

Abstract
Methylprednisolone (MPL), a corticosteroid of intermediate potency, remains an important immunomodulatory agent for autoimmune diseases. Although sex differences in corticosteroid pharmacokinetics/pharmacodynamics (PK/PD) have been documented in humans, comprehensive preclinical assessments of such differences have not been conducted. Limited in vitro evidence indicates possible sex differences in corticosteroid PK and PD. Therefore, it is hypothesized that comparative PK/PD assessments of MPL disposition and selected PD actions in both sexes will provide insights into factors controlling sex differences in steroid responses. This report focused on the plasma and tissue pharmacokinetics of MPL and its adrenal suppressive effects. Because time-dependent (estrous) regulation of sex hormones in females can influence drug responses, female rats were studied in the proestrus (high estradiol/progesterone) and estrus (low estradiol/progesterone) phases of the reproductive cycle. Cohorts of male and female rats were given a 50 mg/kg bolus dose of MPL intramuscularly. Plasma and liver concentrations of MPL as well as plasma corticosterone concentrations were assayed using high-performance liquid chromatography. An enhanced minimal physiologically-based PK/PD model was developed to characterize MPL kinetics and corticosterone dynamics. The clearance of MPL was ∼3-fold higher in males compared with females, regardless of estrous phase, likely attributable to sex-specific hepatic metabolism in males. Strong inhibitory effects on adrenal suppression were observed in all animals. These temporal steroid profiles in plasma and tissues will be used to drive receptor/gene-mediated PD effects of MPL in both sexes, as described in a companion article (Part III).
SIGNIFICANCE STATEMENT
Sex is a relevant factor influencing the pharmacokinetics (PK) and pharmacodynamics (PD) of drugs. Few preclinical PK/PD studies, however, include sex as a variable. Sex differences in the PK and adrenal suppressive effects of the synthetic corticosteroid, methylprednisolone, were assessed in male and female rats as a function of the 4-day rodent reproductive cycle. Drug exposure was 3-fold higher in females, regardless of estrous stage, compared with males. An extended minimal physiologically-based PK/PD model utilizing in vitro and in vivo measurements was developed and applied. These studies provide a framework to account for sex-dependent variability in drug and endogenous agonist (corticosterone) exposures, serving as a prelude to more intricate assessments of sex-related variability in receptor/gene-mediated PD corticosteroid actions.
Introduction
Clinical use of corticosteroids (CS) is frequent and chronic. Therapy with this class of drugs dates back to late 1940s (Burns, 2016), before quantitative methods describing the kinetics and dynamics of drug disposition and actions were considered necessary for drug development and clinical applications. Although highly efficacious in the treatment of autoimmune disorders such as rheumatoid arthritis (Kirwan, 1995), prolonged use of these agents produces numerous undesirable metabolic effects that are manifested as muscle wasting, diabetes, obesity, and osteoporosis (Schäcke et al., 2002), complicating their efficacy–toxicity profile. Despite such issues, CS are among the most widely prescribed drugs in developed countries—taken by up to 1.2% of the adult population (van Staa et al., 2000; Overman et al., 2013).
Methylprednisolone (MPL) exerts both anti-inflammatory and adverse effects by acting through coupled receptor-mediated pharmacogenomic (PG) mechanisms. Briefly, free (unbound) MPL in the tissue interstitial fluid distributes into intracellular spaces, where it binds to cytoplasmic glucocorticoid receptors, leading to glucocorticoid receptor activation, nuclear translocation, and consequent alterations in gene expression. Thus, measurements of mRNA of target genes can be employed as biomarkers of dose- and time-dependent genomic CS actions in tissues (Ayyar et al., 2017). Another well-established biomarker of CS actions is adrenal suppression, in which exogenous CS induces a rapid and robust suppression of circulating glucocorticoid hormones in humans and animals (Mager et al., 2003; Yao et al., 2008).
Sex differences in the clinical pharmacokinetics and pharmacodynamics (PK/PD) of drugs as well as in human or murine biology have been reviewed (Gandhi et al., 2004; Danska, 2014; Kovats, 2015). Efforts have been made by our laboratory to examine sex differences in the disposition and pharmacology of CS in humans, animals, and cell systems. Sex differences in the PK and PD of MPL have been observed in healthy subjects (Lew et al., 1993). Ex vivo assessments of sex-related differences in the sensitivity of prednisolone to inhibit lymphocyte proliferation in whole blood were reported in humans, rabbits, and rats (Ferron and Jusko, 1998). Modest-to-pronounced sex differences were recently observed in collagen-induced arthritis disease progression, in the PK of dexamethasone (DEX), and in the anti-inflammatory efficacy of the drug in collagen-induced arthritis rats (Song et al., 2018). Although these sex differences in PK and PD have been reported, exploration of underlying mechanisms is needed. In particular, possible sex differences in MPL kinetics, adrenal suppressive effects, and receptor/gene-mediated mechanisms of MPL have not been investigated to date.
Major endogenous sex hormones such as testosterone and 17β-estradiol have been shown to influence drug metabolism and protein binding (Brodie, 1956; Gandhi et al., 2004). Sexual dimorphism occurs in the rat hypothalamic-pituitary-adrenal axis (Atkinson and Waddell, 1997), with stronger baseline corticosterone (CST) rhythms during the proestrus stage compared with estrus and in males. Compelling in vitro evidence indicates that 17β-estradiol, the major female sex hormone, can antagonize CS-regulated gene expression in an estrogen receptor–dependent manner at the genomic level (Whirledge et al., 2013; Whirledge and Cidlowski, 2013). Very few preclinical studies to date, however, include sex as a factor in PK/PD. Most studies have been carried out in males, with little information available on potential sex differences in drug action. A major objective of this work was expanding our mechanistic assessments of MPL PK/PD from the intact male rat model (Hazra et al., 2007a) to females with consideration of estrous cycle variations. Characterization of the kinetics and dynamics of MPL and CST in both sexes is an important step prior to the conduct of more mechanistic PD assessments of sex differences in genomic actions of MPL within tissues. These studies are consonant with recent emphasis (Clayton and Collins, 2014; Danska, 2014) that translational studies of sex differences should be expanded.
This second report of a three-part series focused on in vivo assessments of sex differences in MPL disposition and endogenous CST suppression in rats. The studies and modeling efforts described in this work were designed to 1) compare the PK of MPL in plasma and livers of male and female rats, 2) examine the dynamics of CST suppression in both sexes, and 3) support the development of a minimal physiologically-based PK (mPBPK), PD, and PG systems model of MPL, incorporating physiologically-relevant phenomena such as circadian rhythms, receptor dynamics, and the rodent estrous cycle. Experimental studies and mPBPK/PD/PG modeling related to sex differences in receptor-mediated genomic MPL actions are presented in the final companion article (Part III).
Materials and Methods
Reagents and Chemicals
CST, DEX, and 6α-MPL (≥98.5% purity) were purchased from Sigma-Aldrich (St. Louis, MO). High-performance liquid chromatography–grade methylene chloride, heptane, and glacial acetic acid were obtained from Fisher Scientific (Pittsburgh, PA). Rats were dosed with MPL succinate (Solu-Medrol; Pharmacia & Upjohn, Kalamazoo, MI) via the intramuscular route. Milli-Q water was used (Millipore, Bedford, MA).
Animals
Normal male and female Wistar rats were purchased from Envigo (formerly Harlan-Sprague Dawley, Indianapolis, IN). The animals were housed in our University Laboratory Animal Facility and acclimatized under constant temperature (22°C) and humidity with a controlled 12-hour:12-hour light/dark cycle for 1 to 2 weeks, at which point they were weighed and tail-numbered. Three female rats were housed per cage, whereas two males were maintained in each cage. All rats had free access to rat chow and drinking water. In all studies, animals were sacrificed by exsanguination from the abdominal aorta under ketamine/xylazine anesthesia (80 and 10 mg/kg), and blood was harvested using EDTA as anticoagulant (4 mM final concentration) to obtain plasma. Harvested tissues (liver, lung, gastrocnemius, abdominal adipose, tibia, and uterus) were rapidly dissected, frozen in liquid nitrogen, and stored at −80°C until further use. The protocol adhered to the Principles of Laboratory Animal Care (National Institutes of Health publication 85-23, revised 1985) and was approved by the University at Buffalo Institutional Animal Care and Use Committee.
Monitoring of the Estrous Cycle
The stages of the estrous cycle in female rats were monitored by analysis of cell types in vaginal lavages. Lavages were collected daily for 2 to 3 weeks between 9:00 and 10:00 AM (i.e., during the nadir of CST rhythm in rats). Briefly, 100 μl sterile saline was gently released at the opening of the vaginal canal using a disposable pipette, drawn back, expelled three to four times for collection of cells, and then drawn out. Vaginal fluid was placed on glass slides as droplets (in duplicate), allowed to completely dry, stained using methylene blue (1%), and washed by dipping the slides twice in fresh autoclaved water. Three types of cells could be observed in vaginal smears under a light microscope: nucleated epithelial cells, cornified epithelial cells, and leukocytes. Proestrus was defined by moderate numbers of nucleated epithelial cells, and estrus was characterized by the predominance of cornified epithelial cells. Only rats exhibiting at least three consecutive 4-day cycles were used in the main experimental groups. Some female rats in each cohort (∼20%) displayed persistent estrus or diestrus cytology and were classified into a separate noncycling group. From each female rat, a final vaginal smear was collected a few minutes preceding exsanguination to confirm the estrous stage.
Experiments
Cohort 1: Estrus Female Rats (11 Weeks Old).
Two separate batches of 30 normal female Wistar rats were ordered at 6 to 7 weeks of age and acclimated (n = 60 females). Beginning at 8 weeks, vaginal lavages were obtained daily between 9:00 and 10:00 AM. Female rats showing regular cycles up to 11 weeks of age, weighing 220–290 g, were dosed within the estrus stage with 50 mg/kg MPL succinate IM between 1.5 and 3.5 hours after lights on. The animals were euthanized at 0.25, 0.5, 0.75, 1, 2, 4, 5, 6, 7, 8, 12, and 24 hours (n = 3 at each time point except at 4 hours, where n = 2). In addition, a group of 12 estrus-staged female rats was euthanized at 4, 6, 8, and 12 hours after lights on as controls (n = 3 at each time point). The rats in estrus were euthanized at the chosen time points in random across the two batches to reduce any systematic errors in the experiments.
Cohort 2: Proestrus Female Rats (11 Weeks Old).
Two separate batches of 32 normal female Wistar rats were ordered at 6 to 7 weeks of age and acclimated (n = 64 females). Beginning at 8 weeks of age, vaginal lavages were obtained daily between 9:00 and 10:00 AM. Female rats showing regular cycles up to 11 weeks of age, weighing 240–300 g, were dosed within the proestrus stage with 50 mg/kg MPL succinate IM between 1.5 and 3.5 hours after lights on. The animals were euthanized at 0.25, 0.5, 0.75, 1, 2, 4, 5, 6, 7, 8, 12, and 24 hours (n = 3–4 at each time point). In addition, a group of 13 proestrus-staged female rats was euthanized at 2, 4, 6, 8, and 12 hours after lights on as controls (n = 2–4 at each time point). The rats in proestrus were euthanized at the chosen time points in random across the two batches to reduce systematic errors in the experiments.
Cohort 3: Noncycling Female Rats (11 Weeks Old).
Thirteen (13) female rats from Cohort 1 displayed either irregular or absent cycles over 8–11 weeks of age. Similarly, 12 rats from Cohort 2 showed either irregular or absent cycles during the same age. These female rats were considered as noncyclers. Similar to the other cohorts, these rats were dosed with 50 mg/kg MPL succinate IM and euthanized at 0.75, 2, 4, 5, 6, and 12 hours (n = 2–4 at each time point).
Cohort 4: Male Rats (7 Weeks Old).
Samples used for various measurements from this group came from a previously conducted animal study in MPL-dosed male rats (Hazra et al., 2007a). Briefly, 54 normal male Wistar rats weighing 250–325 g were dosed at 7 weeks of age with 50 mg/kg MPL succinate IM. Injections were given at the nadir of their endogenous CST circadian rhythm (1.5–3.5 hours after lights on). The animals were euthanized at 0.25, 0.5, 0.75, 1, 2, 4, 5, 6, 7, 8, 12, 24, 36, 48, 60, 72, 84, and 96 hours (n = 3 at each time point).
Note on Animal Study Design.
Some aspects regarding the experimental design need to be clarified further. Puberty in female rats usually occurs 42 days (6 weeks) after birth, which is associated with vaginal opening and first proestrus (Kei-ichiro et al., 2000). Regular estrous cycles usually begin after 7 weeks. Each animal’s estrous cycle was followed for 2 to 3 weeks starting at 8 weeks of age to ascertain estrous phase at the time of performing the experiment. Hence, 11 weeks of age was the earliest when the female rats could be dosed. Male rats from our previous study (Cohort 4), aged 7 weeks at time of dosing, were comparable in terms of body weight to the females in Cohorts 1–3, aged 11 weeks. However, to account for the possible confounding effect of age between both sexes, a smaller version of the Cohort 4 study was re-enacted, but male rats were dosed at 11 weeks instead. This design facilitated the comparison of females to males by accounting for potential differences in males by age and body weight.
Cohort 5: Male Rats (11 Weeks Old).
Thirty normal male Wistar rats, weighing 360–440 g, were dosed at 11 weeks of age with 50 mg/kg MPL succinate IM between 1.5 and 3.5 hours after lights on. The animals were euthanized at 0.25, 1, 2, 4, 5, 6, 7, 8, 12, 24, and 48 hours (n = 2–3 at each time point). In addition, eight male rats were euthanized at 4, 6, and 12 hours after lights on as controls (n = 3 at each time point except at 12 hours, where n = 2).
Plasma and Tissue Steroid Analysis.
MPL was measured in plasma and tissues with minor modifications in the extraction procedure across matrices. DEX was used as the internal standard for all experiments. Charcoal-stripped plasma was used for constructing standard curves and controls for plasma and filtrate experiments. Tissues from control animals were used for generation of pooled blank tissue homogenate for constructing standards and controls to run against tissue samples. Standards and quality controls were prepared by spiking known quantities of steroids into blank matrices and were run daily for assay calibration. Plasma concentrations of CST were determined simultaneously with MPL. For the extraction procedure, rat plasma samples were thawed and 0.5-ml aliquots were extracted with methylene chloride (MeCl2) in disposable glass tubes (Fisher Scientific). Tubes were shaken for 45 minutes before washing the MeCl2 phase with 0.5 ml 0.1 N sodium hydroxide (NaOH). The NaOH phase was removed following centrifugation, and the MeCl2 phase was washed with 0.5 ml distilled water. The aqueous phase was removed following centrifugation, and the organic phase was dried with anhydrous sodium sulfate powder. The MeCl2 was evaporated off with purified air, leaving a residue that was reconstituted in 120 μl mobile phase. Chromatography conditions involved a mobile phase of 585 ml MeCl2, 350 ml heptane, 10 ml glacial acetic acid, 55 ml ethanol, a Zorbax normal phase column (70 Å Sil, 5 µm, 4.6 × 250 mm), Waters model 1515 isocratic pump, and Waters model 2487 dual-wavelength absorbance detector (Haughey and Jusko, 1988). A sample volume of 100 μl was auto-injected into the system. The lower limit of quantification was 5 ng/ml for CST in plasma, 10 ng/ml for MPL in plasma, and 50 ng/g for MPL in tissues with intra- and interday coefficients of variation of less than 10%. Tissues of interest were weighed and homogenized in five volumes of chilled PBS using a Polytron homogenizer at speed setting 5 with two bursts of 30-second duration with 30-second intervals. Tissue homogenates were then processed identical to plasma samples, with the exception that the final residue was reconstituted in 140 μl mobile phase, transferred into 0.5-ml microfuge tubes, and centrifuged at 13,000g for 5 minutes. An approximate volume of 110–120 μl was then carefully drawn out (from the top) and loaded into injection vials for chromatographic analysis.
Tissue Data Correction for Residual Blood.
Drug concentrations obtained in each tissue were converted from nanograms per milliliter to nanograms per gram tissue by assuming a tissue density of 1 g/ml. Tissue concentrations were corrected for residual trapped blood using the following:
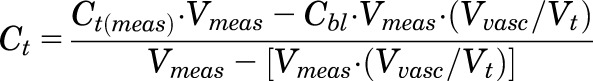 |
(1) |
where Ct and Ct(meas) are the corrected and measured tissue concentrations, 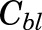 is the measured concentration in blood, Vmeas is the measured volume of collected tissue, and Vvasc/Vt is the fractional vascular volume of blood trapped in tissues as obtained from the literature (Kawai et al., 1998). The ratio of drug concentration in whole blood to that in plasma was assumed to be equal to unity. The corrected tissue concentrations were used for further analysis.
is the measured concentration in blood, Vmeas is the measured volume of collected tissue, and Vvasc/Vt is the fractional vascular volume of blood trapped in tissues as obtained from the literature (Kawai et al., 1998). The ratio of drug concentration in whole blood to that in plasma was assumed to be equal to unity. The corrected tissue concentrations were used for further analysis.
Additional Data Source.
Circadian production of endogenous glucocorticoids, including CST, is well-recognized (Dickmeis, 2009). Therefore, the PK/PD effects of MPL on CST dynamics must be modeled within the context of nonstationary baseline concentrations. Circadian concentrations of CST in male rats have been measured previously in our laboratory (Hazra et al., 2007a). Data from the literature were obtained to account for sex differences in circadian CST concentrations between males, estrus females, and proestrus female rats (Atkinson and Waddell, 1997). Pooling and analysis of baseline CST concentrations were feasible because there was good agreement between their data and our measurements in male rats (Hazra et al., 2007a). The mean values of CST concentrations were extracted from published graphs by computer digitization (WebPlotDigitizer, 4.1, https://automeris.io/WebPlotDigitizer).
PK/PD Modeling and Data Analysis
mPBPK Model for MPL.
The plasma and tissue concentration–time profiles of MPL were characterized using an extended version of the basic mPBPK model (Fig. 1) (Cao and Jusko, 2012).
Fig. 1.

Schematic of the mPBPK/PD model for PK and effects of MPL on adrenal suppression in male and female rats. Parameters are defined in the text and in Tables 2 and 3. The model is described by eqs. 2–11.
The model equations and initial conditions are:
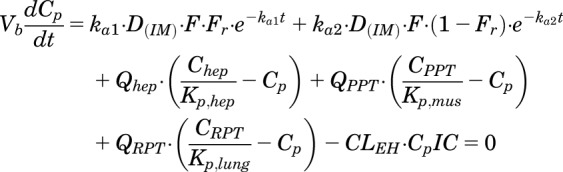 |
(2) |
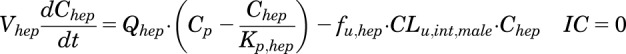 |
(3) |
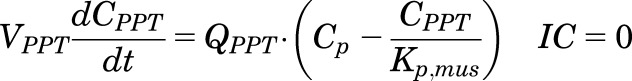 |
(4) |
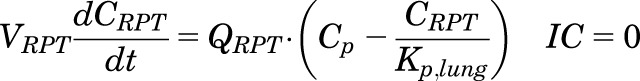 |
(5) |
where  is the dose of MPL,
is the dose of MPL,  is the overall bioavailability,
is the overall bioavailability, 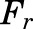 and
and 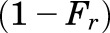 are fractions of dose absorbed through the absorption pathways described by first-order rate constants
are fractions of dose absorbed through the absorption pathways described by first-order rate constants 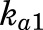 and
and 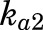 , and
, and 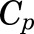 ,
, 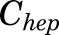 ,
, 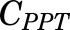 , and
, and 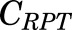 are total MPL concentrations in
are total MPL concentrations in 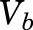 ,
, 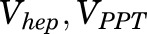 , and
, and 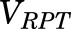 [volumes of blood, liver, poorly perfused tissues (PPT), and rapidly perfused tissues (RPT)]. The
[volumes of blood, liver, poorly perfused tissues (PPT), and rapidly perfused tissues (RPT)]. The 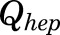 ,
, 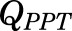 , and
, and 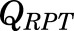 parameters represent organ blood flows to the liver, PPT, and RPT. The
parameters represent organ blood flows to the liver, PPT, and RPT. The 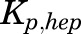 ,
, 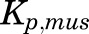 , and
, and 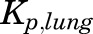 parameters are tissue:plasma partition coefficients for liver, muscle (for PPT), and lung (for RPT). Extrahepatic clearance of total MPL from the plasma is given by
parameters are tissue:plasma partition coefficients for liver, muscle (for PPT), and lung (for RPT). Extrahepatic clearance of total MPL from the plasma is given by 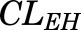 ;
; 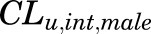 is a male-specific hepatic intrinsic clearance; and
is a male-specific hepatic intrinsic clearance; and 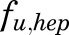 is the fraction of MPL unbound in liver, which is calculated as the ratio of the fraction of MPL unbound in plasma (
is the fraction of MPL unbound in liver, which is calculated as the ratio of the fraction of MPL unbound in plasma (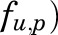 and
and 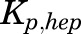 .
.
Certain assumptions were made in developing the model. 1) In the absence of intravenous concentration–time data for MPL in female rats, parameters related to intramuscular bioavailability in females were assumed to be the same as in male rats (Hazra et al., 2007b). 2) Binding of MPL in plasma and tissues is linear (measured), with negligible binding with CS-binding globulin (Ebling et al., 1986). 3) The tissue:plasma partition coefficient (KP) values for rapidly and poorly perfused organs were fixed to measured values in lung and muscle, representing the RPT and PPT groups. Certain physiologic constraints were applied during model development. 1) Blood flows into tissues were calculated using fractional flows of total cardiac output (CO), where cardiac output was assumed to vary by body weight (BW) in males and female rats per the allometric relationship:
| (6) |
2) Muscle, skin, and adipose were assigned as PPT, whereas all other tissues were considered to be RPT. Thus, the total volume of RPT was constrained as:
| (7) |
3) Similarly, the fractional cardiac blood flow into RPT (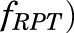 was constrained as:
was constrained as:
| (8) |
where 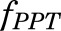 is the fractional cardiac flow into PPT (skin + muscle + adipose) and
is the fractional cardiac flow into PPT (skin + muscle + adipose) and 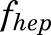 is the fractional hepatic blood flow. Values for fractional blood flows and organ volumes were obtained from previously reported values in rats (Kawai et al., 1998).
is the fractional hepatic blood flow. Values for fractional blood flows and organ volumes were obtained from previously reported values in rats (Kawai et al., 1998).
This model was applied jointly to describe the plasma and liver PK profiles following 50 mg/kg IM MPL in male and female rats. Parameter values estimated during the model fitting were subsequently fixed, and the model-predicted concentration–time profiles were used to drive PD effects.
CST Suppression.
The circadian rhythm of endogenous plasma CST in normal untreated male and female Wistar rats was described by an indirect response model where CST is synthesized by a time-dependent synthesis rate, described by two harmonic functions (Krzyzanski et al., 2000; Yao et al., 2008), and degraded by a first-order loss rate constant, 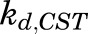 ,
,
| (9) |
The time-dependent synthesis rate of CST, 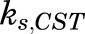 , can be described as:
, can be described as:
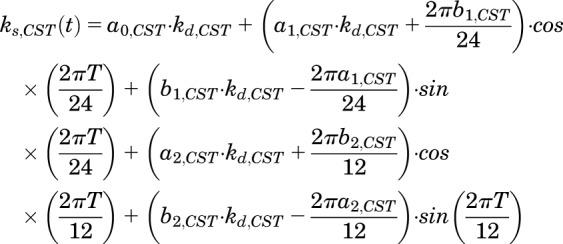 |
(10) |
where a0, a1, a2, b1, and b2 are the Fourier coefficients that were obtained by fitting endogenous plasma CST profiles from untreated rats. Administration of MPL causes suppression of CS production. It is also well known that physical stress such as animal handling and pain stimulates secretion of CS (Hazra et al., 2007a; Yao et al., 2008). The effect of MPL on plasma CS is complicated by both of these factors. Consequently, the inhibition of CST by MPL was described as:
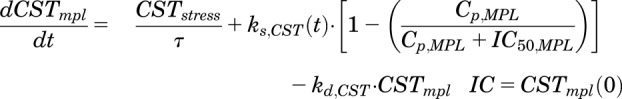 |
(11) |
where 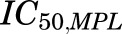 is the concentration of plasma MPL responsible for 50% inhibition of CST. Zero-order secretion of CST
is the concentration of plasma MPL responsible for 50% inhibition of CST. Zero-order secretion of CST 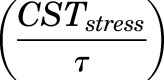 , independent of natural production of plasma CST due to stress, is incorporated to describe early stress-induced secretion of CST, where
, independent of natural production of plasma CST due to stress, is incorporated to describe early stress-induced secretion of CST, where 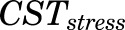 , the maximum concentration of plasma CST due to stress, and
, the maximum concentration of plasma CST due to stress, and 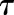 , time when the stress-induced secretion of CST stops, were estimated as model parameters. Thus, the term
, time when the stress-induced secretion of CST stops, were estimated as model parameters. Thus, the term 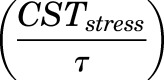 operates when τ ≥ t ≥ tinj (time of injection). The initial conditions for both eqs. 9 and 11 were fixed to 142.7 ng/ml in proestrus-phased females, and 32.0 ng/ml for males and estrus-phased females, which were obtained from the circadian control animals (Atkinson and Waddell, 1997; Hazra et al., 2007a).
operates when τ ≥ t ≥ tinj (time of injection). The initial conditions for both eqs. 9 and 11 were fixed to 142.7 ng/ml in proestrus-phased females, and 32.0 ng/ml for males and estrus-phased females, which were obtained from the circadian control animals (Atkinson and Waddell, 1997; Hazra et al., 2007a).
Data Analysis
Noncompartmental analysis (NCA) of the PK data was performed using Phoenix 8.1 (Certara, Princeton, NJ). The Cmax and the corresponding time when Cmax is reached (Tmax) were directly obtained from the observed data. The areas under the plasma–concentration time curves (AUC(0–Tlast)) were obtained using the linear/log trapezoidal method from time 0 to the last time point of observation (Tlast). The terminal slope (λz) was determined by linear regression of the terminal phase of the log-linear concentration–time profile. The terminal half-life (t1/2_λz) was calculated from ln(2)/λz. AUC(Tlast-∞) from the last quantifiable concentration (Clast) to time infinity was extrapolated using Clast/λz. The total AUC (AUC(0-∞)) was calculated as the sum of AUC(0-Tlast) and AUC(Tlast-∞). Clearance was determined by Dose/AUC(0-∞).
All model fittings and simulations were performed by nonlinear regression using the maximum likelihood algorithm in ADAPT 5 (University of Southern California, Los Angeles, CA) (D’Argenio et al., 2009). Circadian baseline data for CST were modeled by a Fourier harmonics approach using the FOURPHARM algorithm (Krzyzanski et al., 2000). The obtained Fourier coefficients were then fixed during subsequent modeling of the PD data. Replicate measurements from each animal at each time point were employed for model fitting. The mPBPK model code is provided in the Supplemental Materials. The goodness-of-fit was assessed by system convergence, visual inspection of the fitted curves, objective function values such as Akaike Information Criterion, improved likelihood, examination of residuals, and precision (CV%) of the estimated parameters. Separate variance models for fitting PK and PD outputs used:
| (12) |
| (13) |
where V(σ,θ,ti) is the variance for the ith point, Y(θ,ti) is the ith model-predicted value, θ represents the estimated structural parameters, and σ1, σ2 are the variance parameters that were estimated. Animals in the dosing experiments were given MPL between 1.5 and 3 hours after lights on. For modeling the dynamics of CST, the dosing time was assumed at circadian time 2.5 hours to compare the data obtained from both the MPL-dosing and circadian experiments. Hence, all PD profiles are plotted with respect to circadian time, with MPL given at 2.5 hours.
Results
Comparison of MPL PK in Male and Female Rats.
The plasma concentration–time profiles of MPL following a 50 mg/kg IM dose in normal male Wistars aged 7 (Cohort 4) and 11 (Cohort 5) weeks are shown in Fig. 2A. The profile of MPL in animals from Cohort 4 was taken from Hazra et al. (2007b). By visual inspection, no significant difference was observed across both male cohorts. This observation was corroborated by the parameter values from NCA, shown in Table 1. The mean plasma concentration–time profiles of MPL following a 50 mg/kg IM dose in estrus-phased, proestrus-phased, and noncycling female Wistars are shown in Fig. 2B. Differences in the concentration–time profiles between the three female cohorts were minor and nonsignificant, and are reflected by the parameter values from NCA, shown in Table 1. The mean plasma concentration–time profiles of MPL in males (Cohorts 4 and 5) and females (Cohorts 1–3) are shown in Fig. 2C. Plasma MPL exposure was significantly increased in female rats, indicated by an approximate 3.3-fold higher plasma AUC value (males: AUC, 2901 ng h/ml; females: 9751 ng h/ml). Accordingly, total MPL clearance was estimated as 5.1 l/h per kilogram in females and 17.2 l/h per kilogram in males. The plasma Cmax was also higher in females (males: Cmax, 1174 ng/ml; females: 4204 ng/ml). Plasma drug concentrations rose rapidly in all groups by 0.25 hour after dosing and showed a prolonged peak (Tmax ∼0.75–1 hour). The terminal half-lives following intramuscular dosing for males and females were quite similar (2.5 and 2.9 hours). These values were higher in both sexes compared with a previously reported value following intravenous dosing (0.5 hour) in normal male rats (Hazra et al., 2007b), suggesting the existence of flip-flop kinetics. The mean concentration–time profiles of MPL in livers from male (Cohort 4) and female (Cohort 2) rats are shown in Fig. 2D. Liver concentrations of MPL peaked at about 0.75 hour after dosing in both sexes, similar to the plasma Tmax. The AUC in liver was higher in females (males: AUC, 20,285 ± 1231 ng h/g; females: 101,588 ± 5425), consistent with values in plasma. The Cmax of MPL in liver was higher in females (males: Cmax, 8581 ± 1491 ng/g; females: 41,349 ± 7767 ng/g).
Fig. 2.

PK profiles of MPL (50 mg/kg IM) in (A) plasma of 7-week-old (Cohort 4) and 11-week-old (Cohort 5) male rats; (B) plasma of estrus females (Cohort 1), proestrus females (Cohort 2), noncycling (NC) females (Cohort 3), and males (Cohorts 4 and 5); (C) plasma of females (Cohorts 1–3) and males (Cohorts 4 and 5); and (D) livers of females (Cohort 2) and males (Cohort 4). Symbols depict either replicate data from each animal (A) or mean observed data ± S.D. (B–D). Symbols are defined within the figure.
TABLE 1.
PK parameters using NCA for plasma MPL in the study cohorts after intramuscular administration
| Experimental Group (n) | λz (h−1) | T1/2_λz (h) | Cmax (ng/ml) | AUC0-∞ (h × ng/ml)b | CL/F (ml/h per kilogram) |
|---|---|---|---|---|---|
| Cohort 1: estrus female (30) | 0.24 | 2.9 | 5348 ± 488 | 10,489 ± 1054 | 4767 ± 479 |
| Cohort 2: proestrus female (39) | 0.27 | 2.6 | 3859 ± 777 | 9142 ± 646 | 5469 ± 386 |
| Cohort 3: noncycler female (20) | 0.20 | 3.4 | 3796 ± 188 | 8735 ± 601 | 5723 ± 394 |
| Cohort 4: male 7 wk (31) | 0.25 | 2.7 | 1470 ± 109 | 2808 ± 167 | 17,806 ± 1059 |
| Cohort 5: male 11 wk (23) | 0.26 | 2.6 | 1054 ± 94 | 2885 ± 312 | 17,334 ± 1875 |
| All females | 0.24 | 2.9 | 4204 ± 323 | 9751 ± 482 | 5128 ± 252 |
| All males | 0.28 | 2.5 | 1174 ± 194 | 2901 ± 185 | 17,232 ± 1099 |
CL/F, clearance; T1/2, half-life.
Values represent mean ± S.E.
AUC calculated using Bailer’s method for sparse (destructive) sampling.
mPBPK Analysis of MPL.
The final mPBPK/PD model is displayed in Fig. 1. The mPBPK model features utilization of a physiologic structure along with parameters (organ volumes and blood flows) applicable to rats, obtained from the literature (Kawai et al., 1998), and fixed in the model fitting. Measurements of total MPL in plasma and livers of male and female rats permitted estimation of a male-specific hepatic clearance pathway. It was assumed that the nonhepatic route of clearance was the same in both sexes. This physiologically-based clearance structure was based upon multiple sources of evidence: 1) marked differences in the in vitro depletion kinetics of MPL in male and female rat liver homogenates (Ayyar et al., 2019b); 2) sex differences in cytochrome P450 activity in rodents, particularly CYP3A2 (Kato and Yamazoe, 1992); 3) in vitro microsomal and in vivo studies documenting sex differences in DEX metabolism and disposition in rats (Lamiable et al., 1991; Tomlinson et al., 1997); and 4) occurrence of extrahepatic (renal) metabolism of a highly similar steroid, prednisolone, in rats (Rocci et al., 1981). All intramuscular PK data were modeled jointly (see ADAPT5 model code for PK estimation in the Supplemental Materials).
The PK profiles of total MPL in plasma and liver after 50 mg/kg IM doses to female and male rats as well as the model fittings are shown in Fig. 3. The PK parameter estimates are listed in Table 2. Both plasma and tissue profiles of MPL were captured reasonably well with the mPBPK model, with good precision of the estimated parameters. The absorption pattern following intramuscular dosing was similar in both sexes, with a rapid rise in concentrations followed by a slower, sustained absorptive phase. Two first-order absorption processes, one with a faster absorption rate (ka1: male: 0.8 hour−1, 8.8% CV; female: 4.1 hours−1, 24.8% CV) than the other (ka2: 0.17 hour−1, 17.3% CV), were optimal to describe the release of MPL from the injection site. Parameter values for overall bioavailability (F) and fractions absorbed by each route (Fr and 1 − Fr) were fixed in both sexes based on a previous PK analysis in normal male rats (Hazra et al., 2007b). The KP values for PPT and RPT were fixed to values obtained for muscle and lung measured in vitro that were in good agreement with in vivo estimates (Ayyar et al., 2019b). Unbound fractions of MPL in male and female plasma were also fixed to measured values (Ayyar et al., 2019b). The KP of liver was jointly estimated from the liver concentration–time profiles in both sexes as 12.6 (7.6% CV). The unbound intrinsic clearance of MPL in male liver (CLu,int,male) was estimated as 2987 ml/h (12.7% CV). Nonhepatic clearance of MPL in males (347 ml/h, 4.2% CV), assumed to be equal to the overall clearance in female rats, was lower compared with the hepatic clearance of MPL (686 ml/h). Total clearance estimates in both males and females were similar to values obtained by NCA (Table 1). The overall clearance of MPL (1.03 l/h) in males (i.e., CLhep + CLnon-hep) was comparable to previously reported compartmental model estimates in normal (1.2 l/h) and adrenalectomized (ADX) (0.77 l/h) male rats (Sun et al., 1998; Hazra et al., 2007b).
Fig. 3.

Model fittings of the concentration–time profiles of total MPL in plasma (A) and liver (B) of male and female rats that received 50 mg/kg IM doses of MPL. Symbols depict mean observed data ± S.D. Solid lines represent model-fitting results for each sex. Symbols are defined within the figure.
TABLE 2.
PK parameter values for the mPBPK model of MPL
| Parameter | Definition | Estimate (% CV) |
|---|---|---|
| F | Bioavailability | 0.214a |
| Fr | Fraction absorbed by ka1 | 0.725a |
| ka1 (h−1) | Absorption rate constant | 0.8 (8.8)b/4.1 (24.8)c |
| ka2 (h−1) | Absorption rate constant | 0.17 (17.3) |
| CLEH (ml/h) | Extrahepatic clearance | 347 (4.2) |
| CLu,int,male (ml/h) | Hepatic unbound intrinsic clearance (male) | 2987 (12.7) |
| KP, hep | Partition coefficient—liver | 12.6 (7.6) |
| KP, PPT (muscle) | Partition coefficient—poorly perfused tissue | 1.4d |
| KP, RPT (lung) | Partition coefficient—richly perfused tissue | 2.1d |
| fu,p | MPL fraction unbound in plasma | 0.4d |
Parameter values fixed from Hazra et al. (2007b).
Male.
Female.
Fixed to in vitro estimates from Ayyar et al. (2019).
Simulations of mean concentrations of MPL following 10 and 50 mg/kg IV bolus dosing in male rats using the final mPBPK model are shown in Fig. 4. Replicate data from two separate studies in normal male rats (Hazra et al., 2007b; Jin and Jusko, 2009) are also shown to examine the predictability of our model. The model performed reasonably well in simulating the disposition kinetics of MPL following intravenous dosing in males.
Fig. 4.

Model-based simulations of the PK profiles of MPL following 10 mg/kg (dashed line) and 50 mg/kg (solid line) IV doses in normal male rats. Triangles are low-dose data obtained from Jin and Jusko (2009), whereas open circles are high-dose data taken from Hazra et al. (2007b). Parameter values used in the simulations are listed in part in Table 2.
CST Suppression.
Plasma CST concentrations after IM dosing of 50 mg/kg MPL in males are shown in Fig. 5. The profile of CST in control and treated animals in Cohort 4 was taken from Hazra et al. (2007b). Also shown are the simulated circadian baseline concentrations of CST in control animals. Data for characterizing the circadian rhythm of CST in females during estrus and proestrus were obtained from Atkinson and Waddell (1997). No discernable differences in CST dynamics were observed between males aged 7 and 11 weeks (Cohorts 4 and 5; data not shown). Although differences in the mesor of the basal rhythm of CST were modest between males (102 ± 21 ng/ml) and estrus-phased females (129 ± 11 ng/ml), rats in proestrus displayed greatly elevated rhythms (mesor 246 ± 14 ng/ml). Therefore, sex- and estrous phase-specific baseline rhythms were incorporated for modeling the PD effects of MPL on CST.
Fig. 5.

Model fittings of plasma CST dynamics in estrus female rats (left), proestrus female rats (middle), and male rats (right) that received 50 mg/kg IM doses of MPL. Symbols depict mean observed data ± S.D. Solid lines represent model-fitting results for each group after MPL dosing, and broken line depicts simulated profile of CST rhythm in control animals. Dark (shaded) and light (unshaded) time periods are indicated.
Significantly higher CST concentrations were observed 15 minutes postdosing in all animal groups compared with nondosed controls, although MPL was given at the nadir of CST. This is consistent with previous observations in male rats (Yao et al., 2008) and is likely attributable to stress during dosing. An empirical zero-order secretion rate of CST independent of daily CST production rate was able to capture these early high concentrations of CST in all groups. The latter averaged 535.1 ng/ml (18.1% CV) in males, 1292 ng/ml (13.3% CV) in estrus females, and 1397 ng/ml (12.9% CV) in proestrus females over a period of 20 minutes (12.8% CV). This indicates a higher initial stress effect in female rats. Following the brief spike, MPL elicited marked adrenal suppression in all groups, which persisted up to 8 hours postdosing, and CST slowly began to return to its daily rhythm by 24 hours. Even after 24 hours, CST concentrations were visibly lower compared with the controls. The indirect response model describing the inhibitory effect of MPL on the endogenous secretion rate of CST (eq. 11) was able to capture the CST profile across all groups reasonably well. The Fourier coefficients as well as estimated model parameters are listed in Table 3. The IC50,CST values of 3.4 ng/ml (47.6% CV) in males, 41.6 ng/ml (48% CV) in estrus females, and 12.9 ng/ml (45.6% CV) in proestrus females indicated strong inhibitory effects of MPL on CST, with some differences in drug sensitivity across the groups. The turnover rate constant of 1.38 hours−1 (12.6% CV) translates to a plasma half-life for total CST of ∼30 minutes. This is in close agreement with a previously reported value (25 minutes) based on the elimination kinetics of CST after a 5 mg/kg IV bolus (Sainio et al., 1988).
TABLE 3.
PD parameter estimates for plasma CST suppression by MPL
| Parameter | Definition | Estimate (% CV) |
|---|---|---|
| Circadian Rhythm Parameters for CST | ||
| T (h) | Biorhythmic period | 24a,b,c |
| a0 | Fourier coefficient | 115.7a,b/254.4b |
| a1 | −120.9a,b/−168.7b | |
| a2 | 38.8a,b/56.9b | |
| b1 | −41.1a,b/−95.6b | |
| b2 | 1.6a,b/29.1b | |
| Pharmacodynamic Parameters | ||
| kdeg,CST (h−1) | Loss rate constant for CST | 1.38 (12.6)a,b,c |
| τ (h) | Time to CSTstress | 0.34 (12.8)a,b,c |
| CSTstress (ng/ml) | Maximal CST due to stress | 535 (18.0)a/1292 (13.3)b/1397 (12.9)c |
| IC50,CST (ng/ml) | Half-maximal inhibition concentration | 3.4 (47.6)a/41.6 (48.0)b/12.9 (45.6)c |
Male.
Estrus female.
Proestrus female.
Discussion
This study extends previous assessments of CS from male to female rats, a seemingly more physiologically complex, however, relevant animal model in preclinical research. Consideration of drug kinetics, correction for protein binding, attention to drug distribution disequilibria, and examination of endogenous agonist concentrations are important steps prior to evaluating the determinants of PD variability (Levy, 1998). The studies reported in this work (Part II) as well as in Part I serve as a prelude to our PD studies of sex-dependent variability in the receptor/gene-mediated actions of MPL in tissues (Part III).
MPL Pharmacokinetics.
Comparison of MPL PK following intramuscular dosing in male and female rats revealed pronounced differences in systemic and liver exposures (Fig. 2; Table 1). To our knowledge, this is the first study to characterize the PK of MPL in female rats and to document related sex differences. The findings are consistent with a previous study in which a significant (∼3-fold) increase of DEX clearance was observed in males compared with ovariectomized female rats with and without estradiol replacement therapy (Lamiable et al., 1991). Pronounced effects of estradiol and testosterone have been documented on hepatic drug metabolism (Brodie, 1956; Kato, 1974). We examined whether PK differed as a function of natural variations in plasma sex hormone concentrations. No significant differences were observed in MPL exposure between the female groups. This is consistent with previous findings of no difference in plasma AUC of DEX for female ovariectomized rats given DEX alone and those given DEX with estradiol replacement therapy (Lamiable et al., 1991). Similarly, ketamine disposition in female rats dosed within diestrus and proestrus phases showed no difference (Saland and Kabbaj, 2018). Sex-specific forms of cytochrome P450 have been isolated in the rat and are important in determining sex-dependent steroid metabolism (Zaphiropoulos et al., 1989). For instance, hepatic content of CYP3A2 was about 128 ± 6.6 pmol/mg protein in males and was undetectable in females (Kato and Yamazoe, 1992; Ghosal et al., 1996) by 7 to 8 weeks of age. DEX metabolism in microsomes from male and female rat livers produced distinct chromatographic profiles, with all detectable metabolites formed in much greater quantities in male microsomes (Tomlinson et al., 1997). During our assessments of MPL binding in liver homogenates (Ayyar et al., 2019b), marked differences in MPL loss kinetics were observed in male versus female liver homogenates. These findings collectively indicate that sex-specific hepatic metabolism can explain the lower exposures of MPL observed in vivo in male rats. The lack of hepatic metabolism of MPL in female rats prompts the consideration of other clearance routes for MPL. Urinary excretion of unchanged MPL is negligible (0.5%) in rats (Kong and Jusko, 1991). The role of extrahepatic reversible metabolism of MPL by 11β-hydroxysteroid dehydrogenase enzymes has been assessed in the rabbit (Ebling et al., 1985), but occurs to a much lesser extent in rats (Haughey and Jusko, 1988). Prednisolone undergoes renal biotransformation to form 20β-hydroxylated metabolites in isolated perfused kidneys from the rat and rabbit (Rocci et al., 1981; Rocci and Jusko, 1981). All of these assessments were, however, made in male animals. To prevent overparameterization, the mPBPK model assumed that nonhepatic metabolism was the only clearance route in females, and that the extent of extrahepatic metabolism was same in both sexes. Male-specific hepatic clearance was estimated as 686 ml/h, which was slightly lower compared with clearance in perfused male rat livers (858 ml/h) (Kong and Jusko, 1991). The liver KP of MPL in liver was 12.6, indicating a greater degree of steroid uptake compared with muscle (1.4) and lung (2.1). The total volume of distribution at steady state (1.96 l/kg), calculated per the Gillette equation (Gillette, 1971), was comparable to previous compartmental model estimates from male rats (∼1.7 l/kg) (Hazra et al., 2007b; Jin and Jusko, 2009). The model fitting somewhat underpredicted the plasma Cmax in females, despite estimation of a higher ka1 in that sex. Sex differences in the drug distribution volume and/or release from the gastrocnemius injection site are possible (Mierzejewska-Krzyżowska et al., 2011). Although the developed model reasonably captured the intramuscular PK in both sexes (Fig. 3) and well predicted the intravenous kinetics of MPL in male rats (Fig. 4), additional studies examining MPL metabolism and intravenous kinetics in females would further strengthen our findings. Prominent species differences in CS PK exist between rodents and humans. A previous study in healthy volunteers indicated that women exhibited a greater MPL clearance and a shorter elimination half-life than men (Lew et al., 1993). Increased activity of human CYP3A in females (Hunt et al., 1992) was posed as the major contributing factor. In contrast, CYP3A2 activity is male-specific in rats. Thus, although sex differences in MPL disposition may be influenced by a common pathway (viz. hepatic metabolism), pronounced quantitative differences across species can produce differing kinetic behavior. No sex difference existed in MPL plasma binding in rodents and humans, but the unbound fractions were quantitatively different (40% vs. 24%) (Lew et al., 1993; Ayyar et al., 2019b).
mPBPK Model.
An enhanced version (Fig. 1) of the basic mPBPK model (Cao and Jusko, 2012) was developed and applied to characterize MPL concentration–time profiles. The mPBPK models provide a realistic basis for describing plasma PK data and differ from traditional mammillary models in ways of initial distribution space, physiologic assignments, and restrictions, as well as flexibilities in handling different clearance mechanisms (Cao and Jusko, 2012). The previous generation model maintained mass-balance by constraining the sum of each tissue compartment volume to the drug distribution space in the body (i.e., VT1 + VT2 + Vblood = BW or extracellular fluid volume). Under this configuration, the volume of one tissue compartment (VT1) was estimated during model fitting, whereas the volume of the other compartment (VT2) was constrained as BW − (VT1 + Vblood). As such, tissue compartment volumes could vary based on qualitative aspects of the PK data. In addition, estimation of a fd parameter, which represented a composite of the fractional blood flow as well as drug permeability, was also necessary. The present model builds upon this framework by incorporating two physiologically lumped tissue spaces; a poorly perfused tissue space VPPT (defined as Vadipose + Vmuscle + Vskin); and a rapidly perfused space VRPT [defined as BW − (VPPT + Vblood)]. In addition, blood flow through VPPT was fixed as Qadipose + Qmuscle + Qskin (QPPT), and blood flow for VRPT was constrained as QCO − QPPT. Tissues of particular interest can be readily separated from the lumped spaces and assigned with independent parameters, as was done with liver in this study (Fig. 1). Organ-specific clearance mechanisms can also be accounted for as necessary. Although, in this case, in vitro values of KP in muscle and lung (Ayyar et al., 2019b) were fixed for modeling in vivo MPL PK (Table 2), our general model structure permits estimation of one or two KP values, depending on the plasma PK profiles and data available. The proposed mPBPK model is expected to show applicability for characterizing the concentration–time profiles of other small-molecule drugs as well.
CST Suppression.
A stress-related surge in CST, which peaked around the first sampling point of 15 minutes, was apparent in all animal groups due to handling of the rats during dosing. This surge at peak was evidently higher in females (1300–1400 ng/ml) compared with males (∼500 ng/ml), consistent with naturally heightened basal CST concentrations in control female rats (Atkinson and Waddell, 1997). An empirical zero-order stress response (based on observed data) suitably captured this phenomenon. Exogenous CS inhibits endogenous CST secretion rapidly by a negative feedback loop in the hypothalamic-pituitary-adrenal axis in humans as well as in rodents (Yamada and Satoh, 1985; Lew et al., 1993). Modest sex differences existed in the sensitivity of adrenal suppression to MPL, as indicated by slightly higher mean IC50 values in estrus and proestrus females (42 and 13 ng/ml) compared with males (3.4 ng/ml). The percentage of CV for this parameter was, however, relatively large (40%–50%). The estimated IC50 value in male rats was very similar to a previously reported value (2.4 ng/ml) (Jin and Jusko, 2009). The return profile of CST after suppression by MPL was similar across the groups despite higher MPL exposure in females, likely attributable to the higher IC50 in females. The time for return to normal rhythm in proestrus rats is also influenced by a significantly higher baseline compared with the estrus and male animals. The kdeg,CST was jointly estimated as 1.4 hours−1, which was equal to a previously reported estimate (Jin and Jusko, 2009).
In summary, sex differences in the PK of MPL were observed following intramuscular dosing in rats, attributable, at least in part, to sex-dependent hepatic clearance. Modest sex differences were also observed in the sensitivity of adrenal suppression to MPL. A mPBPK/PD model was developed, which included physiologic and pharmacologic factors that accounted for the sex differences in MPL PK and dynamics of CST suppression. This work serves as the foundation toward more intricate assessments of sex differences in the receptor/gene-mediated PD of MPL in tissues (Ayyar et al., 2019a).
Acknowledgments
We thank Piotr Gawel for the technical assistance with animal experiments.
Abbreviations
- λz
terminal slope
- AUC
area under the plasma–concentration time curve
- BW
body weight
- CS
corticosteroid
- CST
corticosterone
- DEX
dexamethasone
- KP
tissue:plasma partition coefficient
- mPBPK
minimal physiologically-based PK
- MPL
methylprednisolone
- NCA
noncompartmental analysis
- PD
pharmacodynamic
- PG
pharmacogenomic
- PK
pharmacokinetic
- PPT
poorly perfused tissues
- RPT
rapidly perfused tissues
- Tmax
corresponding time when Cmax is reached
Authorship Contributions
Participated in research design: Ayyar, DuBois, Almon, Jusko.
Conducted experiments: Ayyar, DuBois.
Performed data analysis: Ayyar, Nakamura, Jusko.
Wrote or contributed to the writing of the manuscript: Ayyar, DuBois, Nakamura, Almon, Jusko.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Grants GM24211 and GM131800].
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Atkinson HC, Waddell BJ. (1997) Circadian variation in basal plasma corticosterone and adrenocorticotropin in the rat: sexual dimorphism and changes across the estrous cycle. Endocrinology 138:3842–3848. [DOI] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2017) Mechanistic multi-tissue modeling of glucocorticoid-induced leucine zipper regulation: integrating circadian gene expression with receptor-mediated corticosteroid pharmacodynamics. J Pharmacol Exp Ther 363:45–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, DuBois DC, Almon RR, Jusko WJ. (2019a) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part III: estrous cycle and estrogen receptor-dependent antagonism of GILZ enhancement by corticosteroids. J Pharmacol Exp Ther 370:327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyar VS, Song D, DuBois DC, Almon RR, Jusko WJ. (2019b) Modeling corticosteroid pharmacokinetics and pharmacodynamics, part I: determination and prediction of dexamethasone and methylprednisolone tissue binding in the rat. J Pharmacol Exp Ther 370:318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie BB. (1956) Pathways of drug metabolism. J Pharm Pharmacol 8:1–17. [DOI] [PubMed] [Google Scholar]
- Burns CM. (2016) The history of cortisone discovery and development. Rheum Dis Clin North Am 42:1–14, vii. [DOI] [PubMed] [Google Scholar]
- Cao Y, Jusko WJ. (2012) Applications of minimal physiologically-based pharmacokinetic models. J Pharmacokinet Pharmacodyn 39:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton JA, Collins FS. (2014) Policy: NIH to balance sex in cell and animal studies. Nature 509:282–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danska JS. (2014) Sex matters for mechanism. Sci Transl Med 6:258fs40. [DOI] [PubMed] [Google Scholar]
- D’Argenio D, Schumitzky A, Wang X. (2009) ADAPT 5 User’s Guide: Pharmacokinetic/Pharmacodynamic Systems Analysis Software, BioMedical Simulations Resource, Los Angeles, CA. [Google Scholar]
- Dickmeis T. (2009) Glucocorticoids and the circadian clock. J Endocrinol 200:3–22. [DOI] [PubMed] [Google Scholar]
- Ebling WF, Milsap RL, Szefler SJ, Jusko WJ. (1986) 6 alpha-Methylprednisolone and 6 alpha-methylprednisone plasma protein binding in humans and rabbits. J Pharm Sci 75:760–763. [DOI] [PubMed] [Google Scholar]
- Ebling WF, Szefler SJ, Jusko WJ. (1985) Methylprednisolone disposition in rabbits: analysis, prodrug conversion, reversible metabolism, and comparison with man. Drug Metab Dispos 13:296–304. [PubMed] [Google Scholar]
- Ferron GM, Jusko WJ. (1998) Species- and gender-related differences in cyclosporine/prednisolone/sirolimus interactions in whole blood lymphocyte proliferation assays. J Pharmacol Exp Ther 286:191–200. [PubMed] [Google Scholar]
- Gandhi M, Aweeka F, Greenblatt RM, Blaschke TF. (2004) Sex differences in pharmacokinetics and pharmacodynamics. Annu Rev Pharmacol Toxicol 44:499–523. [DOI] [PubMed] [Google Scholar]
- Ghosal A, Sadrieh N, Reik L, Levin W, Thomas PE. (1996) Induction of the male-specific cytochrome P450 3A2 in female rats by phenytoin. Arch Biochem Biophys 332:153–162. [DOI] [PubMed] [Google Scholar]
- Gillette JR. (1971) Factors affecting drug metabolism. Ann N Y Acad Sci 179:43–66. [DOI] [PubMed] [Google Scholar]
- Haughey DB, Jusko WJ. (1988) Analysis of methylprednisolone, methylprednisone and corticosterone for assessment of methylprednisolone disposition in the rat. J Chromatogr A 430:241–248. [DOI] [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007a) Modeling receptor/gene-mediated effects of corticosteroids on hepatic tyrosine aminotransferase dynamics in rats: dual regulation by endogenous and exogenous corticosteroids. J Pharmacokinet Pharmacodyn 34:643–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazra A, Pyszczynski N, DuBois DC, Almon RR, Jusko WJ. (2007b) Pharmacokinetics of methylprednisolone after intravenous and intramuscular administration in rats. Biopharm Drug Dispos 28:263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CM, Westerkam WR, Stave GM. (1992) Effect of age and gender on the activity of human hepatic CYP3A. Biochem Pharmacol 44:275–283. [DOI] [PubMed] [Google Scholar]
- Jin JY, Jusko WJ. (2009) Pharmacodynamics of glucose regulation by methylprednisolone. II. Normal rats. Biopharm Drug Dispos 30:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato R. (1974) Sex-related differences in drug metabolism. Drug Metab Rev 3:1–32. [DOI] [PubMed] [Google Scholar]
- Kato R, Yamazoe Y. (1992) Sex-specific cytochrome P450 as a cause of sex- and species-related differences in drug toxicity. Toxicol Lett 64–65:661–667. [DOI] [PubMed] [Google Scholar]
- Kawai R, Mathew D, Tanaka C, Rowland M. (1998) Physiologically based pharmacokinetics of cyclosporine A: extension to tissue distribution kinetics in rats and scale-up to human. J Pharmacol Exp Ther 287:457–468. [PubMed] [Google Scholar]
- Kei-ichiro M, Satoshi O, Hiroko T. (2000) Physiology of reproduction, in The Handbook of Experimental Animals: The Laboratory Rat (Krinke GJ. ed) pp 145–177, Academic Press, New York. [Google Scholar]
- Kirwan JR, The Arthritis and Rheumatism Council Low-Dose Glucocorticoid Study Group (1995) The effect of glucocorticoids on joint destruction in rheumatoid arthritis. N Engl J Med 333:142–146. [DOI] [PubMed] [Google Scholar]
- Kong AN, Jusko WJ. (1991) Disposition of methylprednisolone and its sodium succinate prodrug in vivo and in perfused liver of rats: nonlinear and sequential first-pass elimination. J Pharm Sci 80:409–415. [DOI] [PubMed] [Google Scholar]
- Kovats S. (2015) Estrogen receptors regulate innate immune cells and signaling pathways. Cell Immunol 294:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzyzanski W, Chakraborty A, Jusko WJ. (2000) Algorithm for application of Fourier analysis for biorhythmic baselines of pharmacodynamic indirect response models. Chronobiol Int 17:77–93. [DOI] [PubMed] [Google Scholar]
- Lamiable D, Vistelle R, Fay R, Bensussan B, Millart H, Wiczewski M, Choisy H. (1991) Influence of sex and oestrogen replacement on the disposition of dexamethasone in rats. Fundam Clin Pharmacol 5:733–740. [DOI] [PubMed] [Google Scholar]
- Levy G. (1998) Predicting effective drug concentrations for individual patients: determinants of pharmacodynamic variability. Clin Pharmacokinet 34:323–333. [DOI] [PubMed] [Google Scholar]
- Lew KH, Ludwig EA, Milad MA, Donovan K, Middleton E, Jr, Ferry JJ, Jusko WJ. (1993) Gender-based effects on methylprednisolone pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 54:402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mager DE, Lin SX, Blum RA, Lates CD, Jusko WJ. (2003) Dose equivalency evaluation of major corticosteroids: pharmacokinetics and cell trafficking and cortisol dynamics. J Clin Pharmacol 43:1216–1227. [DOI] [PubMed] [Google Scholar]
- Mierzejewska-Krzyżowska B, Drzymała-Celichowska H, Celichowski J. (2011) Gender differences in the morphometric properties of muscle fibres and the innervation ratio of motor units in rat medial gastrocnemius muscle. Anat Histol Embryol 40:249–255. [DOI] [PubMed] [Google Scholar]
- Overman RA, Yeh JY, Deal CL. (2013) Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken) 65:294–298. [DOI] [PubMed] [Google Scholar]
- Rocci ML, Jusko WJ. (1981) Analysis of prednisone, prednisolone and their 20β-hydroxylated metabolites by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl 224:221–227. [Google Scholar]
- Rocci ML, Jr, Szefler SJ, Acara M, Jusko WJ. (1981) Prednisolone metabolism and excretion in the isolated perfused rat kidney. Drug Metab Dispos 9:177–182. [PubMed] [Google Scholar]
- Sainio EL, Lehtola T, Roininen P. (1988) Radioimmunoassay of total and free corticosterone in rat plasma: measurement of the effect of different doses of corticosterone. Steroids 51:609–622. [DOI] [PubMed] [Google Scholar]
- Saland SK, Kabbaj M. (2018) Sex differences in the pharmacokinetics of low-dose ketamine in plasma and brain of male and female rats. J Pharmacol Exp Ther 367:393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäcke H, Döcke WD, Asadullah K. (2002) Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther 96:23–43. [DOI] [PubMed] [Google Scholar]
- Song D, DuBois DC, Almon RR, Jusko WJ. (2018) Modeling sex differences in anti-inflammatory effects of dexamethasone in arthritic rats. Pharm Res 35:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YN, DuBois DC, Almon RR, Jusko WJ. (1998) Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm 26:289–317. [DOI] [PubMed] [Google Scholar]
- Tomlinson ES, Maggs JL, Park BK, Back DJ. (1997) Dexamethasone metabolism in vitro: species differences. J Steroid Biochem Mol Biol 62:345–352. [DOI] [PubMed] [Google Scholar]
- van Staa TP, Leufkens HG, Abenhaim L, Begaud B, Zhang B, Cooper C. (2000) Use of oral corticosteroids in the United Kingdom. QJM 93:105–111. [DOI] [PubMed] [Google Scholar]
- Whirledge S, Cidlowski JA. (2013) Estradiol antagonism of glucocorticoid-induced GILZ expression in human uterine epithelial cells and murine uterus. Endocrinology 154:499–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whirledge S, Xu X, Cidlowski JA. (2013) Global gene expression analysis in human uterine epithelial cells defines new targets of glucocorticoid and estradiol antagonism. Biol Reprod 89:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Satoh T. (1985) In vivo effects of glucocorticoids on serum corticosterone levels in rats. Res Commun Chem Pathol Pharmacol 47:441–444. [PubMed] [Google Scholar]
- Yao Z, DuBois DC, Almon RR, Jusko WJ. (2008) Pharmacokinetic/Pharmacodynamic modeling of corticosterone suppression and lymphocytopenia by methylprednisolone in rats. J Pharm Sci 97:2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaphiropoulos PG, Mode A, Norstedt G, Gustafsson JA. (1989) Regulation of sexual differentiation in drug and steroid metabolism. Trends Pharmacol Sci 10:149–153. [DOI] [PubMed] [Google Scholar]