

Abstract
It is well recognized that many cancers are addicted to a constant supply of fatty acids (FAs) and exhibit brisk de novo FA synthesis. Upregulation of a key lipogenic enzyme, fatty acid synthase (FASN), is a near-universal feature of human cancers and their precursor lesions, and has been associated with chemoresistance, tumor metastasis, and diminished patient survival. FASN inhibition has been shown to be effective in killing cancer cells, but progress in the field has been hindered by off-target effects and poor pharmaceutical properties of candidate compounds. Our initial hit (compound 1) was identified from a high-throughput screening effort by the Sanford-Burnham Center for Chemical Genomics using purified FASN thioesterase (FASN-TE) domain. Despite being a potent inhibitor of purified FASN-TE, compound 1 proved highly unstable in mouse plasma and only weakly cytotoxic to breast cancer (BC) cells in vitro. An iterative process of synthesis, cytotoxicity testing, and plasma stability assessment was used to identify a new lead (compound 41). This lead is more cytotoxic against multiple BC cell lines than tetrahydro-4-methylene-2S-octyl-5-oxo-3R-furancarboxylic acid (the literature standard for inhibiting FASN), is stable in mouse plasma, and shows negligible cytotoxic effects against nontumorigenic mammary epithelial cells. Compound 41 also has drug-like physical properties based on Lipinski’s rules and is, therefore, a valuable new lead for targeting fatty acid synthesis to exploit the requirement of tumor cells for fatty acids.
SIGNIFICANCE STATEMENT
An iterative process of synthesis and biological testing was used to identify a novel thioesterase domain FASN inhibitor that has drug-like properties, is more cytotoxic to breast cancer cells than the widely used tetrahydro-4-methylene-2S-octyl-5-oxo-3R-furancarboxylic acid, and has negligible effects on the growth and proliferation of noncancerous mammary epithelial cells. Our studies have confirmed the value of using potent and selective FASN inhibitors in the treatment of BC cells and have shown that the availability of exogenous lipoproteins may impact both cancer cell FA metabolism and survival.
Introduction
Cancer cells exhibit fundamental metabolic alterations that drive and maintain the malignant phenotype. Since the 1920s it has been known that, in contrast to most normal tissues, cancer cells avidly take up glucose and convert it to lactate through the glycolytic pathway, irrespective of whether oxygen is present (aerobic glycolysis, the Warburg effect) (Warburg, 1956). Aerobic glycolysis not only provides cancer cells with energy, but also diverts carbon to the synthesis of cellular building blocks, including lipids (Zaidi et al., 2013). Heightened de novo lipid synthesis, mediated through upregulation of the requisite enzymes, including fatty acid synthase (FASN), is considered a near-universal hallmark of most human tumor cells and their precursor lesions (Menendez and Lupu, 2007; Hanahan and Weinberg, 2011; Ward and Thompson, 2012; Benjamin et al., 2015; Gonzalez-Guerrico et al., 2016). Increased FASN expression is associated with cancer progression, higher risk of recurrence and metastasis, reduced response to classic chemotherapeutic agents, multidrug resistance, and diminished patient survival (Rysman et al., 2010; Liu et al., 2013; Wu et al., 2014; Jung et al., 2015; Heuer et al., 2017; Menendez and Lupu, 2017).
FASN is a multifunctional, homodimeric enzyme. Each monomer is a large polypeptide comprised of seven catalytic domains working in concert to synthesize the 16-carbon saturated fatty acid (FA) palmitate from acetyl-CoA and malonyl-CoA (Buckley et al., 2017). Palmitate and palmitate-derived lipids have integral roles in cell metabolism, membrane architecture, protein localization, and intracellular signaling (Menendez and Lupu, 2007; Flavin et al., 2010; Daniëls et al., 2014; Gonzalez-Guerrico et al., 2016). Studies of the mechanistic role of FASN have revealed that it fuels cancer cell proliferation and malignant progression by: 1) generating FA precursors required for membrane synthesis and ATP production (Buckley et al., 2017; Luo et al., 2017; Jafari et al., 2019); 2) altering membrane composition and fluidity to confer resistance to endogenous and exogenous insults, including chemotherapy and membrane peroxidative damage (Liu et al., 2008; Rysman et al., 2010); 3) modulating the composition of lipid raft membrane microdomains, which house the membrane receptor tyrosine kinases that modulate key cellular processes including signal transduction, intracellular trafficking, cell polarization, and migration (Swinnen et al., 2003; Menendez et al., 2005); 4) providing palmitate for the covalent post-translational modification (S-acylation and palmitoylation) of tumor-promoting signaling proteins (e.g., Wnt, tubulin, and H/N/K-RAS) (Levental et al., 2010; Anderson and Ragan, 2016; Heuer et al., 2017); 5) regulating the formation of structures that drive metastasis and invasion (Zaytseva et al., 2012; Benjamin et al., 2015; Singh et al., 2015; Ventura et al., 2015; Gonzalez-Guerrico et al., 2016; Wang et al., 2016; Jafari et al., 2019); and 6) generating oncogenic signaling lipids, such as diacylglycerol and phosphatidylinositol (Benjamin et al., 2015; Heuer et al., 2017; Wagner et al., 2017). In these various ways, FASN modulates cancer cell proliferation, survival, extracellular matrix organization, migration and invasion, and the expression and activity of signaling networks required for maintaining the malignant phenotype (Menendez and Lupu, 2017).
Participation of FASN in this plethora of cancer-promoting pathways positions the enzyme as an attractive therapeutic target. Pharmacologic or RNA interference–mediated inhibition of FASN has been shown to attenuate cancer cell growth, induce apoptosis, and diminish metastatic potential of cancer cells in many different models, while exerting no direct effects on the growth and survival of normal cells (Kuhajda et al., 1994; Pizer et al., 1996; De Schrijver et al., 2003; Kridel et al., 2004; Menendez and Lupu, 2007, 2017; Zaytseva et al., 2012, 2014; Li and Cheng, 2014; Ventura et al., 2015; Röhrig and Schulze, 2016; Buckley et al., 2017). Clinical efficacy of FASN inhibitors (FASNis) alone or in combination therapy has begun to be realized with 4-(1-(4-cyclobutyl-2-methyl-5-(5-methyl-4H-1,2,4-triazol-3-yl)benzoyl)piperidin-4-yl)benzonitrile (TVB-2640), a compound developed by 3-V Biosciences (https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2016-01710&r=1) (Dean et al., 2016). However, progress in the field continues to be slowed by off-target effects and poor pharmaceutical properties of candidate FASN inhibitors, as reviewed in Menendez and Lupu (2007, 2017), Flavin et al. (2010), Liu et al. (2010), Kinlaw et al. (2016), and Röhrig and Schulze (2016). The generation of more potent and selective FASN inhibitors has potential application to a wide range of cancer types, and is therefore an important goal.
Imidazole-4,5-dicarboxylic acid is a useful scaffold for drug discovery (Baures, 2006) and has been used to create inhibitors of human immunodeficiency virus protease (Baures, 1999) to target HL-60 promyelocytic leukemia cells (Perchellet et al., 2005) and in the design of inhibitors of protein-protein interactions (VanCompernolle et al., 2003). The ability to readily derivatize the scaffold and the conformational behavior resulting from a strong intramolecular hydrogen bond (Baures et al., 2002; Rush et al., 2005) were features that enabled the inclusion of these compounds in the Molecular Library Small Molecule Repository for screening within the National Institutes of Health Molecular Libraries Initiative (Austin et al., 2004). A high-throughput bioassay was used to identify inhibitors of the FASN thioesterase (FASN-TE) domain. This initial screening effort and a series of confirmatory screens identified compound 1 as the most potent inhibitor against this domain from the library of 362,050 compounds. Compound 1 and several closely related derivatives (Fig. 1) were found to be highly unstable in mouse plasma and demonstrated limited cytotoxicity against breast cancer (BC) cells in vitro. Multiple rounds of rational modification were used to overcome the stability issues of compound 1 and generate stable, potent, and selective FASNis. The new lead compound reported herein is a structurally unique FASN-TE inhibitor that is more cytotoxic across a panel of BC cell lines than literature standards, such as (−)-4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75), and has displayed selectivity for BC cells. These studies have additionally yielded insights into the different pathways that cancer cells use to acquire the FAs that they require.
Fig. 1.

Structure-activity relationship summary of N,N′-disubstituted imidazole-4,5-dicarboxamides active in confirmatory screens against FASN-TE.
Materials and Methods
General.
Reagents were obtained from commercial suppliers and used without additional purification. Solvents were purchased in the highest grade available from Fisher Scientific and passed through a MBRAUN MB-SPS-800 purification system prior to use. Thin layer chromatography was performed on 250 µm silica gel plates and compounds were visualized using UV light. Column chromatography was performed on silica gel (grade 60, 230–400 mesh; Fisher Scientific). Columns were prepared in plastic syringe bodies (130 × 25 mm) filled to two-thirds capacity with silica gel. Fractions containing only the desired product were pooled and concentrated under vacuum. 1H NMR spectra were recorded at 400 MHz in CDCl3 with CHCl3 as the internal reference (δ 7.24) or DMSO-d6 with DMSO as the internal reference (δ 2.49). The 13C NMR spectrum of compound 41 was recorded at 100.5 MHz in CDCl3 with CHCl3 as the internal reference (δ 77.00).
Synthesis.
Synthesis of compound 1.4 from compound 1.1 was performed as shown in Scheme 1, as previously described (Solinas et al., 2008). Briefly, freshly prepared compound 1.2 was used in the reaction with lysine derivative 1.3 to give compound 1.4 as a white or off-white solid following extraction and precipitation. This intermediate can be stored at room temperature indefinitely. Two equivalents of glycine benzyl ester hydrochloride and compound 1.5 were added to compound 1.4 in dichloromethane along with two equivalents of diisopropylethylamine, and the reaction was stirred at room temperature for 16–48 hours. The progress of each reaction was followed with thin layer chromatography. Purification by column chromatography using a gradient of hexanes and ethyl acetate afforded compound 1 as a film with 1H NMR and liquid chromatography (LC)/mass spectrometry data consistent with that previously reported for this compound (Solinas et al., 2009). Compounds 11–43 were prepared from compound 1.4 using the same approach and the corresponding amino acid or amine to replace compound 1.5. Compounds 15–24 required the synthesis of derivatives of compound 1.5 that were subsequently used in the reaction. Compounds 11–43 were purified by column chromatography using a gradient of hexanes/ethyl acetate or dichloromethane with 3% methanol (v/v). The purity and identity of the final products were determined by LC, liquid chromatography–tandem mass spectrometry (LC-MS/MS), and 1H NMR spectroscopy. Additional details regarding the synthesis and characterization of these derivatives are provided in the Supplemental Material.
Scheme 1.

Synthesis of compound 1 from compound 1.1.
LC and LC-MS/MS Supporting Compound Purity and Identity.
Compounds were analyzed for purity and identity by LC-MS/MS using a Dionex Ultimate 3000 component LC system and Thermo Scientific TSQ Vantage triple quadrupole mass spectrometer (LC-MS/MS) with HESI-II probe operating in positive ion mode. Solutions of the compounds were injected onto a 1.9 μm Hypersil GOLD (50 × 2.1 mm) high-performance liquid chromatography (HPLC) column with a guard column (10 × 2.1 mm) attached at 40°C. Compounds were eluted over 6 minutes with a gradient of 30%–90% CH3CN and 70%–10% H2O, each containing 0.1% CH3CO2H at a 0.75 ml/min flow rate. The column was washed with 90% CH3CN for 1.5 minutes at the end of each injection, and then equilibrated with 30% CH3CN for 1.5 minutes before each new injection. The mass spectrum was recorded using electrospray ionization detection from 50 to 800 (mass-to-charge ratio). In addition, the samples were analyzed for purity on a Polaris 5 μm C18-A (50 × 2.0 mm) HPLC column and eluted with a gradient of CH3CN/H2O containing 0.1% CH3CO2H at a 0.2 ml/min flow rate. Compounds were detected at 218 nm. The mobile phase gradient was as follows: 0 minutes, 4:6 CH3CN/H2O → 1 minute, 4:6 CH3CN/H2O → 12 minutes, 10:0 CH3CN/H2O → 13 minutes, 10:0 CH3CN/H2O → 14 minutes, 4:6 CH3CN/H2O → 20 minutes, 4:6 CH3CN/H2O.
Cell Lines and Tissue Culture.
MCF-7, MDA-MB-231, BT-474, DU4475, SKBR3, and T47-D BC cells, and HeLa cervical cancer cells were obtained from American Type Culture Collection and routinely grown in phenol red–containing HyClone RPMI 1640 media with 10% (v/v) heat-inactivated FBS (GE Healthcare Life Sciences) and 1% penicillin-streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. MCF10A mammary epithelial cells were cultured in Dulbecco’s modified Eagle’s medium/F12 growth media (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml epidermal growth factor (Peprotech), 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin, 10 µg/ml insulin (Sigma), and 1% penicillin-streptomycin. Cell line characteristics are listed in Table 1. Lipoprotein-depleted and matched control FBS were attained from Kalen Biomedical.
TABLE 1.
Immortalized human cell lines used in this study and their respective biomarker status and subtype, where applicable
| Cell Line | Source | Tumor Type | Subtype | ER | PR | HER2 |
|---|---|---|---|---|---|---|
| BT-474 | Primary breast | Ductal carcinoma | Luminal B | + | + | + |
| DU4475 | Mammary gland | Epithelial cell | TNBC, IM subtype | − | − | − |
| HeLa | Cervix | Adenocarcinoma | N/A | |||
| MCF-7 | Pleural effusion | Invasive ductal carcinoma | Luminal A | + | + | − |
| MCF10A | Primary breast | Epithelial cell | Basal | − | − | − |
| MDA-MB-231 | Pleural effusion | Adenocarcinoma | Claudin-low or basal-like | − | − | − |
| LiSa-2 | Pleomorphic liposarcoma | Liposarcoma | N/A | + | ||
| SKBR3 | Pleural effusion | Adenocarcinoma | Luminal | − | − | + |
| T47-D | Pleural effusion | Invasive ductal carcinoma | Luminal A | + | + | − |
ER, estrogen receptor; HER2, ErbB2; IM, immunomodulatory; N/A, not applicable; PR, progesterone receptor; TNBC, triple-negative breast cancer.
Cell Viability Assays.
FASN and lipase inhibitors were purchased from Cayman Chemical (Ann Arbor, MI), diluted in DMSO, and stored in the dark at −20°C until use. These included: (−)-tetrahydro-4-methylene-2S-octyl-5-oxo-3R-furancarboxylic acid (20 mM stock), cerulenin (20 mM), N-[2-methyl-5-(1-piperidinylsulfonyl)-3-furanyl]-N′-phenyl-urea (GSK264220A) (50 mM), and orlistat (20 mM). Inhibitors were diluted in tissue culture medium immediately before use; control cells were cultured in medium containing the same concentration of DMSO as the highest treatment condition.
The seeding densities for each cell line were determined for the treatment durations to ensure that cells were both capable of logarithmic growth and remained subconfluent at the experimental endpoint. Cells were seeded into 96-well plates, allowed to adhere overnight, treated with freshly diluted drugs on day 2, and cultured for 72 hours prior to assessment. Initial cytotoxicity screens were conducted with MCF-7 BC cells, with cells being treated for 72 hours in standard media at concentrations ranging from 5 to 100 µM of inhibitor. Cell viability was assessed using alamarBlue Cell Viability Reagent (Invitrogen), according to the manufacturer’s guidelines. Extended viability screens were carried out using the ATP-based CellTiter-Glo 2.0 Assay (Promega). Luminescence was read using a LMAX II luminometer (Molecular Devices). Background luminescence was calculated from blank/CellTiter-Glo–alone wells. Luminescence values (relative light units) were normalized to vehicle and compiled as percentage of DMSO control. Half-maximal inhibitory concentration (IC50) determination was performed using a nonlinear regression curve-fitting algorithm: log(inhibitor) versus response-variable slope (four parameter) with Graph Pad Prism 6.0.
Cell Death and Apoptosis Assay.
Apoptosis was assessed using a Dead Cell Apoptosis Kit with Annexin V Alexa Fluor 488 and propidium iodide (PI) (ThermoFisher Scientific), according to the manufacturer’s instructions. Flow cytometry data were read with the 8-Color MACSQuant-10 (Miltenyi Biotec) and analyzed using FlowJo software. The percentage of apoptosis is shown by the percentage of annexin V+ cells in the population, where total cell death may be calculated as the sum of the annexin V+/PI+, annexin V+/PI−, and annexin V−/PI+ populations.
Statistics.
Statistical significance was evaluated using two-tailed unpaired student’s t tests with correction for multiple comparisons (the Holm-Sidak method), where applicable, on the mean values of at least three independent in vitro experiments. Values of P < 0.05 were deemed significant: *P < 0.05, **P < 0.01, ***P < 0.001. Error is presented as mean ± S.D.
Plasma Stability Assay.
Each FASN inhibitor (200 nM in a total volume of 50 μl) was tested for stability both in HPLC solvent (70% acetonitrile) and in mouse plasma following 1-hour incubation under several conditions (on ice, at 25°C, and at 37°C). Immediate recovery (no incubation) of compound from plasma at 25°C was also tested. After incubation, 200 µl of acetonitrile containing 80 nM of a stable internal standard (compound ILP-III-15) (Supplemental Fig. 1) was added to each sample. Samples were immediately vortexed and centrifuged at 12,000 rpm for 5 minutes. Supernatants were dried under nitrogen at 45°C, resuspended in 50 µl 70% acetonitrile, and transferred to the autosampler where 5 µl was injected for LC-MS/MS analysis.
Stability Analysis by LC-MS/MS.
Samples were injected onto a 50 × 2.1 mm, 1.9 μm Hypersil GOLD column fitted with a 10 × 2.1 mm C18 guard at 40°C. Isocratic elutions were achieved with 60% acetonitrile, 40% water, and 0.1% acetic acid at a rate of 0.75 ml/min on the Dionex/Thermo LC-MS/MS system. Mass spectrometry parameters were: spray voltage 3750 V, vaporizer temperature 474°C, capillary temperature 202°C, sheath gas pressure 30, and auxiliary gas pressure 25. Selected reaction monitoring, collision energy, and S-Lens parameters were tuned selectively for each compound.
Analysis of Compound Stability.
We defined stability as recovery of ≥70% of compound from mouse plasma after 1-hour incubation at 37°C. Recovery was considered with respect to drug in solvent or plasma on ice (presumed to be the most stable conditions) versus plasma at 37°C. Some compounds may have been subjected to enzymatic degradation in the plasma, while others may have bound covalently to plasma proteins.
Malonyl-CoA LC-MS/MS Assay.
BT-474 BC cells were treated with FASNi (50 μM) for 4 hours in standard RPMI 1640 media, washed with PBS, harvested with trypsin, and frozen as cell pellets at −20°C. Cell pellets were resuspended at 10,000 cells/μl in cold acetonitrile:methanol:water (40:40:20) containing 1 μg/ml 13C-malonyl-CoA internal standard. Calibrator and quality control samples were made in acetonitrile:methanol:water (40:40:20) containing internal standard. All samples were vortexed and centrifuged 4300g for 20 minutes at 4°C. Supernatants were collected and 25 μl was injected onto a Dionex Ultimate 3000 HPLC system in tandem with a TSQ Vantage mass spectrometer. HPLC separation was achieved with a gradient of 5% → 95% methanol and 95% → 5% 5 mM ammonium acetate over 3 minutes at a flow rate of 0.3 ml/min on a 40°C Phenomenex Luna C18 2.1 × 50 mm, 1.6 μm column with 2.1 × 10 mm C18 guard cartridge. The mass spectrometer was operated in positive ion mode using a HESI-II probe, with spray voltage 3750 V, vaporizer and capillary temperatures at 380°C and 241°C, respectively, and sheath and auxiliary gases at 20 and 5, respectively. Single reaction monitoring mode was used for the following ion transitions: malonyl-CoA 854.055 → 303.040 mass-to-charge ratio with S-Lens 191 and collision energy 35 and 13-C-malonyl-CoA 857.061 → 350.090 mass-to-charge ratio with S-Lens 187 and collision energy 29. The calibration curve was linear over the concentration range 0.01–5 μg/ml, with all quality controls having a CV < 15%.
His6-Tagged FASN-TE Protein Expression and Purification.
A pET15b plasmid coding an N-terminal His6-tagged FASN-TE, with a thrombin recognition sequence separating the His6 tag from the FASN-TE domain, was generously provided by W. Todd Lowther (Wake Forest University, Winston-Salem, NC). This was used for the protein expression and purification of FASN-TE with slight modification from a previously described procedure (Pemble et al., 2007). We used a solution of Bacterial Protein Extraction Reagent (Thermo Scientific) with lysozyme and DNase I at a ratio of 5 ml/g of cells to lyse the cells. Following the addition of streptavidin-agarose (Thermo Scientific) to remove the biotinylated-thrombin, the protein solution was passed through Ni2+-sepharose and the cleaved protein was collected in the flow-through. This step on Ni2+-sepharose ensured that no residual His6-tagged FASN-TE remained in the protein solution. The FASN-TE domain was dialyzed into 20 mM HEPES, pH 7.5, 100 mM NaCl, and 1 mM DTT. For the enzyme assays, the protein was diluted in H2O and dialyzed against 25 mM HEPES, pH 7.5, 12.5 mM NaCl, 0.001% Brij-35, and 125 mM sarcosine, resulting in a stock protein concentration of 2.6 mg/ml (75 μM). A 0.71 M tris-(2-carboxyethyl)phosphine stock solution was used to yield a 1 mM tris-(2-carboxyethyl)phosphine concentration to protect cysteines from oxidation.
FASN-TE Inhibition Experiments.
Prior to each experiment, stock protein was diluted to 7.5 μM with 200 mM Tris, pH 7.5, 100 mM NaCl, 0.01% Brij-35, and 1.0 M sarcosine. The FASN-TE domain was used to conduct IC50 determinations using 4-methylumbelliferyl heptanoate (Sigma Aldrich) as the substrate with enzyme alone versus solutions of varying concentrations of compounds 1 and 41. A stock solution of compound 41 was prepared in 25 mM in DMSO, while a stock solution of compound 1 was prepared at 6.25 mM in DMSO. Serial dilutions of the stocks were prepared, and 1.0 μl of each was added to 155 μl of the pooled enzyme solution. A DMSO control was prepared analogously. These solutions were incubated for 30 minutes at room temperature before adding 50 μl from each into three wells of a 96-well plate (Costar black polystyrene, round bottom). A substrate stock concentration was prepared at 100 mM in DMSO and diluted to 60 μM in water containing 10% DMSO prior to the assay. To each well containing enzyme with or without an inhibitor 50 μl of the substrate solution was then added, giving a final substrate concentration of 30 μM. The final enzyme concentration in the assay was 3.75 μM. The final concentrations of compound 1 in the enzyme assay were 20, 10, 5, 2.5, 1.25, 0.625, 0.313, 0.156, 0.078, and 0.039 μM, respectively. The final concentrations of compound 41 in the enzyme assay were 80, 40, 20, 10, 5, 2.5, 1.25, 0.625, 0.313, and 0.156 μM. The plate was shaken for 10 seconds and fluorescence was recorded (360/40 ex, 460/40 em), in which the shaking was repeated and read every minute for 4 hours. In the absence of inhibitors, the fluorescence reached a maximum in approximately 60 minutes. The background signal resulting from the nonenzymatic hydrolysis of the substrate was subtracted from the enzyme plus the substrate alone control solution and all of the wells that contained compound 1 or 41. Initial velocities were determined at or prior to the point that 10% substrate hydrolysis was evident as determined by the percentage of maximum fluorescence intensity. Data for each concentration were collected in triplicate, and three experiments were used to determine an average IC50 curve for compounds 1 and 41. Half-maximal inhibitory concentration (IC50) determination was performed using a nonlinear regression curve-fitting algorithm: log(inhibitor) versus response-variable slope (four parameter) with Graph Pad Prism 6.0.
Results
Synthesis
The methodology for synthesizing compounds 1–10 has been previously reported (Perchellet et al., 2005; Solinas et al., 2008, 2009); these methods were also used in the synthesis of compounds 11–43 (Scheme 1). The millimolar scale for the synthesis of compounds 11–43, the thin layer chromatography eluant, and Rf value, as well as the yield of purified compounds are shown in Supplemental Table 1. The synthetic procedures and details for reaction intermediates, the reaction scheme, structures, 1H NMR spectra, and LC/mass spectrometry analysis are also provided (Supplemental Figs. 2–18; Supplemental Scheme 1; Supplemental Table 2). Compounds 11–43 were characterized by 1H NMR spectroscopy, 13C NMR spectroscopy (compound 41 only), and LC/mass spectrometry using the total ion count (Supplemental Figs. 19–85). Additional HPLC traces with UV detection at 218 nm to assess for purity are shown in Supplemental Figs. 86–118. (The confirmatory bioassays can be viewed in PubChem by searching AID 624326 or AID 624327 under PubChem Bioassay. The SID number for compound 1 is 49733438. Direct links to the confirmatory bioassay data for compound 1 are as follows: https://pubchem.ncbi.nlm.nih.gov/bioassay/624326#sid=49733438 and https://pubchem.ncbi.nlm.nih.gov/bioassay/624327#sid=49733438.)
Rational Design of Structurally Related Compounds to Improve the Stability and In Vitro Cytotoxicity of Compound 1
Modifications of the Benzyl Ester Moiety.
Compound 1 was weakly cytotoxic to MCF-7 BC cells (IC50 = 117 μM) (Supplemental Fig. 120) and was unstable in mouse plasma (0% recovery, <5 minutes). Heat inactivation of mouse plasma abrogated the instability of compound 1, suggesting the presence of esterase or protease activity. We hypothesized that the lability in mouse plasma was due to the benzyl ester bond based on this evidence and the fact that amino acid derivatives like compound 2, containing tert-butyl esters, were recovered at higher percentages (30%–59%) from mouse plasma after 1 hour at 37°C (data not shown). Replacing the glycine benzyl ester moiety in compound 1 for a leucine tert-butyl ester resulted in complete recovery of the compound (ILP-III-15) from mouse plasma. This leucine derivative was thus used as the internal standard for our compound stability assay (Supplemental Fig. 1).
Our design, synthesis, and testing efforts started with the most active hit, compound 1, and focused on improving the stability of the benzyl ester bond. This led to derivatives 11–14 that replaced part or all of the benzyl ester (Fig. 2A). Each of these derivatives was substantially more cytotoxic against MCF-7 BC cells in vitro than compound 1 (with IC50 values ranging from 8 to 25 µM), but only compound 13 showed marked improvement in plasma stability (40% recovery after 1 hour at 37°C) (Fig. 2, B and C). The less than complete recovery of compound 13 suggested that another activity such as hydrolysis of the glycine amide bond in compound 1 was possible. A second approach to blocking the action of the protease/esterase activity against these compounds was thus employed. The benzyl ester was modified with an amide bond (compound 15) and coupled to steric bulk at the amide nitrogen (compound 16), the benzyl carbon (compounds 17 and 18), or the α-carbon (compounds 19 and 20) of the amino acid (Fig. 3). Compound 16 was recovered at only 29% from 37°C plasma after 1 hour, and the addition of steric bulk did not improve the cytotoxicity of the compounds (data not shown). Combining a methyl group at the amide nitrogen with one at the α-carbon of the amino acid (compounds 21 and 22) or the benzyl carbon (compounds 23 and 24) likewise showed no improvement in the cytotoxicity (data not shown), leading us to abandon this series.
Fig. 2.

Structure, cytotoxicity, and mouse plasma stability data for compounds 11–14. (A) Structure of derivatives that modify the ester bond of compound 1. (B and C) MCF-7 BC cells were treated with FASNi for 72 hours in standard media and assessed using the alamarBlue cell viability assay. (B) Concentration-response curves for compounds 11–14. Data are presented as mean ± S.D. (N = 3–5 experiments). Here and in all ensuing figures, the IC50 values were generated using a nonlinear regression curve-fitting algorithm: log(inhibitor) vs. response-variable slope (four parameters) with Graph Pad Prism 6.0. (C) Compiled IC50 and mouse plasma stability results, represented by percentage of compound remaining after 1 hour incubation in mouse plasma at 37°C (N.D., not determined).
Fig. 3.

Structure of ester-to-amide derivatives of 1 along with methyl groups that add steric bulk.
The Benzylamine Series.
Benzylamine derivative 25 was flagged as active in the initial high-throughput screen with purified FASN-TE, and also formed the base structure of compound 13, which demonstrated improved cytotoxicity in our assays. (The original bioassay can be viewed in PubChem by searching AID 602261 under PubChem Bioassay. The SID number for compound 15 is 24833677. A direct link to the bioassay data for compound 15 is as follows: https://pubchem.ncbi.nlm.nih.gov/bioassay/602261#sid=24833677§ion=Top.) Compounds based on a substituted benzylamine (compounds 26–37) were thus prepared and tested. Substituents on the aromatic ring impacted both the cytotoxicity and the plasma stability; however, even the more potent of these compounds was not sufficiently stable to justify further investigation (Fig. 4).
Fig. 4.

Structure, cytotoxicity, and mouse plasma stability data for compounds 25–37. (A) Benzylamine derivatives 25–37: structure, compiled IC50 values, and mouse plasma stability results, represented by percentage of compound remaining after 1-hour incubation in mouse plasma at 37°C. (B and C) Cytotoxicity concentration-response curves for compounds 25–37 against MCF-7 BC cells following 72-hour continuous treatment in standard media, assessed using the alamarBlue cell viability assay, N = 2–5 experiments. Data shown as mean ± S.D.
Cyclohexylamine Derivatives.
Compound 9 demonstrated activity against purified FASN-TE in initial confirmatory assays (FASN-TE IC50 = 1.56 μM). While the potency of compound 9 was approximately 10-fold less than compound 1 (FASN-TE IC50 < 0.16 μM), this compound demonstrated greater plasma stability than other initial hits and was, therefore, used as the inspiration for a series of cyclohexylamine derivatives (Fig. 5). A boost in the cytotoxicity (IC50 ∼16–24 μM) and plasma stability (90% recovery after 1 hour at 37°C) was observed for compound 38 when the substituent was just a cyclohexylamine. The improvement in cytotoxicity observed for the benzylamine series when an alkyl group was added at the 4-position (compounds 34–36) led us to synthesize the racemic cis- and trans-4-tert-butylcyclohexylamine derivative, compound 39. This modification further improved cytotoxicity (IC50 ∼7–16 μM), with plasma stability of 80%. The diastereomeric cis- and trans-isomers, compounds 40 and 41, were prepared for comparison. Both diastereomers were equally cytotoxic to BC cells (∼14–18 μM); however, the cis-isomer (compound 40) was recovered at 62%, while the trans-isomer (compound 41) was 98% recovered from mouse plasma. Diastereomers 42 and 43 were comparable in cytotoxicity to compound 41 but demonstrated far less stability in mouse plasma (Fig. 5).
Fig. 5.

Structure, cytotoxicity, and mouse plasma stability data for compounds 9 and 38–43. (A) Structural comparison of cyclohexylamine derivatives based on compound 9. (B) Cytotoxicity of compounds 38–43 against MCF-7 BC cells following 72-hour continuous treatment in standard media, assessed using the alamarBlue cell viability assay, N = 4–6 experiments. (C) Compiled IC50 and mouse plasma stability results, represented by percentage of compound remaining after 1-hour incubation in mouse plasma at 37°C.
The four most promising compounds from this series (compounds 38, 39, 41, and 42) were directly compared in terms of cytotoxicity and plasma stability (Fig. 6). It was on the bases of these cytotoxicity and stability results, and structural considerations, that compound 41 was chosen as the new lead compound for extended studies.
Fig. 6.

Cytotoxicity and stability of promising cyclohexylamine derivatives (compounds 38, 39, 41, and 42). (A) Cell viability was assessed using the ATP-based CellTiter-Glo assay following 72-hour continuous treatment with FASNi in standard media. A comparison of the concentration-response trends generated for each FASNi in three different BC cell lines (MCF-7, MDA-MB-231, and BT-474), N = 2–5 experiments per compound. (B) Compiled IC50 values for each of the four FASNi. (C) Viability of noncancerous MCF10A mammary epithelial cells following 72-hour treatment with FASNi in standard media. Data not fitted to a nonlinear curve. (D) Compound stability represented as percentage of recovery compared with control compound (ILP-III-15).
Extended Characterization of a New Lead, Compound 41.
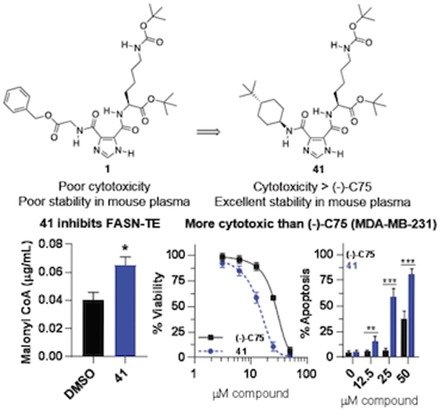
The ability of compound 41 to directly bind and inhibit FASN was assessed using a malonyl-CoA LC-MS/MS assay. Treatment of BT-474 BC cells with compound 41 (4 hours, 50 μM) resulted in significant accumulation of the committed substrate of de novo lipogenesis, malonyl-CoA (Fig. 7A). The identity of compound 41 as a FASNi targeting the thioesterase domain was next confirmed using a FASN-TE enzyme activity assay. Here, both compounds 1 and 41 displayed concentration-dependent FASN-TE enzyme inhibition compared with hydrolysis observed with substrate alone (with data corrected for nonenzymatic hydrolysis of the substrate) (Fig. 7B). Together, these data show that compound 41 binds and inhibits FASN-TE, resulting both in reduced thioesterase enzyme activity and accumulation of the cytotoxic substrate, malonyl-CoA.
Fig. 7.

On-target inhibition of FASN by compound 41. (A) Treatment of BT-474 lipogenic, HER2+ BC cells with compound 41 (4 hours, 50 μM) directly inhibited FASN activity, as shown by significant accumulation of the committed substrate for de novo lipogenesis, malonyl-CoA (*P < 0.05, one-way ANOVA; biologic duplicate and technical triplicate samples). (B) The ability of compounds 1 and 41 to specifically target the thioesterase domain of FASN was assessed using a FASN-TE substrate hydrolysis assay (N = 3 experiments). Under these conditions, the FASN-TE IC50 value of compound 1 was 1.47 ± 0.17 μM with a Hill slope of 0.773, whereas that of compound 41 was 14.58 ± 1.16 μM with a Hill slope of 0.824.
Cytotoxicity of compound 41 was compared with the FASNi (−)-C75 following 72-hour continuous treatment in standard media (Fig. 8). The cytotoxic effects of compound 41 exceed those observed with treatment of (−)-C75 across BC cell lines (Fig. 8, A and B). The FASNi-mediated changes in cell viability could be attributable to a mix of growth inhibition or cytostasis and cell death. Cells were thus stained with Annexin V Alexa Fluor 488 and PI and assessed by flow cytometry following 72-hour continuous treatment to quantify FASNi-induced apoptosis, more specifically. FASN inhibition resulted in significant concentration-dependent apoptosis in BC cells, with the impact of compound 41 exceeding that of (−)-C75 at equimolar concentration (Fig. 8, C and D).
Fig. 8.

Cytotoxicity of compound 41 exceeds that of (−)-C75 across a range of cancer cell lines. Cells were treated with FASNi continuously for 72 hours in standard 10% FBS RPMI 1640 and assessed using the ATP-based CellTiter-Glo viability assay (A, B, E, and F) or annexin V/PI cell death/apoptosis assay (C and D). (A) Concentration-response curves for each BC cell line. (B) IC50 values for compound 41 vs. (−)-C75. (C and D) FASN inhibition causes concentration-dependent apoptosis in MDA-MB-231 and BT-474 BC cells, as detected by annexin V/PI flow cytometry. The apoptosis observed with compound 41 treatment significantly exceeds that of (−)-C75 at several concentrations, as indicated. Two-tailed, unpaired t tests were performed to compare the effects of (−)-C75 at equimolar concentrations, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (E) Compound 41 displays minimal cytotoxicity against MCF10A mammary epithelial cells. (F) Similar cytotoxicity trends observed across cancer cell lines; negligible impact on MCF10A mammary epithelial cell viability. N ≥ 5 experiments for all treatments.
Compound 41 exhibited minimal cytotoxic effects against nontumorigenic MCF10A mammary epithelial cells, creating the potential for a large therapeutic window (Fig. 8, E and F). This is in contrast to other FASNis, including (−)-C75 and cerulenin, where substantial cytotoxicity was observed against MCF10A cells at high concentrations. Effects of compound 41 and other FASNis were validated using several different measures of cell viability including the CellTiter-Glo ATP-based assessment (reported here), the DNA-based Hoechst assay, the protein-based sulforhodamine B assay, and the alamarBlue redox indicator (used for initial profiling studies, data not shown) (Figs. 2–5).
The cytotoxic effects of FASNi treatment are concentration and time dependent, (Supplemental Fig. 121) and are impacted by the availability of exogenous lipoproteins in the media (Fig. 9). This latter observation conforms with previous publications (Kuemmerle et al., 2011; Zaidi et al., 2012, 2013; Daniëls et al., 2014; Schug et al., 2015; Svensson et al., 2016; Cao et al., 2017) and underscores the importance of both endogenous and exogenous FA sources for cancer cell survival and proliferation.
Fig. 9.

Availability of media lipoproteins impacts cytotoxicity and cell death from FASNi treatment. Increased cytotoxicity was observed with 72-hour treatment of compound 41 in RPMI 1640 media containing 10% lipoprotein-depleted serum (LPDS) vs. the matched serum control. This trend was observed in all cell lines tested and reflected in both assessments of (A) cell viability (CellTiter-Glo) and (B) apoptosis (Annexin/PI flow cytometry). No significant differences in cell viability were observed in cells grown for 72 hours in matched or LPDS media alone (data not shown). N = 3–7 experiments per assay. Error expressed as S.D. from the mean, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
With this in mind, we next investigated the potential of targeting FA synthesis and exogenous FA uptake simultaneously using our FASNi compound 41 and GSK264220A, a urea-based inhibitor of lipoprotein lipase (LPL) and endothelial lipase (Keller et al., 2008; Nomura and Casida, 2016). LPL is the major enzyme for extracellular hydrolysis of triglyceride transported in lipoprotein particles, and thus renders this source of FAs accessible to cells (Kuemmerle et al., 2011; Kinlaw et al., 2016). Concentration-dependent cytotoxicity was observed when BC cells were treated with GSK264220A continuously for 72 hours, with most effects on viability observed at concentrations greater than 25 µM (Fig. 10A). Combined treatment with GSK264220A and compound 41 caused increased cytotoxicity over either drug alone (Fig. 10B). We hypothesized that the cytotoxic effects of lipogenesis and lipolysis inhibition might additionally be augmented by removing lipoprotein substrates from the media, and found that this was, in fact, the case. MDA-MB-231 cells cultured in 10% lipoprotein-depleted FBS media and treated with a combination of compound 41 and GSK264220A (100 µM) showed increased cytotoxicity over compound 41 alone, or the combination in media containing lipoproteins (Fig. 10C).
Fig. 10.

Simultaneous inhibition of FA synthesis and uptake (via lipolysis) results in increased cytotoxicity of BC cells in vitro. (A) Continuous treatment with lipase inhibitor, GSK264220A (GSK), for 72 hours resulted in concentration-dependent cytotoxicity in MDA-MB-231 and BT-474 BC cells, and is impacted by the presence of lipoproteins in the media at high concentrations (˃50 μM). (B) Simultaneously targeting FA synthesis and uptake using compound 41 and GSK264220A causes increased cytotoxicity in MDA-MB-231 and BT-474 BC cells. The effects of continuous combined treatments are compared with that of compound 41 alone (supplemented with DMSO to match the highest vehicle exposure). (C) The impact of media lipoproteins on the cytotoxic effects of compound 41 and combined treatments of compound 41 and GSK at 100 µM. Viability was measured using the ATP-based CellTiter-Glo assay. (B and C) Data points at a concentration of 0 µM of compound 41 represent the relative viability of the vehicle control condition or the effect of GSK alone (without FASNi present). N = 3–5 experiments for all treatments.
Discussion
Here, we have summarized the stepwise process through which compound 41 was rationally designed, established its efficacy against cancer cell lines in vitro, and highlighted the therapeutic potential of targeting fatty acid synthesis in BC cells through the FASN-TE domain. Initial screening data that yielded hits 1–10 was useful, both in providing basic structure-activity relationship data for inhibition of purified FASN-TE and in offering clues for how to modify compound 1 to improve cytotoxicity and plasma stability. The fact that heat-denatured plasma led to improved stability of compound 1 pointed to the action of an esterase or protease. We hypothesized that the lability in mouse plasma was predominantly due to the benzyl ester bond and showed that replacing the glycine benzyl ester moiety in compound 1 for a leucine tert-butyl ester (compound ILP-III-15) resulted in complete recovery of the compound from mouse plasma.
An important goal at the start of this project was to obtain a compound in this structural class worthy of investigating in a mouse model of BC, and thereby gauge the suitability of FASN-TE inhibitors for further development. Therefore, the plasma stability of compound 1 was an important issue to solve and led us to prepare derivatives 11–14, which replaced all or part of the benzyl ester. Each of these derivatives was substantially more cytotoxic against MCF-7 BC cells than compound 1 (IC50 = 117 μM) (Supplemental Fig. 120); however, only compound 13 showed improvement in plasma stability. The stability of compound 13 was similar to that of compound 2, further supporting the hypothesis that lack of metabolic stability results in part from hydrolysis of the glycine amide bond of compound 1. The biphenyl substructure in compounds 13 and 14 is a privileged scaffold found both in compounds that bind multiple proteins (Hajduk et al., 2000) and in compounds known to be FA mimetics (Proschak et al., 2017). Thus, while compound 13 was a valuable intermediary in testing the significance of the benzyl ester bond to plasma stability, we did not think it represented an optimal structure to move forward.
We simultaneously prepared derivatives 15–24 with the dual goal of blocking the action of the protease/esterase activity by adding methyl groups as steric bulk and potentially improving the stability and drug-like properties of the compounds—effects that have been tied to the N-methylation of peptides (Biron et al., 2008). No improvements in cytotoxicity were observed with derivatives 15–24, leading us to discontinue this series.
Benzylamine derivative 25 was flagged as active in the initial high-throughput screen with the purified thioesterase domain, and since it also formed the base structure of compound 13, a series of benzylamine derivatives (25–37) were prepared. The plasma stability of derivative 25 was comparable to compound 13 at room temperature or on ice, but lower at 37°C (Supplemental Table 2). Addition of a methyl group at the amide nitrogen (derivative 26) improved recovery from plasma to 65% at 37°C, but derivative 26 was still not as cytotoxic as compound 13. Either the cytotoxicity or the plasma stability, or both, were reasons enough for derivatives 27–37 to be uninteresting, and this series was subsequently abandoned.
Compound 9 was active in the confirmatory assays, albeit approximately 10 times less potent (IC50 = 1.56 μM) at the purified thioesterase domain compared with compound 1 (IC50 < 0.16 μM). However, compound 9 showed improved plasma stability compared with other initial hits; therefore, it was used as the inspiration for a series of cyclohexylamine derivatives (Fig. 5). (The SID number for compound 9 is 26724152. Direct links to the confirmatory bioassay data for compound 9 are as follows: https://pubchem.ncbi.nlm.nih.gov/bioassay/624326#sid=26724152 and https://pubchem.ncbi.nlm.nih.gov/bioassay/624327#sid=26724152.) A boost in the cytotoxicity (IC50 ∼16–24 μM) and plasma stability (90% recovery after 1 hour at 37°C) was observed for compound 38, when the substituent was a cyclohexylamine alone. The racemic cis- and trans-4-tert-butylcyclohexylamines were both commercially available and relatively inexpensive; therefore, they were used to synthesize compound 39 to assess whether cytotoxicity gains could be made, as observed in the benzylamine series between compounds 25 and 35. This was indeed the case, and the mixture of diastereomers only demonstrated a small decrease in plasma stability compared with compound 39. We reasoned the cytotoxicity and plasma stability should be different for the two diastereomers in compound 39; therefore, we next synthesized individual diastereomers 40 and 41 to make the comparison. The trans-stereoisomer (compound 41) had superior plasma stability and cytotoxicity against BC cells than that of the cis-stereoisomer (compound 40). A second pair of diastereomeric cyclohexylamine derivatives, diastereomers 42 and 43, were prepared to further probe for any increase in cytotoxicity; however, both diastereomers 42 and 43 were comparable in cytotoxicity to compound 41 and were far less stable in mouse plasma.
Compound 41 was selected for further investigation on the basis of its promising stability and cytotoxicity parameters (Figs. 6 and 8), the fact that substituted cyclohexylamines are not readily available or prepared, and that there was no obvious structure-activity information from another series to suggest what modifications could be made to pursue additional gains in cytotoxicity. Compound 41 was verified as having on-target inhibitory effects against the FASN-TE domain using a malonyl-CoA accumulation assay and a FASN-TE enzyme activity assay (Fig. 7). To further evaluate compound 41, we used a range of BC cell lines that varied by tumor type and biomarker status (estrogen receptor, progesterone receptor, and HER2) (Table 1). Importantly, compound 41 demonstrated concentration-dependent cytotoxicity across this panel of BC cell lines, was more potent than (−)-C75, and also more selective than classic FASNi, as evidenced by negligible cytotoxicity against the nontumorigenic mammary epithelial MCF10A cell line.
This new lead compound represents an improved FASNi to investigate the importance of this target in BC. We have shown that compound 41 is stable in mouse plasma and more cytotoxic across a wide array of BC cell lines than other widely used FASNis. Compound 41 has drug-like properties and structure, making it a promising lead for continued development toward a clinical therapeutic. Valuable guidelines for drug discovery include the rule-of-five set of properties (Lipinski et al., 1997; Lipinski, 2000, 2004), the number of rotatable bonds (Veber et al., 2002), and cell permeability (Egan et al., 2000). Lagorce et al., 2008 used these rules and others to create an online screening tool for filtering lead compounds against the adsorption, distribution, metabolism, excretion, and toxicity properties, thereby facilitating decision making at early stages of drug discovery (http://fafdrugs4.mti.univ-paris-diderot.fr). Importantly, compound 41 has no major issues related to these filters (Fig. 11). Only the rotatable bonds are outside of the parameters favored for small-molecule drug discovery, while the intramolecular hydrogen bond formed in this scaffold is recognized as a beneficial feature in the design of orally available drugs (Doak et al., 2014).
Fig. 11.

Physical-chemical parameters for compound 41 (intersects of the blue line) over the drug-like filter area for a lead compound (blue space). Abbreviations clockwise from top are as follows: HBA, number of hydrogen bond acceptors; tPSA, topological polar surface area; ratioH_C, ratio of hydrogen atoms to carbon atoms; n_hetero, total number of heteroatoms; n_carbon, total number of carbon atoms; n_SystemRing, smallest number of rings; logP, the logarithm of the partition coefficient between water and n-octanol; TotalCharge, formal charge of the compound; RotatableBonds, number of single nonring bonds; RigidBonds, number of nonflexible bonds; NumCharges, number of charged groups; SizeSystemRing, number of atoms in the largest ring; MW, molecular weight; and HBD, number of hydrogen bond donors.
In addition to the insights into the inhibition of the FASN-TE domain provided by this work, important pathophysiological information regarding the role of lipogenesis in breast cancer biology has also emerged. The addiction of several types of cancer cells to fatty acid synthesis is well recognized (Menendez and Lupu, 2007; Baenke et al., 2013; Röhrig and Schulze, 2016). We, and others, have previously reported that in addition to de novo FA synthesis, the uptake of exogenous lipids may provide an alternative source of FA to cancer cells (Ruby et al., 2010; Kuemmerle et al., 2011; Zaidi et al., 2013; Rozovski et al., 2015, 2016, 2018; Cao et al., 2017), and that this process is facilitated by the expression of LPL and the cell surface FA uptake channel, CD36. This prompts the hypothesis that cancer cells may occupy various positions along a spectrum of dependence upon FASN-mediated lipogenesis and LPL-driven lipolysis to satisfy their requirement for FA. In the case of prostate cancer cells, the recent findings of Daniëls et al. (2014) further demonstrate that the balance between the two modes of lipid acquisition may be fluid, with the removal of exogenous lipoproteins eliciting increased expression of genes involved in FA synthesis, including FASN. Our current finding that sensitivity to FASN-TE inhibition is enhanced by either inhibition of lipase activity and/or removal of lipoproteins from tissue culture media strongly supports the concept that FA synthesis and uptake both support BC cell survival. This notion has begun to be realized across a variety of different cancer types, including not only BC (Kuemmerle et al., 2011; Song et al., 2012; Slebe et al., 2016) but also colorectal cancer (Notarnicola et al., 2012; Tang et al., 2012), prostate cancer (Wang et al., 1995; Cerhan et al., 1997; Connolly et al., 1997; Fair et al., 1997; Tokuda et al., 2003; Brown et al., 2006; Gazi et al., 2007; Narita et al., 2008), hepatocellular carcinoma (Li et al., 2016; Cao et al., 2017), lung cancer (Sundaram et al., 2018), and pancreatic cancer (Vasseur and Guillaumond, 2016).
Several groups have linked obesity or high-fat diets to cancer prognosis, as well as implicated tumor-adjacent adipocytes in altering the metabolism, growth, and progression of solid tumors (Tokuda et al., 2003; Gazi et al., 2007; Notarnicola et al., 2012; Nieman et al., 2013; Abel et al., 2014; Massa et al., 2016; Cozzo et al., 2017; Gonzalez-Reyes et al., 2018; Luo et al., 2018). It thus appears that, at least for some tumors, simultaneous targeting of both lipogenesis and uptake/lipolysis will provide the largest anticancer effect. The importance of interactions between these complementary pathways will come into sharper focus as increasingly potent and specific FASN inhibitors, such as compound 41 and its derivatives, become available.
Acknowledgments
The authors thank W. Todd Lowther (Wake Forest University, Winston-Salem, NC) for the plasmid coding of the N-terminal His6-tagged thioesterase domain of FASN.
Abbreviations
- BC
breast cancer
- C75
4-methylene-2-octyl-5-oxotetrahydrofuran-3-carboxylic acid
- FA
fatty acid
- FASN
fatty acid synthase
- FASNi
fatty acid synthase inhibitor
- FASN-TE
fatty acid synthase thioesterase
- GSK264220A
N-[2-methyl-5-(1-piperidinylsulfonyl)-3-furanyl]-N′-phenyl-urea
- HPLC
high-performance liquid chromatography
- LC
liquid chromatography
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- LPL
lipoprotein lipase
- PI
propidium iodide
Authorship Contributions
Participated in research design: Lupien, Lewis, Pooler, Kinlaw, Baures.
Conducted experiments: Lupien, Dunkley, Maloy, Lehner, Foisey, Ouellette, Pooler, Baures.
Contributed new reagents or analytic tools: Lewis.
Performed data analysis: Lupien, Lewis, Pooler, Baures.
Wrote or contributed to the writing of the manuscript: Lupien, Lewis, Pooler, Kinlaw, Baures.
Footnotes
This research was made possible by the Institutional Development Award Program of the National Institutes of Health National Institute of General Medical Sciences [Grant P20GM103506] (to P.W.B. and W.B.K.) as well as funding from the National Institutes of Health National Cancer Institute [Grant R01CA58961] (to W.B.K.). This research was also supported by the National Institutes of Health National Cancer Institute [Grant P30CA023108] (to L.D.L. and D.B.P.) Support for the Keene State College NMR Facility [National Science Foundation 1337206] (to P.W.B.) is appreciated, as is support from The Friends of Norris Cotton Cancer Center (to P.W.B. and W.B.K.).
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Abel S, Riedel S, Gelderblom WCA. (2014) Dietary PUFA and cancer. Proc Nutr Soc 73:361–367. [DOI] [PubMed] [Google Scholar]
- Anderson AM, Ragan MA. (2016) Palmitoylation: a protein S-acylation with implications for breast cancer. NPJ Breast Cancer 2:16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin CP, Brady LS, Insel TR, Collins FS. (2004) NIH molecular libraries initiative. Science 306:1138–1139. [DOI] [PubMed] [Google Scholar]
- Baenke F, Peck B, Miess H, Schulze A. (2013) Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech 6:1353–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baures PW. (1999) Heterocyclic HIV-1 protease inhibitors. Org Lett 1:249–252. [DOI] [PubMed] [Google Scholar]
- Baures PW. (2006) Imidazole-4,5-dicarboxylic acid: a versatile scaffold for drug discovery and materials research. Trends Heterocyclic Chem 11:1–22. [Google Scholar]
- Baures PW, Rush JR, Wiznycia AV, Desper J, Helfrich BA, Beatty AM. (2002) Intramolecular hydrogen bonding and intermolecular dimerization in the crystal structures of imidazole-4,5-dicarboxylic acid derivatives. Cryst Growth Des 2:653–664. [Google Scholar]
- Benjamin DI, Li DS, Lowe W, Heuer T, Kemble G, Nomura DK. (2015) Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS Chem Biol 10:1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, et al. (2008) Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew Chem Int Ed Engl 47:2595–2599. [DOI] [PubMed] [Google Scholar]
- Brown MD, Hart CA, Gazi E, Bagley S, Clarke NW. (2006) Promotion of prostatic metastatic migration towards human bone marrow stoma by omega 6 and its inhibition by omega 3 PUFAs. Br J Cancer 94:842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D, Duke G, Heuer TS, O’Farrell M, Wagman AS, McCulloch W, Kemble G. (2017) Fatty acid synthase—modern tumor cell biology insights into a classical oncology target. Pharmacol Ther 177:23–31. [DOI] [PubMed] [Google Scholar]
- Cao D, Song X, Che L, Li X, Pilo MG, Vidili G, Porcu A, Solinas A, Cigliano A, Pes GM, et al. (2017) Both de novo synthetized and exogenous fatty acids support the growth of hepatocellular carcinoma cells. Liver Int 37:80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerhan JR, Torner JC, Lynch CF, Rubenstein LM, Lemke JH, Cohen MB, Lubaroff DM, Wallace RB. (1997) Association of smoking, body mass, and physical activity with risk of prostate cancer in the Iowa 65+ Rural Health Study (United States). Cancer Causes Control 8:229–238. [DOI] [PubMed] [Google Scholar]
- Connolly JM, Coleman M, Rose DP. (1997) Effects of dietary fatty acids on DU145 human prostate cancer cell growth in athymic nude mice. Nutr Cancer 29:114–119. [DOI] [PubMed] [Google Scholar]
- Cozzo AJ, Fuller AM, Makowski L. (2017) Contribution of adipose tissue to development of cancer. Compr Physiol 8:237–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniëls VW, Smans K, Royaux I, Chypre M, Swinnen JV, Zaidi N. (2014) Cancer cells differentially activate and thrive on de novo lipid synthesis pathways in a low-lipid environment. PLoS One 9:e106913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean EJ, Falchook GS, Patel MR, Brenner AJ, Infante JR, Arkenau HT, Borazanci EH, Lopez JS, Pant S, Schmid P, et al. (2016) Preliminary activity in the first in human study of the first-in-class fatty acid synthase (FASN) inhibitor, TVB-2640. J Clin Oncol 34:2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Schrijver E, Brusselmans K, Heyns W, Verhoeven G, Swinnen JV. (2003) RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res 63:3799–3804. [PubMed] [Google Scholar]
- Doak BC, Over B, Giordanetto F, Kihlberg J. (2014) Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem Biol 21:1115–1142. [DOI] [PubMed] [Google Scholar]
- Egan WJ, Merz KM, Jr, Baldwin JJ. (2000) Prediction of drug absorption using multivariate statistics. J Med Chem 43:3867–3877. [DOI] [PubMed] [Google Scholar]
- Fair WR, Fleshner NE, Heston W. (1997) Cancer of the prostate: a nutritional disease? Urology 50:840–848. [DOI] [PubMed] [Google Scholar]
- Flavin R, Peluso S, Nguyen PL, Loda M. (2010) Fatty acid synthase as a potential therapeutic target in cancer. Future Oncol 6:551–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazi E, Gardner P, Lockyer NP, Hart CA, Brown MD, Clarke NW. (2007) Direct evidence of lipid translocation between adipocytes and prostate cancer cells with imaging FTIR microspectroscopy. J Lipid Res 48:1846–1856. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Guerrico AM, Espinoza I, Schroeder B, Park CH, Kvp CM, Khurana A, Corominas-Faja B, Cuyàs E, Alarcón T, Kleer C, et al. (2016) Suppression of endogenous lipogenesis induces reversion of the malignant phenotype and normalized differentiation in breast cancer. Oncotarget 7:71151–71168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Reyes C, Marcial-Medina C, Cervantes-Anaya N, Cortes-Reynosa P, Salazar EP. (2018) Migration and invasion induced by linoleic acid are mediated through fascin in MDA-MB-231 breast cancer cells. Mol Cell Biochem 443:1–10. [DOI] [PubMed] [Google Scholar]
- Hajduk PJ, Bures M, Praestgaard J, Fesik SW. (2000) Privileged molecules for protein binding identified from NMR-based screening. J Med Chem 43:3443–3447. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. (2011) Hallmarks of cancer: the next generation. Cell 144:646–674. [DOI] [PubMed] [Google Scholar]
- Heuer TS, Ventura R, Mordec K, Lai J, Fridlib M, Buckley D, Kemble G. (2017) FASN inhibition and taxane treatment combine to enhance anti-tumor efficacy in diverse xenograft tumor models through disruption of tubulin palmitoylation and microtubule organization and FASN inhibition-mediated effects on oncogenic signaling and gene expression. EBioMedicine 16:51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari N, Drury J, Morris AJ, Onono FO, Stevens PD, Gao T, Liu J, Wang C, Lee EY, Weiss HL, et al. (2019) De novo fatty acid synthesis-driven sphingolipid metabolism promotes metastatic potential of colorectal cancer. Mol Cancer Res 17:140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YY, Kim HM, Koo JS. (2015) Expression of lipid metabolism-related proteins in metastatic breast cancer. PLoS One 10:e0137204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller PM, Rust T, Murphy DJ, Matico R, Trill JJ, Krawiec JA, Jurewicz A, Jaye M, Harpel M, Thrall S, et al. (2008) A high-throughput screen for endothelial lipase using HDL as substrate. J Biomol Screen 13:468–475. [DOI] [PubMed] [Google Scholar]
- Kinlaw WB, Baures PW, Lupien LE, Davis WL, Kuemmerle NB. (2016) Fatty acids and breast cancer: make them on site or have them delivered. J Cell Physiol 231:2128–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kridel SJ, Axelrod F, Rozenkrantz N, Smith JW. (2004) Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res 64:2070–2075. [DOI] [PubMed] [Google Scholar]
- Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM, et al. (2011) Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther 10:427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. (1994) Fatty acid synthesis: a potential selective target for antineoplastic therapy. Proc Natl Acad Sci USA 91:6379–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagorce D, Sperandio O, Galons H, Miteva MA, Villoutreix BO. (2008) FAF-Drugs2: free ADME/tox filtering tool to assist drug discovery and chemical biology projects. BMC Bioinformatics 9:396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Lingwood D, Grzybek M, Coskun U, Simons K. (2010) Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci USA 107:22050–22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cheng JX. (2014) Direct visualization of de novo lipogenesis in single living cells. Sci Rep 4:6807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Che L, Tharp KM, Park HM, Pilo MG, Cao D, Cigliano A, Latte G, Xu Z, Ribback S, et al. (2016) Differential requirement for de novo lipogenesis in cholangiocarcinoma and hepatocellular carcinoma of mice and humans. Hepatology 63:1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA. (2000) Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods 44:235–249. [DOI] [PubMed] [Google Scholar]
- Lipinski CA. (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol 1:337–341. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu JY, Wu X, Zhang JT. (2010) Biochemistry, molecular biology, and pharmacology of fatty acid synthase, an emerging therapeutic target and diagnosis/prognosis marker. Int J Biochem Mol Biol 1:69–89. [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu Y, Zhang JT. (2008) A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Ther 7:263–270. [DOI] [PubMed] [Google Scholar]
- Liu H, Wu X, Dong Z, Luo Z, Zhao Z, Xu Y, Zhang JT. (2013) Fatty acid synthase causes drug resistance by inhibiting TNF-α and ceramide production. J Lipid Res 54:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, He Y, Yu X. (2018) Bone marrow adipocyte: an intimate partner with tumor cells in bone metastasis. Front Endocrinol (Lausanne) 9:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Cheng C, Tan Z, Li N, Tang M, Yang L, Cao Y. (2017) Emerging roles of lipid metabolism in cancer metastasis. Mol Cancer 16:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa M, Gasparini S, Baldelli I, Scarabelli L, Santi P, Quarto R, Repaci E. (2016) Interaction between breast cancer cells and adipose tissue cells derived from fat grafting. Aesthet Surg J 36:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7:763–777. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Lupu R. (2017) Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin Ther Targets 21:1001–1016. [DOI] [PubMed] [Google Scholar]
- Menendez JA, Vellon L, Lupu R. (2005) Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses 64:997–1001. [DOI] [PubMed] [Google Scholar]
- Narita S, Tsuchiya N, Saito M, Inoue T, Kumazawa T, Yuasa T, Nakamura A, Habuchi T. (2008) Candidate genes involved in enhanced growth of human prostate cancer under high fat feeding identified by microarray analysis. Prostate 68:321–335. [DOI] [PubMed] [Google Scholar]
- Nieman KM, Romero IL, Van Houten B, Lengyel E. (2013) Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta 1831:1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura DK, Casida JE. (2016) Lipases and their inhibitors in health and disease. Chem Biol Interact 259 (Pt B):211–222. [DOI] [PubMed] [Google Scholar]
- Notarnicola M, Miccolis A, Tutino V, Lorusso D, Caruso MG. (2012) Low levels of lipogenic enzymes in peritumoral adipose tissue of colorectal cancer patients. Lipids 47:59–63. [DOI] [PubMed] [Google Scholar]
- Pemble CW, IV, Johnson LC, Kridel SJ, Lowther WT. (2007) Crystal structure of the thioesterase domain of human fatty acid synthase inhibited by Orlistat. Nat Struct Mol Biol 14:704–709. [DOI] [PubMed] [Google Scholar]
- Perchellet EM, Perchellet JP, Baures PW. (2005) Imidazole-4,5-dicarboxamide derivatives with antiproliferative activity against HL-60 cells. J Med Chem 48:5955–5965. [DOI] [PubMed] [Google Scholar]
- Pizer ES, Jackisch C, Wood FD, Pasternack GR, Davidson NE, Kuhajda FP. (1996) Inhibition of fatty acid synthesis induces programmed cell death in human breast cancer cells. Cancer Res 56:2745–2747. [PubMed] [Google Scholar]
- Proschak E, Heitel P, Kalinowsky L, Merk D. (2017) Opportunities and challenges for fatty acid mimetics in drug discovery. J Med Chem 60:5235–5266. [DOI] [PubMed] [Google Scholar]
- Röhrig F, Schulze A. (2016) The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer 16:732–749. [DOI] [PubMed] [Google Scholar]
- Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z, Wu JY, Jain P, Wierda W, Burger J, et al. (2015) Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol Cancer Res 13:944–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovski U, Harris DM, Li P, Liu Z, Jain P, Ferrajoli A, Burger J, Thompson P, Jain N, Wierda W, et al. (2018) STAT3-activated CD36 facilitates fatty acid uptake in chronic lymphocytic leukemia cells. Oncotarget 9:21268–21280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovski U, Hazan-Halevy I, Barzilai M, Keating MJ, Estrov Z. (2016) Metabolism pathways in chronic lymphocytic leukemia. Leuk Lymphoma 57:758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby MA, Goldenson B, Orasanu G, Johnston TP, Plutzky J, Krauss RM. (2010) VLDL hydrolysis by LPL activates PPAR-α through generation of unbound fatty acids. J Lipid Res 51:2275–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JR, Sandstrom SL, Yang J, Davis R, Prakash O, Baures PW. (2005) Intramolecular hydrogen bond strength and pKa determination of N,N′-disubstituted imidazole-4,5-dicarboxamides. Org Lett 7:135–138. [DOI] [PubMed] [Google Scholar]
- Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, Van Veldhoven PP, Waltregny D, Daniëls VW, Machiels J, et al. (2010) De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res 70:8117–8126. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015) Acetyl-coA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27:57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Yadav V, Kumar S, Saini N. (2015) MicroRNA-195 inhibits proliferation, invasion and metastasis in breast cancer cells by targeting FASN, HMGCR, ACACA and CYP27B1. Sci Rep 5:17454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slebe F, Rojo F, Vinaixa M, García-Rocha M, Testoni G, Guiu M, Planet E, Samino S, Arenas EJ, Beltran A, et al. (2016) FoxA and LIPG endothelial lipase control the uptake of extracellular lipids for breast cancer growth. Nat Commun 7:11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas R, DiCesare JC, Baures PW. (2008) Parallel synthesis of an imidazole-4,5-dicarboxamide library bearing amino acid esters and alkanamines. Molecules 13:3149–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas R, DiCesare JC, Baures PW. (2009) Parallel synthesis of a library of symmetrically- and dissymmetrically-disubstituted imidazole-4,5-dicarboxamides bearing amino acid esters. Molecules 14:352–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Sneddon AA, Heys SD, Wahle KW. (2012) Regulation of fatty acid synthase (FAS) and apoptosis in estrogen-receptor positive and negative breast cancer cells by conjugated linoleic acids. Prostaglandins Leukot Essent Fatty Acids 87:197–203. [DOI] [PubMed] [Google Scholar]
- Sundaram S, Žáček P, Bukowski MR, Mehus AA, Yan L, Picklo MJ. (2018) Lipidomic impacts of an obesogenic diet upon Lewis lung carcinoma in mice. Front Oncol 8:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, et al. (2016) Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med 22:1108–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, Verhoeven G. (2003) Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem Biophys Res Commun 302:898–903. [DOI] [PubMed] [Google Scholar]
- Tang FY, Pai MH, Chiang EP. (2012) Consumption of high-fat diet induces tumor progression and epithelial-mesenchymal transition of colorectal cancer in a mouse xenograft model. J Nutr Biochem 23:1302–1313. [DOI] [PubMed] [Google Scholar]
- Tokuda Y, Satoh Y, Fujiyama C, Toda S, Sugihara H, Masaki Z. (2003) Prostate cancer cell growth is modulated by adipocyte-cancer cell interaction. BJU Int 91:716–720. [DOI] [PubMed] [Google Scholar]
- VanCompernolle SE, Wiznycia AV, Rush JR, Dhanasekaran M, Baures PW, Todd SC. (2003) Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology 314:371–380. [DOI] [PubMed] [Google Scholar]
- Vasseur S, Guillaumond F. (2016) LDL receptor: an open route to feed pancreatic tumor cells. Mol Cell Oncol 3:e1033586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623. [DOI] [PubMed] [Google Scholar]
- Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, Buckley D, Kemble G, Heuer TS. (2015) Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine 2:808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Stübiger G, Veigel D, Wuczkowski M, Lanzerstorfer P, Weghuber J, Karteris E, Nowikovsky K, Wilfinger-Lutz N, Singer CF, et al. (2017) Multi-level suppression of receptor-PI3K-mTORC1 by fatty acid synthase inhibitors is crucial for their efficacy against ovarian cancer cells. Oncotarget 8:11600–11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xi Q, Wu G. (2016) Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med 5:1599–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Corr JG, Thaler HT, Tao Y, Fair WR, Heston WD. (1995) Decreased growth of established human prostate LNCaP tumors in nude mice fed a low-fat diet. J Natl Cancer Inst 87:1456–1462. [DOI] [PubMed] [Google Scholar]
- Warburg O. (1956) On the origin of cancer cells. Science 123:309–314. [DOI] [PubMed] [Google Scholar]
- Ward PS, Thompson CB. (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21:297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Qin L, Fako V, Zhang JT. (2014) Molecular mechanisms of fatty acid synthase (FASN)-mediated resistance to anti-cancer treatments. Adv Biol Regul 54:214–221. [DOI] [PubMed] [Google Scholar]
- Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. (2013) Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 52:585–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi N, Royaux I, Swinnen JV, Smans K. (2012) ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther 11:1925–1935. [DOI] [PubMed] [Google Scholar]
- Zaytseva YY, Elliott VA, Rychahou P, Mustain WC, Kim JT, Valentino J, Gao T, O’Connor KL, Neltner JM, Lee EY, et al. (2014) Cancer cell-associated fatty acid synthase activates endothelial cells and promotes angiogenesis in colorectal cancer. Carcinogenesis 35:1341–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaytseva YY, Rychahou PG, Gulhati P, Elliott VA, Mustain WC, O’Connor K, Morris AJ, Sunkara M, Weiss HL, Lee EY, et al. (2012) Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res 72:1504–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]