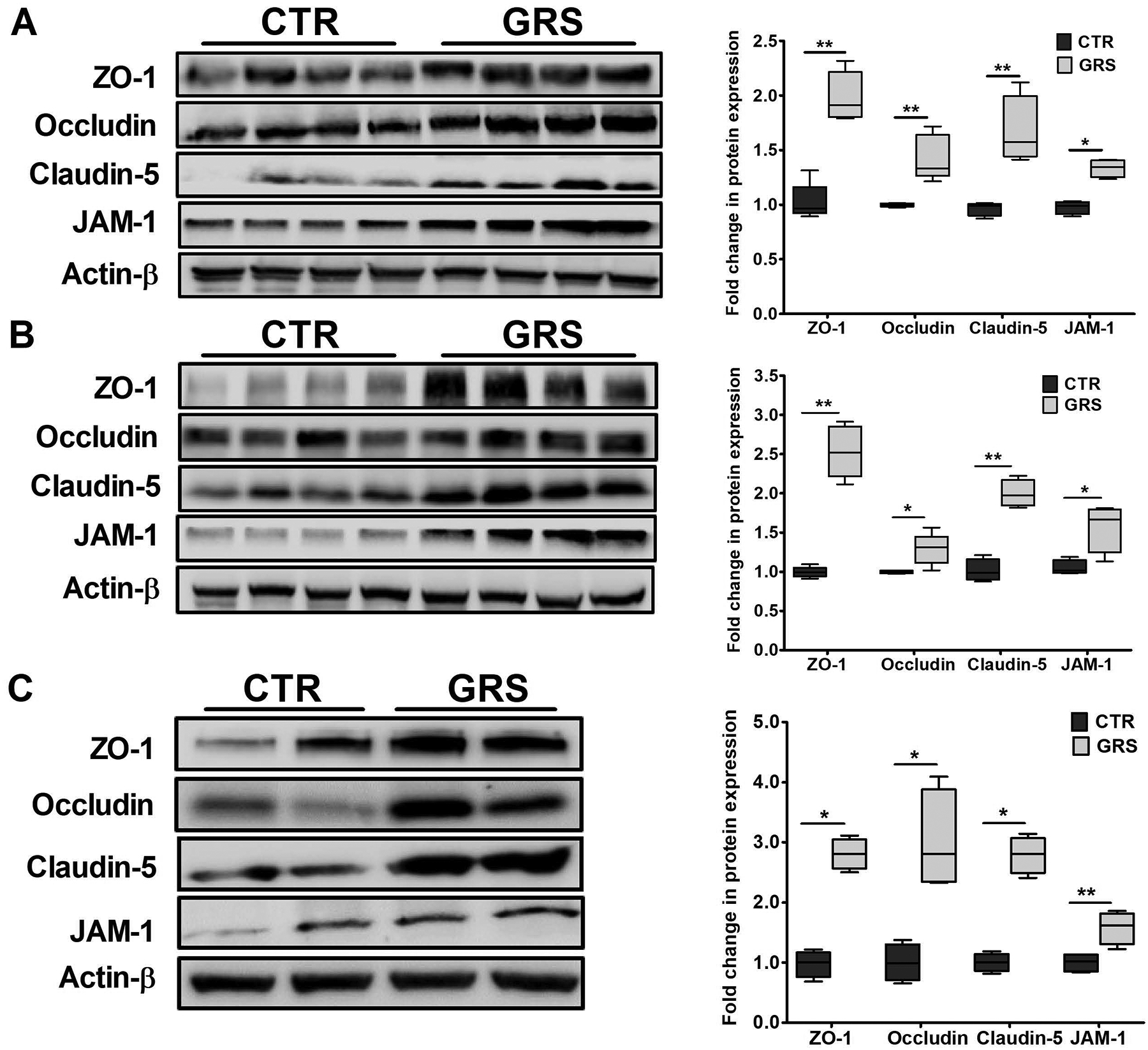
Representative western blots and densitometry analysis of the BBB tight junction proteins ZO-1, occludin, claudin-5 and JAM-1 in brain microvessels isolated from young (A), aged (B) wild type mice, and (C) TgSwDI mice. Statistical analysis was determined by Student’s t-test (n=5–7 mice). All values were normalized to CTR (1.0). Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. *P < 0.05, and **P < 0.01.
In vitro studies on the effect of granisetron on bEnd3 cells tightness and calcium intracellular levels in bEnd3 and SH-SY5Y-APP695 cells
Mouse brain endothelial cells (bEnd3) treatment with 10 μM granisetron for 72 h significantly increased expression of tight junction proteins (Fig. 3A). Granisetron increased ZO-1, occludin, claudin-5, and JAM-1 by 3.5, 1.3, 3.4 and 1.3-fold. The effect of granisetron on intracellular Ca+2 levels was evaluated by testing the inhibitory effect of granisetron on calcium flux. For this, bEnd3 cells and SH-SY5Y-APP transfected cells were treated with either granisetron (10 and 20 μM) or media for 24 h. The media was removed and replaced with HBSS buffer (Ca+2/Mg+2 free), and incubated with exact volume of the sensitive Ca+2 fluorescence indicator Fluo-4 direct as described in the methods section. As shown in (Fig. 3B and C), intracellular Ca+2 was significantly reduced in both bEnd3 (at 10 and 20 μM) and SH-SY5Y-APP (at 10 μM) cells. Interestingly, however, in SH-SY5Y-APP cells, granisetron at 20 μM did not reduce intracellular Ca+2 levels suggesting that in these cells, at higher concentrations, granisetron could exhibit non-specific targeting, thus altering the overall effect. In another separate experiment, granisetron effect on neuronal Ca+2 flux induced by Aβ42 (2 μM) acute exposure was also evaluated. Granisetron significantly reduced Aβ42-induced Ca+2 flux by ~40% (Fig. 3D).
Figure 3. Granisetron (GRS) increased tight junction proteins in bEnd3 cells and reduced intracellular calcium levels in vitro.
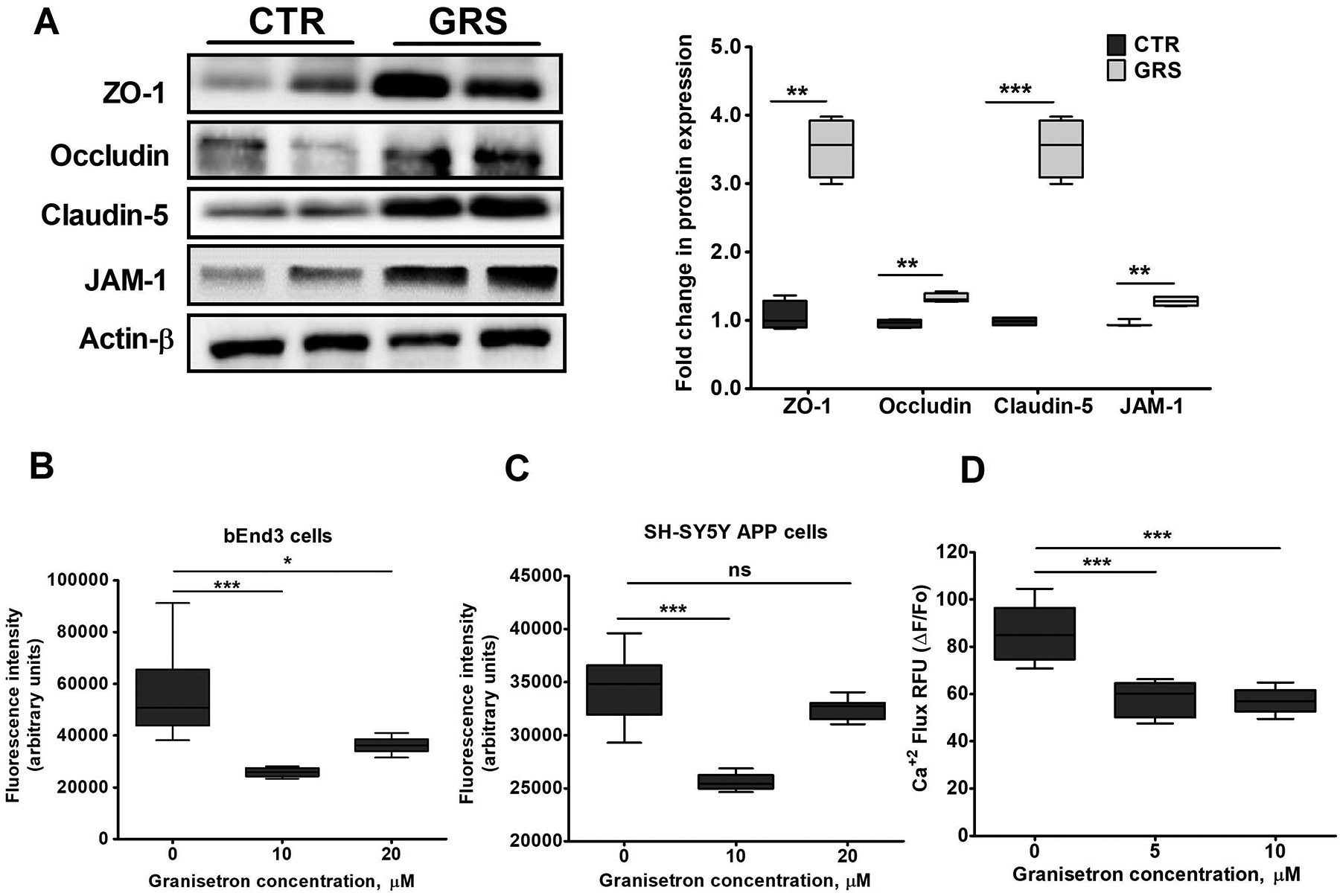
(A) Granisetron (10 μM) treatment for 72 h upregulated the expression of tight and adherent junction proteins in bEnd3 cells as shown in the representative blots and their densitometry analysis. All values were normalized to CTR (1.0). (B) Comparison of the effect of granisetron on intracellular Ca+2 level measured using the Fluo-4 assay in (B) bEnd3 cells, and (C) SH-SY5Y-APP cells after 24 h treatment with 10 and 20 μM. (D) SH-SY5Y-APP cells were additionally pre-treated with granisetron 5 and 10 μM for 24 h and were then acutely exposed to freshly prepared Aβ42 (2 μM) to measure Ca+2 flux kinetics using Fluo-4 assay. Ca+2 flux assays were performed in HBSS buffer devoid of Ca+2 and Mg+2 to determine the source of Ca+2 moving into the cytosol. Each treatment was tested in a minimum of eight replicate wells per assay and the experiments repeated at a minimum of three times over separate days. Statistical analysis was determined by Student’s t-test for (A), and one-way ANOVA test for (B-D). Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. ns = not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 versus vehicle treated cells (CTR, control).
Effect of granisetron treatment on total Aβ load in TgSwDI mice brains
Compared to vehicle treated mice, granisetron significantly reduced total Aβ immunostaining by 75% (Fig. 4A–C). In addition, granisetron significantly reduced Thio-S staining of Aβ plaques by 80% compared to control group (Fig. 4D–F). ELISA quantification of soluble Aβ40 and Aβ42 in brain homogenates from mice treated with granisetron also confirmed a significant reduction in both peptides brains levels (Fig. 4G–H). To explain Aβ reduction, effect of granisetron on Aβ production and clearance proteins were evaluated by western blot. For Aβ production proteins, granisetron treatment reduced soluble APPβ (sAPPβ) and increased soluble APPα (sAPPα) expressions without altering BACE1 and nicastrin, a subunit of γ-secretase enzyme (Fig. 5A–B). In addition to reduced Aβ production, granisetron enhanced Aβ clearance by inducing Aβ major transport protein P-gp without altering LRP-1 expression, and reduced the receptor for advanced glycation end products (RAGE) at the BBB as determined in isolated brain microvessels (Fig. 5C–D). Furthermore, granisetron significantly increased the expression of Aβ degrading enzymes IDE and NEP in mice brains homogenate when compared to control group (Fig. 5E–F).
Figure 4. Granisetron (GRS) reduced Aβ burden in the brain and hippocampus of TgSwDI mice.
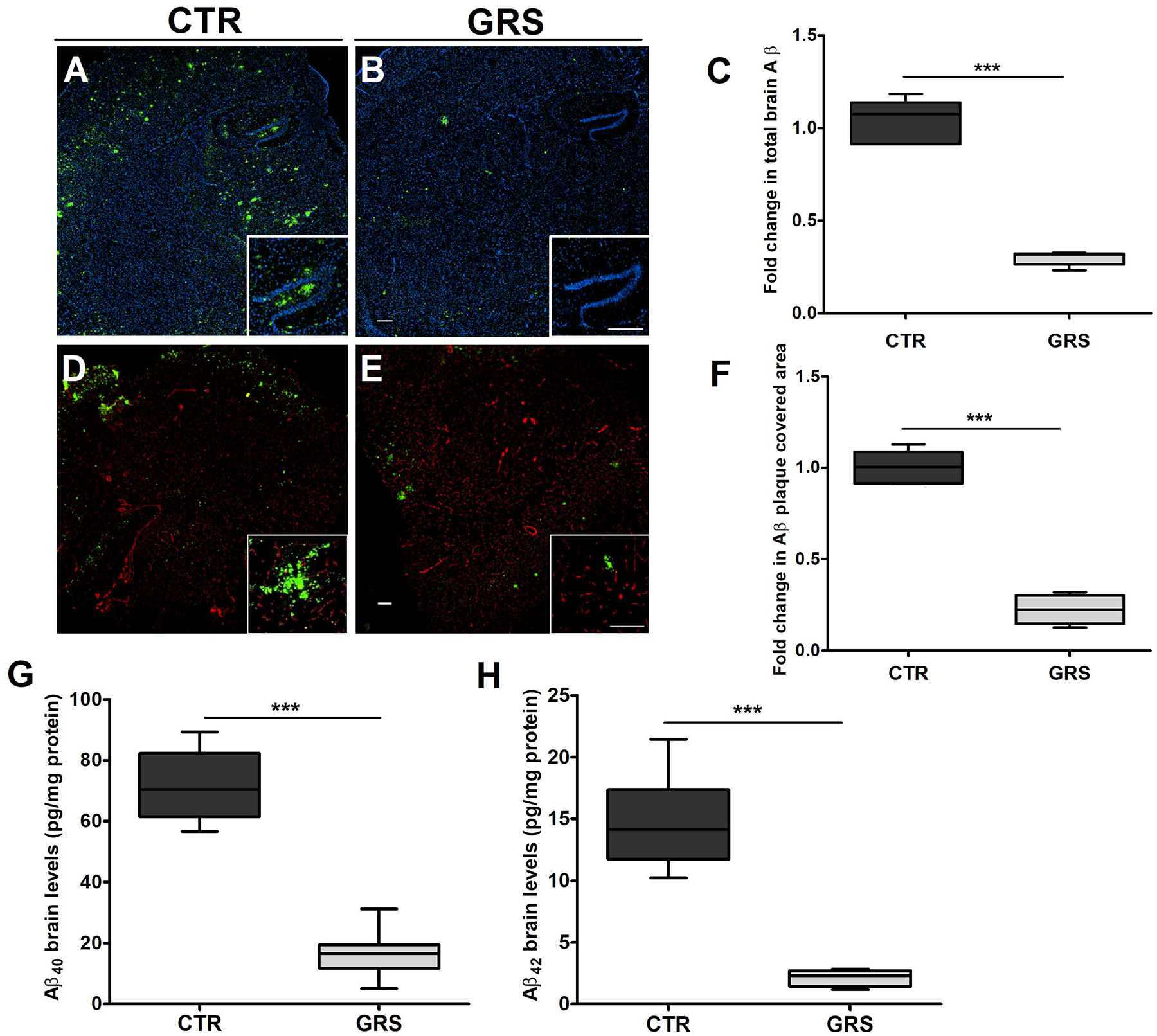
(A-C) Representative brain sections stained with 6E10 (green) antibody against Aβ to detect total Aβ load and anti-collagen IV (blue) to stain microvessels. Hippocampus is seen at higher magnification in the closed inserts; (A) control (CTR) group, (B) granisetron group, (C) quantification of total Aβ deposition. Values were normalized to CTR (1.0). (D-F) Representative hippocampus sections stained with Thio-S (green) to detect Aβ plaques load and anti-collagen IV (red) to stain microvessels; (D) control (CTR) group, (E) granisetron group, (F) quantification of area covered with Aβ plaques. Values were normalized to CTR (1.0). Individual red and green channels for Panels D and E can be found under supplementary material (Fig. S3). Brain levels of both soluble human Aβ40 (G) and soluble human Aβ42 (H) levels were determined by ELISA. Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. Statistical analysis was determined by Student’s t-test (n=5 mice per group). ***P < 0.001 versus control (CTR) group. Scale bar, 200 μm.
Figure 5. Granisetron (GRS) reduced Aβ brain load by reducing Aβ production and increasing Aβ clearance in TgSwDI mice brains.
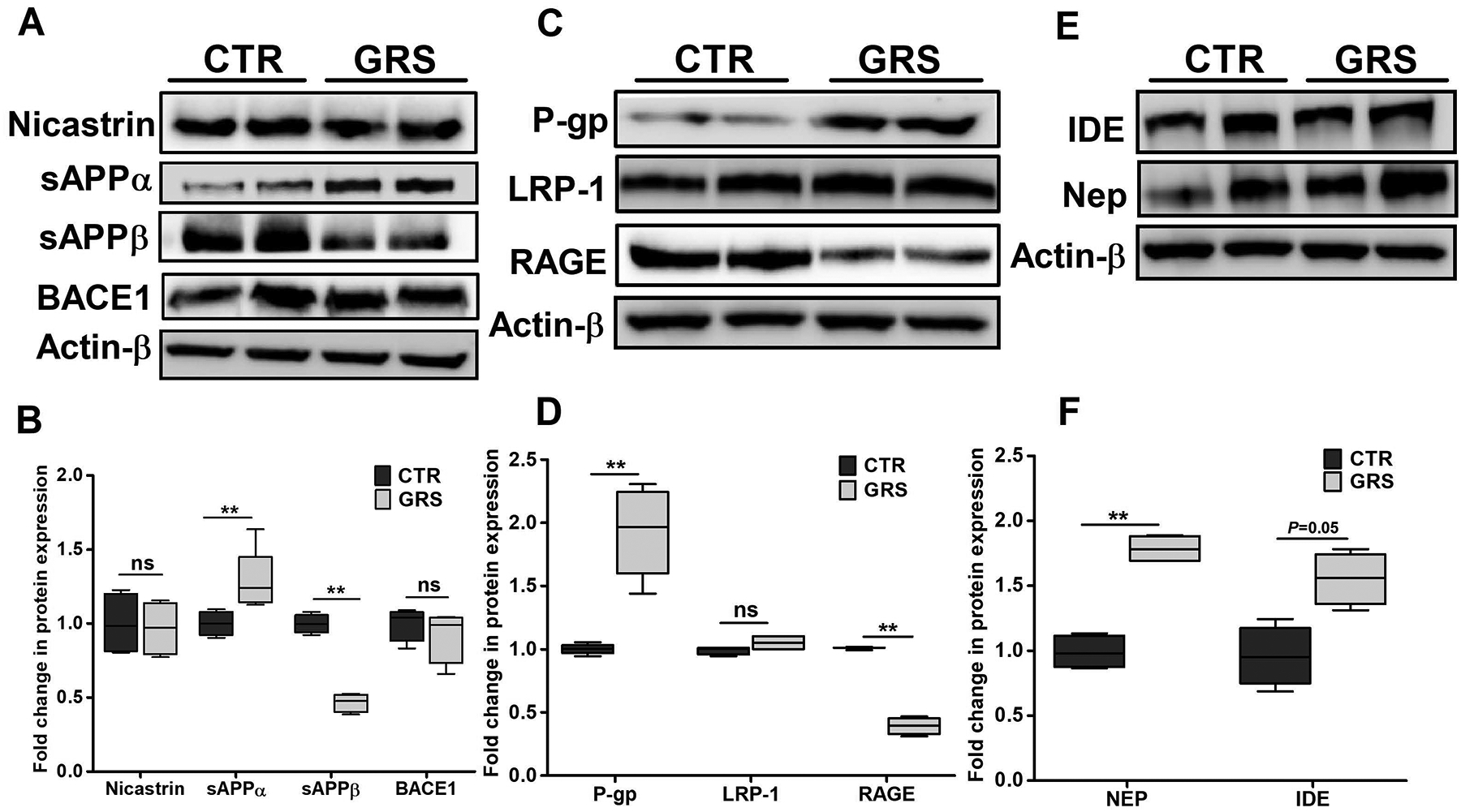
(A) Representative blots, and (B) densitometry analysis showed a significant increase in sAPPα and reduction in sAPPβ levels in granisetron treated group, with no significant effect on either BACE-1 enzyme or the nicastrin subunit of the γ-secretase enzyme in mice brains homogenates of TgSwDI mice. (C) Representative blots, and (D) densitometry analysis showed granisetron treatment significantly increased P-gp expression, a major Aβ clearance protein across the BBB, and reduced RAGE responsible for Aβ transport from blood to brain, but has no effect on LRP1. (E) Representative blots, and (F) densitometry analysis of major Aβ degrading enzymes IDE and neprilysin (NEP). Statistical analysis was determined by Student’s t-test (n=5 mice per group). Values were normalized to CTR (1.0). Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. ns=not significant, *P < 0.05, and **P < 0.01 versus control (CTR) group.
Effect of granisetron on brain inflammatory markers in TgSwDI mice brains
The anti-inflammatory effect of granisetron against chronic inflammatory diseases in animal models has been reported previously [20,32], however, its anti-inflammatory effect on Aβ-induced brain inflammation is not known. One of the earliest markers of astrocyte inflammatory activation is the increased levels of glial fibrillary acidic protein (GFAP) with elongated shape and thick branches. Our results demonstrated granisetron to reduce astrocytes activation and ameliorated the astrocytes shape when compared to the control group (Fig. 6A). Moreover, granisetron treatment significantly reduced brain levels of both the microglial marker Iba-1 by 30%, and metalloproteinase-9 (MMP-9) enzyme by 47% (Fig. 6C and F).
Figure 6. Granisetron (GRS) treatment reduced neuroinflammation, up-regulated neuro-synaptic markers, reduced apoptosis and increased acetylcholine levels in the brains of TgSwDI mice.
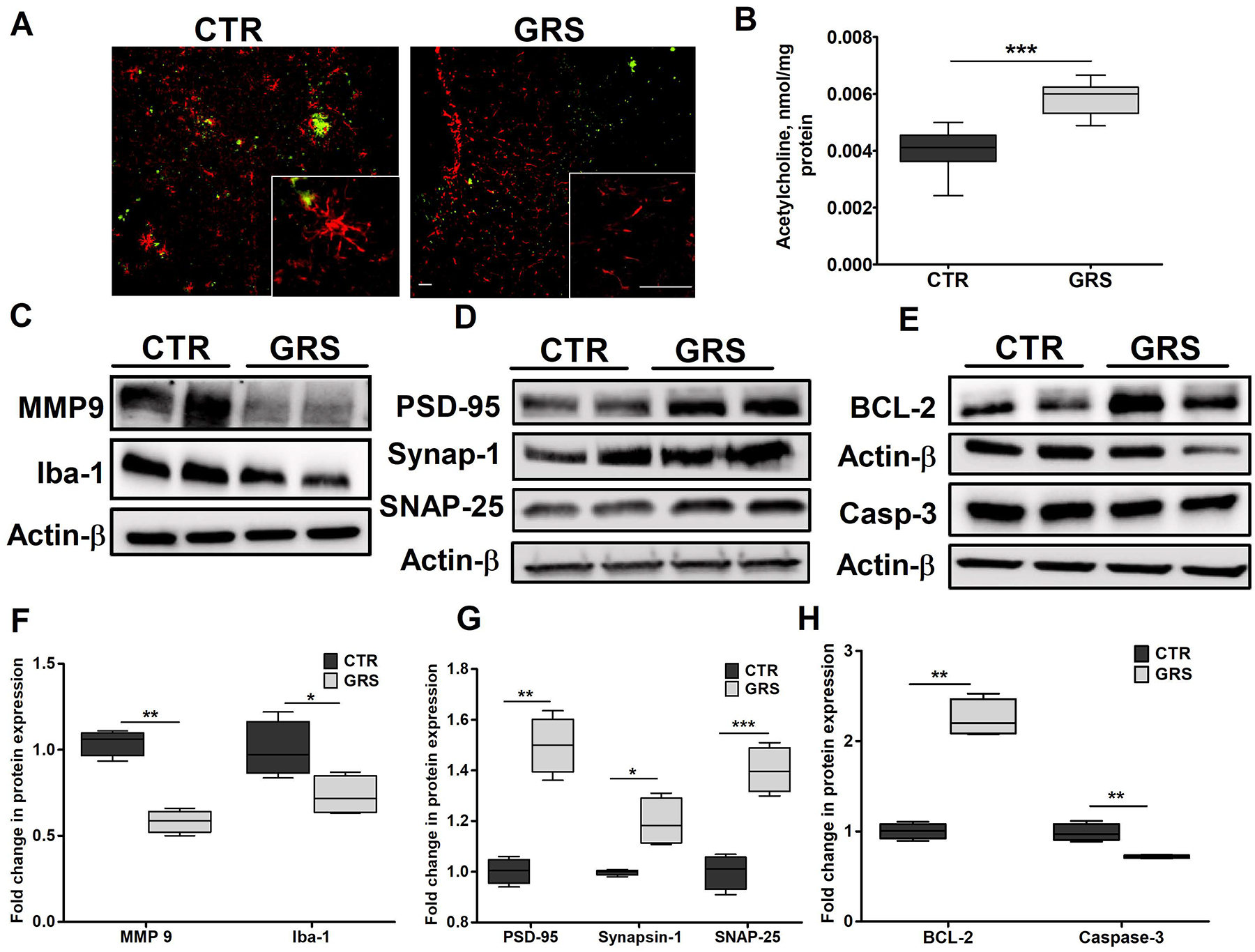
Neuroinflammation was assessed by measuring astrocytes activation monitored by astrocytes morphology, MMP-9 and the microglial marker Iba-1 expressions where granisetron treatment significantly reduced their levels. (A) Representative brain sections stained with anti-GFAP (red) antibody to detect activated astrocytes and 6E10 (green) antibody to detect Aβ in control (CTR) and granisetron (GRS) groups (seen at higher magnification in the closed inserts). Scale bar, 200 μm. Individual red and green channels for Panel A can be found under supplementary material (Fig. S4). (B) Acetylcholine level in the brain homogenates of both control and granisetron treated mice was measured using ELISA. (C) Representative blots, and (F) densitometry analysis of MMP-9 and Iba-1 in TgSwDI mice brains homogenates. (D) Representative blots, and (G) densitometry analysis of the neuro-synaptic markers PSD-95, Synapsin-1 and SNAP-25 in mice brains homogenates of TgSwDI mice. (E) Representative blots, and (H) densitometry analysis of the anti-apoptotic marker Bcl-2 and the apoptotic marker caspase-3 in brain homogenates of TgSwDI mice. Values were normalized to CTR (1.0). Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. Statistical analysis was determined by Student’s t-test (n=5 mice per group). *P < 0.05, ** P <0.01, and ***P < 0.001 versus vehicle treated (CTR) group.
Effect of granisetron on the expression of synaptic markers and acetylcholine brain levels
Granisetron significantly increased brain acetylcholine levels by 35% compared to vehicle treated group (Fig. 6B). In addition, three synaptic markers were evaluated, the pre-synaptic marker synaptosomal-associated protein (SNAP-25), the post-synaptic marker postsynaptic density protein-95 (PSD-95) and Synapsin-1. As shown in Fig. 6D and G, granisetron treatment significantly increased expression of the three evaluated synaptic markers, PSD-95, synapsin-1 and SNAP-25 by 1.5-, 1.2- and 1.4-fold, respectively. This increase was associated with a significant reduction in levels of the apoptotic marker activated caspase-3 by 30% and increase in the anti-apoptotic marker Bcl-2 by 40% (Fig. 6E and H).
Effect of granisetron on calcium signaling pathway in brains of TgSwDI mice
To investigate the effect of granisetron on modulating proteins associated with Ca+2 pathway, IP3-receptor (responsible for Ca+2 efflux from the endoplasmic reticulum (ER) storage), phosphorylated-IP3R (Ser1756), Ca+2/calmodulin-dependent protein kinase II (CaMKII) and phosphorylated-CaMKII (α, β, γ) were analyzed by Western blot. As shown in Fig. 7, granisetron significantly reduced the expressions of IP3R and phosphorylated-IP3R (Ser1756) by 50% without affecting the ratio suggesting reduced levels of phosphorylated-IP3R is due to reduced total IP3R (Fig. 7A–B). In addition, the treatment significantly increased CREB and p-CREB by 2.8 and 1.5-fold, respectively, increased PKAc by 50%, and BDNF by 70% (Fig. 7A–B). Another important key player in maintaining intracellular Ca+ 2 homeostasis is the calcium buffering protein calbindin-D28K, which was also significantly increased by granisetron treatment by 2.3-fold (Fig. 7C–D). Besides, granisetron significantly reduced CaMKII expression while increased the three phosphorylated-CaMKII α, β, γ isoforms (Fig. 7E, F and G).
Figure 7. Granisetron (GRS) restored Ca+2 homeostasis by modulating calcium pathway and significantly improved memory as determined by Morris Water Maze test in TgSwDI mice.
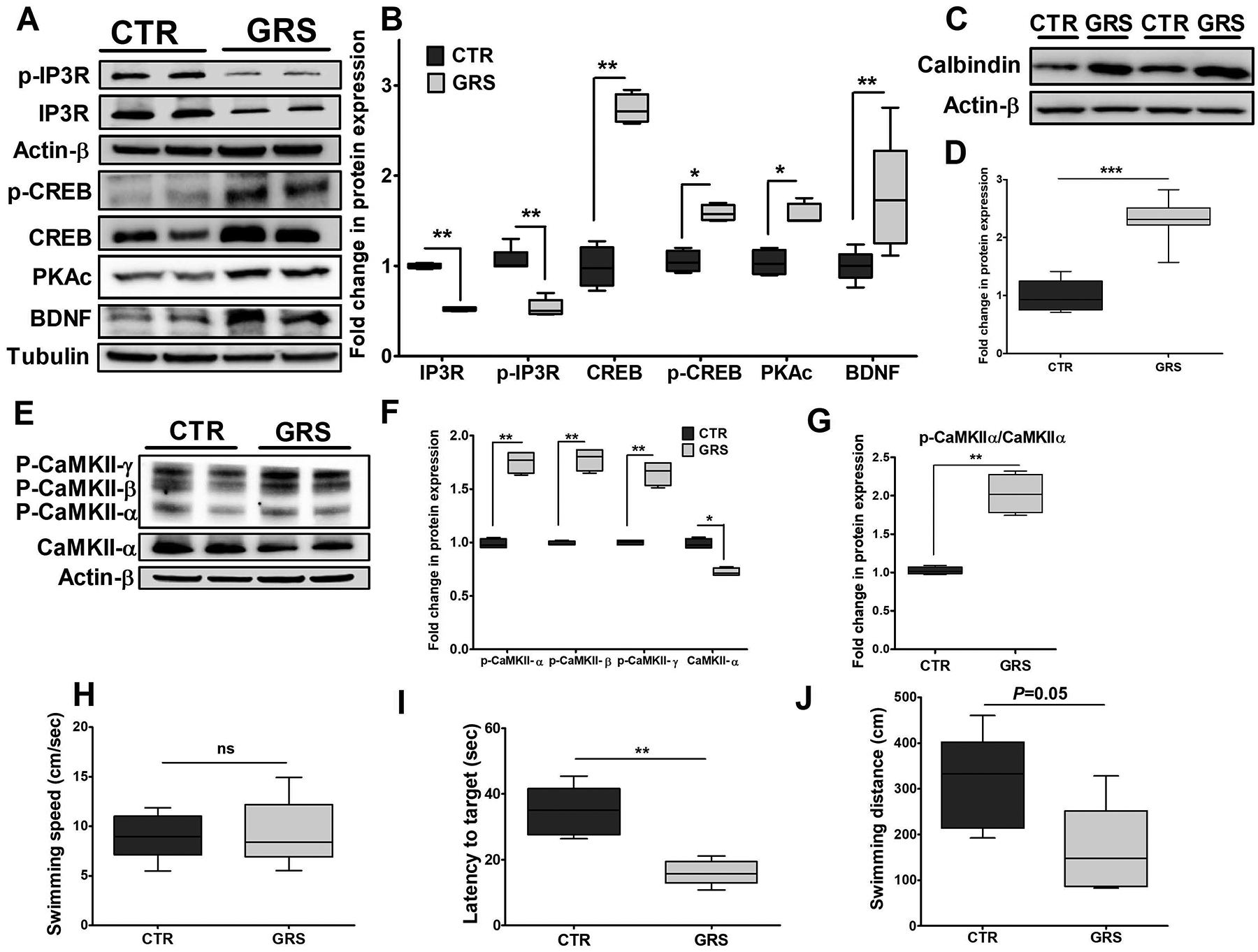
(A) Representative blots, and (B) densitometry analysis showed granisetron treatment significantly reduced total IP3R and p-IP3R expressions, and increased PKAc, BDNF, CREB and p-CREB expressions, which play major role in enhancing LTP. (C) Representative blots, and (D) densitometry analysis showed significant increase in the expression of the Ca+2 buffering protein calbindin D28K. (E, F, G) Representative blots and densitometry analysis showed significant reduction in CaMKII expression with increased levels of phosphorylated-CaMKII α, β, γ isoforms. Values were normalized to CTR (1.0). The effect of granisetron treatment on (H) swimming speed, (I) latency to target, and (J) swimming distance of the mice. Granisetron significantly decreased latency and swimming distance compared to the vehicle treated TgSwDI mice (control, CTR). Data are presented as box-and-whisker plots representing median and IQR, with minimum and maximum values. Statistical analysis was determined by Student’s t-test (n=5 mice per group). ns = not significant, *P<0.05, **P<0.01, and ***P<0.001 versus control group.
Effect of granisetron on Morris Water Maze performance
The MWM test was performed to assess granisetron effect on learning and memory functions. The following parameters were analyzed, the time a mouse takes to find the platform (latency, s), swimming speed (cm/s) and swimming distance (cm). As shown in Fig. 7H, I and J, mice received granisetron found the platform faster than vehicle treated mice (16.02 ± 1.69 vs 34.71 ± 3.38 s, respectively, P<0.01); and granisetron reduced mice swimming distance from 312.8 ± 46.6 cm (control) to 164.9 ± 44.3 cm (P=0.05). These results suggest granisetron improved behavioral outcomes and enhanced memory function. There were no differences between the two groups in swimming speed thus excluding motor changes as a cause of the observed improvement.
Identification and quantification of calcium pathway-associated proteins in response to granisetron, and IPA analysis
TMT-labeled relative quantitative proteomics were used to identify and quantify differentially expressed proteins between granisetron treatment in both young and aged groups and their control groups. Due to effect similarity between young and aged mice, data was pooled. These proteins were further being used in IPA analysis. For combined data set, a total of 434 differentially expressed proteins in response to treatment were identified using Mann-Whitney Test with significance level P<0.05 corrected by Benjamini-Hochberg.
To better understand the role of granisetron in the brain, differentially expressed proteins were further subjected to IPA core analysis. Among 434 differentially expressed proteins between treatment and control groups, 422 proteins were mapped with IPA (155 down and 267 up) with increased or decreased more than 2-fold change. IPA revealed that 11 proteins (ATP2B1 (PMCA), CACNA2D2 (VGCC), CACNG8, CAMK4, CAMK2G, CAMKK1, GRIA2 (AMPAR), HDAC6 ITPR1 (IP3R), PPP3CA, RAP2B) involved in calcium signaling pathway (P = 0.002, Z score = 0.632; Fig. S1, Table S1). Also, 15 proteins (CACNA2D2, CACNG8, CAMK4, CAMK2G, GNA13, GNAL, GNB1, GRIA2, GRID2, ITPR1, PLCB3, PLCB4, PRKCA, PRKCD, RPS6KA1) were found in CREB signaling pathway (P = 1.35E-05, Z score = −0.632; Fig. 8, Table S2). The activity of molecules in both pathways were assessed with the molecule activity predictor in IPA. Blue colored molecules are predicted to be inhibited (decreased activity/expression) and orange colored molecules to be activated (increased activity/expression) by granisetron treatment. These predictions are based on Ingenuity Knowledge Base findings (i.e. literature).
Figure 8. CREB signaling in neurons in response to granisetron treatment with the IPA molecule activity predictor.
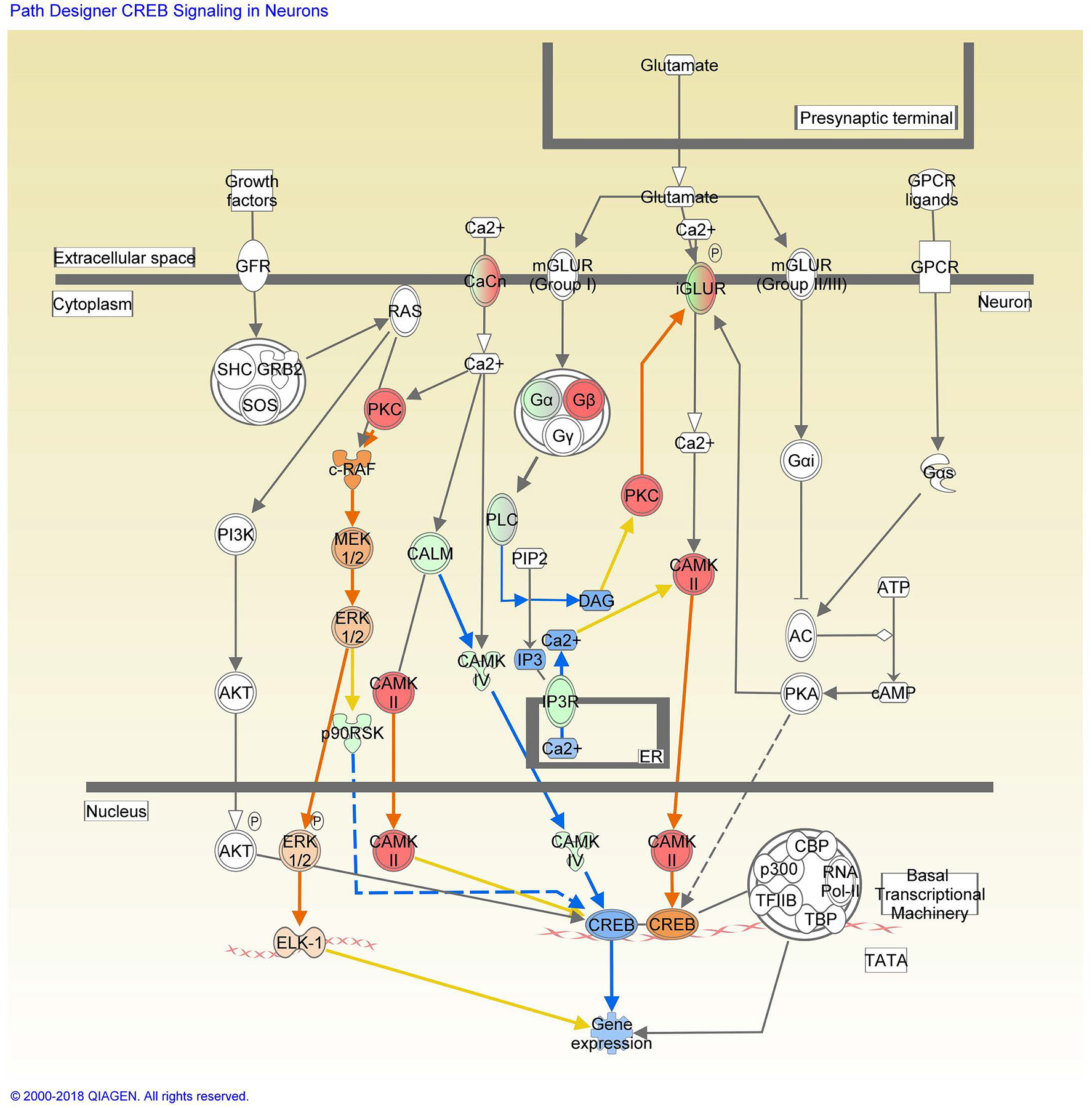
Red and green symbols indicate proteins up- and down-regulation in granisetron treated mice, respectively. Orange and blue nodes indicate predictions of to be activated or inhibited in granisetron treated mice, respectively. The color intensity is proportional to the degree of fold change. Edges between the nodes are colored orange when leading to the activation of downstream proteins, blue when inhibiting downstream proteins. Yellow edges indicate that the states of downstream genes are inconsistent with the prediction based on previous findings.
Selected proteins from aged mice brain homogenates were analyzed by Western blot for confirmation. These proteins include IP3R, CaMKII, calbindin and CREB. Results from Western blot revealed consistent pattern with LC/MS analyzed proteins IP3R, calbindin and CaMKII; CREB, on the other hand, was increased that is inconsistent with the pathway prediction (Fig. S2).
Discussion
Recent studies have reported that 5-HT3 receptor antagonists have neuroprotective properties in aged mice, and in neurotoxicity- and memory impairment-induced animals [33–36]. For example, granisetron improved memory performance in scopolamine-induced memory impaired mice [21]. In another study, granisetron enhanced learning and memory in wild-type mice after footshock [34]. In an AD mouse model, tropisetron, another 5-HT3 receptor antagonist, improved spatial and working memory in PDAPP mice [24]. Similarly, in the current study, our data revealed that granisetron reversed AD pathology in an AD mouse model, suggesting a therapeutic role for 5HT3 receptor in the disease. However, the mechanism(s) these drugs produce such effects are not fully investigated.
Findings from our HTS assay identified granisetron as a potent drug to rectify Aβ-induced disruption of an in vitro BBB model, an effect that was not observed with other screened 5-HT3 antagonists [15]. To confirm these results, in this work, we aimed to evaluate granisetron effect in wild-type young and aged mice and in a mouse model of AD, and investigate the mechanism(s) by which granisetron exhibits such effect. Mice were daily administered with granisetron for 28 days; a treatment period that is longer than other studies evaluated granisetron effect at a single dose up to one-week given every other day [20–24]. The 28 days treatment period was selected in order to test the therapeutic potential of long-term dosed granisetron on the progression of AD pathology in a mouse model of AD. Our findings demonstrated granisetron to restore the BBB integrity and function, reduce Aβ accumulation and neuroinflammation, decrease sustained calcium flux and apoptosis in addition to enhance the cognitive function in TgSwDI, a mouse model of AD and CAA. In addition, we showed for the first time that granisetron effect is mediated, at least in part, by CaMKII/CREB pathway that is perturbed by aging and AD.
Available in vitro studies have demonstrated 5-HT3 receptor antagonists including granisetron to inhibit extracellular calcium influx into cells [37–40]. Based on its mechanism, granisetron directly decrease depolarization and elevation of calcium by blocking 5-HT3 receptor [41,42]. A recent study has reported the treatment of Aβ-challenged cortical neurons isolated from rats brains with 5-HT3 receptor antagonists MDL 72222 and Y 25130 significantly reduced Aβ-caused calcium elevation, caspase-3 activity and apoptosis, an effect that was mediated by 5-HT3 receptor antagonism [43]. Similarly, our in vitro studies demonstrated treatment of endothelial and neuronal cells with granisetron for 24 h significantly reduced Aβ induced-intracellular calcium levels, suggesting granisetron reduced calcium influx by blocking 5-HT3 receptor. This effect could explain the positive outcome observed in vivo as blocking 5HT3 receptor by granisetron is expected to reduce intracellular calcium levels, and thus restore CaMKII/CREB pathway.
Preliminary results from the proteomics analysis demonstrated granisetron to modulate calcium and CREB pathways associated proteins including IP3R, CaMK proteins, calcium voltage-gated channels and other proteins important to maintain Ca+2 homeostasis that are altered in AD [44–46]. Increased Ca+2 influx and disrupted intracellular Ca+2 homeostasis caused by calcium abnormal and excessive release from the ER through IP3R [47,48], and ryanodine receptor (RYR) [49,50] have been reported as key players in the pathology of AD that are associated with cognitive dysfunction [51]. To confirm the proteomics data, LC/MS measured and IPA predicted proteins were analyzed by Western blotting. For this, a selected number of proteins from CREB pathway in aged mice brains including IP3R, CaMKII, calbindin, and IPA predicted CREB protein were analyzed. Findings demonstrated a comparable effect between proteomics-measured data and Western blot-analyzed proteins (i.e. reduced IP3R, and increased CaMKII and calbindin) in aged mice. However, the IPA analysis that predicts effect based on the Ingenuity Knowledge Base from literature, predicted two opposing effects on CREB, activation via CaMKII pathway or inactivation via CALM/CaMKIV pathway (Fig. 8). Western blot findings in aged and TgSwDI mice brains, however, determined granisetron to up-regulate/activate CREB, which is consistent with CaMKII/CREB pathway shown in Fig. 8.
Similarly, Western blot data from TgSwDI mice brain homogenates confirmed reduced expression of IP3R and p-IP3R with a significant increase in the level of calcium buffering protein calbindin D28K, which plays important role in restricting the amplitude of Ca+2 signals [52]. Our findings also indicated that besides CREB activation through αCaMKII, 5HT3-recepotor blocking by granisetron activated PKA/CREB pathway. Both pathways play a major role in memory formation through regulating the long-term potentiation (LTP), which are dysregulated in AD [53,54]. The nature of αCaMKII dysregulation in AD is complex and not well understood, however reduced activation of this enzyme was associated with synaptic loss [55]. Consistent with its effect on CaMKII activation in aged mice brains observed from proteomics data and Western blot, in TgSwDI mice brains, granisetron significantly increased p-CaMKII levels where the treatment was able to activate αCaMKII by phosphorylation at Thr286 site. Phosphorylation of αCaMKII at Thr286 amino acid switches the subunit activity within αCaMKII from a Ca+2/CaM-dependent to independent state, which is important for NMDAR-dependent LTP at hippocampal CA1 synapses and essential for spatial memory formation [53]. In addition, it has been shown that Aβ inhibits PKA/CREB pathway [54]. The association of PKA/CREB pathway downregulation with Ca+2 dyshomeostasis has been reported [56]. CREB phosphorylation by CaMKII and PKA is essential for synaptic strengthening and memory formation [46,57]. Available in vivo studies in AD mouse models showed a significant reduction in CREB activation in the nuclear fraction [44,46], and subsequent BDNF downregulation that was associated with memory impairment [54,58]. CREB is a transcriptional regulator of BDNF that plays important role in learning and memory processes. BDNF transcription is initiated through the phosphorylation of CREB at Ser133 by CaMKII and PKA activation [57,59]. In our studies, in addition to CaMKII activation, granisetron significantly increased PKAc (the free and active form of PKA) and CREB, which collectively contributed to increased levels of p-CREB (S133) and BDNF. This positive effect of granisetron on calcium and CREB pathways and other pathological hallmarks of AD was ultimately translated into a positive outcome on memory in TgSwDI mice as determined by MWM behavioral studies. Granisetron improved memory function when compared to vehicle treated mice. This enhanced cognitive function could be an overall consequence of reduced sustained increase in intracellular Ca+2 levels that sequentially directly and/or indirectly enhanced BBB integrity and function, reduced Aβ levels and associated neuroinflammation, increased ACh levels and synaptic markers, and reduced neurotoxicity.
The precise mechanism leading to BBB dysfunction remains unclear, however, in AD, Aβ could, directly or indirectly, disrupts tight junction proteins by disrupting calcium homeostasis, leading to an overall BBB dysfunction [60,61]. Measurements of exogenous and/or endogenous plasma-derived molecules extravasation in brain parenchyma has been widely used as a method to detect BBB disruption [62]. Findings from aged mice confirmed granisetron to enhance the BBB tightness as measured by reduced permeability of the endogenous BBB permeability marker IgG (150KDa) and exogenous florescent-tagged dextran of 10 and 40 KDa. These results were supported with increased expression of tight junction proteins. In young mice, however, while granisetron increased tight junction proteins expression, no changes in dextran or IgG extravasation was observed suggesting intact BBB in young mice (5 months at end of treatment). When tested in TgSwDI mice, granisetron significantly enhanced BBB integrity and function as measured by reduced IgG extravasation and increased expression of tight junction proteins, which is consistent with our previously reported in vitro finding in the CAA-cell based BBB model [15], and reduced brain Aβ accumulation. This reduction in Aβ load was concomitant with a significant increase in the expression of P-gp, Aβ major transport protein across the BBB, in isolated microvessels, and Aβ degrading enzymes NEP and IDE in brain homogenates. Reduced levels of P-gp and degrading enzymes with aging and in AD have been previously reported and were related to brain Aβ accumulation [63,64], where their up-regulation reduced brain Aβ levels and related pathology [65,66]. Besides increasing Aβ clearance, granisetron significantly reduced sAPPβ and increased sAPPα levels collectively contributing to reduced Aβ brain load. Several studies reported sAPPα possesses neurotrophic and neuroprotective activities [67], which could impacted the enhanced memory observed in TgSwDI mice, a finding that is consistent with that reported with tropisetron [24].
In addition, granisetron successfully reduced neurotoxicity and neuroinflammation as determined by increased expressions of synaptic markers, reduced apoptosis as measured by reduced caspase 3 and increased BCL-2, reduced MMP-9 and glial activation. Mice treated with granisetron exhibited significant reduction in glial activation markers Iba-1 and GFAP. GFAP-expressing hypertrophic astrocytes exhibited a widespread pattern in the brain of vehicle treated TgSwDI mice. These activated astrocytes were readily identified around the senile plaques by their thicker, star-shape and bigger cell bodies. This greater increase in GFAP-positive cells was significantly reduced by granisetron. These findings add to existing reports on the potential anti-inflammatory effect of granisetron [32], and tropisetron [68]. This anti-inflammatory effect by 5HT3-recptors antagonists could be related to calcineurin inhibition [69]; calcineurin is a Ca+2/CaM-dependent enzyme that upregulates numerous cytokines and pro-inflammatory factors [70].
In addition, granisetron treatment significantly increased acetylcholine levels in TgSwDI mice brains. The inhibitory role of 5-HT3 receptor agonists on acetylcholine release has been reported. For example, 2-methyl-5-HT, a selective 5-HT3 agonist, inhibited acetylcholine release from the cerebral cortex of freely moving guinea-pigs, an effect that was blocked by ICS205–930, a selective 5-HT3 antagonist [71]. Likewise, granisetron produced a concentration-dependent increase in acetylcholine release in rat entorhinal cortex and striatal slices, an effect that was calcium dependent [72]. Collectively, these findings suggest granisetron effect on acetylcholine-increased levels is mediated, at least in part, by blocking 5HT3 receptor.
Several studies linked BBB disruption [60], Aβ accumulation [50], neuroinflammation [73], and memory impairment to calcium dyshomeostasis. Indeed, further studies are required to answer the question of whether granisetron observed positive effect on Aβ brain load and other associated pathological hallmarks is through its ability to restore calcium homeostasis or independent of calcium pathway. However, data from the in vitro studies and aged mice that do not express Aβ pathology suggest granisetron demonstrated its effect, at least in part, by restoring calcium homeostasis through blocking 5HT3-recepotor. Thus, our findings support granisetron repurposing to slow, hold the progression and/or treat AD and related disorders. Furthermore, based on its positive effect to restore the BBB function, in addition to AD, our findings suggest granisetron could be useful for other neurological diseases characterized by disrupted BBB function such as amyotrophic lateral sclerosis, Parkinson disease, multiple sclerosis [74].
Granisetron is used primarily in the treatments of nausea and vomiting caused by cancer treatments, and post-operative nausea and vomiting [16]. In humans, oral granisetron is administered in 1 to 2 mg dose up to one week, or subcutaneously for extended release in 10 mg dose. Within this regimen, in most human studies, granisetron has had little effect on blood pressure, heart rate or ECG [75–77], however, pending further studies, precautions have recently been issued of possible QT prolongation especially in patients with cardiac diseases [78]. Nevertheless, a recent interesting study evaluated the efficacy of granisetron as an add-on therapy in the treatment of negative symptoms of patients with stable schizophrenia [22] Granisetron add-on administered at 1 mg twice daily in addition to risperidone for eight weeks proved safe and effectively reduced primary negative symptoms of patients with schizophrenia. In our study, the anti-AD effect of granisetron was observed at a dose of 3 mg/kg administered daily for 28 days, a regimen that is higher and longer than prescribed for humans. While mice treatment for 28 days with 3 mg/kg daily dose didn’t demonstrate visible side effects, the consequences of translating the chronic administration of such dose to humans are unknown that necessitate further evaluation. Without a doubt, testing granisetron in AD mouse models at doses that corresponds to human dose or less, in parallel with toxicity studies, are necessary. However, as a proof-of-concept study with the tested dosage regimen, granisetron demonstrated a therapeutic potential against AD that prompts further evaluations.
Conclusions
Our findings, summarized in scheme 1, demonstrated granisetron enhanced the BBB integrity and function that was associated with reduced Aβ-related pathology and enhanced memory in TgSwDI, a mouse model of AD and CAA. The observed effect of granisetron was mediated, at least in part, by activating CaMKII/CREB pathway that is perturbed by aging and AD. While further studies are essential to test the effect of long-term administration of granisetron in dose-dependent studies, our findings support the repurposing of granisetron as a therapeutic drug for AD, and possibly other neurological diseases associated with BBB disruption.
Scheme 1.
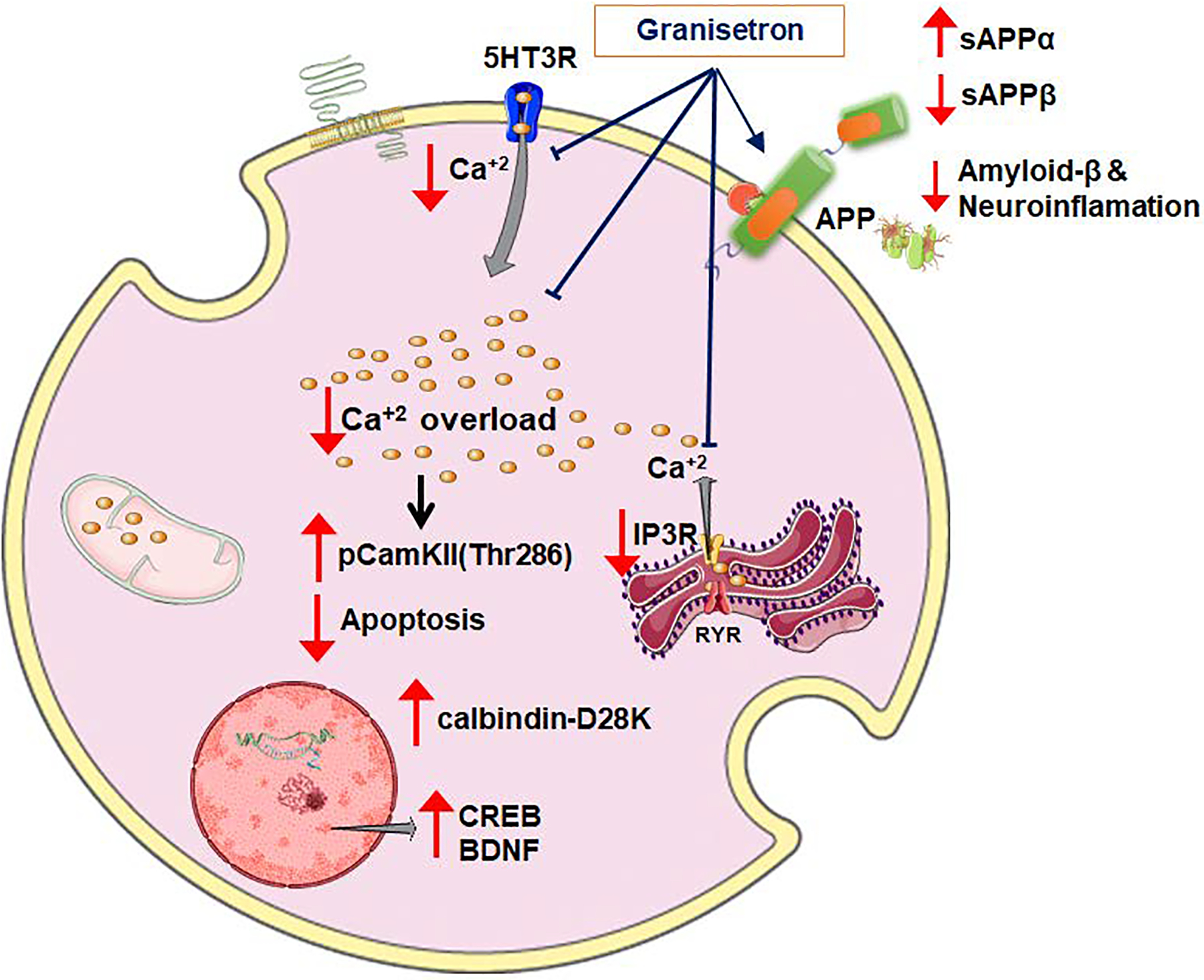
Proposed mechanism of granisetron effect to reduce Aβ-related pathology in TgSwDI mice via CaMKII/CREB pathway.
Supplementary Material
Fig. S1. Calcium signaling pathway in response to granisetron treatment with the IPA molecule activity predictor. Red and green symbols indicate proteins up- and down-regulation in granisetron treated mice, respectively. Orange and blue nodes indicate predictions of to be activated or inhibited in granisetron treated mice, respectively. The color intensity is proportional to the degree of fold change. Edges between the nodes are colored orange when leading to the activation of downstream proteins, blue when inhibiting downstream proteins. Yellow edges indicate that the states of downstream genes are inconsistent with the prediction based on previous findings.
Fig. S2. Granisetron restored Ca+2 homeostasis by modulating CREB pathway in the brains of C57Bl/6J aged mice. Statistical analysis was determined by Student’s t-test (n=5 mice per group). *P<0.05 and **P<0.01 versus control group.
Fig. S3. Individual red and green channels for Panels D and E of Figure 4. These images are representative hippocampus sections stained with Thio-S (green) to detect Aβ plaques load and anti-collagen IV (red) to stain microvessels from control and granisetron treated groups.
Fig. S4. Individual red and green channels for Panel A of Figure 6. These images are representative brain sections stained with 6E10 (green) antibody to detect Aβ, and anti-GFAP (red) antibody to detect activated astrocytes in control and granisetron treated groups.
Table S1. A selection of significantly changed proteins in calcium pathway (P<0.05; fold change > 2) for granisetron effect in C57Bl/6J wild-type mice as determined by IPA analysis.
Table S2. A selection of significantly changed proteins in CREB pathway (P<0.05; fold change > 2) for granisetron effect in C57Bl/6J wild-type mice as determined by IPA analysis.
Acknowledgements
This research work was funded by the National Institute of Neurological Disorders and Stroke (NIH/NINDS) under grant number R15NS091934 (A.K.), and UAMS Proteomics Core supported by NIH grants P20GM121293 and S10OD018445.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Alzheimer’s Association (2018) 2018 Alzheimer’s Disease Facts and Figures. Alzheimers Dement 14, 367–429. [Google Scholar]
- [2].Alzheimer’s Disease International (2018) World Alzheimer Report 2018 - The state of the art of dementia research: New frontiers. 1–48.
- [3].Thies W and L B (2012) 2012 Alzheimer’s disease facts and figures. Alzheimer’s Dement 8, 131–168. [DOI] [PubMed] [Google Scholar]
- [4].Uzuki BKS, Wata AI, Watsubo TI (2017) Review The past, present, and future of disease-modifying therapies for Alzheimer’s disease 93, 757–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV (2016) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta - Mol Basis Dis 1862, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Disterhoft JF, Moyer JR, Thompson LT (1994) The calcium rationale in aging and Alzheimer’s disease. Evidence from an animal model of normal aging. Ann N Y Acad Sci 747, 382–406. [DOI] [PubMed] [Google Scholar]
- [7].Brown RC, Davis TP (2002) Calcium modulation of adherens and tight junction function: A potential mechanism for blood-brain barrier disruption after stroke. Stroke 33, 1706–1711. [DOI] [PubMed] [Google Scholar]
- [8].Gibson G, Cotman C, Lynch G, Blass J (2017) Calcium Hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimer’s Dement 13, 178–182.e17. [DOI] [PubMed] [Google Scholar]
- [9].Yarlagadda A, Kaushik S, Clayton AH (2007) Blood brain barrier: the role of calcium homeostasis. Psychiatry 4, 55–59. [PMC free article] [PubMed] [Google Scholar]
- [10].Raza M, Deshpande LS, Blair RE, Carter DS, Sombati S, DeLorenzo RJ (2007) Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci Lett 418, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yamazaki Y, Kanekiyo T (2017) Blood-brain barrier dysfunction and the pathogenesis of alzheimer’s disease. Int J Mol Sci 18, pii: E1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Di Domenico F, Barone E, Perluigi M, Butterfield DA (2017) The Triangle of Death in Alzheimer’s Disease Brain: The Aberrant Cross-Talk Among Energy Metabolism, Mammalian Target of Rapamycin Signaling, and Protein Homeostasis Revealed by Redox Proteomics. Antioxid Redox Signal 26, 364–387. [DOI] [PubMed] [Google Scholar]
- [13].Montagne A, Barnes SR, Sweeney MD, Halliday MR, Abhay P, Zhao Z, Toga AW, Jacobs RE, Liu CY, Harrington MG, Chui HC, Law M, Zlokovic BV (2016) Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 85, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kang HM, Sohn I, Jung J, Jeong JW, Park C (2016) Age-related changes in pial arterial structure and blood flow in mice. Neurobiol Aging 37, 161–170. [DOI] [PubMed] [Google Scholar]
- [15].Qosa H, Mohamed LA, Al Rihani SB, Batarseh YS, Duong QV, Keller JN, Kaddoumi A (2016) High-Throughput Screening for Identification of Blood-Brain Barrier Integrity Enhancers: A Drug Repurposing Opportunity to Rectify Vascular Amyloid Toxicity. J Alzheimer’s Dis 53, 1499–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yarker YE, McTavish D (1994) Granisetron. An update of its therapeutic use in nausea and vomiting induced by antineoplastic therapy. Drugs 48, 761–793. [DOI] [PubMed] [Google Scholar]
- [17].Nayak SV, Rondé P, Spier AD, Lummis SCR, Nichols RA (1999) Calcium changes induced by presynaptic 5-hydroxytryptamine-3 serotonin receptors on isolated terminals from various regions of the rat brain. Neuroscience 91, 107–117. [DOI] [PubMed] [Google Scholar]
- [18].Gunn AP, Wong XBX, Johanssen XT, Griffith XJC, Masters CL, Bush AI, Barnham KJ, Duce JA, Cherny RA (2016) Amyloid-Peptide A-3pE-42 Induces Lipid Peroxidation, Membrane Permeabilization, and Calcium Influx in Neurons. J Biol Chem 291, 6134–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Davis J, Xu F, Deane R, Romanov G, Previti M Lou, Zeigler K, Zlokovic BV, Van Nostrand WE (2004) Early-onset and Robust Cerebral Microvascular Accumulation of Amyloid β-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant Form of Amyloid β-Protein Precursor. J Biol Chem 279, 20296–20306. [DOI] [PubMed] [Google Scholar]
- [20].Fakhfouri G, Rahimian R, Daneshmand A, Bahremand A, Rasouli MR, Dehpour AR, Mehr SE, Mousavizadeh K (2010) Granisetron ameliorates acetic acid-induced colitis in rats. Hum Exp Toxicol 29, 321–328. [DOI] [PubMed] [Google Scholar]
- [21].Javadi-Paydar M, Zakeri M, Norouzi A, Rastegar H, Mirazi N, Dehpour AR (2012) Involvement of nitric oxide in granisetron improving effect on scopolamine-induced memory impairment in mice. Brain Res 1429, 61–71. [DOI] [PubMed] [Google Scholar]
- [22].Khodaie-Ardakani MR, Seddighi S, Modabbernia A, Rezaei F, Salehi B, Ashrafi M, Shams-Alizadeh N, Mohammad-Karimi M, Esfandiari GR, Hajiaghaee R, Akhondzadeh S (2013) Granisetron as an add-on to risperidone for treatment of negative symptoms in patients with stable schizophrenia: Randomized double-blind placebo-controlled study. J Psychiatr Res 47, 472–478. [DOI] [PubMed] [Google Scholar]
- [23].Aithal S, Geetha S, Swetha ES, Balaji V, Chethan Kumar S (2014) Evaluation of antidepressant activity of granisetron in albino mice. Int J Pharm Sci Rev Res 27, 345–348. [Google Scholar]
- [24].Spilman P, Descamps O, Gorostiza O, Peters-Libeu C, Poksay KS, Matalis A, Patent A, Rao R, John V, Bredesen DE (2014) The multi-functional drug tropisetron binds APP and normalizes cognition in a murine Alzheimer’s model. Brain Res 1551, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mohamed LA, Qosa H, Kaddoumi A (2015) Age-Related Decline in Brain and Hepatic Clearance of Amyloid-Beta is Rectified by the Cholinesterase Inhibitors Donepezil and Rivastigmine in Rats. ACS Chem Neurosci 6, 725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Egawa G, Nakamizo S, Natsuaki Y, Doi H, Miyachi Y, Kabashima K (2013) Intravital analysis of vascular permeability in mice using two-photon microscopy. Sci Rep 3: 1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gage GJ, Kipke DR, Shain W (2012) Whole Animal Perfusion Fixation for Rodents. J Vis Exp pii: 3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Batarseh YS, Bharate SS, Kumar V, Kumar A, Vishwakarma RA, Bharate SB, Kaddoumi A (2017) Crocus sativus Extract Tightens the Blood-Brain Barrier, Reduces Amyloid β Load and Related Toxicity in 5XFAD Mice. ACS Chem Neurosci 8, 1756–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kruyer A, Soplop N, Strickland S, Norris EH (2015) Chronic hypertension leads to neurodegeneration in the TgSwDI mouse model of Alzheimer’s disease. Hypertension 66, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang C, Zhao C, Jiang G, Gu X, Feng J, Jiang J (2016) The Role of Posttraumatic Hypothermia in Preventing Dendrite Degeneration and Spine Loss after Severe Traumatic Brain Injury. Sci Rep 6, 37063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Coleman HN, Greenfield WW, Stratton SL, Vaughn R, Kieber A, Moerman-Herzog AM, Spencer HJ, Hitt WC, Quick CM, Hutchins LF, Mackintosh SG, Edmondson RD, Erickson SW, Nakagawa M (2016) Human papillomavirus type 16 viral load is decreased following a therapeutic vaccination. Cancer Immunol Immunother. 65, 563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Maleki-Dizaji N, Eteraf-Oskouei T, Fakhrjou A, Maljaie SH, Garjani A (2010) The effects of 5HT3receptor antagonist granisetron on inflammatory parameters and angiogenesis in the air-pouch model of inflammation. Int Immunopharmacol 10, 1010–1016. [DOI] [PubMed] [Google Scholar]
- [33].Rahimian R, Fakhfouri G, Ejtemaei Mehr S, Ghia J-E, Genazzani AA, Payandemehr B, Dehpour AR, Mousavizadeh K, Lim D (2013) Tropisetron attenuates amyloid-beta-induced inflammatory and apoptotic responses in rats. Eur J Clin Invest 43, 1039–1051. [DOI] [PubMed] [Google Scholar]
- [34].Chugh Y, Saha N, Sankaranarayanan A, Sharma PL (1991) Memory enhancing effects of granisetron (BRL 43694) in a passive avoidance task. Eur J Pharmacol 203, 121–123. [DOI] [PubMed] [Google Scholar]
- [35].Ohno M, Watanabe S (1997) Differential effects of 5-HT3 receptor antagonism on working memory failure due to deficiency of hippocampal cholinergic and glutamatergic transmission in rats. Brain Res 762, 211–215. [DOI] [PubMed] [Google Scholar]
- [36].Boast C, Bartolomeo AC, Morris H, Moyer JA (1999) 5HT antagonists attenuate MK801-impaired radial arm maze performance in rats. Neurobiol Learn Mem 71, 259–271. [DOI] [PubMed] [Google Scholar]
- [37].Homma K, Kitamura Y, Ogawa H, Oka K (2006) Serotonin induces the increase in intracellular Ca2+ that enhances neurite outgrowth in PC12 cells via activation of 5-HT3 receptors and voltage-gated calcium channels. J Neurosci Res 84, 316–325. [DOI] [PubMed] [Google Scholar]
- [38].Hutchinson TE, Zhong W, Chebolu S, Wilson SM, Darmani NA (2015) L-type calcium channels contribute to 5-HT3-receptor-evoked CaMKIIalpha and ERK activation and induction of emesis in the least shrew (Cryptotis parva). Eur J Pharmacol 755, 110–118. [DOI] [PubMed] [Google Scholar]
- [39].Ronde P, Nichols RA (1997) 5-HT3 receptors induce rises in cytosolic and nuclear calcium in NG108–15 cells via calcium-induced calcium release. Cell Calcium 22, 357–365. [DOI] [PubMed] [Google Scholar]
- [40].Takenouchi T, Munekata E (1998) Serotonin increases cytoplasmic Ca2+ concentration in PC12h cells: effect of tachykinin peptides. Neurosci Lett 246, 141–144. [DOI] [PubMed] [Google Scholar]
- [41].Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D (1991) Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 254, 432–437. [DOI] [PubMed] [Google Scholar]
- [42].Derkach V, Surprenant A, North RA (1989) 5-HT3 receptors are membrane ion channels. Nature 339, 706–709. [DOI] [PubMed] [Google Scholar]
- [43].Ban JY, Seong YH (2005) Blockade of 5-HT3 receptor with MDL72222 and Y25130 reduces ??-amyloid protein (25–35)-induced neurotoxicity in cultured rat cortical neurons. Eur J Pharmacol 520, 12–21. [DOI] [PubMed] [Google Scholar]
- [44].Hardingham GE, Arnold FJ, Bading H (2001) Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4, 261–267. [DOI] [PubMed] [Google Scholar]
- [45].Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, Dalton ND, Peterson KL, Chen J, Bers D, Brown JH (2009) Requirement for Ca 2 + / calmodulin – dependent kinase II in the transition from pressure overload – induced cardiac hypertrophy to heart failure in mice. J Clin Invest 119, 1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Teich AF, Nicholls RE, Puzzo D, Fiorito J, Purgatorio R, Fa’ M, Arancio O (2015) Synaptic Therapy in Alzheimer’s Disease: A CREB-centric Approach. Neurotherapeutics 12, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang W, Tingare A, Ng DC-H, Johnson HW, Schell MJ, Lord RL, Chawla S (2012) IP3-dependent intracellular Ca2+ release is required for cAMP-induced c-fos expression in hippocampal neurons. Biochem Biophys Res Commun 425, 450–5. [DOI] [PubMed] [Google Scholar]
- [48].Stutzmann GE (2005) Calcium dysregulation, IP3 signaling, and Alzheimer’s disease. Neuroscientist 11, 110–115. [DOI] [PubMed] [Google Scholar]
- [49].Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: Minding the store. Aging Cell 6, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Oulès B, Del Prete D, Greco B, Zhang X, Sevalle J, Moreno S, Paterlini-bréchot P, Checler F, Benfenati F, Chami M (2013) Ryanodine receptors blockade reduces Amyloid-beta load and memory impairmnet of Tg2576 mouse model of Alzheimer’s disease. J Neurosci 32, 11820–11834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Egorova PA, Bezprozvanny IB (2018) Inositol 1,4,5-trisphosphate receptors and neurodegenerative disorders. FEBS J 1–19. [DOI] [PubMed] [Google Scholar]
- [52].Palop J, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer’s disease-related cognitive deficits. Proc Natl Acad Sci U S A 100, 9572–9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yamagata Y, Kobayashi S, Umeda T, Inoue A, Sakagami H, Fukaya M, Watanabe M, Hatanaka N, Totsuka M, Yagi T, Obata K, Imoto K, Yanagawa Y, Manabe T, Okabe S (2009) Kinase-Dead Knock-In Mouse Reveals an Essential Role of Kinase Activity of Ca2+/Calmodulin-Dependent Protein Kinase II in Dendritic Spine Enlargement, Long-Term Potentiation, and Learning. J Neurosci 29, 7607–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M (2002) Amyloid-peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci 99, 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Gu Z, Liu W, Yan Z (2009) beta-amyloid impairs AMPA receptor trafficking and function by reducing Ca2+/calmodulin-dependent protein kinase II synaptic distribution. J Biol Chem 284, 10639–10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Liang Z, Liu F, Grundke-Iqbal I, Iqbal K, Gong C-X (2007) Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J Neurochem 103, 2462–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Rosa E, Fahnestock M (2018) CREB expression mediates amyloid β-induced basal BDNF downregulation. Neurobiol Aging 36, 2406–2413. [DOI] [PubMed] [Google Scholar]
- [58].Walton MR, Dragunow M (2018) Is CREB a key to neuronal survival? Trends Neurosci 23, 48–53. [DOI] [PubMed] [Google Scholar]
- [59].Yan X, Liu J, Ye Z, Huang J, He F, Xiao W, Hu X, Luo Z (2016) CaMKII-Mediated CREB Phosphorylation Is Involved in Ca(2+)-Induced BDNF mRNA Transcription and Neurite Outgrowth Promoted by Electrical Stimulation. PLoS One 11, e0162784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kook S-Y, Hong HS, Moon M, Ha CM, Chang S, Mook-Jung I (2012) Amyloid 1–42-RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier Via Ca2+-Calcineurin Signaling. J Neurosci 32, 8845–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE (1992) beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci 12, 376–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wisniewski HM, Vorbrodt AW, Wegiel J (1997) Amyloid Angiopathy and Blood–Brain Barrier Changes in Alzheimer’s Disease. Ann N Y Acad Sci 826, 161–172. [DOI] [PubMed] [Google Scholar]
- [63].Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, Jiang H, Prior JL, Sagare A, Bales KR, Paul SM, Zlokovic BV, Piwnica-Worms D, Holtzman DM (2005) P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest 115, 3285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Carty N, Nash KR, Brownlow M, Cruite D, Wilcock D, Selenica M-LB, Lee DC, Gordon MN, Morgan D (2013) Intracranial Injection of AAV Expressing NEP but Not IDE Reduces Amyloid Pathology in APP+PS1 Transgenic Mice. PLoS One 8, e59626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Qosa H, Abuznait AH, Hill RA, Kaddoumi A (2012) Enhanced brain amyloid-beta clearance by rifampicin and caffeine as a possible protective mechanism against Alzheimer’s disease. J Alzheimers Dis 31, 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Mohamed LA, Keller JN, Kaddoumi A (2016) Role of P-glycoprotein in mediating rivastigmine effect on amyloid-β brain load and related pathology in Alzheimer’s disease mouse model. Biochim Biophys Acta 1862, 778–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Mattson MP, Chan SL, Duan W (2002) Modification of Brain Aging and Neurodegenerative Disorders by Genes, Diet, and Behavior. Physiol Rev 82, 637–672. [DOI] [PubMed] [Google Scholar]
- [68].Rahimian R, Daneshmand A, Mehr SE, Barzegar-Fallah A, Mohammadi-Rick S, Fakhfouri G, Shabanzadeh AP, Dehpour AR (2011) Tropisetron ameliorates ischemic brain injury in an embolic model of stroke. Brain Res 1392, 101–109. [DOI] [PubMed] [Google Scholar]
- [69].Rahimian R, Dehpour AR, Fakhfouri G, Khorramizadeh MR, Ghia J-E, Seyedabadi M, Caldarelli A, Mousavizadeh K, Forouzandeh M, Mehr SE (2011) Tropisetron upregulates cannabinoid CB1 receptors in cerebellar granule cells: possible involvement of calcineurin. Brain Res 1417, 1–8. [DOI] [PubMed] [Google Scholar]
- [70].Medyouf H, Alcalde H, Berthier C, Guillemin MC, dos Santos NR, Janin A, Decaudin D, de The H, Ghysdael J (2007) Targeting calcineurin activation as a therapeutic strategy for T-cell acute lymphoblastic leukemia. Nat Med 13, 736–741. [DOI] [PubMed] [Google Scholar]
- [71].Bianchi C, Siniscalchi A, Beani L (1990) 5-HT1A agonists increase and 5-HT3 agonists decrease acetylcholine efflux from the cerebral cortex of freely-moving guinea-pigs. Br J Pharmacol 101, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Ramirez MJ, Cenarruzabeitia E, Lasheras B, Del Rio J (1996) Involvement of GABA systems in acetylcholine release induced by 5-HT3 receptor blockade in slices from rat entorhinal cortex. Brain Res 712, 274–280. [DOI] [PubMed] [Google Scholar]
- [73].González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L (2017) Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front Mol Neurosci 10, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sweeney MD, Sagare AP, Zlokovic BV (2018) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14, 133–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tricco AC, Soobiah C, Blondal E, Veroniki AA, Khan PA, Vafaei A, Ivory J, Strifler L, Ashoor H, MacDonald H, Reynen E, Robson R, Ho J, Ng C, Antony J, Mrklas K, Hutton B, Hemmelgarn BR, Moher D, Straus SE (2015) Comparative safety of serotonin (5-HT3) receptor antagonists in patients undergoing surgery: a systematic review and network meta-analysis. BMC Med 13, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Tricco AC, Blondal E, Veroniki AA, Soobiah C, Vafaei A, Ivory J, Strifler L, Cardoso R, Reynen E, Nincic V, Ashoor H, Ho J, Ng C, Johnson C, Lillie E, Antony J, Roberts DJ, Hemmelgarn BR, Straus SE (2016) Comparative safety and effectiveness of serotonin receptor antagonists in patients undergoing chemotherapy: a systematic review and network meta-analysis. BMC Med 14, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Kim JE, Hong YS, Lee J-L, Kim K-P, Park SJ, Sym SJ, Shin DB, Lee J, Park YS, Ahn JS, Kim TW (2015) A randomized study of the efficacy and safety of transdermal granisetron in the control of nausea and vomiting induced by moderately emetogenic chemotherapy in Korean patients. Support Care Cancer 23, 1769–1777. [DOI] [PubMed] [Google Scholar]
- [78].Brygger L, Herrstedt J (2014) 5-Hydroxytryptamine3 receptor antagonists and cardiac side effects. Expert Opin. Drug Saf 13, 1407–1422. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Calcium signaling pathway in response to granisetron treatment with the IPA molecule activity predictor. Red and green symbols indicate proteins up- and down-regulation in granisetron treated mice, respectively. Orange and blue nodes indicate predictions of to be activated or inhibited in granisetron treated mice, respectively. The color intensity is proportional to the degree of fold change. Edges between the nodes are colored orange when leading to the activation of downstream proteins, blue when inhibiting downstream proteins. Yellow edges indicate that the states of downstream genes are inconsistent with the prediction based on previous findings.
Fig. S2. Granisetron restored Ca+2 homeostasis by modulating CREB pathway in the brains of C57Bl/6J aged mice. Statistical analysis was determined by Student’s t-test (n=5 mice per group). *P<0.05 and **P<0.01 versus control group.
Fig. S3. Individual red and green channels for Panels D and E of Figure 4. These images are representative hippocampus sections stained with Thio-S (green) to detect Aβ plaques load and anti-collagen IV (red) to stain microvessels from control and granisetron treated groups.
Fig. S4. Individual red and green channels for Panel A of Figure 6. These images are representative brain sections stained with 6E10 (green) antibody to detect Aβ, and anti-GFAP (red) antibody to detect activated astrocytes in control and granisetron treated groups.
Table S1. A selection of significantly changed proteins in calcium pathway (P<0.05; fold change > 2) for granisetron effect in C57Bl/6J wild-type mice as determined by IPA analysis.
Table S2. A selection of significantly changed proteins in CREB pathway (P<0.05; fold change > 2) for granisetron effect in C57Bl/6J wild-type mice as determined by IPA analysis.