

Abstract
Cells depend on robust DNA damage recognition and repair systems to maintain genomic integrity for survival in a mutagenic environment. In the pathogenic yeast Candida albicans, a subset of genes involved in the response to DNA damage-induced genome instability and morphological changes has been found to regulate virulence. To better understand the virulence-linked DNA repair network, we screened for methyl methane sulfonate (MMS) sensitivity within the GRACE conditional expression collection and identified 56 hits. One of these potential DNA damage repair-associated genes, a HOF1 conditional mutant, unexpectedly had a previously characterized function in cytokinesis. Deletion of HOF1 resulted in MMS sensitivity and genome instability, suggesting Hof1 acts in the DNA damage response. By probing genetic interactions with distinct DNA repair pathways, we found that Hof1 is genetically linked to the Rad53 pathway. Furthermore, Hof1 is down-regulated in a Rad53-dependent manner and its importance in the MMS response is reduced when Rad53 is overexpressed or when RAD4 or RAD23 is deleted. Together, this work expands our understanding of the C. albicans DNA repair network and uncovers interplay between the cytokinesis regulator Hof1 and the Rad53-mediated checkpoint.
INTRODUCTION
To remain viable, cells must maintain genomic integrity, but both external and internal factors such as radiation, replication errors, and reactive oxygen species can cause DNA damage that compromises this integrity (Hoeijmakers, 2009). By creating chromosomal abnormalities, DNA damage can disrupt cell function, potentially resulting in cancer or cell death (Hoeijmakers, 2009). To defend against such events, cells have evolved molecular mechanisms to recognize and repair damaged DNA, improving the fidelity of genetic information transfer between generations (Zhou and Elledge, 2000; Wood et al., 2001).
Past studies have applied a well-developed genetic and molecular toolkit to characterize the DNA damage response in the ascomycete yeast Saccharomyces (Schwartz et al., 2002; Boiteux and Jinks-Robertson, 2013). Experimentally, DNA damage is often induced with methyl methane sulfonate (MMS), a methylating agent that modifies both guanine and adenine, causing base mispairing and ultimately double-strand breaks (Lundin et al., 2005; Shi et al., 2007). Once DNA is damaged, a cell employs diverse set of proteins to recognize the damage, pause the cell cycle and DNA replication, activate the proper repair machinery, and restart the cell cycle after the repair (Sirbu and Cortez, 2013).
Activation of signaling pathways termed checkpoints pauses cellular processes, allowing time for DNA repair (Zhou and Elledge, 2000). In particular, the activation pathway of the checkpoint kinase Rad53 has been well studied (Pellicioli and Foiani, 2005; Sweeney et al., 2005; Branzei and Foiani, 2006; Conde et al., 2010; Chen et al., 2015). First, sensors such as Tel1 and Mec1 receive the signal from damaged DNA (Sanchez et al., 1996; Baroni et al., 2004) and, with the help of adaptor proteins Mrc1 or Rad9 (Murakami-Sekimata et al., 2010; Berens and Toczyski, 2012; Ohouo et al., 2013; Bacal et al., 2018), phosphorylate Rad53. Phosphorylated Rad53 then auto-phosphorylates to generate the active form of the kinase (Heideker et al., 2007). Activated Rad53 interrupts the cell cycle and regulates the downstream target proteins required to repair the damage. Rad53 has also been implicated in modulating morphology, potentially through interactions with septins (Smolka et al., 2006). Deletion of Rad53, or other checkpoint kinases like Rad9 and Mrc1, prevents coordination of the repair process and therefore results in a strong sensitivity to DNA-damaging reagents like MMS (Weinert et al., 1994; Hanway et al., 2002; Kitanovic and Wolfl, 2006).
After checkpoint activation, distinct pathways are involved in repairing the DNA damage. The base excision repair (BER) pathway, the Rad52-related recombination epistasis group (Kwon and Sung, 2017), and the Rad6 epistasis group take part in the response to MMS-induced DNA damage (Somasagara et al., 2017). The BER pathway, initiated by a DNA glycosylase that recognizes and removes the damaged or abnormal base, leaving an abasic site, is mainly used to repair damage that creates minor disturbances in the DNA helix (Memisoglu and Samson, 2000). The abasic site is further processed by either short-patch BER (for replacement of 1 nucleotide) or long-patch BER (for replacement of 2–13 nucleotides). The RAD6 group represents a postreplication repair pathway and includes genes encoding specialized translesion synthesis polymerases, able to replicate through DNA damage (Lawrence, 1994). Furthermore, mutants lacking some components of other repair pathways, like the Rad14-related nucleotide excision repair (NER) process (Prakash and Prakash, 2000), are also MMS sensitive, highlighting the diversity of proteins required to recognize and correct MMS-induced damage. After DNA repair, the checkpoint kinase must be deactivated to allow the cell to resume the cell cycle. This process is mediated by protein phosphatases including Pph3 and Ptc2/Ptc3 (Leroy et al., 2003; O’Neill et al., 2007; Kim et al., 2011), which are important for the recovery from, or adaptation to, MMS-induced DNA damage.
Candida albicans is one of the most common fungal pathogens, and its pathogenicity is related to characteristics such as adhesion to and invasion of host cells, the secretion of hydrolases, the yeast-to-hypha transition, contact sensing/thigmotropism, and biofilm formation (Whiteway and Bachewich, 2007). DNA damage in C. albicans causes genome instability and abnormal growth (Loll-Krippleber et al., 2014). C. albicans yeast cells treated with either the DNA-replication inhibitor hydroxyurea (HU) or the DNA methylation agent MMS exhibit activation of the checkpoint kinase Rad53 accompanied by significant filamentous growth (Shi et al., 2007). Similarly, deletion of RAD52 causes a strong sensitivity to MMS and activates filamentous growth (Andaluz et al., 2006), and blocking the deactivation of Rad53 by deletion of PPH3 promotes filamentous growth and increased virulence (Sun et al., 2011; Feng et al., 2013, 2017). Deletion of RTT109, which plays critical roles in maintaining genome stability, results in fungal cells that are significantly less pathogenic in mice and more susceptible to killing by macrophages in vitro than wild-type (WT) cells (Lopes da Rosa et al., 2010). While foundational C. albicans DNA damage response studies have been performed, there is a need for more systematic studies of the DNA damage response given its known role in virulence.
To enhance our understanding of DNA damage repair in C. albicans, we screened the GRACE collection of tet-repressible conditional mutations (Roemer et al., 2003) for MMS sensitivity. We identified 56 strains that were sensitive after repression of the regulated gene. Among these strains, we were intrigued that loss of Hof1, a protein previously shown to play a critical function in cytokinesis, led to MMS sensitivity. To better understand the potential function of Hof1 in the DNA damage pathway, we used genetic approaches to characterize the function of Hof1. This work suggests there is interplay between Hof1 and the Rad53-mediated checkpoint.
RESULTS
Large-scale identification of MMS-sensitive strains in C. albicans
To identify MMS-sensitive C. albicans strains, the GRACE strains (Roemer et al., 2003) were conditionally repressed on tetracycline-containing (100 μg/ml) YPD plates with MMS (0.01% vol/vol) and grown at 30°C for 72 h. We identified 56 strains from the 2357 single mutants of the GRACE collection that were sensitive to MMS on repression (Figure 1). Close to half of those sensitive mutants identified were components of the cell division machinery, with roles in DNA replication, DNA damage repair, and cytokinesis. Genes involved in transcription and RNA processing accounted for a further one-third of the sensitive strains while the remaining genes were involved in translation, protein processing, cellular transport, and biosynthetic pathways.
FIGURE 1:
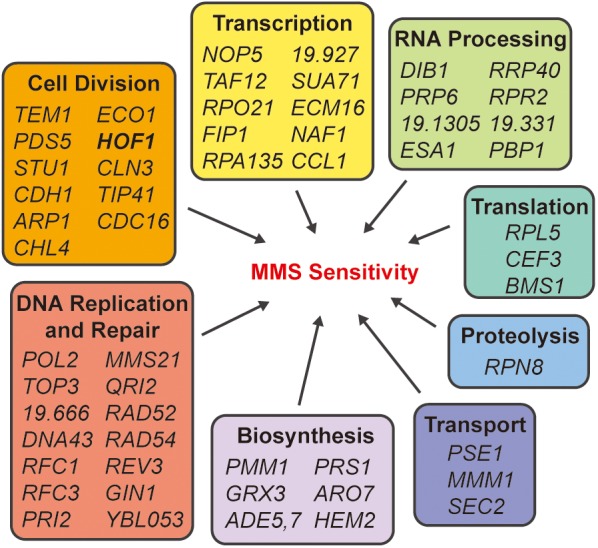
Identification of MMS-sensitive mutants from GRACE library screening. The 56 mutants that were scored as sensitive to MMS under tetracycline shutoff are shown, clustered by their predicted cellular processes.
HOF1 deletion results in sensitivity to genotoxic stress
The GRACE library screening showed conditional inactivation of Hof1-rendered cells sensitive to MMS. In S. cerevisiae, Hof1 is a well-studied protein implicated in regulating actin organization, cytokinesis, and secretory vesicle trafficking (Vallen et al., 2000; Blondel et al., 2005; Meitinger et al., 2011; Wang et al., 2018). In C. albicans, previous research reported a similar function for Hof1 in cytokinesis (Li et al., 2006). Since Hof1 is predicted to be cytokinesis-related, MMS sensitivity on loss of HOF1 was unexpected. To investigate the DNA damage–related function of Hof1 in C. albicans, we deleted HOF1 in the SN148 background using a CRISPR/Cas9 system.
Cells lacking HOF1 had an abnormal morphology consistent with the role of Hof1 in cytokinesis. Most hof1/hof1 cells were elongated and connected (Figure 2A). Indeed, quantification of cell chains with three or more cells revealed that 80% of Hof1 deletion cells were in chains versus 5% in WT cells (Figure 2B). On plates, HOF1 deletion colonies showed irregular surfaces and edges (Figure 2A). Deletion of HOF1 also reduced the growth rate (Figure 2C).
FIGURE 2:

Strains lacking HOF1 display cell division defects. (A) Wild-type and hof1/hof1 strains were grown in YPD media and imaged with DIC optics (left images) or grown on YPD plates to image colony morphology (right images). (B) Percentage of cells in chains was determined by imaging cells grown in YPD. N ≥ 200/condition. (C) Growth curves of wild-type and hof1/hof1 strains were made by measuring OD600 at the indicated time points. For each strain, three isolates were tested and the average is displayed.
Consistent with our screen, hof1/hof1 cells were MMS sensitive and this phenotype could be complemented (Figure 3A). Furthermore, the HOF1 deletion strain was sensitive to other DNA replication stresses including HU and to UV light (Figure 3A). These results suggest Hof1 plays a general role in the response to genotoxic stress.
FIGURE 3:

Deletion of HOF1 causes sensitivity to genotoxic stress and increases genome instability. (A) hof1/hof1 cells are sensitive to genotoxic stresses MMS, HU, and UV light. Growth assays with WT (WT+CIP10), HOF1 deletion (hof1/hof1+CIP10), and HOF1 deletion-complemented (hof1/hof1+CIP10-HOF1) strains. (B) The frequency of losing a heterozygous URA3 marker was assessed by comparing the number of colonies on YNB+ 5-FOA and YPD plates; n = 3. (C) WT and HOF1 deletion strains were treated with 0.02% MMS or 50 mM HU and imaged after 3 h (MMS) or 6 h (HU). Bud length was measured by ImageJ software; >30 cells/condition. (D) Growth assays in the presence of H2O2, rapamycin, fluconazole, and Congo red.
To test the function of Hof1 in mediating genomic stability, we designed a heterozygous URA3+ strain to investigate loss of heterozygosity through a 5-fluoroorotic acid (5-FOA) resistance assay. We found that the WT strain has a relatively low frequency of losing the URA3 marker (3.7 × 10-5), while the HOF1 deletion strain has a roughly 20-fold higher frequency of loss (6.7 × 10-4), indicating Hof1 plays a role in maintaining genome stability (Figure 3B).
As Hof1-modulated morphological changes may be associated with the DNA damage response, we treated WT and HOF1 deletion cells with 0.02% MMS and 50 mM HU for 6 h before checking the cell morphology. Both WT and hof1/hof1 cells were elongated after the treatments (Figure 3C). However, compared with the WT strain, the HOF1 deletion cells were substantially longer, suggesting loss of Hof1 exacerbates morphological defects associated with genotoxic stress.
Given that genome instability may be associated with nuclear segregation defects, we compared nuclear segregation in WT and HOF1 deletion cells with or without MMS treatment. To facilitate the comparison, 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI)-stained cells were binned into categories of metaphase, anaphase/telophase, abnormal binucleate, noncompact nuclei, and other, which captured poorly stained and G1/S/G2 mononucleate cells. In the absence of MMS, only 2% of WT cells showed abnormal binucleate or noncompact nuclei structures while hof1/hof1 cells were highly abnormal, with 34.5% of cells either binucleate or containing noncompact nuclei (Table 1). Notably, only 23% of the hof1/hof1 cells were visibly undergoing mitosis, which was a substantial reduction relative to both WT (38%) and suggests slower progression through G1/S/G2. When cells were treated with MMS, WT cells largely arrested in metaphase (58.5%), whereas hof1/hof1 cells (18%) did not arrest effectively and the binucleate/noncompact nuclei phenotypes persisted. Thus, hof1/hof1 cells display impeded cell cycle progression, show high levels of binucleate cells consistent with defective cytokinesis, and are poorly competent for MMS-triggered metaphase arrest.
TABLE 1:
Nuclear segregation in HOF1 and/or RAD53 deletion cells.
 | |||||
| Untreated | |||||
| WT | 16.5 ± 4.5 | 0.5 ± 0.5 | 1.5 ± 0.5 | 21.5 ± 4.5 | 60.0 ± 8.0 |
| rad53/rad53 | 19.5 ± 3.5 | 3.0 ± 1.0 | 5.0 ± 5.0 | 20.5 ± 8.5 | 51.5 ± 5.5 |
| hof1/hof1 | 8.0 ± 3.0 | 25.5 ± 5.5 | 9.0 ± 0 | 15.0 ± 1.0 | 41.5 ± 2.5 |
|
rad53/rad53 hof1/hof1 |
10.0 ± 3.0 | 10.5 ± 2.5 | 5.0 ± 2.0 | 6.5 ± 1.5 | 67.5 ± 1.5 |
| +0.02% MMS | |||||
| WT | 58.5 ± 6.5 | 0 | 2.0 ± 2.0 | 10.5 ± 3.5 | 29.0 ± 1.0 |
| rad53/rad53 | 13.0 ± 1.0 | 14.0 ± 2.0 | 15.0 ± 1.0 | 17.0 ± 3.0 | 41.0 ± 1.0 |
| hof1/hof1 | 18.0 ± 2.0 | 15.0 ± 0.0 | 7.0 ± 0 | 10.0 ± 1.0 | 50.0 ± 1.0 |
|
rad53/rad53 hof1/hof1 |
8.5 ± 2.5 | 14.0 ± 1.0 | 13.5 ± 1.5 | 27.0 ± 2.0 | 37.0 ± 1.0 |
MMS-treated or untreated cells were stained with DAPI to show nuclear structure. Cells with nuclear structures that fell into one of four categories (metaphase, anaphase/telophase, binucleate, or noncompact nuclei) were identified, and the relative fraction of each category was determined; n = 2, ≥52 cells/condition per replicate.
We further checked whether Hof1 plays a role in the response to other cellular stresses. We found HOF1 deletion renders cells sensitive to 2.5 mM H2O2, 15 ng/ml rapamycin, 4 μg/ml fluconazole, and 200 mg/ml Congo red (Figure 3D). These results suggest Hof1 may be involved in responding to a variety of stresses.
Functional analysis of Hof1 domains
In S. cerevisiae, the N terminus of Hof1 is implicated in control of cell size and actin cable levels, while the C terminus controls actin cable organization via regulation of the formin Bnr1 (Kamei et al., 1998; Graziano et al., 2014). Alignments show that the amino acid sequence of CaHof1 has only 15% identity with ScHof1. However, the proteins have a similar domain structure (Figure 4A): an N-terminal F-BAR domain (11–266 amino acids), containing an FCH domain (13–102 amino acids), and a C-terminal SH3 domain (539–605 amino acids). To further investigate how Hof1 may function in the DNA damage response, we checked cell morphology and sensitivity to genotoxic stress in strains lacking a single Hof1 domain each.
FIGURE 4:

Functional analysis of the Hof1 F-BAR and SH3 domains. (A) Schematics of C. albicans Hof1 and the Hof1ΔF-BAR and Hof1ΔSH3 mutants. (B) Cell morphologies of WT and Hof1 mutant strains grown in YPD imaged with DIC optics. (C) Growth assays of WT and Hof1 mutant strains in the presence of genotoxic stresses. (D) Western blot of samples from log phase Hof1-myc and Hof1ΔF-BAR-myc cultures showing expression levels in the absence and presence of MMS (0.02% for 90 min). b-Tubulin is a loading control, and normalized band intensities are shown below. (E) Expression of Hof1-HA tested by Western blotting after treating log phase cells with 40 mM HU, 2.5 mM H2O2, 10 nM rapamycin, 100 μg/ml Congo red, 5 μg/ml fluconzole, 1.5 M NaCl, or 200 mM CaCl2 for 90 min before lysis. Control cells were untreated and Hof1 was probed using an anti-HA antibody. b-Tubulin acts as a loading control and normalized band intensities are shown below. Two-tailed t test; n = 3; **P < 0.01; *P < 0.05.
Deletion of the F-BAR domain, but not the SH3 domain, resulted in defects of similar severity to a hof1/hof1 mutant. Cells with a SH3 domain deletion were morphologically similar to the WT, while deletion of the F-BAR domain resulted in numerous cell chains and morphology similar to HOF1 deletion cells (Figure 4B). Furthermore, the F-BAR deletion, but not the SH3 deletion, caused sensitivity to both MMS and HU, suggesting the F-BAR domain is required for both the response to DNA damage and the maintenance of cell morphology while the SH3 domain is dispensable (Figure 4C).
To test whether the F-BAR deletion (Hof1ΔF-BAR) may cause the above phenotypes indirectly by destabilizing Hof1, we checked the expression of Hof1 and Hof1ΔF-BAR by Western blotting (Figure 4D). Deletion of the F-BAR domain strongly reduced Hof1 levels. Furthermore, the levels of both Hof1 and Hof1ΔF-BAR decreased significantly on 0.02% MMS treatment (Figure 4D), and the levels were further reduced at higher MMS concentrations (unpublished data). Thus, the dysfunction caused by the F-BAR deletion can be partially explained by the destabilization of Hof1. Since the Hof1 levels decreased in cells treated with MMS, we also checked Hof1 levels in response to other stresses. Hof1 levels were down-regulated in response to HU, H2O2, rapamycin, and NaCl (Figure 4E). By contrast, a significant up-regulation of Hof1 protein level was observed in response to Congo red, which may reflect a targeted response by Hof1 to cell wall stress.
Genetic interaction analysis of HOF1 with DNA damage–related genes
In S. cerevisiae, MMS sensitivity of a HOF1 deletion strain has been reported based on high throughput screening results (Chang et al., 2002; Svensson et al., 2011), but no specific function in the DNA damage response pathway was assigned to Hof1. To better understand the role of Hof1 in response to DNA damage in C. albicans, a genetic interaction analysis was performed.
To do so, a series of DNA damage repair gene deletions and double deletions, also lacking HOF1, were made. Rad53, a main checkpoint kinase in C. albicans (Shi et al., 2007), was chosen to address a potential role of Hof1 in DNA damage signal transduction. We also deleted PPH3, a catalytic subunit of protein phosphate 4, which controls Rad53 deactivation during the recovery from or adaption to DNA damage (Sun et al., 2011; Feng et al., 2017). Rad18, of the Rad6 epistasis group, was chosen as a component of postreplication repair (Bailly et al., 1997). Mms22, a putative adaptor subunit of an E3 ubiquitin ligase complex, represented the replication repair pathway (Yan et al., 2015). Mms21, a potential SUMO E3 ligase, was chosen to check the potential function of Hof1 in the SUMO-related DNA repair pathway, and Rad52 represented the homologous DNA recombination group (Lisby et al., 2001). Siz1 is a potential homologue of the ScSiz1 SUMO E3 ligase, which is functionally distinct from Mms21 and helps address DNA damage in the absence of Nfi1 (Takahashi et al., 2006). Rad14, Rad4, and Rad23 represented the NER group where Rad14 is part of the NER1 complex and Rad4 and Rad23 are part of NER2.
Removal of any of Rad18, Mms21, Mms22, Pph3, Rad52, or Rad14 resulted in an MMS-sensitive phenotype, and the sensitivity to MMS of double mutants of Hof1 with Rad18, Mms21, Mms22, Rad52, Pph3, or Rad14 was increased relative to the single mutants (Figure 5A), suggesting Hof1 acts independent of those proteins in the MMS response. By contrast, while the RAD53 deletion strain was MMS sensitive, additional deletion of HOF1 did not result in increased MMS sensitivity, suggesting Rad53 and Hof1 may act in a similar pathway (Figure 5B). The SIZ1 deletion strain was not MMS sensitive even at high concentrations (unpublished data), nor did SIZ1 deletion affect hof1/hof1 sensitivity, indicating Siz1 does not play a role in responding to MMS (Figure 5C). Neither the RAD4 nor the RAD23 deletion strains showed sensitivity to MMS, but strikingly both deletions were able to rescue hof1/hof1 MMS sensitivity (Figure 5D). This indicates hof1/hof1 MMS sensitivity is suppressed by loss of RAD4 or RAD23. Taken together, these results suggest that Hof1 shows a role in MMS response independent of Rad18, Mms21, Mms22, Rad52, Rad14, and Pph3, but appears to be part of the Rad53 checkpoint kinase-related circuit, and the requirement of Hof1 in this circuit for repair of MMS damage is greatly reduced on RAD4 or RAD23 deletion.
FIGURE 5:

Genetic epistasis analysis of HOF1. Growth assays show MMS sensitivity increases on double deletion of HOF1 and MMS22, PPH3, MMS21, RAD18, RAD52, or RAD14 (A), but not in the double deletions of HOF1 with RAD53 (B) or SIZ1 (C). (D) Deletion of RAD23 or RAD4 rescues hof1/hof1 MMS sensitivity.
Interplay between Hof1 and the Rad53 circuit
Our genetic interaction data suggest Hof1 is associated with the Rad53 circuit. In C. albicans, Rad53 is a main checkpoint kinase; it is activated by phosphorylation in response to DNA damage and inactivated by dephosphorylation after repair (Yao et al., 2017). Blocking activation or inactivation of the checkpoint leads to DNA damage sensitivity (O’Neill et al., 2007; Fiorani et al., 2008). Given the Hof1–Rad53 association, we checked Rad53 behavior in a HOF1 deletion background. Previously, we reported that Rad53 is phosphorylated in response to MMS, resulting in a slower migration rate in SDS–PAGE (Feng et al., 2013). Here, we found that Rad53 was phosphorylated after MMS exposure in both WT and HOF1 deletion strains (Figure 6A). After removal of MMS, Rad53 could be dephosphorylated in both backgrounds. Taken together, HOF1 deletion has no dramatic impact on the activation or deactivation of Rad53.
FIGURE 6:

Checkpoint kinase Rad53 modulates Hof1 levels. (A) Anti-HA Western blot of WT and hof1/hof1 strains with Rad53-HA that were incubated with 0.02% MMS for 90 min (MMS), resuspended in YPD, and further incubated for indicated times (2 and 6 h). Untreated cells (0) were included as controls. (B) Western blot of Hof1-HA in WT and a RAD53 deletion strain with and without a 90-min 0.02% MMS treatment. Hof1-HA was probed using an anti-HA antibody, and b-tubulin was a loading control. Band intensities relative to WT untreated are shown. Two-tailed t test; n = 3; **P < 0.01.
We next asked whether Rad53 modulates the behavior of Hof1. As the level of the Hof1 protein is down-regulated in response to MMS (Figure 4D), we tested whether this could be influenced by Rad53. While RAD53 deletion has no impact on the level of Hof1 in untreated cells, RAD53 deletion stabilizes Hof1 levels after MMS treatment (Figure 6B). Thus, RAD53 deletion blocked the MMS-induced reduction of Hof1 levels. RAD53 deletion also exhibited a similar stabilizing effect on Hof1 levels in the presence of HU (Supplemental Figure S1). Rad53 may directly stabilize Hof1 in the presence of these genotoxic stresses, or increased Hof1 levels may be a result of disabling the Rad53-mediated DNA repair checkpoint, releasing cells from a phase where Hof1 levels are normally depressed. To differentiate between these possibilities, WT cells expressing Hof1-HA and GFP-tagged tubulin (Tub2-GFP) were synchronized by MMS exposure, released, and both Hof1-HA levels and spindle length were tracked (Supplemental Figure S2). After MMS exposure, Hof1-HA levels are depressed, but they recover within 3 h as the cell cycle progresses, denoted by increasing spindle length. Taking the results together, Hof1 levels are modified by Rad53, though the effect may be an indirect effect of halting the cell cycle at a phase with lowered Hof1 levels.
We further investigated the influence of Rad53 and Hof1 on checkpoint-mediated, MMS-induced cell cycle arrest by microscopy. As previously noted, DAPI-stained cells were binned into categories of metaphase, anaphase/telophase, abnormal binucleate, noncompact nuclei, and other, which captured poorly stained and G1/S/G2 mononucleate cells. As shown in Table 1, treatment of WT cells with 0.02% MMS caused efficient arrest (58.5% metaphase). In contrast, while the RAD53 deletion has almost no effect on the distribution of untreated cells, MMS-treated cells do not arrest efficiently (only 13% metaphase), similar to previous findings (Shi et al., 2007), and there is a substantial population of abnormal cells (29% binucleate and noncompact nuclei). Likewise, the HOF1 deletion strain did not show a substantial arrest on MMS exposure (18% metaphase). When a RAD53 HOF1 double deletion was imaged there was no indication of cell cycle arrest after MMS treatment (8.5% metaphase), though there were somewhat fewer abnormal cells when untreated (8% vs. 34.5% for hof1/hof1 mutants), suggesting RAD53 deletion had a moderating influence on the hof1/hof1 strain. Notably, many more RAD53 HOF1 double deletion cells were observed in anaphase/telophase than in either single deletion (27% vs. 17% or 10%) on MMS treatment, suggesting possible defects in late stage mitosis. Overall, HOF1 and RAD53 deletions have remarkably similar effects on MMS-induced metaphase arrest, consistent with their association in the DNA damage response.
Rad23 rescues MMS sensitivity of HOF1 deletions
Through the genetic interaction assays (Figure 5), we noted RAD23 deletion rescued hof1/hof1 MMS sensitivity. To confirm this phenotype, we performed a quantitative survival assay by counting viable cells after 2 h MMS treatment (Figure 7A). As previously observed, deletion of HOF1 decreased MMS survival, deletion of RAD23 increased MMS survival, and the HOF1 RAD23 double deletion strain had MMS resistance similar to the RAD23 deletion strain. This further demonstrates that loss of Rad23 rescues MMS sensitivity caused by loss of Hof1.
FIGURE 7:

RAD23 deletion rescues hof1/hof1 MMS sensitivity. (A) The indicated strains were treated with either 0.01% MMS or 0.02% MMS for 2 h prior to dilution and dispersion on YPD plates to measure survival. Colonies were counted and compared with untreated control cells. (B) Growth assays measuring hof1/hof1 and rad23/rad23 sensitivity to HU and UV. (C) Images of the indicated strains in YPD media (top) or on YPD plates (bottom). (D) RAD23 deletion can elevate Rad53 levels. Anti-HA Western blot of the indicated deletion strains with Rad53-HA. b-Tubulin is a loading control. Rad53 band intensities were measured and normalized to WT cells. Two-tailed t test; n = 3; *P < 0.05. (E) Growth assay showing that elevated expression of Rad53 suppresses hof1/hof1 sensitivity to MMS and HU.
To establish whether this rescue is specific for MMS, we also checked the phenotype of the above mutants under other genotoxic stresses. Loss of RAD23 rescued hof1/hof1 HU sensitivity, suggesting RAD23 deletion may suppress the sensitivity of hof1/hof1 cells to a range of stresses. In contrast, the HOF1 and RAD23 double deletion strain showed nearly the same sensitivity as the RAD23 deletion strain to UV light indicating there is some specificity in the Rad23-mediated rescue of HOF1 deletion phenotypes (Figure 7B). Similar results were obtained for a HOF1 and RAD4 double deletion strain (unpublished data). Since Hof1 is a cytokinesis-related protein, we asked whether loss of Rad23 could rescue the observed defects of HOF1 strains in cell division. We found that the HOF1 RAD23 double deletion strain showed similar cellular and colony morphology to that of the HOF1 deletion strain (Figure 7C). Thus, the ability of RAD23 deletion to suppress hof1 mutant phenotypes is limited to repressing DNA damage sensitivity and does not extend to the cytokinesis defect.
In S. cerevisiae, Rad23 is critical for NER, is part of a ubiquitin/proteasome pathway, and has been shown to repress DNA damage response genes (Reed and Gillette, 2007; Zhou et al., 2015). Owing to its regulatory role of DNA damage response genes and since decreased proteasome function enables increased DNA repair (Lommel et al., 2000), we tested whether RAD23 deletion increases Rad53 levels (Figure 7D). Indeed, we found elevated Rad53 levels in a RAD23 deletion and a RAD23 HOF1 double deletion strain, but not in a HOF1 deletion strain. Thus, the RAD23 deletion-mediated rescue of a HOF1 deletion strain could be caused by increased repair efficiency resulting from elevated Rad53 levels. To test whether high Rad53 levels can rescue hof1 mutant MMS sensitivity, we substituted the RAD53 promoter with the strong MET3 promoter and measured MMS sensitivity. As shown in Figure 7E, RAD53 overexpression partially rescued hof1 mutant sensitivity to MMS and HU.
DISCUSSION
Our screen of the GRACE library for MMS sensitivity has uncovered many elements of the C. albicans DNA damage response network. By pursuing one unexpected hit, cytokinesis regulator Hof1, we have characterized a link between cytokinesis and DNA damage response. HOF1 deletion causes MMS sensitivity and genome instability, and Hof1 is not only genetically associated with the Rad53 pathway but also its levels drop in a Rad53-dependent manner in response to MMS. The role of Hof1 appears to be coordinated with other aspects of the DNA damage response, given that the importance of Hof1 in the MMS response is reduced in Rad23 or Rad4 mutant strains and when Rad53 is overexpressed.
Overall, our screen identified several genes, including HOF1, that do not appear to be directly connected to DNA repair. Establishing the connection of these genes to sensitivity to MMS treatment should expand our understanding of the complex relationships between cellular repair and functions linked to processes such as metabolism and cell cycle control.
In S. cerevisiae, the physical interactions of Hof1 with septins and actin during septum formation and the role of Hof1 in cytokinesis have been well characterized (Vallen et al., 2000; Blondel et al., 2005). In C. albicans, Hof1 is also reported to function in cytokinesis (Li et al., 2006). We confirmed HOF1 deletion causes cell division defects and explored a novel role for Hof1 in MMS tolerance. Since cytokinesis occurs late in the cell cycle, typically after DNA replication, the function of Hof1 in response to DNA damage may be distinct from its role from cytokinesis. We found Hof1 protein levels decrease in response to the stressors MMS, HU, H2O2, and rapamycin. RNA sequence analysis indicates MMS lowers HOF1 expression (log2 fold change = –1.48, P < 0.001; unpublished data), suggesting Hof1 levels are transcriptionally regulated to some extent. In contrast, Hof1 levels increased in response to the cell wall–damaging agent Congo red. This contrasting impact on Hof1 levels may be due to a link between cytokinesis and wall remodeling. Furthermore, the ability of rad4 and rad23 mutants to suppress hof1 mutant MMS sensitivity, but not cell division defects, supports the possibility of distinct roles for Hof1 in damage repair and in cytokinesis.
While Hof1 may have two distinct functions, the cell wall integrity and DNA repair pathways may also modulate Hof1 indirectly. For example, DNA damage could halt the cell cycle earlier, resulting in lowering Hof1 expression, whereas damage to the cell wall may necessitate an increase in Hof1 levels, or greater time preparing for cell division, to ensure cell wall integrity. In this model, loss of Hof1 may decrease control of the cell cycle, increasing the likelihood of irreparable damage in the presence of either type of stressor. In any case, there is a logic to linking the DNA damage checkpoint to regulation of cytokinesis, and though the roles of Hof1 in damage regulation and cytokinesis might not overlap completely, they may be connected.
The MMS-induced decrease in Hof1 levels does appear to be checkpoint related given its dependence on the main DNA damage checkpoint kinase Rad53. In the initial response to MMS, Rad53 is activated while Hof1 levels are suppressed. However, the reduction of Hof1 levels is not as persistent; after a 3 h MMS treatment followed by a 2-h wash out, Rad53 remains in its active phosphorylated form, while Hof1 protein levels have recovered (unpublished data). This suggests Hof1 depression is more transient during DNA damage. Importantly, we also found that Hof1 levels fluctuate during the cell cycle and appear to be depressed in S phase when the Rad53-mediated repair checkpoint occurs (Supplemental Figure S1). This suggests indirect regulation of Hof1 by Rad53, whose deletion may lead to abnormal cell cycle regulation as the cell compensates for its loss. While there is evidence that the Hof1–Rad53 interaction is indirect, the fact that our cell synchronization assay depended on MMS for synchronization is a confounding factor as it may have also driven more direct down-regulation of Hof1. To better tease apart this relationship, new approaches to cell synchronization in C. albicans are required. Even if Rad53 regulation of Hof1 is primarily indirect, we cannot exclude the role of Hof1 in the checkpoint-related DNA damage response.
We found that deletion of the NER components, Rad4 and Rad23, partially rescued hof1 mutant MMS sensitivity. In S. cerevisiae, Rad4 and Rad23 can bind the promoters of a subset of DNA damage-responsive genes, repressing their expression (Zhou et al., 2015). The slightly increased Rad53 protein levels observed in both rad23 and hof1 rad23 mutants could be explained by the derepression of RAD53 transcription caused by loss of Rad23. Thus, one explanation for suppression of MMS sensitivity by RAD4 or RAD23 deletions is that their loss increases Rad53 levels, elevating the rate of DNA repair and cell survival under MMS stress. Alternatively, Rad4 and Rad23 have been shown to heavily down-regulate Dun1, a G2/M checkpoint kinase downstream of Rad53 required for DNA damage-induced cell cycle arrest and previously shown to down-regulate Hof1 in S. cerevisiae (Zhou et al., 2015). The elevated levels of Dun1 resulting from loss of Rad4/23 may itself be enough to stall the cell cycle, allowing DNA repair independent of a direct action of Rad4/23 on Rad53. In either case, loss of Rad4/23 accelerates DNA repair or stalls the cell cycle, which likely proceeds independent of Hof1, allowing suppression of MMS sensitivity.
We propose a speculative model to explain the connection of Hof1 to MMS damage repair (Figure 8). In this model Rad53 is activated in response to DNA damage and lowers Hof1 levels indirectly or directly, blocking cell cycle progression to ensure time to repair damaged DNA. Here Hof1 may act as a core component of the cytokinesis machinery to link DNA damage repair to cytokinesis through its responsiveness to the checkpoint kinase Rad53 either directly, or indirectly, through Rad53 effectors such as Dun1 (Jaehnig et al., 2013). Previously, both Enserink et al. (2006) and Smolka et al. (2006) have demonstrated a potential close association between Rad53 and the septin machinery required for cytokinesis in S. cerevisiae. Hof1 may play a role in this linkage or act in a parallel pathway to restrict cytokinesis in the presence of DNA damage. In either case, deletion of HOF1 induces MMS sensitivity through poor regulation of cytokinesis, potentially exacerbated by weakening the Rad53-mediated checkpoint. Poorly controlled cytokinesis may either take longer, allowing the accumulation of further mutations, or proceed rapidly, before mutations can be repaired. The need for Hof1 can be bypassed by accelerating DNA repair by overexpressing Rad53 or deleting Rad4/23.
FIGURE 8:
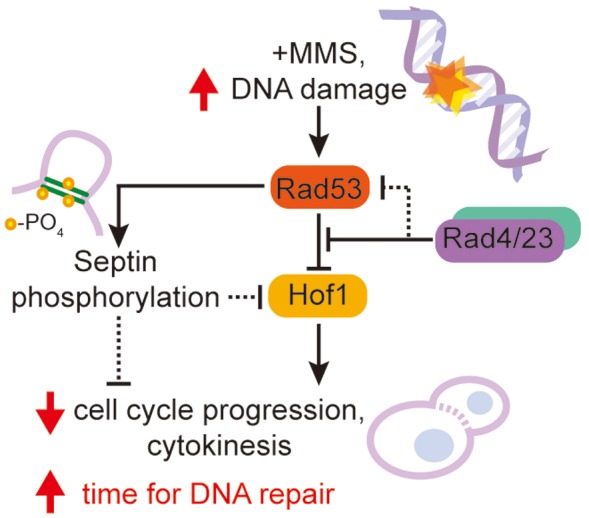
Hypothetical model of the Hof1, Rad53, and Rad4/Rad23 regulatory network in C. albicans. A potential regulatory network that explains how a DNA damage signal propagates through Rad53 and subsequently Hof1 to regulate the cell cycle. Note that Rad53 may also act through septin phosphorylation according to work in S. cerevisiae and Rad4/23 may act to depress signal transduction either downstream of Rad53 or of Rad53 itself. Solid lines indicate interactions (direct or indirect) previously identified in S. cerevisiae while dotted arrows are hypothetical. Arrows represent activation, while bars represent inhibition.
Potentially, Hof1 could act as a necessary additional signal for Rad53 activation, though its presence at the cell periphery and its cell cycle-dependent expression make it an unlikely candidate to detect DNA damage. Here, deletion of Hof1 or inactivation of Rad53 leads to similar levels of damage sensitivity, and the rad53 hof1 double mutant has an equivalent sensitivity to either single mutant. This would position Hof1 as a timer for the checkpoint, eventually shutting off the Rad53-induced checkpoint and resetting the normal cell cycle.
In this study, we establish that the cytokinesis regulator Hof1 is genetically linked to the Rad53 checkpoint kinase in C. albicans. Loss of Hof1 leads to enhanced DNA damage sensitivity, yet the Rad53 response to MMS in part acts by decreasing Hof1 levels, likely through an indirect pathway. Hof1 thus appears to be a potential component linking the DNA damage checkpoint and cytokinesis. Further work is needed to fully unravel the details of Hof1 molecular regulation and the link to the Rad53 checkpoint.
MATERIALS AND METHODS
Strains, media, and reagents
The C. albicans strains were grown in yeast extract peptone dextrose (YPD) medium with 50 mg uridine/l as described (MacPherson et al., 2005). Strains and primers used in this study are listed in Supplemental Tables S1 and S2. MMS was purchased from Sigma (USA). HU and other chemicals were purchased from Bioshop (Canada). The yeast nitrogen base (YNB) medium was supplemented with appropriate nutrients for plasmids selection and maintenance. Solid media contained 2% agar.
GRACE library MMS sensitivity assay
The GRACE library was transferred from frozen glycerol stocks into 96-well plates containing liquid YPD with 200 μg/ml nourseothricin using sterile 96 pin replicators and incubated overnight at 30°C. Overnight cultures were spotted, using a 96 pin replicator, onto YPD agar, YPD agar with tetracycline (100 μg/ml), and YPD agar with tetracycline-added MMS (0.01% vol/vol). Plates were incubated at 30°C for 3 d, and strains that were growing well on tetracycline but not on plates with both tetracycline and MMS were scored as sensitive. Sensitivity of those strains was further confirmed by making 10-fold dilutions of overnight cultures in sterile distilled water and then plating 5 μl of each dilution on tetracycline and tetracycline plus MMS plates. Images of plates and colonies were scanned at 300 dots per inch using an Epson Perfection v500 photo scanner.
Gene deletion, rescue, and epitope tagging of proteins
To construct a HOF1 deletion strain in C. albicans, we used a CRISPR/Cas9 system (Vyas et al., 2015). The small guide RNA (sgRNA) of HOF1 was formed by annealing primers HOF1-sg-F and HOF1-sg-R and cloning the resulting ds fragment into the BsmBI site of pV1093 to generate plasmid pV1093-Hof1-sgRNA. A repair DNA fragment, containing the HIS1 marker, was amplified with primers HOF1-Re-F and HOF1-Re-R using pFA-HIS1 as a template. The pV1093-Hof1-sgRNA was then linearized with SacI and KpnI and transformed into C. albicans strain SN148 together with the HOF1 repair DNA. Transformants were selected on YNB-his plus 200 μg/ml nourseothricin. The knockout strains were confirmed with primers HOF1-Te-F and HOF1-Te-R. To rescue the phenotype of HOF1 deletion strain, a 3031-base pair DNA fragment containing the full-length HOF1 gene was amplified with primers HOF1-Te-F and HOF1-Te-R and cloned into the KpnI site of plasmid CIP10 (Murad et al., 2000), generating pCIP10-HOF1. Then, pCIP10-HOF1 was linearized by StuI and integrated into the RP10 locus of the genome. The integration was confirmed using primers URA-TF and RPS-F.
Similarly, a transient CRISPR/Cas9 system (Min et al., 2016) was used to knock out MMS22, RAD18, RAD52, RAD53, RAD14, RAD4, RAD23, and SIZ1 in SN148 or the HOF1 deletion strain. Generally, a Cas9 gene was amplified with common primers P7 and P8. The sgRNA was amplified by annealing PCR with primers P5 and P6, using the products of two separate PCR reactions containing the sgRNA sequence, and repair DNA was amplified from pFA-ARG4. Finally, the Cas9, sgRNA and repair DNA products were transformed into either the WT or the Hof1 deletion strain and selected on SD-Arg plates. The correct knockout strains were confirmed by PCR.
To construct the PPH3 HOF1 double deletion strain, a previous PPH3 deletion strain (Feng et al., 2013) was used as a background strain and a similar CRISPR/Cas9 system for HOF1 was used. Similarly, a MMS21 deletion strain constructed before (Islam et al., 2019) was used as a background strain to construct the MMS21 HOF1 double deletion strain.
To construct the RAD53 overexpression strain, a MET3 promoter was amplified from pFA-URA3-MET3 to substitute the original promoter of RAD53 gene.
To test the expression of the Hof1 protein, a MYC or HA tag was amplified from the pFA-MYC-URA or pFA-HA-URA plasmids and integrated at the 3′ end of the HOF1 gene using the CRISPR/Cas9 system, generating a homozygous HOF1-MYC or HOF1-HA strain. Similarly, a HA tag was integrated into the 3′ end of the RAD53 gene using the CRISPR/Cas9 system. The integration was confirmed by both PCR and by Western blotting.
To check the spindle morphology, a GFP tag was amplified from the pFA-GFP-URA plasmid and integrated at the 3′ end of the TUB2 gene in SN148 background.
Protein extraction, Western blot, and immunoprecipitation (IP)
Exponentially growing cells were harvested by centrifugation and ∼100 mg of cells were suspended in 200 μl of ice-cold RIPA buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, EDTA-free protease inhibitor mix [Roche], and phosphatase inhibitor cocktail [Roche]) (Gao et al., 2014). After an equal volume of acid-washed glass beads was added, cells were broken by five rounds of 60 s of beating, with 5 min of cooling on ice between rounds, using a FastPrep-24 Disruptor. The supernatant was collected after centrifugation of the cell lysate at 12,000 rpm for 10 min at 4°C. Protein concentrations were determined using a BCA protein assay (Feng et al., 2013). Western blot analysis was carried out as described (Feng et al., 2017).
For the IP assays, anti-MYC beads (QED Bioscience) (10 μl) were washed twice with RIPA buffer and incubated with 5 mg total protein extract in 1 ml extraction buffer at 4°C overnight. Next morning, beads were harvested and washed three times with 1 ml RIPA buffer. Proteins were subjected to Western blot analysis as above using rabbit monoclonal anti-MYC antibody (QED Bioscience) to detect Hof1-MYC.
To detect Rad53-HA, a rabbit monoclonal anti-HA antibody (QED Bioscience) and the goat anti-rabbit immunoglobulin G secondary antibody were purchased from ODSEY and processed as described above for the anti-MYC antibody.
Growth assay
Cells were grown overnight in liquid YPD medium at 30°C, serially diluted by 10-fold, and spotted onto solid plates containing stress chemicals. Plates were incubated for 2–3 d at 30°C before scanning by an Epson Perfection v500 photo scanner.
Genome stability assay
To assess genome stability (Kumaran et al., 2013), the CIP10 plasmid (Murad et al., 2000) containing a URA marker was linearized by StuI and integrated into the RP10 locus of both WT and HOF1 deletion strains, generating heterozygous URA3 strains. The strains were streaked on YNB dextrose-ura twice and inoculated in YPD overnight at 30°C. Next day, cells were harvested and washed twice with distilled water; 10-fold dilutions were made before spotting. For each strain, 10-1 cells (100 μl) were spotted on YNB dextrose 5-FOA plates while 10-4 cells (20 μl) were spotted on YPD plates to check the cell numbers. All the strains were grown as duplicates in three independent samples. Finally, the average numbers of cells on YNB dextrose 5-FOA plates and YPD plates were calculated.
Microscopy
Overnight cells were diluted down to an OD600 of 0.2 in YPD with and without 0.02% MMS and incubated for 6 h. To visualize DNA, cells were fixed in fresh 70% ethanol for 20 min, washed with sterile water, incubated in 1.0 μg/ml DAPI (Sigma-Aldrich) for 20 min, washed twice with sterile water, and mounted on slides. Cells were imaged on a Leica DM6000B microscope (Leica Microsystems Canada, Richmond Hill, ON, Canada) equipped with a Hamamatsu-ORCA ER camera (Hamamatsu Photonics, Hamamatsu City, Japan) and the HCX PL APO 63× NA 1.40-0 oil or the HCX PLFLUO TAR 100× NA 1.30–0.6 oil objectives. Differential interference contrast (DIC) optics or epifluorescence with DAPI (460 nm) filters were utilized. Images were captured with Volocity software (Improvision, Perkin-Elmer, Waltham, MA).
Supplementary Material
Acknowledgments
We thank all the members of Whiteway lab for the helpful discussion, and we thank Merck for access to the Grace collection of regulated gene disruptions. This work was supported by the Natural Science Foundation of Nantong City to J.F. (No. JC2018052), Natural Sciences and Engineering Research Council of Canada (NSERC) grants to M.W. (CRC 950-228957 and discovery RGPIN/4799), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and a Jiangsu government scholarship for overseas studies, China.
Abbreviations used:
- BER
base excision repair
- DAPI
4′, 6′-diamidino-2-phenylindole dihydrochloride
- DIC
differential interference contrast
- HU
hydroxyurea
- IP
immunoprecipitation
- MMS
methyl methane sulfonate
- NER
nucleotide excision repair
- WT
wild type
- YNB
yeast nitrogen base.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-06-0316) on January 15, 2020.
REFERENCES
- Andaluz E, Ciudad T, Gomez-Raja J, Calderone R, Larriba G. (2006). Rad52 depletion in Candida albicans triggers both the DNA-damage checkpoint and filamentation accompanied by but independent of expression of hypha-specific genes. Mol Microbiol , 1452–1472. [DOI] [PubMed] [Google Scholar]
- Bacal J, Moriel-Carretero M, Pardo B, Barthe A, Sharma S, Chabes A, Lengronne A, Pasero P. (2018). Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J , e99319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly V, Lauder S, Prakash S, Prakash L. (1997). Yeast DNA repair proteins Rad6 and Rad18 form a heterodimer that has ubiquitin conjugating, DNA binding, and ATP hydrolytic activities. J Biol Chem , 23360–23365. [DOI] [PubMed] [Google Scholar]
- Baroni E, Viscardi V, Cartagena-Lirola H, Lucchini G, Longhese MP. (2004). The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol Cell Biol , 4151–4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berens TJ, Toczyski DP. (2012). Colocalization of Mec1 and Mrc1 is sufficient for Rad53 phosphorylation in vivo. Mol Biol Cell , 1058–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel M, Bach S, Bamps S, Dobbelaere J, Wiget P, Longaretti C, Barral Y, Meijer L, Peter M. (2005). Degradation of Hof1 by SCF(Grr1) is important for actomyosin contraction during cytokinesis in yeast. EMBO J , 1440–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boiteux S, Jinks-Robertson S. (2013). DNA repair mechanisms and the bypass of DNA damage in Saccharomyces cerevisiae. Genetics , 1025–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M. (2006). The Rad53 signal transduction pathway: Replication fork stabilization, DNA repair, and adaptation. Exp Cell Res , 2654–2659. [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. (2002). A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA , 16934–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Donnianni RA, Handa N, Deng SK, Oh J, Timashev LA, Kowalczykowski SC, Symington LS. (2015). Sae2 promotes DNA damage resistance by removing the Mre11-Rad50-Xrs2 complex from DNA and attenuating Rad53 signaling. Proc Natl Acad Sci USA , E1880–E1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde F, Ontoso D, Acosta I, Gallego-Sanchez A, Bueno A, San-Segundo PA. (2010). Regulation of tolerance to DNA alkylating damage by Dot1 and Rad53 in Saccharomyces cerevisiae. DNA Repair (Amst) , 1038–1049. [DOI] [PubMed] [Google Scholar]
- Enserink JM, Smolka MB, Zhou H, Kolodner RD. (2006). Checkpoint proteins control morphogenetic events during DNA replication stress in Saccharomyces cerevisiae. J Cell Biol , 729–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Duan Y, Qin Y, Sun W, Zhuang Z, Zhu D, Jiang L. (2017). The N-terminal pY33XL motif of CaPsy2 is critical for the function of protein phosphatase 4 in CaRad53 deactivation, DNA damage-induced filamentation and virulence in Candida albicans. Int J Med Microbiol , 471–480. [DOI] [PubMed] [Google Scholar]
- Feng J, Zhao Y, Duan Y, Jiang L. (2013). Genetic interactions between protein phosphatases CaPtc2p and CaPph3p in response to genotoxins and rapamycin in Candida albicans. FEMS Yeast Res , 85–96. [DOI] [PubMed] [Google Scholar]
- Fiorani S, Mimun G, Caleca L, Piccini D, Pellicioli A. (2008). Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle , 493–499. [DOI] [PubMed] [Google Scholar]
- Gao XJ, Feng JX, Zhu S, Liu XH, Tardieux I, Liu LX. (2014). Protein phosphatase 2C of Toxoplasma gondii interacts with human SSRP1 and negatively regulates cell apoptosis. Biomed Environ Sci , 883–893. [DOI] [PubMed] [Google Scholar]
- Graziano BR, Yu HY, Alioto SL, Eskin JA, Ydenberg CA, Waterman DP, Garabedian M, Goode BL. (2014). The F-BAR protein Hof1 tunes formin activity to sculpt actin cables during polarized growth. Mol Biol Cell , 1730–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanway D, Chin JK, Xia G, Oshiro G, Winzeler EA, Romesberg FE. (2002). Previously uncharacterized genes in the UV- and MMS-induced DNA damage response in yeast. Proc Natl Acad Sci USA , 10605–10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heideker J, Lis ET, Romesberg FE. (2007). Phosphatases, DNA damage checkpoints and checkpoint deactivation. Cell Cycle , 3058–3064. [DOI] [PubMed] [Google Scholar]
- Hoeijmakers JH. (2009). DNA damage, aging, and cancer. N Engl J Med , 1475–1485. [DOI] [PubMed] [Google Scholar]
- Islam A, Tebbji F, Mallick J, Regan H, Dumeaux V, Omran RP, Whiteway M. (2019). Mms21: a putative SUMO E3 ligase in Candida albicans that negatively regulates invasiveness and filamentation, and is required for the genotoxic and cellular stress response. Genetics , 579–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehnig EJ, Kuo D, Hombauer H, Ideker TG, Kolodner RD. (2013). Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep , 174–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei T, Tanaka K, Hihara T, Umikawa M, Imamura H, Kikyo M, Ozaki K, Takai Y. (1998). Interaction of Bnr1p with a novel Src homology 3 domain-containing Hof1p. Implication in cytokinesis in Saccharomyces cerevisiae. J Biol Chem , 28341–28345. [DOI] [PubMed] [Google Scholar]
- Kim JA, Hicks WM, Li J, Tay SY, Haber JE. (2011). Protein phosphatases pph3, ptc2, and ptc3 play redundant roles in DNA double-strand break repair by homologous recombination. Mol Cell Biol , 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanovic A, Wolfl S. (2006). Fructose-1,6-bisphosphatase mediates cellular responses to DNA damage and aging in Saccharomyces cerevisiae. Mutat Res , 135–147. [DOI] [PubMed] [Google Scholar]
- Kumaran R, Yang SY, Leu JY. (2013). Characterization of chromosome stability in diploid, polyploid and hybrid yeast cells. PLoS One , e68094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Sung P. (2017). Rad52, maestro of inverse strand exchange. Mol Cell , 1–3. [DOI] [PubMed] [Google Scholar]
- Lawrence C. (1994). The RAD6 DNA repair pathway in Saccharomyces cerevisiae: what does it do, and how does it do it? Bioessays , 253–258. [DOI] [PubMed] [Google Scholar]
- Leroy C, Lee SE, Vaze MB, Ochsenbein F, Guerois R, Haber JE, Marsolier-Kergoat MC. (2003). PP2C phosphatases Ptc2 and Ptc3 are required for DNA checkpoint inactivation after a double-strand break. Mol Cell , 827–835. [DOI] [PubMed] [Google Scholar]
- Li WJ, Wang YM, Zheng XD, Shi QM, Zhang TT, Bai C, Li D, Sang JL, Wang Y. (2006). The F-box protein Grr1 regulates the stability of Ccn1, Cln3 and Hof1 and cell morphogenesis in Candida albicans. Mol Microbiol , 212–226. [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH. (2001). Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA , 8276–8282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loll-Krippleber R, d’Enfert C, Feri A, Diogo D, Perin A, Marcet-Houben M, Bougnoux ME, Legrand M. (2014). A study of the DNA damage checkpoint in Candida albicans: uncoupling of the functions of Rad53 in DNA repair, cell cycle regulation and genotoxic stress-induced polarized growth. Mol Microbiol , 452–471. [DOI] [PubMed] [Google Scholar]
- Lommel L, Chen L, Madura K, Sweder K. (2000). The 26S proteasome negatively regulates the level of overall genomic nucleotide excision repair. Nucleic Acids Res , 4839–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes da Rosa J, Boyartchuk VL, Zhu LJ, Kaufman PD. (2010). Histone acetyltransferase Rtt109 is required for Candida albicans pathogenesis. Proc Natl Acad Sci USA , 1594–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundin C, North M, Erixon K, Walters K, Jenssen D, Goldman AS, Helleday T. (2005). Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res , 3799–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson S, Akache B, Weber S, De Deken X, Raymond M, Turcotte B. (2005). Candida albicans zinc cluster protein Upc2p confers resistance to antifungal drugs and is an activator of ergosterol biosynthetic genes. Antimicrob Agents Chemother , 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Boehm ME, Hofmann A, Hub B, Zentgraf H, Lehmann WD, Pereira G. (2011). Phosphorylation-dependent regulation of the F-BAR protein Hof1 during cytokinesis. Genes Dev , 875–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memisoglu A, Samson L. (2000). Base excision repair in yeast and mammals. Mutat Res , 39–51. [DOI] [PubMed] [Google Scholar]
- Min K, Ichikawa Y, Woolford CA, Mitchell AP. (2016). Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere , e00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murad AM, Lee PR, Broadbent ID, Barelle CJ, Brown AJ. (2000). CIp10, an efficient and convenient integrating vector for Candida albicans. Yeast , 325–327. [DOI] [PubMed] [Google Scholar]
- Murakami-Sekimata A, Huang D, Piening BD, Bangur C, Paulovich AG. (2010). The Saccharomyces cerevisiae RAD9, RAD17 and RAD24 genes are required for suppression of mutagenic post-replicative repair during chronic DNA damage. DNA Repair (Amst) , 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB. (2013). DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature , 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill BM, Szyjka SJ, Lis ET, Bailey AO, Yates JR, 3rd, Aparicio OM, Romesberg FE. (2007). Pph3-Psy2 is a phosphatase complex required for Rad53 dephosphorylation and replication fork restart during recovery from DNA damage. Proc Natl Acad Sci USA , 9290–9295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicioli A, Foiani M. (2005). Signal transduction: how rad53 kinase is activated. Curr Biol , R769–R771. [DOI] [PubMed] [Google Scholar]
- Prakash S, Prakash L. (2000). Nucleotide excision repair in yeast. Mutat Res , 13–24. [DOI] [PubMed] [Google Scholar]
- Reed SH, Gillette TG. (2007). Nucleotide excision repair and the ubiquitin proteasome pathway—do all roads lead to Rome? DNA Repair (Amst) , 149–156. [DOI] [PubMed] [Google Scholar]
- Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Sillaots S, Marta C, et al (2003). Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol , 167–181. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ. (1996). Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science , 357–360. [DOI] [PubMed] [Google Scholar]
- Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. (2002). Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell , 1055–1065. [DOI] [PubMed] [Google Scholar]
- Shi QM, Wang YM, Zheng XD, Lee RT, Wang Y. (2007). Critical role of DNA checkpoints in mediating genotoxic-stress-induced filamentous growth in Candida albicans. Mol Biol Cell , 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Cortez D. (2013). DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol , a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolka MB, Chen SH, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, Desai A, Kolodner RD, Zhou H. (2006). An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J Cell Biol , 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasagara RR, Spencer SM, Tripathi K, Clark DW, Mani C, Madeira da Silva L, Scalici J, Kothayer H, Westwell AD, Rocconi RP, et al (2017). RAD6 promotes DNA repair and stem cell signaling in ovarian cancer and is a promising therapeutic target to prevent and treat acquired chemoresistance. Oncogene , 6680–6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LL, Li WJ, Wang HT, Chen J, Deng P, Wang Y, Sang JL. (2011). Protein phosphatase Pph3 and its regulatory subunit Psy2 regulate Rad53 dephosphorylation and cell morphogenesis during recovery from DNA damage in Candida albicans. Eukaryot Cell , 1565–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson JP, Pesudo LQ, Fry RC, Adeleye YA, Carmichael P, Samson LD. (2011). Genomic phenotyping of the essential and non-essential yeast genome detects novel pathways for alkylation resistance. BMC Syst Biol , 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. (2005). Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol , 1364–1375. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Yong-Gonzalez V, Kikuchi Y, Strunnikov A. (2006). SIZ1/SIZ2 control of chromosome transmission fidelity is mediated by the sumoylation of topoisomerase II. Genetics , 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallen EA, Caviston J, Bi E. (2000). Roles of Hof1p, Bni1p, Bnr1p, and myo1p in cytokinesis in Saccharomyces cerevisiae. Mol Biol Cell , 593–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas VK, Barrasa MI, Fink GR. (2015). A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv , e1500248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Nishihama R, Onishi M, Pringle JR. (2018). Role of the Hof1-Cyk3 interaction in cleavage-furrow ingression and primary-septum formation during yeast cytokinesis. Mol Biol Cell , 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert TA, Kiser GL, Hartwell LH. (1994). Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev , 652–665. [DOI] [PubMed] [Google Scholar]
- Whiteway M, Bachewich C. (2007). Morphogenesis in Candida albicans. Annu Rev Microbiol , 529–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RD, Mitchell M, Sgouros J, Lindahl T. (2001). Human DNA repair genes. Science , 1284–1289. [DOI] [PubMed] [Google Scholar]
- Yan L, Xiong J, Lu H, Lv QZ, Ma QY, Cote P, Whiteway M, Jiang YY. (2015). The Role of Mms22p in DNA damage response in Candida albicans. G3 (Bethesda) , 2567–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G, Wan J, Liu Q, Mu C, Wang Y, Sang J. (2017). Characterization of Pph3-mediated dephosphorylation of Rad53 during methyl methanesulfonate-induced DNA damage repair in Candida albicans. Biochem J , 1293–1306. [DOI] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ. (2000). The DNA damage response: putting checkpoints in perspective. Nature , 433–439. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Humphryes N, van Eijk P, Waters R, Yu S, Kraehenbuehl R, Hartsuiker E, Reed SH. (2015). UV induced ubiquitination of the yeast Rad4-Rad23 complex promotes survival by regulating cellular dNTP pools. Nucleic Acids Res , 7360–7370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.