

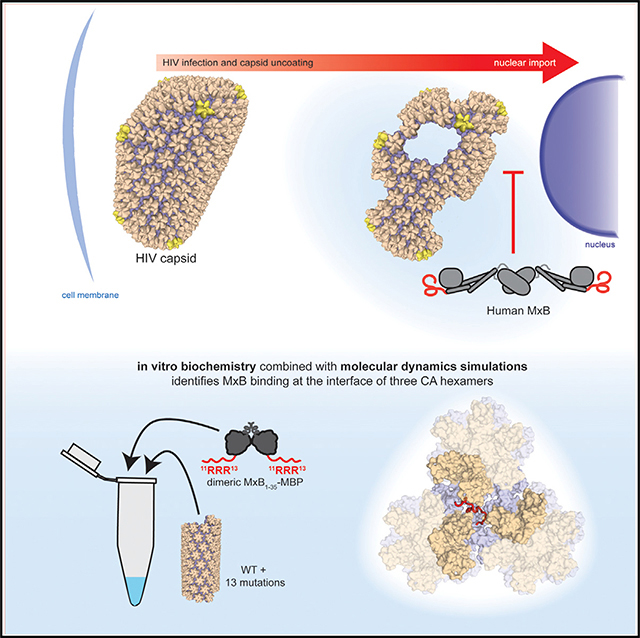
SUMMARY
The human antiviral protein MxB is a restriction factor that fights HIV infection. Previous experiments have demonstrated that MxB targets the HIV capsid, a protein shell that protects the viral genome. To make the conical-shaped capsid, HIV CA proteins are organized into a lattice composed of hexamer and pentamer building blocks, providing many interfaces for host proteins to recognize. Through extensive biochemical and biophysical studies and molecular dynamics simulations, we show that MxB is targeting the HIV capsid by recognizing the region created at the intersection of three CA hexamers. We are further able to map this interaction to a few CA residues, located in a negatively charged well at the interface between the three CA hexamers. This work provides detailed residue-level mapping of the targeted capsid interface and how MxB interacts. This information could inspire the development of capsid-targeting therapies for HIV.
In Brief
In this paper Smaga et al. determine that the restriction factor MxB binds to the HIV capsid at regions where three CA hexamers meet. This work reports residue-level information about the MxB-CA interaction, providing important insights about how MxB senses the HIV capsid lattice.
Graphical Abstract
INTRODUCTION
The myxovirus resistance (Mx) proteins are dynamin-like GTPases with antiviral activity. The two Mx proteins found in humans, MxA and MxB, originated from an ancient gene duplication. MxA is a well-characterized restriction factor of influenza and a wide range of other viruses (reviewed in Haller et al., 2015). MxB was recently identified as an interferon-inducible restriction factor of HIV-1 and herpesviruses, as well as murine cytomegaloviruses (MCMVs) (Crameri et al., 2018; Goujon et al., 2013; Jaguva Vasudevan et al., 2018; Kane et al., 2013; Liu et al., 2013; Schilling et al., 2018). In cells, MxB localizes to the nuclear periphery, potentially near nuclear pores (Busnadiego et al., 2014; King et al., 2004). MxB appears to restrict HIV approximately 10-fold, and block infection between the steps of reverse transcription and integration (Busnadiego et al., 2014; Goujon et al., 2013; Kane et al., 2013; Liu et al., 2013).
MxB is similar to MxA in both sequence and structure but with different viral restriction activities. Each of the Mx proteins is composed of three conserved domains: the stalk domain, the GTPase domain and the bundling signaling element (BSE) domain. The stalk domain is responsible for the oligomerization of Mx proteins. In MxA (and MxB restriction of herpesviruses and MCMVs), the GTPase domain is essential for restriction activity (Crameri et al., 2018; Goujon et al., 2013; Jaguva Vasudevan et al., 2018; Kane et al., 2013; Schilling et al., 2018). The BSE transmits information between domains for viral inhibition (Gao et al., 2011). The atomic resolution structures of MxA and MxB (Fribourgh et al., 2014; Gao et al., 2011; Xu et al., 2015) have shed light on the overall architecture of Mx proteins. The structure of individual monomers is very similar, but substantial differences occur in the higher-order dimerization and oligomerization of MxA and MxB (Fribourgh et al., 2014; Gao et al., 2011) Furthermore, MxB contains an N-terminal tail (43 amino acids) that is absent in MxA, and this N terminus is missing or disordered in the available MxB structures. The unstructured N terminus of MxB is essential for its restriction activity. Deleting the N-terminal region of MxB, or replacing it with the shorter N terminus of MxA, abolishes MxB’s restriction activity against HIV-1 (Fribourgh et al., 2014; Fricke et al., 2014; Goujon et al., 2015; Schulte et al., 2015). Importantly, the restriction activity of MxB can be abolished by mutating a triple arginine motif 11RRR13 to 11AAA13 (Goujon et al., 2015).
The HIV-1 restriction activity of MxB is correlated to its ability to bind the viral capsid. It has been reported that certain mutations in the HIV-1 capsid protein (CA) reduce the potency of MxB restriction, suggesting that capsid plays a role in the restriction activity of MxB (Goujon et al., 2013; Kane et al., 2013; Liu et al., 2013; Matreyek et al., 2014; Wei et al., 2016). We and others observed that MxB binds to capsid in vitro by using helical assemblies of purified CA and in cells by using the viral capsid core (Fribourgh et al., 2014; Fricke et al., 2014; Kong et al., 2014; Wei et al., 2016). However, MxB does not bind to the individual capsid building blocks: CA dimers, hexamers, or pentamers (Fribourgh et al., 2014; Fricke et al., 2014; Kong et al., 2014). This suggests that MxB is recognizing a feature present only in the higher-order CA lattice on capsid, potentially at an interface between hexamer building blocks. Further evidence to support this hypothesis was provided in an in vitro evolution experiment that found two capsid mutations located in the interface between three capsid hexamers, G208R and T210K, that escaped MxB restriction. Other mutations, including P207S, M10I, P90T, and M185L escaped MxB restriction to lesser degrees (Busnadiego et al., 2014). However, the direct role of these residues in CA-MxB interactions is not clear. These experiments were performed by passaging the virus in cell culture, where a capsid mutation that generates a defect in assembly, stability, or interaction with cofactors could affect viral sensitivity to MxB indirectly.
In the present work, we describe how MxB recognizes the HIV capsid by defining the MxB-CA-binding interface. Using capsid co-pelleting assays with highly pure MxB and CA components, we performed comprehensive mutagenesis screening of capsid residues that potentially interact with MxB. Our data suggest that MxB recognizes the interface between three capsid hexamers, particularly through interactions between its basic 11RRR13 motif and acidic CA residues. Our crystal structures show that the MxB-resistant mutations at the tri-hexamer interface do not affect the structures of CA hexamers or the capsid lattice, although they do reduce MxB binding. In addition, results from long timescale molecular dynamics (MD) simulations demonstrate binding of MxB to the tri-hexamer interface of higher-order capsid assemblies. Furthermore, our simulations establish key residues in CA that mediate interactions with MxB’s N-terminal domain and are in agreement with the biochemical and biophysical experiments.
RESULTS
The N Terminus of MxB Binds Directly to CA
We produced a minimal MxB construct with good solution behavior and containing the established capsid-binding motif. Previous in vitro studies of MxB binding were limited by low yield and poor behavior of full-length MxB protein. Inspired by reports that the N terminus of MxB dictates restriction and includes the positively charged RRR residues that potentially target capsid (Fribourgh et al., 2014; Goujon et al., 2014, 2015; Kong et al., 2014), we focused our study on the N-terminal 35 residues of MxB. To mimic the extended dimeric architecture of the native protein, which has the two N termini located at the opposite ends, we fused the N-terminal 35 residues of MxB (MxB1–35) to maltose binding protein (MBPs). The MxB1–35-MBP fusion is further linked to a GCN4 dimerization helix to enforce a dimeric geometry (Figure 1A). The MBP acts as a spacer, creating a separation between the two MxB N termini, and at the same time serves as a solubility enhancer. This construct, MxB1–35-MBPdi, provides a similar geometry to the extended MxB dimer, while allowing us to focus our study on the interactions of its N terminus without the solution behavior complications of full-length MxB protein.
Figure 1. An MxB-MBP Dimer Construct Binds CA Helical Tubes In Vitro.

(A) Construct schematic of MxB1–35-MBPdi.
(B) Cartoon model of MxB1–35-MBPdi (left), which mimics the extended dimer architecture of the full-length MxB protein (right).
(C) Top: diagram of the co-pelleting assay. Purified CA tubes and MxB constructs are incubated (“total”); after centrifugation, “soluble” and “pellet” fractions are analyzed. Bottom: MxB1–35-MBPdi binds CA tubes. MxB1–35AAA-MBPdi, in which the RRR CA-binding motif is mutated to AAA binds significantly less. MBP and MBP-CCCyp are used as negative and positive controls, respectively. Full gels can be found in Figure S1.
(D) MxB1–35-MBPdi binding to CA tubes is salt dependent and is abolished at above 300 mM NaCl. Full gels can be found in Figure S1.
(E) Quantification of the co-pelleting assay in (D). The fraction of MxB bound was computed for each sample by subtracting the soluble from the total and normalized to the total. Error bars represent the standard error of the mean from three independent experiments. Full gels for each replicate can be found in Figure S1.
The MxB1–35-MBPdi construct binds directly to CA tubes in vitro in a co-pelleting assay, where a soluble capsid cofactor is found in the pellet only on binding to insoluble polymerized CA (Henning et al., 2014; Matreyek et al., 2013; Stremlau et al., 2006). We chose to use A14C/E45C disulfide-crosslinked CA tubes in our assay, which mimic the lattice seen in the capsid and tolerate a range of experimental conditions (Pornillos et al., 2009). To quantify MxB binding, we compared the reduction of MxB in the soluble fraction, instead of the amount pelleted, relative to the total input. This approach produces more accurate quantification than attempting to measure the MxB bound to the washed or unwashed CA pellet. The results show that over 90% of MxB1–35-MBPdi co-pellets with CA at 3 μM MxB concentration, indicating efficient binding under the experimental conditions and validating our MxB construct, which may be useful in future studies of MxB-capsid interactions (Figure 1B). The negative control, MBP alone, has a background level of less than 10% binding. As expected, MxB1–35AAA-MBPdi, in which the triple arginine motif known to bind CA is mutated to 11AAA13, showed much weaker binding than wild type (WT) at 20%. This further validates the construct and the residual binding may indicate that additional residues outside of 11RRR13 play a minor role in capsid interaction.
The binding of MxB1–35-MBPdi to CA tubes decreases on increasing salt concentration, suggesting that the interaction is driven by electrostatic interactions. At 75 mM NaCl, over 90% of MxB1–35-MBPdi binds to CA tubes (Figures 1C and 1D). At the near-physiological salt concentration of 150 mM NaCl, MxB1–35-MBPdi retains around 80% of binding. However, at above 300 mM NaCl, the binding of MxB1–35-MBPdi is reduced to the ~20% residual level of binding observed in MxB1–35AAA-MBPdi. The low-level binding of MxB1–35AAA-MBPdi is consistent within error across concentrations of NaCl, suggesting that any remaining interactions may not be charge-driven. To investigate the intrinsic interaction property and more easily distinguish potential subtle mutagenesis effects, we performed subsequent assays at 75 mM NaCl, where binding is at its maximum.
MxB Interacts with CA at the Interface between Three Hexamers
We predicted that the positively charged 11RRR13 motif of MxB would interact with negatively charged regions of the CA lattice surface based on its charge character and our observation that MxB binding to CA tubes is salt dependent. We identified three regions of negative charge on the CA lattice from its calculated surface electrostatic potential distribution (Figure 2A). The residues that contribute to these negatively charged regions include: E71, E75, E212, and E213 at the tri-hexamer interface, E180 and E187 at the di-hexamer interface, and E98 in the CypA-binding loop (Figure 2B). Furthermore, based on the previous observation that individual CA hexamers are not sufficient to confer MxB binding, we posit that the tri-hexamer or di-hexamer interface residues are important for MxB binding. To test these hypotheses, we mutated each of these glutamates, as well as residues in the CypA-binding loop, to alanine and tested their ability to interact with MxB1–35-MBPdi in our co-pelleting assay. Our binding results pinpoint the MxB-interacting site at the tri-hexamer interface.
Figure 2. The MxB Binding Site on Capsid Maps to the Tri-hexamer Interface.

(A) Surface electrostatic potential distribution of three CA hexamers (PDB: 4XFX) shows the negative charge at the interfaces between the hexamers (major), and in the CypA-binding loop (minor). A cartoon (bottom) shows the overall arrangement of hexamers and the symmetry axes at the di-hexamer (cat eyes) and tri-hexamer (triangle) interfaces.
(B) Location of CA residues selected for mutation. CA hexamers (surface) are shown in light blue, light yellow, and light green. Residues selected for mutation are shown in red sticks.
(C) MxB1–35-MBPdi binding to CA tube variants in co-pelleting assays. The total, soluble, and pellet fractions were analyzed by SDS-PAGE. For simplicity, only the MxB1–35-MBPdi band is shown. MxB1–35AAA-MBPdi is used as a negative control. A notable reduction in MxB binding is observed for CAE213A, CAE71A, CAE75A, and CA212/213 EE->AA. Positive and negative controls performed with MBP-CCCyp and MBP, respectively, can be found in Figure S3. Full gels can be found in Figure S2.
(D) Quantification of data in (C), performed as in Figure 1. Error bars represent the standard error of the mean from three independent experiments. Full gels can be found in Figure S1 (one replicate of “No CA” and “CA”), Figure S2, and Figure S3. Quantification of controls can be found in Figure S3.
Mutations in tri-hexamer interface residues had the greatest effect on MxB binding. This region of the CA lattice contains the densest concentration of negative charge, with 12 glutamate residues (three copies of E71, E75, E212, and E213) creating a deep well of negative charge. In the tri-hexamer interface, three sets of CA CTD residues E212 and E213 from three adjacent symmetry-related monomers form the floor of this negatively charged well. MxB binding to CAE213A was significantly reduced (~40% of WT), and CAE212A/E213A drastically reduced binding to the background level (~15% of WT) (Figures 2C and 2D). The CA NTD residues E71 and E75 form the wall of the negatively charged well and mutations at these sites (CAE71A and CAE75A) also substantially reduced (~60% of WT) MxB binding (Figures 2C and 2D).
Mutation of residues in the di-hexamer interface has a mild effect on MxB binding. This interface is present in the WT CA dimer, which showed no binding to MxB in solution in previous experiments (Fribourgh et al., 2014). We identified glutamates E180 and E187 on helix 9 at the CTD-CTD dimerization interface as responsible for the patch of negative charge between two CA hexamers. Both CAE180A and CAE187A modestly affected MxB binding (~73%–83% of WT) (Figures 2C and 2D). This suggests they do not contribute substantially to the MxB interaction.
In contrast to the effect from perturbations at the hexamer interfaces, mutation of residues around the CypA-binding loop on the surface of the CA hexamer had no effect on MxB binding. The most prominent region of negative charge on the hexamer surface is contributed by residue E98 near the CypA-binding loop, which is not near any lattice interfaces. Consistent with our interface-binding hypothesis, MxB1–35-MBPdi bound CAE98A tubes at a level similar to CA tubes (Figures 2C and 2D). We also examined the interactions of MxB1–35-MBPdi with CA tubes having mutations in the CypA-binding loop. Since the G89V and P90A mutations have been reported to affect MxB restriction in vivo and G89V has been reported to reduce MxB co-pelleting of MxB in cell lysates with CANC tubes (Fricke et al., 2014; Goujon et al., 2013; Kane et al., 2013), we tested these two mutations in our in vitro system using purified MxB and CA components. Our previous work demonstrated that G89V had no effect on full-length MxB pelleting (Fribourgh et al., 2014). Using our minimal MxB-MBPdi construct, we confirmed that neither CAG89V nor CAP90A reduce the binding (Figures 2C and 2D).
Tri-hexamer Interface Mutations that Escape MxB Restriction Also Abolish MxB1–35-MBPdi Binding
Three mutations at the tri-hexamer interface (P207S, G208R, and T210K) (Figure 4A) were previously identified in an in vitro evolution experiment to escape MxB restriction to varying degrees (Busnadiego et al., 2014). The mutation P207S is relatively conservative, while both G208R and T210K introduce bulky, positively charged residues to the predominantly negatively charged environment described above (Figures 2A and 3A). Based on our interaction mapping results, we predicted that both G208R and T210K, which drastically alter the electrostatic environment of the tri-hexamer interface, would decrease MxB binding to the capsid. To test this hypothesis, we introduced these mutations into CA tubes and performed co-pelleting assays with MxB1–35-MBPdi.
Figure 4. All-Atom Molecular Simulations Identify MxB Interactions at the Tri-hexamer Interface.

(A) The all-atom model of MxB1–35 at the tri-hexamer interface. Three dimers from different hexamers are in blue, yellow, and green (left). The MxB1–35 peptide in red is placed above the middle of the tri-hexamer region before simulations (right).
(B) The dynamics of MxB1–35 in tri-hexamer interface. The evolution of MxB secondary structure (top) during the simulation, computed by the STRIDE program in VMD and the root-mean-square deviation (RMSD) plot of the MxB Cα atoms (bottom).
(C) MxB1–35 (red) binds in the tri-hexamer interface. The MxB 11RRR13 motif is shown in magenta sticks.
(D) Molecular contacts between the MxB 11RRR13 motif and experimentally tested CA residues are shown for R11 (left), R12 (middle), and R13 (right), with corresponding contact occupancies.
(E) In a second simulation, the MxB peptide was initially placed at the interface between two CA hexamers (blue/green). The di- and tri-hexamer interfaces are marked by an oval and a triangle, respectively.
(F) Still images from the second simulation at the beginning and after 15 μs reveal that the MxB peptide again migrates to the tri-hexamer interface. The CA-binding 11RRR13 motif is shown in red sticks (right).
See also, Figure S5.
Figure 3. CA Mutations that Escape MxB Restriction Severely Reduce MxB1–35-MBPdi Binding.

(A) P207, G208, and T210 (red sticks) are located at the interface between three CA hexamers (light blue, light green, and light yellow surface).
(B) MxB1–35-MBPdi binding to CA tubes and CA tubes containing the mutations P207S, G208R in co-pelleting assays. The total, soluble, and pellet fractions were analyzed by SDS-PAGE. For simplicity, only the MxB1–35-MBPdi band is shown. Full gels and positive and negative controls performed with MBP-CCCyp and MBP, respectively, are shown in Figure S4.
(C) Quantification of data in (B), performed as in Figure 1. Error bars represent the standard error of the mean from three independent experiments. Quantification of the positive and negative controls can be found in Figure S4.
(D) Alignment of P207S (red), G208R (blue), and T210K (green) crystal structures to the structure of WTCA (white, PDB: 4XFX) at the tri-hexamer interface (top) and within a monomer (bottom). All of the mutations preserve the wild-type lattice structure, with only minor differences.
The CA mutations G208R and T210K drastically reduced MxB binding (Figure 3B). MxB1–35-MBPdi binding to CAG208R was indistinguishable from the MxB1–35-AAA-MBPdi background signal. Binding to CAT210K was also substantially reduced. By neutralizing the strong negative charge at the tri-hexamer interface, these mutations likely disrupt charge-charge interactions that are required for MxB binding. It is also possible that the bulkiness of these residues occludes the well at the tri-hexamer interface and sterically blocks MxB access. The relatively modest mutation P207S had a minor effect on MxB interaction (Figure 3B). These data lend strong support to our mapping results that identify the MxB targeting site to the tri-hexamer interface on capsid.
Hexamer Interface Mutations that Affect MxB Interaction Do Not Change the Conformation of CA Lattice
We validated that the reduction in MxB binding observed for the hexamer interface mutations was because of a direct disruption in the MxB-capsid interface and not from indirect changes in the CA lattice by examining the structure. Large conformational changes in the overall CA lattice could potentially lead to the loss of MxB binding at other sites. We first validated the overall morphology of the CA mutants tested by examining crosslinked CA tubes with negative stain electron microscopy. Morphologically, the CA mutant tubes appear similar to crosslinked tubes without mutation (Figures S2 and S4). The CA tubes mimic the lattice seen in the capsid (Pornillos et al., 2009). Interestingly, the E180A mutant produced more cone-shaped structures reminiscent of the viral capsid. In any case, no obvious defects in CA assembly were observed for the mutants. We further determined the crystal structures of those CA mutants that affect MxB restriction—P207S, G208R, and T210K—at resolutions ranging from 3.0 to 3.3 Å (Table 1). These mutants crystalized in the same crystal form as that of the WT CA, where the crystal packing recapitulates the CA lattice on the viral capsid (Gres et al., 2015). The crystal structures of the mutants closely align to the structure of WT CA, both at the individual CA monomer level (root-mean-square deviation [RMSD] = 0.2–0.3 Å) and lattice level (RMSD = 0.3–1.2 Å) for the six CA molecules surrounding 3-fold axis between three hexamers (Figures 3D and 3E). These results are consistent with the idea that mutations at the tri-hexamer interface directly disrupt MxB binding.
Table 1.
Crystal Structures of CA Mutants P207S, G208R, T210K
| P207S | G208R | T210K | |
|---|---|---|---|
| PDB | 6MQA | 6MQO | 6MQP |
| Data Collection | |||
| Space group | P6 | P6 | P6 |
| Cell dimensions | |||
| a, b, c (Å) | 90.34, 90.34, 57.62 | 92.01, 92.01, 57.19 | 92.04, 92.04, 57.35 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 |
| Molecules/ASU | 1 | 1 | 1 |
| Resolution (Å) | 3.20 (3.39–3.20) | 3.00 (3.18–3.00) | 3.30 (3.49–3.30) |
| Rmerge | 12.8 | 10.4 | 13.9 |
| I/σ | 6.6 (1.4) | 10.7 (2.7) | 7.6 (1.7) |
| Completeness (%) | 98.9 (99.1) | 98.3 (97.3) | 99.3 (99.4) |
| Redundancy | 2.6 | 3.1 | 3.5 |
| Unique reflections | 8,591 (1,377) | 8,809 (1,407) | 8,116 (1,320) |
| Refinement | |||
| No. of non-hydrogen atoms | 1,666 | 1,693 | 1,644 |
| Rwork/Rfree (%) | 18.6/21.9 | 20.6/27.1 | 20.7/24.0 |
| Average B factor | 73.6 | 76.0 | 87.6 |
| Root-mean-square deviation | |||
| Bond lengths (Å) | 0.005 | 0.017 | 0.01 |
| Bond angles (°) | 1.0 | 1.8 | 1.1 |
| Ramachandran analysis (%) | |||
| Preferred regions | 97.14 | 97.64 | 98.11 |
| Allowed regions | 2.86 | 2.36 | 1.42 |
| Outliers | 0 | 0 | 0.47 |
Data collection and refinement statistics for CAP207S, CAG208R, CAT210K. Values shown in parentheses are for highest-resolution shell.
During purification, we observe that CAG208R and CAT210K formed hyperstable CA assemblies. These proteins have substantially lower solubility and visibly polymerize on concentration and freeze/thaw. We believe this is due to the introduction of positive charge at a negatively charged region of the CA lattice, resulting in charge neutralization that increases the stability of the CA lattice. In WT CA, it is likely that ideal lattice stability is maintained through balanced repulsion of negative charges at this interface. However, in the G208R and T210K mutants, the total negative charge at this interface is reduced, resulting in less lattice destabilizing repulsion force. A hyperstable capsid deviating from a fine balance could have deleterious effects for the virus, which may explain why these mutations are rarely seen in patient samples (De Luca et al., 2017).
MD Simulations Identify the Tri-hexamer Interface as the Site of MxB-CA Interaction
To investigate how MxB targets CA at an atomistic level, we constructed a CA tri-hexamer interface model and placed the 35 N-terminal residues of MxB (MxB1–35) in close proximity (Figure 4A). The model is composed of three dimers from adjacent CA hexamers, a structural motif commonly referred to as a trimer-of-dimers. Subsequently, the model was subjected to 30 ms of all-atom MD simulation without any experimental restraints (Video S1). The canonical MD simulations permit MxB’s N terminus and CA trimer-of-dimers to sample several physical conformations.
Results from our MD analysis show that MxB1–35 binds persistently in the tri-hexamer region throughout the entirety of the simulation, with extensive molecular contacts between MxB N-terminal region and CA residues (Table 2). Importantly, CA residues in the tri-hexamer interface, E71, E75, and E213, interact with the 11RRR13 motif of MxB with high occupancies (Figure 4D; Table 2), which is in good agreement with our co-pelleting assay data (Figures 2C and 2D). In fact, there is a remarkable agreement between the MD results with the experimental binding data obtained independently. For example, contacts between E212 and the MxB N terminus were observed less frequently, which is consistent with the experimental finding that the E212A mutation has a smaller disruption of MxB binding than other glutamate mutations do at the tri-hexamer interface (Figure 2D). Moreover, we observe CA residues T210, G208, and P207 interacting with the 11RRR13 motif of MxB, which accounts for the importance of these sites in MxB binding (Figure 3B) and sensitivity (Busnadiego et al., 2014). In addition, we found contacts between CA and residues outside of the 11RRR13 motif (Table 2; Figure S5), for example, the cation-π interaction between CA R82 and MxB W8 residue, which may contribute to the residual level of binding observed with MxB1–35AAA-MBPdi.
Table 2.
Top 20 MxB-CA Contacts from 30 μs All-Atom MD Simulation
| No. | MxB Residue | CA Residue | Occupancy (%) |
|---|---|---|---|
| 1 | ARG12 | GLU213 | 95.2 |
| 2 | TRP8 | ARG82 | 94.1 |
| 3 | ARG11 | THR210 | 91.1 |
| 4 | TYR10 | GLU75 | 90.6 |
| 5 | ARG12 | PRO207 | 88.8 |
| 6 | ARG12 | LEU205 | 87.4 |
| 7 | ARG12 | GLY208 | 84.5 |
| 8 | TYR10 | ALA78 | 83.9 |
| 9 | ARG11 | GLY208 | 74.8 |
| 10 | ARG11 | GLU71 | 68.8 |
| 11 | LYS6 | THR210 | 68.5 |
| 12 | ARG13 | GLU213 | 66.9 |
| 13 | ARG13 | THR210 | 65.2 |
| 14 | LYS6 | GLU75 | 63.5 |
| 15 | ARG11 | PRO207 | 61.4 |
| 16 | ARG11 | GLU75 | 60.1 |
| 17 | PRO9 | GLY208 | 59.6 |
| 18 | ARG12 | ALA209 | 59.6 |
| 19 | PRO7 | THR210 | 56.8 |
| 20 | HIS5 | ALA78 | 53.0 |
MxB and CA residues that were experimentally mutated are shown in italic.
The MxB-binding pocket revealed by the simulation is primarily formed by key CA residues located in adjacent CA hexamers (Table 2; Figures 4D and S5). The MxB 11RRR13 motif is anchored to the bottom of the pocket, primarily interacting with the negatively charged CA E213 residues, while the rest of the MxB1–35 peptide exhibits significantly higher flexibility (Figure 4D; Video S1). This observation is supported by the experimental finding that the E213A mutation shows the largest effect on MxB binding (Figure 2D). Notably, the significant difference in effect between E212A and E213A on MxB binding is consistent experimentally and computationally. This is likely due to their relative positions at the interface: E213 is closer to the 3-fold symmetry axis (Figure 2B) and other E213 residues from adjacent hexamers, such that MxB can bind E213 from multiple CA monomers simultaneously (Figure 4D). Bioinformatics tools pre dict the MxB N-terminal region to be disordered by itself, and it is either disordered or not present in the available 3D structures of MxB (Alvarez et al., 2017; Fribourgh et al., 2014). Our secondary structure analysis (Figure 4B) shows that, while the bound MxB1–35 still contains large disordered areas, the region near 11RRR13 motif maintains a stable conformation during the simulation (Video S1), which further supports the importance of the 11RRR13 motif in MxB-capsid interaction.
To further confirm that the initial placement of MxB does not bias the simulation result, we conducted a second simulation by placing MxB directly above the di-hexamer interface away from the predicted binding site (Figure 4E) and still captured MxB binding to the tri-hexamer interface (Figure 4F). In this simulation, MxB1–35 was initially placed above the di-hexamer interface so that the 11RRR13 motif was positioned above residues E180 and E187, whose mutations did not affect MxB binding substantially in our biochemical assays. The MxB peptide diffuses away from the di-hexamer interface during the first 200 ns of the 15-μs simulation. Subsequently, MxB’s 11RRR13 moved toward the tri-hexamer interface and formed an interaction with E213, observed for 7 μs of the simulation (Figure S5D; Video S2). The MxB peptide demonstrated more persistent contacts with residues in the tri-hexamer interface, similar to that obtained in our first simulation, and resided there for a longer period of time than at the di-hexamer interface, further supporting that the tri-hexamer interface is the preferred MxB binding site.
In addition, the MD simulation provides insight into the structure of the MxB1–35 on binding to capsid and its influence on the ionic environment. The simulation shows that binding of MxB1–35 in the tri-hexamer interface decreases the sodium ion occupancies in the CA lattice. Based on the trajectory analysis, when the MxB N-terminal region occupies one of the three grooves between hexamers at the tri-hexamer interface, the calculated occupancy of sodium ions in the groove proximal to the bound region are lower than corresponding regions in the other two unoccupied grooves (Figure S5). On the other hand, the chloride ion occupancy does not show an appreciable difference in three grooves. Together with the high-occupancy molecular contacts between the positively charged 11RRR13 and the CA glutamates in the tri-hexamer region, these data agree with the experimental observation that MxB binding is salt dependent, the interaction may lead to displacement of sodium ions at both the binding interface and nearby regions.
DISCUSSION
MxB is a potent restriction factor of HIV-1 infection. It restricts the nuclear import step of the viral life cycle. The inhibition requires an interaction with the HIV-1 capsid. Previous studies suggested that MxB recognized features present only in assembled capsid: interfaces between CA hexamers forming a lattice. Despite extensive work to identify CA mutations which affect MxB activity (Busnadiego et al., 2014; Fribourgh et al., 2014; Fricke et al., 2014; Goujon et al., 2013; Kane et al., 2013; Liu et al., 2013, 2015; Matreyek et al., 2014), the interaction site and mechanism of interaction remain unclear. While cellular assays are efficient ways to identify mutations, they often fall short of disentangling the pleomorphic effects of CA mutations, where a functional defect in MxB sensitivity may not be directly related to MxB binding to capsid. Instead, it could be due to altered capsid stability or affecting other cellular factors that are also important in the MxB restriction pathway. Our biochemical and biophysical experimental system provides a stringent test to identify and verify mutations that have a direct effect on the MxB-capsid interaction. Combined with all-atom MD simulations, our results demonstrate the molecular basis of how capsid mutations evade MxB restriction and provide insight into how MxB recognizes the capsid (Figure 5A).
Figure 5. MxB Recognizes CA Features that Are Potentially Conserved in Lentiviruses but not in Retroviruses.

(A) Model of MxB binding to the CA lattice, generated using existing structural data of the MxB dimer (gray, PDB: 4WHJ) and HIV CA (light gray/tan, PDB: 4XFX) and MD simulation data of the MxB N-terminal peptide binding to CA (red).
(B) Alignment of CA structures available from different lentiviruses: HIV (PDB: 4XFX, red), equine infectious anemia virus (EIAV) (PDB: 2EIA, blue), and feline immunodeficiency virus (FIV) (PDB: 5NA2, yellow); residue E213 shown in sticks located in the same position in each CA.
(C) CA protein sequence alignment of lentiviruses. Gray box denotes primate lentiviruses, which are restricted by MxB. Residue E213 and its homologous residues (red) are conserved in the lentiviral family.
(D) Surface electrostatic potential distributions of diverse retroviral capsids show few shared characteristics at the tri-hexamer interface. The following structures are used in comparison: HIV-1 (PDB: 4XFX), Rous sarcoma virus (RSV) (PDB: 4TIR), and bovine leukemia virus (BLV) (PDB: 4PH0).
Our results paint a clear picture of MxB targeting the HIV capsid at the interface between CA hexamers. To map the site of the MxB-capsid interaction, we performed systematic alanine scanning guided by an analysis of the electrostatic surface of the CA lattice, as well as previously reported MxB escape mutations (Braaten et al., 1997; Busnadiego et al., 2014; Franke et al., 1994). Mutations of the negatively charged residues surrounding the 3-fold axis on capsid, including E71/E75/E212/E213, had a substantial deleterious effect on MxB interaction. These residues line a negatively charge well at the tri-hexamer interface, which is likely the binding site for the positively charged 11RRR13 motif in MxB that is critical for capsid interaction and HIV-1 restriction (Goujon et al., 2015). Consistently, we found that the tri-hexamer interface mutations that escape MxB restriction, G208R and T210K, also significantly disrupted the binding of MxB, likely due to a combined result of steric clash and charge repulsion with the 11RRR13 motif. In contrast, mutations in the di-hexamer interface and around the CypA-binding loop on the hexamer surface had little effect on MxB binding. Our structural studies showed that these mutations had no apparent effect on the structure of CA lattice assemblies, supporting that the mutagenesis results revealed direct binding of MxB at the tri-hexamer interface.
Though our mutant CA tubes are similar in morphology to that of WT CA, many of these mutations have deleterious effects in the virus, which may explain why MxB escape mutations are not commonly seen. Notably, it has been reported that the mutation E213A leads to decreased infectivity in cellular assays, and while Gag processing and viral cores appear normal, the core remains stable longer than WT on entry into the target cell (Byeon et al., 2009). This is consistent with our observation during protein purification that E213A appeared to produce more stable CA assemblies and prone to precipitation. Viruses containing the CA mutations E71A and E180A had reduced infectivity relative to WT (von Schwedler et al., 2003; Yufenyuy and Aiken, 2013). The latter suggests that these two mutations are not tolerated well by the virus, and perhaps coping with MxB restriction is a lower-cost measure than evolving resistance.
Previous reports have suggested that residues in the CypA loop play an important role in MxB restriction. In this paper, we are able to directly test the effect of the G89V and P90A mutations in a simplified system containing only HIV CA and MxB1–35. We observed that neither mutation has any effect on the direct MxB-CA interaction, in agreement with previous in vitro biochemical studies (Fribourgh et al., 2014; Fricke et al., 2014). However, HIV with CA G89V or P90A (or in one case, P90T) mutation is insensitive or less sensitive to MxB in cellular studies (Busnadiego et al., 2014; Goujon et al., 2013; Kane et al., 2013; Matreyek et al., 2014). These CypA loop mutations may escape MxB restriction in cells through their effect on cyclophilin A, which has been shown to play a role in MxB restriction (Kane et al., 2018; Liu et al., 2013). Further work is necessary to untangle the precise function of CypA and other factors in MxB restriction.
Our all-atom MD simulations identified the same important tri-hexamer interface independent of the experimental biochemistry. In the simulation, the bound MxB peptide interacts with many CA residues that were tested biochemically to be critical for MxB interaction (E71, E75, E212, and E213) and shown to be functionally important for MxB sensitivity (P207, G208, and T210). The extensive contacts between the MxB peptide and CA residues illustrate where and how MxB targets the CA lattice. Moreover, the glutamates in the tri-hexamer region, i.e., E213, create inter-hexamer repulsion and undermine the tri-hexamer interface structure and thus the CA lattice (Byeon et al., 2009). Therefore, these MxB-CA contacts imply that the MxB N terminus may stabilize the CA lattice by screening charge repulsions between inter-hexamer glutamates, introducing additional contacts connecting neighboring hexamers and reinforcing the tri-hexamer region in the CA lattice. The inter-hexamer interactions give the capsid structural plasticity, which affects its stability (Gres et al., 2015). It is possible that CA stabilization is related to how MxB restricts HIV, as altering the capsid stability likely disrupts the timing of uncoating and capsid-modulated processes such as nuclear import. Further study is necessary to test this hypothesis.
The interface recognized by MxB appears to be a conserved feature of lentiviral capsid structure. Structure-based sequence alignments show that the residues that make up the acidic grooves of the HIV capsid are conserved among lentiviruses, but not retroviruses more generally. Both bovine leukemia virus and Rous sarcoma virus, two divergent retroviruses not restricted by MxB, have surface charge and geometry drastically different from those of HIV (Bailey et al., 2012; Obal et al., 2015). In the lentiviral family, available structures of equine infectious anemia virus and feline immunodeficiency virus capsid overlay well with the WT HIV capsid structure (Figure 5B) (Folio et al., 2017; Jin et al., 1999). Although non-primate lentiviruses appear to have the necessary capsid surface for MxB biding, they are not restricted by MxB, perhaps because they use different nuclear import pathways (Lee et al., 2010). These observations suggest that the presence of a negatively charged lattice interface may be necessary for MxB restriction, but that restriction may be dependent on other factors such as nuclear import pathways.
The work presented herein advances the mechanistic understanding of MxB restriction in molecular detail. We defined the HIV-1 capsid site recognized by the MxB 11RRR13 motif to be the interface between three CA hexamers. This represents a residue-level mapping of an HIV-1 capsid lattice-sensing restriction factor. Further work is needed to structurally characterize the interaction of the MxB N terminus and its interaction with CA. In addition, a better understanding of the binding affinity and behavior of the MxB oligomer will shed more light on its restriction. Due to the conservation of this interface among retroviruses, the tri-hexamer interface may represent a general binding site for host factors and a fruitful target for novel therapeutics.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yong Xiong (yong.xiong@yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Plasmids encoding each protein of interest were chemically transformed into E. coli BL21 (DE3) cells (Lucigen). Overnight cultures were diluted 1000X in Terrific Broth and grown with shaking at 37°C to an OD600 of 0.4–0.6, briefly cooled to room temperature, and induced with 0.5mM IPTG. MxB proteins and MBP-CCCyp were expressed at 18°C overnight. CA proteins were expressed at 25°C overnight.
METHOD DETAILS
Purification of Recombinant MxB Fusion Proteins
Plasmids encoding MxB1–35-MBPdi and MxB1–35AAA-MBPdi were transformed in E. coli BL21(DE3), grown and induced with IPTG, and MxB proteins were expressed at 18C overnight. Cells were centrifuged and pellets were resuspended in 40mL lysis buffer (50mM Tris pH8, 300mM NaCl, 0.2mM TCEP) with one complete EDTA-free protease inhibitor cocktail tablet (Sigma) and lysed using a microfluidizer. Lysate was cleared by centrifugation at 13,500RPM in an SS-34 rotor at 4C for 35 min. Cleared lysate was loaded onto a 10mL bed of Ni-NTA agarose beads (Qiagenn) pre-equilibrated in lysis buffer. Then, the column was washed with 3 volumes of lysis buffer, and protein was eluted in 3 volumes of Nickel B (50mM Tris pH8, 300mM NaCl, 400mM Imidazole, 0.2mM TCEP). The elute was concentrated to a volume of 5–10mL and diluted 10-fold with buffer SA (50mM HEPES pH7, 0.2mM TCEP). The sample was loaded onto a 5mL HiTrap S HP column (GE Healthcare) and eluted using a gradient of buffer SB (SA + 1M NaCl). Peak fractions were concentrated to a volume of <2mL and loaded onto a HiLoad Superdex 200 16/600 prep grade size exclusion column (GE Healthcare) equilibrated in 50mM Tris pH 8 and 300mM NaCl. Peak fractions were collected, concentrated, and flash frozen in liquid nitrogen. Purity of the sample was checked by SDS-PAGE at each step. At each step, phenylmethane sulfonyl fluoride (PMSF) was added to a final concentration of 1mM to reduce proteolysis.
Purification of Recombinant MBP-CCCyp Protein
6xHis-MBP-CCCyp was expressed in E coli BL21(DE3) cells (Lucigen) grown in Terrific Broth to an OD600 of 0.6–0.8 and induced with 0.5mM IPTG. Protein expression occurred overnight at 16°. Cells were pelleted by centrifugation and resuspended in lysis buffer (50mM Tris pH8, 300mM NaCl, 0.2mM TCEP) with one complete EDTA-free protease inhibitor cocktail (Sigma) and lysed using a microfluidizer. Lysate was cleared by centrifugation at 13,500 RPM for 35 minutes at 4°C using an SS-34 rotor and loaded onto a 10mL gravity column of Ni-NTA agarose beads (Qiagen) equilibrated in lysis buffer. After collecting the flow-through for analysis, the column was washed with 3 volumes of lysis buffer and MBP-CCCyp was eluted with 3 volumes of Nickel B (lysis buffer + 400mM imidazole). This sample was concentrated and diluted 10-fold in QA (50mM Tris pH 8, 0.2mM TCEP) before loading onto a HiTrap Q HP 5mL column (GE Healthcare). MBP-CCCyp was eluted with a gradient of QB (QA + 1M NaCl). Peak fractions were collected, concentrated to 1–2mL and run on a HiPrep Superdex 200 16/600 Prep Grade (GE Healthcare). The peak fractions were collected, concentrated, and flash frozen in liquid nitrogen for later use.
Purification of Recombinant Capsid Proteins and MBP
To improve solubility, we expressed all variants of our 14C/45C/WM proteins as CA-Mpro-MBP-6xHis fusions, with a SARS Mpro protease cleavage site and 6xHis tag to aid in purification. BL21(DE3) cells containing the plasmid encoding variants of CA14C/45C-mpro-MBP6xHis were grown in Terrific Broth at 37°C to an OD600 of 0.4–0.6, then protein expression was induced with 0.5mM IPTG at 25°C and allowed to progress overnight. Cells were pelleted and pellets were resuspended in 40mL lysis buffer (50mM Tris pH8, 300mM NaCl, 0.2mM TCEP) and lysed using a microfluidizer. Lysate was cleared (13,500RPM in an SS-34 rotor at 4°C for 35 min) and loaded onto a 10mL gravity column of Ni-NTA agarose beads (Qiagen) equilibrated in lysis buffer. The column was washed with 3 volumes of lysis buffer, and protein was eluted in 30mL of Nickel B buffer (lysis buffer + 400mM imidazole). The elute was concentrated to a volume of 5–10mL and diluted 10-fold with buffer QA (50mM Tris pH8, 0.2mM TCEP). This was loaded onto 2 stacked 5mL HiTrap Q HP columns (GE Healthcare) and eluted using a gradient of buffer QB (QA+ 1M NaCl). All proteins eluted between 5 and 15% buffer QB. Mpro protease (Yang et al., 2003) was added directly to the peak fractions and allowed to incubate at 4°C. After 2 days, this protein was diluted with buffer SA (25mM HEPES pH 7, 0.2mM TCEP) and loaded onto 1 or 2 HiTrap SP columns (GE Healthcare) and eluted using a gradient of buffer SB (SA + 1M NaCl). Proteins eluted between 5–10% buffer SB. Peak fractions were concentrated to a protein concentration of 10–15mg/ml and dialyzed using Slide-A-Lyzer casettes (ThermoFisher) for 4 hours at 4°C into storage buffer (50mM Tris, 75mM NaCl, 30mM BME). Purity of sample was checked by SDS-PAGE at each purification step. Flow-through fractions containing MBP from the SP column were collected, checked for purity, concentrated, and flash frozen.
Wild-type CA proteins containing the P207S, G208R, and T210K mutations were purified using a modification of previously reported protocols (Hung et al., 2013). Briefly, BL21(DE3) cells (Lucigen) containing the plasmid encoding CAP207S, CAG208R, and CAT210K were grown in Terrific Broth at 37°C to an OD600 of 0.4–0.6, then protein expression was induced with 0.5mM IPTG at 25°C overnight. Cell pellets were resuspended in lysis buffer and lysed using a microfluidizer. Lysate was cleared by centrifugation at 13,500RPM for 35 minutes at 4°C. Solid ammonium sulfate was added to a final concentration of 27% with stirring at 4°C for 30 minutes. Precipitated protein (including CA) was collected via centrifugation at 13,500 RPM for 15 minutes at 4°C and resuspended in 40mL of buffer SA. This sample was loaded into pre-treated RC Tubing (Spectrum Labs) and dialyzed overnight in buffer SA· Then, the sample was diluted to 150mL with buffer SA, loaded on a HiTrap SP HP column (GE Healthcare), and eluted with increasing concentrations of buffer SB. Peak fractions were concentrated and flash-frozen in liquid nitrogen. Purity was checked by SDS-PAGE.
Co-pelleting Assay
CA tubes were assembled by dialysis for 2 nights at high salt (50mM Tris pH 8, 1M NaCl) and subsequently one night at no salt (50mM Tris pH8). Concentrations were normalized through quantification of the CA bands on SAD-PAGE gels. Co-pelleting assays were performed with 3uM of MxB1–35-MBPdi or control proteins, and 100uM CA. Stock solutions of capsid tubes (200uM) and MxB1–35-MBPdi, MBP, or CC-Cyp proteins (6uM) were prepared. CA tubes were diluted in CA buffer (50mM Tris pH 8). Test proteins were diluted with twice the final concentration of salt in buffer containing 50mM Tris pH 8 and 150/300/600mM NaCl. Samples were prepared in 1.5mL Eppendorf tubes by combining 24ul of MxB1–35-MBPdi or control protein and 24ul of CA (or 50mM Tris pH8 for CA-controls) and incubated for 30 minutes at room temperature on a Nutator mixer. After 30 minutes, a 14ul “total” sample was removed and the remaining 28ul were centrifuged for 15 minutes at 4°C, 20,000RPM. The soluble fraction was subsequently removed and the pellet resuspended in 28ul of 50mM Tris pH8. Gel samples of the soluble fraction were made by taking 14ul of the soluble fraction and adding loading dye. 4ul of each gel sample was analyzed by SDS-PAGE (Invitrogen) and stained using SimplyBlue stain (Thermo Fisher).
Molecular Dynamics (MD) Simulations
The 3D structure of first 35 residues of MxB (MxB1–35) was built de novo in Rosetta3 (Leaver-Fay et al., 2011) and then equilibrated in a water box for 500 ns. The structure of native HIV-1 CA monomer (PDB:4XFX) was used to build the CA tri-hexamer model (Figures 4A and 4B). The equilibrated MxB1–35 was initially placed above the tri-hexamer region. The combined structure was then solvated with TIP3P water (Jorgensen and Jenson, 1998) and neutralized by NaCl at 150 mM concentration. The resulting model had a total atom number of about 150,000 (Figure 4B), included protein, ions and water, in a simulation dimension of 125 × 121 × 138 Å3.
After model building, the systems were initially subjected to a minimization and thermalization step. During thermalization the system was heated from 50K to 310K in 20K increments. Subsequently, the whole systems were equilibrated for over 20 ns, while the carbon alpha of the peripheral helices of CA monomer and one end of MxB1–35 were restrained. The minimization, thermalization and equilibration steps were completed in NAMD2.12 (Phillips et al., 2005).
This equilibrated model was then run on a special purpose computer Anton2 (Shaw et al., 2014) in the Pittsburgh supercomputing center for a total simulation time of 30 μs. To mimic the CA hexamer lattice in the simulation, the Cα atoms of three sets of CA α-helices on the peripheries of the tri-hexamer model were applied with a harmonic restraint of 1 Kcal/mol Å2 in x, y and z directions. Also, the Cα atoms of MxB residue 35 was applied a harmonic restraint of 1 Kcal/mol Å2 at 40Å away from the COM (center of mass) of CA in z direction. CHARMM 36 (Best et al., 2012) force field was employed for all simulations. During the simulation, the temperature (310 K) and pressure (1 atm) was maintained by employing the Multigrator integrator (Lippert et al., 2013) and the simulation time step was set to 2.5 fs/step, with short-range forces evaluated at every time step, and long-range electrostatics evaluated at every second time step. Short-range non-bonded interactions were cut off at 17 Å; long range electrostatics were calculated using the k-Gaussian Split Ewald method (Shan et al., 2005).
A second simulation was performed similarly, with the MxB peptide initially placed at above the di-hexamer interface. The resulting system contained 246,000 atoms, including protein, ions and water molecules. The salt concentration was set to 150mM NaCl. The system was minimized and equilibrated using NAMD2.13. Subsequently, unbiased MD was performed for a total of 15 μs. During the simulation, the C-terminus of MxB1–35 was positionally restrained (1.0 Kcal/mol Å2) in the Z direction, and freely to diffuse in the x-y plane.
MD Trajectory Analysis
The trajectory analyses were performed in VMD (Humphrey et al., 1996). The MxB-CA contacts and their occupancy scores were calculated by a homemade Tcl script. The secondary structure of MxB and ion occupancies were calculated with VMD plugins TimeLine and VolMap Tool, respectively.
Crystallization and Crystal Structure Analysis
CA crystals were obtained using the microbatch under oil method (Chayen et al., 1992). Purified CA P207S, G208R, or T210K was diluted to 1mg/ml (P207S) or 0.5 mg/ml (G208R or T210K) in 50mM Tris pH 8 buffer. 1ul of protein solution was mixed with 1ul precipitant solution (15% PEG3350, 0.1M Nal, 0.1M buffer) under oil (2:1 paraffin: silicon). P207S crystals were grown with BisTrisPropane pH 6.5 while G208R and T210K were grown with sodium cacodylate pH 6.6 and 5.8 respectively. Crystals were harvested after several weeks of growth, cryo-protected in paratone oil and flash frozen in liquid nitrogen. Data were collected at the Advanced Photon Source, NE-CAT beamline 24-ID-C. Data processing was performed in XDS (Kabsch, 2010a, b). The structure of WT CA (PDB: 4XFX) was used as a model for molecular replacement with the CCP4 program Phaser (Collaborative Computational Project, 1994; Gres et al., 2015; McCoy et al., 2007; Winn et al., 2011). Iterative refinement and manual rebuilding were performed using Refmac5, Phenix, and Coot (Adams et al., 2010; Emsley and Cowtan, 2004; Murshudov et al., 1997). Molecular graphics were generated using PyMol (Schrodinger, 2015) and the MxB-capsid model was generated using Chimera (Pettersen et al., 2004).
Electron Microscopy
Assembled CA tubes were diluted to 10uM in 50mM Tris pH 8 and negative-stained with uranyl acetate onto holey carbon grids after glow-discharge. Grids were examined on an FEI Talos L120C microscope and images at 57,000X magnification were collected using TEM Imaging and Analysis software (Gatan).
QUANTIFICATION AND STATISTICAL ANALYSIS
All co-pelleting assays were performed in triplicate. ImageStudioLite was used to quantify proteins of interest in the total and soluble fractions. The fraction of protein bound to CA was calculated by subtracting the ratio of soluble to total protein of interest from 1. The data were graphed with standard error of the mean using Prism 7.
DATA AND SOFTWARE AVAILABILITY
The structures of CAP207S, CAG208R and CAT210K have been deposited in the PDB under accession numbers 6MQA, 6MQO, and 6MQP.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | Lucigen | Cat#60401 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete EDTA-free protease inhibitor cocktail | Sigma | Cat#11873580001 |
| Recombinant MxB1–35-MBPdi | This paper | N/A |
| Recombinant MxB1–35AAA-MBPdi | This paper | N/A |
| Recombinant MBP-CCCyp | This paper | N/A |
| Recombinant MBP | This paper | N/A |
| Recombinant CA14C/45C | This paper | N/A |
| Recombinant CA14C/45C/E71A | This paper | N/A |
| Recombinant CA14C/45C/E75A | This paper | N/A |
| Recombinant CA14C/45C/G89V | This paper | N/A |
| Recombinant CA14C/45C/P90A | This paper | N/A |
| Recombinant CA14C/45C/E98A | This paper | N/A |
| Recombinant CA14C/45C/E180A | This paper | N/A |
| Recombinant CA14C/45C/E187A | This paper | N/A |
| Recombinant CA14C/45C/E212A | This paper | N/A |
| Recombinant CA14C/45C/E213A | This paper | N/A |
| Recombinant CA 14C/45C/EE212/213AA | This paper | N/A |
| Recombinant CA14C/45C/P207S | This paper | N/A |
| Recombinant CA14C/45C/G208R | This paper | N/A |
| Recombinant CA14C/45C/T210K | This paper | N/A |
| Recombinant CAP207S | This paper | N/A |
| Recombinant CAG208R | This paper | N/A |
| Recombinant CAT210K | This paper | N/A |
| Recombinant Mpro protease | (Yang et al., 2003) | N/A |
| Critical Commercial Assays | ||
| KOD polymerase (for site-directed mutagenesis) | Toyobo/Novagen | Cat#71086-3 |
| Deposited Data | ||
| Crystal structure of CAP207S | This paper | PDB: 6MQA |
| Crystal structure of CAG208R | This paper | PDB: 6MQO |
| Crystal structure of CAT210K | This paper | PDB: 6MQP |
| Crystal structure of WT CA | (Gres et al., 2015) | PDB: 4XFX |
| Crystal structure of RSV capsid | (Bailey et al., 2012) | PDB: 4TIR |
| Crystal structure of BLV capsid | (Obal et al., 2015) | PDB: 4PH0 |
| Oligonucleotides | ||
| See Table S1 for primers used to generate CA mutations | ||
| Recombinant DNA | ||
| Plasmid: MBP-CCCyp | Laboratory collection | N/A |
| Plasmid: MxB1–35-MBPdi in pETDuet | Laboratory collection | N/A |
| Plasmid: MxB1–35AAA-MBPdi in pETDuet | Laboratory collection | N/A |
| Plasmid: CA14C/45C-Mpro-MBP in pET11a | Laboratory collection | N/A |
| Plasmid: CA14C/45C/E71A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/E75A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/G89V-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/P90A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/E98A-Mpro -MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/E180A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/E187A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/E212A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid CA14C/45C/E213A-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/EE212/213AA-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/P207S-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/G208R-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: CA14C/45C/T210K-Mpro-MBP in pET11a | This paper | N/A |
| Plasmid: WT CA in pET11a | Laboratory collection | N/A |
| Plasmid: CAP207S in pET11a | This paper | N/A |
| Plasmid: CAG208R in pET11a | This paper | N/A |
| Plasmid: CAT210K in pET11a | This paper | N/A |
| Software and Algorithms | ||
| ImageStudioLite | LI-COR | https://www.licor.com/bio/products/software/image_studio_lite/ |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Rosetta3 | (Leaver-Fay et al., 2011) | https://www.rosettacommons.org/forums/rosetta-3 |
| NAMD2.12 | (Phillips et al., 2005) | http://www.ks.uiuc.edu/Research/namd/ |
| CHARMM 36 | (Best et al., 2012) | https://www.charmm.org/charmm/ |
| VMD | (Humphrey et al., 1996) | https://www.ks.uiuc.edu/Research/vmd/ |
| XDS | (Kabsch, 2010a, b) | http://xds.mpimf-heidelberg.mpg.de/ |
| CCP4 | (Collaborative Computational Project, 1994; Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| Phaser | (McCoy et al., 2007) | http://www.ccp4.ac.uk/html/phaser.html |
| Refmac5 | (Murshudov et al., 1997) | http://www.ccp4.ac.uk/dist/html/refmac5.html |
| Phenix | (Adams et al., 2010) | https://www.phenix-online.org/ |
| Coot | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL (v1.6.0.0) | (Schrodinger, 2015) | https://pymol.org/2/ |
| Chimera (v13.0) | (Pettersen et al., 2004) | https://www.cgl.ucsf.edu/chimera/ |
| TEM Image and Analysis | Gatan | http://www.gatan.com/products/tem-analysis/gatan-microscopy-suite-software |
| Other | ||
| Ni-NTA Agarose | Qiagen | Cat#30230 |
| HiTrap Q HP 5mL | GE Healthcare | Cat#17115401 |
| HiTrap SP HP 5mL | GE Healthcare | Cat#17115201 |
| HiLoad 16/600 Superdex 200 PG | GE Healthcare | Cat#28990945 |
| Pre-treated RC Tubing MWCO 10kD | Spectrum Labs | Cat#132120 |
| Slide-A-Lyzer Dialysis Cassettes, 10K MWCO | Thermo Fisher | Cat#PI66383 |
| NuPAGE 4–12% Bis-Tris Midi gels | Invitrogen | Cat#WG1403B0X |
| SimplyBlue SafeStain | Thermo Fisher | Cat#LC6060 |
Highlights.
The HIV restriction factor MxB targets the CA tri-hexamer interface on viral capsid
Positively charged MxB N terminus interacts with negatively charged CA residues
Interface mutations that disrupt restriction also reduce binding
Molecular dynamics simulations show specific binding of MxB at the interface
ACKNOWLEDGMENTS
This work was supported by NIH grants P50GM082251 and P30GM110758, and the Collaboration Development Pilot Program award from the Pittsburgh Center for HIV Protein Interactions (to Y.X. and J.R.P.), the Yale Predoctoral Training Program in Virology Training Program NIH T32 AI055403 (to S.S.S.), the Predoctoral Program in Biophysics NIH T32 GM008283 (to B.J.S.), and K.M.D. was supported by the Interdisciplinary Immunology Training Program NIH T32 AI007019.
This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH (P41 GM103403). We are grateful to the staff at the Advanced Photon Source beamline 24-ID-C. The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Anton computer time was provided by the Pittsburgh Supercomputing Center (PSC) through grant R01-GM116961 from the NIH. The Anton machine at PSC was generously made available by D.E. Shaw Research.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2019.04.015.
REFERENCES
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJD, He S, Perilla JR, Jang S, Schulten K, Engelman AN, Scheres SHW, and Zhang P (2017). CryoEM structure of MxB reveals a novel oligomerization interface critical for HIV restriction. Sci. Adv 3, e1701264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey GD, Hyun JK, Mitra AK, and Kingston RL (2012). A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J. Mol. Biol 417, 212–223. [DOI] [PubMed] [Google Scholar]
- Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, and Mackerell AD Jr. (2012). Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. J. Chem. Theory Comput 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, Ansari H, and Luban J (1997). The hydrophobic pocket of cyclophilin is the binding site for the human immunodeficiency virus type 1 Gag polyprotein. J. Virol 71, 2107–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, et al. (2014). Host and viral determinants of Mx2 antiretroviral activity. J. Virol 88, 7738–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byeon IJ, Meng X, Jung J, Zhao G, Yang R, Ahn J, Shi J, Concel J, Aiken C, Zhang P, et al. (2009). Structural convergence between cryo-EM and NMR reveals intersubunit interactions critical for HIV-1 capsid function. Cell 139, 780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chayen NE, Shaw Stewart PD, and Blow DM (1992). Microbatch crystallization under oil — a new technique allowing many small-volume crystallization trials. J. Cryst. Growth 122, 176–180. [Google Scholar]
- Collaborative Computational Project (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Crameri M, Bauer M, Caduff N, Walker R, Steiner F, Franzoso FD, Gujer C, Boucke K, Kucera T, Zbinden A, et al. (2018). MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun 9, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Sidumo ZJ, Zanelli G, Magid NA, Luhanga R, Brambilla D, Liotta G, Mancinelli S, Marazzi MC, Palombi L, et al. (2017). Accumulation of HIV-1 drug resistance in patients on a standard thymidine analogue-based first line antiretroviral therapy after virological failure: implications for the activity of next-line regimens from a longitudinal study in Mozambique. BMC Infect. Dis 17, 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Folio C, Sierra N, Dujardin M, Alvarez G, and Guillon C (2017). Crystal structure of the full-length feline immunodeficiency virus capsid protein shows an N-terminal beta-hairpin in the absence of N-terminal proline. Viruses 9, 10.3390/v9110335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke EK, Yuan HE, and Luban J (1994). Specific incorporation of cyclophilin A into HIV-1 virions. Nature 372, 359–362. [DOI] [PubMed] [Google Scholar]
- Fribourgh JL, Nguyen HC, Matreyek KA, Alvarez FJ, Summers BJ, Dewdney TG, Aiken C, Zhang P, Engelman A, and Xiong Y (2014). Structural insight into HIV-1 restriction by MxB. Cell Host Microbe 16, 627–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nunez A, and Diaz-Griffero F (2014). MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 11, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, and Daumke O (2011). Structure of myxovirus resistance protein a reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35, 514–525. [DOI] [PubMed] [Google Scholar]
- Goujon C, Greenbury RA, Papaioannou S, Doyle T, and Malim MH (2015). A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J. Virol 89, 4676–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, and Malim MH (2014).Transfer of the amino-terminal nuclear envelope targeting domain of human MX2 converts MX1 into an HIV-1 resistance factor. J. Virol 88, 9017–9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, and Malim MH (2013). Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, and Sarafianos SG (2015). Structural virology. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller O, Staeheli P, Schwemmle M, and Kochs G (2015). Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol. 23, 154–163. [DOI] [PubMed] [Google Scholar]
- Henning MS, Dubose BN, Burse MJ, Aiken C, and Yamashita M (2014). In vivo functions of CPSF6 for HIV-1 as revealed by HIV-1 capsid evolution in HLA-B27-positive subjects. PLoS Pathog. 10, e1003868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, and Schulten K (1996). VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
- Hung M, Niedziela-Majka A, Jin D, Wong M, Leavitt S, Brendza KM, Liu X, and Sakowicz R (2013). Large-scale functional purification of recombinant HIV-1 capsid. PLoS One 8, e58035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaguva Vasudevan AA, Bahr A, Grothmann R, Singer A, Haussinger D, Zimmermann A, and Munk C (2018). MXB inhibits murine cytomegalovirus. Virology 522, 158–167. [DOI] [PubMed] [Google Scholar]
- Jin Z, Jin L, Peterson DL, and Lawson CL (1999). Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol 286, 83–93. [DOI] [PubMed] [Google Scholar]
- Jorgensen WL, and Jenson C (1998). Temperature dependence of TIP3P, SPC, and TIP4P water from NPT Monte Carlo simulations: seeking temperatures of maximum density. J. Comput. Chem 19, 1179–1186. [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Rebensburg SV, Takata MA, Zang TM, Yamashita M, Kvaratskhelia M, and Bieniasz PD (2018). Nuclear pore heterogeneity influences HIV-1 infection and the antiviral activity of MX2. Elife 7, 10.7554/eLife.35738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, et al. (2013). MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502, 563–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, Raposo G, and Lemmon MA (2004). Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proc. Natl. Acad. Sci. U S A 101, 8957–8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Xu B, Wei W, Wang X, Xie W, and Yu XF (2014). Characterization of the amino-terminal domain of Mx2/MxB-dependent interaction with the HIV-1 capsid. Protein Cell 5, 954–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith CA, Sheffler W, et al. (2011). ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ambrose Z, Martin TD, Oztop I, Mulky A, Julias JG, Vandegraaff N, Baumann JG, Wang R, Yuen W, et al. (2010). Flexible use of nuclear import pathways by HIV-1. Cell Host Microbe 7, 221–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert RA, Predescu C, lerardi DJ, Mackenzie KM, Eastwood MP, Dror RO, and Shaw DE (2013). Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys 139, 164106. [DOI] [PubMed] [Google Scholar]
- Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, and Liang C (2013). The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14, 398–410. [DOI] [PubMed] [Google Scholar]
- Liu Z, Pan Q, Liang Z, Qiao W, Cen S, and Liang C (2015). The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 12, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Wang W, Serrao E, Singh PK, Levin HL, and Engelman A (2014). Host and viral determinants for MxB restriction of HIV-1 infection. Retrovirology 11, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matreyek KA, Yucel SS, Li X, and Engelman A (2013). Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity. PLoS Pathog. 9, e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Obal G, Trajtenberg F, Carrion F, Tome L, Larrieux N, Zhang X, Pritsch O, and Buschiazzo A (2015). Structural virology. Conformational plasticity of a native retroviral capsid revealed by X-ray crystallography. Science 349, 95–98. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005). Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, and Yeager M (2009). X-ray structures of the hexameric building block of the HIV capsid. Cell 137, 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling M, Bulli L, Weigang S, Graf L, Naumann S, Patzina C, Wagner V, Bauersfeld L, Goujon C, Hengel H, et al. (2018). Human MxB protein is a pan-herpesvirus restriction factor. J. Virol 92, 10.1128/JVI.01056-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger, L.L.C. (2015). The PyMOL Molecular Graphics System, Version 1.8.
- Schulte B, Buffone C, Opp S, Di Nunzio F, De Souza Aranha Vieira DA, Brandariz-Nunez A, and Diaz-Griffero F (2015). Restriction of HIV-1 requires the N-terminal region of MxB as a capsid-binding motif but not as a nuclear localization signal. J. Virol 89, 8599–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Klepeis JL, Eastwood MP, Dror RO, and Shaw DE (2005). Gaussian split Ewald: a fast Ewald mesh method for molecular simulation. J. Chem. Phys 122, 54101. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Grossman JP, Bank JA, Batson B, Butts JA, Chao JC, Deneroff MM, Dror RO, Even A, Fenton CH, et al. (2014). Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis (New Orleans, Louisana, IEEE Press), pp. 41–53. [Google Scholar]
- Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, and Sodroski J (2006). Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U S A 103, 5514–5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stray KM, Garrus JE, and Sundquist WI (2003). Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol 77, 5439–5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Guo H, Ma M, Markham R, and Yu XF (2016). Accumulation of MxB/Mx2-resistant HIV-1 capsid variants during expansion of the HIV-1 epidemic in human populations. EBioMedicine 8, 230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Kong J, Wang X, Wei W, Xie W, and Yu XF (2015). Structural insight into the assembly of human anti-HIV dynamin-like protein MxB/Mx2. Biochem. Biophys. Res. Commun 456, 197–201. [DOI] [PubMed] [Google Scholar]
- Yang H, Yang M, Ding Y, Liu Y, Lou Z, Zhou Z, Sun L, Mo L, Ye S, Pang H, et al. (2003). The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U S A 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yufenyuy EL, and Aiken C (2013). The NTD-CTD intersubunit interface plays a critical role in assembly and stabilization of the HIV-1 capsid. Retrovirology 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structures of CAP207S, CAG208R and CAT210K have been deposited in the PDB under accession numbers 6MQA, 6MQO, and 6MQP.