

Abstract
Stage 4 neuroblastoma (NB) is a devastating childhood cancer whose poor outcome has remained essentially unchanged in the last 20 years. Receptor tyrosine kinases have important roles in the control of proliferation, differentiation and apoptosis of NB cells. Thus, we tested the activity of second generation tyrosine kinase inhibitor Dasatinib in human NB cell lines in vitro and in an orthotopic mouse model. Dasatinib inhibited cell viability with an IC50 in the submicromolar range in 7 of 10 tested cell lines. In sensitive cells, Dasatinib reduced anchorage-independent growth and, in some instances, induced senescence and apoptosis. In HTLA-230 cells, Dasatinib treatment caused down-regulation of c-Kit and c-Src phosphorylation in conjunction with strong inhibition of Erk1/2 and Akt activity. To test the efficacy of Dasatinib in vivo, HTLA-230 and SY5Y cells were orthotopically injected in the adrenal gland of nude mice and drug treatments carried out until day 40. In mice injected with HTLA-230 cells, tumour growth was significantly inhibited at the dose of 30 mg/Kg/day when treatment was started 7 days after injection. In animals injected with SY5Y cells that were exquisitely sensitive in vitro (IC50= 92 nM), the antitumour effect of Dasatinib was observed at the dose of 60mg/Kg/day but only when treatment was started 1 day after injection. However, the anti-tumour effect of Dasatinib in vivo was partial in both orthotopic models, emphasizing the importance of testing candidate new drugs in animal environments closely mimicking the human tumour.
Keywords: neuroblastoma, Dasatinib, orthotopic model
Introduction
Neuroblastoma (NB) is the most common childhood extra-cranial tumour1. NB derives from precursor cells of the sympatoadrenal lineage and it can develop anywhere in the sympathetic system1. About 40% of NBs at diagnosis are localized tumours which, in general, respond to chemotherapy and have an outcome that spans from favourable (stages 1, 2 and 3 with no MYCN amplification) to intermediate/poor (stages 2 and 3 with MYCN amplification)2. However, the greatest clinical challenge is represented by stage 4 in children older than 18 months which accounts for approximately 50% of NB cases at diagnosis. Stage 4 NB is characterized by metastases to distant sites such as cortical bone, bone marrow, and lymph nodes non-contiguous to the primary tumour2. Outcome for these patients is generally very poor, a 5-year survival not exceeding 30%1. A striking clinical phenotype of NB is stage 4S (S= special), that occurs in about 8% of cases2. These infants have a small primary tumour with widespread involvement of liver, skin, and/or bone marrow, which spontaneously regress in a substantial number of cases. Outcome of 4S tumours is similar to stage 1 and 2 unless other unfavourable prognostic markers are present3.
Beside tumour stage, many clinical and genetic features are utilized in NB to modulate appropriate treatment and accurately define prognosis. Age > 18 months4 and histology5 are potent indicators of poorer outcome. The most commonly used genetic marker is, by far, the amplification of MYCN oncogene that defines an aggressive subset of NBs6. Cytogenetic features such as 1p deletion, 11q loss and 17q gain are also associated with poor outcome (reviewed in2). Expression of neurotrophin receptor TrKA is associated with favourable outcome7, in marked contrast to TrkB expression8. Recently, germ-line activating mutations of the ALK (Anaplastic Lymphoma Kinase) gene were found to be strongly associated with hereditary NB9. Of interest, somatic ALK mutations were also found in 6–12 % of sporadic NBs9,10. ALK inhibition resulted in profound inhibition of growth in all cell lines harbouring mutant or amplified ALK, pointing to this cell-surface kinase as a possible novel target for NB treatment11.
The development of less toxic therapies is a compelling priority in paediatric oncology since the young age of patients and the likelihood of long-term side effects pose an additional problem of clinical management. At present, stage 4 NB patients are treated with an aggressive multimodal treatment12 that causes severe or life-threatening acute side effects and it is only partially effective in achieving long term survival (reviewed in2).
Targeted therapies by selective protein kinase inhibitors have had a substantial impact in the treatment of several human malignancies, in which these agents elicit major clinical responses with considerable reduction of side effects compared with conventional cytotoxic chemotherapy13. We14,15 and others16–18 demonstrated Imatinib Mesylate activity in NB cells in vitro and in vivo. In this paper we have utilized Dasatinib, a second generation tyrosine kinase inhibitor19, to treat NB cells in vitro and in an orthotopic mouse model. Our data show that Dasatinib is effective in reducing NB growth and highlight the importance of using an appropriate in vivo model to validate in vitro results.
Materials and methods
Chemical compound.
Dasatinib powder was provided by Bristol Myers Squibb (Princeton, NJ). Dasatinib was dissolved in DMSO (10 mM) for in vitro studies or in 80 mM citric acid (pH 2.1) to make 10 mg/ml stock solution and then diluted to 1 mg/ml with 80 mM citric acid (pH 3.1) for in vivo applications.
Cell lines.
Human NB cell lines ACN, GI-CA-N, KCNR, RN-GA, SH-EP, HTLA-230, LAN-5, SK-N-AS, SK-N-BE2c, were kept in culture as previously described14. Human NB cell line SY5Y20 was cultured in D-MEM (Euroclone, Paignton, Devon, UK) supplemented by 10 % fetal calf serum (FCS) (Hyclone, Logan, UT, USA), penicillin and streptomycin (100 μg/ml each), and 2mM L-glutamine at 37°C, 5% CO2.
Cell proliferation, senescence and apoptosis.
Cell growth was evaluated by seeding cells in triplicate in complete medium (medium supplemented by 10% FCS). After 18 hours, fresh medium or medium containing Dasatinib (10, 100 and 1000 nM) was added. Cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) assay21 after 72 hours. Expression of beta-galactosidase activity as a marker of cellular senescence was carried out as described22. Anchorage-independent growth was performed in semi-solid agar as follows: base agar (0.5% agar, 1x RPMI 1640 and 10% FCS) was added in each well and let solidify; an equal volume of top agar (0.35% agar, 1x RPMI 1640 and 10% FCS) in which cells were present at the concentration of 103 cells/cm2 was added to the same well. Base agar and top agar were prepared by including Dasatinib (10, 100 and 1000 nM) in the formulation where needed. Plates were incubated at 37°C, 5% CO2 in a humidified incubator for 11–13 days and stained with 0.005% Crystal Violet for >1 hour. Single cells (defined as cells or aggregates of ≤20 cells) and clones (aggregates of >20 cells) were scored in the plates counting at least 300 elements in each plate. Each experimental point was carried out in duplicate. Experiments were repeated twice with reproducible results. Cell cycle analysis and evaluation of the sub G1 peak were carried out using propidium iodide staining (50 μg/ml) followed by flow cytometric analysis (FACSCalibur, Becton Dickinson, Bedford, MA).
Caspase-3 and −7 activities were measured using the luminescent Caspase-Glo™ 3/7 kit (Promega, Madison, WI) based on detection of a luminescent caspase 3/7 substrate following caspase cleavage. In each well, 1.5×104 cells were seeded in RPMI 1640 medium supplemented with 10% FCS. Dasatinib at concentrations 20% above the calculated IC50 of each cell line was added where needed.
Tumour invasion in Matrigel-coated chambers.
Cells were pretreated in complete medium supplemented with the increasing (10, 100, 1000 nM) Dasatinib concentrations for 24 hours before plating (1.25 × 105 cell/well) in the BD Matrigel® invasion chambers (BD Biosciences, Bedford, MA). Mock treatments were carried out pre-treating the cells in the same medium without Dasatinib. Medium in the upper chamber was supplemented with 5% FCS. In the lower chamber, FCS concentration was 20%. After 24 hours, cells migrated into the lower chamber were stained and counted. Experiments were carried out in triplicate and repeated twice.
Flow cytometry:
1 × 106 cells were harvested and pellets were washed twice with PBS. Cells were then fixed in cold 70% ethanol added dropwise while vortexing gently. Fixed cells were kept overnight at 4° C. Cells were centrifuged and pellets were resuspended in 1 ml of PI/RNase Staining Buffer (BD Biosciences). Reactions were incubated for 20 min at 4°C, protected from the light. Samples were analyzed by flow cytometry using a FACScalibur flow cytometer. For each sample, at least 2×104 cells were analyzed. Cell cycle distribution was calculated by Cell Quest software (BD Biosciences).
Protein analysis.
Cellular proteins were extracted, separated on SDS-polyacrylamide gels and Western blot analyses were carried out as previously described23. Antibodies used were: Anti-HSP-70 (SPA-820) (Stressgen, Ann Arbour, MI), , anti-c-Kit (C-14) and anti-Bax (N-20) (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Bad (#9292), anti-phospho-c-Kit (Tyr 721) (#3391), anti-Akt (#9272), anti-Phospho-Akt (Ser 473)(#9271), anti-p44/42 MAP Kinase (#9102), anti-Phospho-p44/42 MAP Kinase (Thr202/Tyr204)(#9101) all from Cell Signaling Technology, Inc. (Danvers, MA), anti-c-Src (Calbiochem, La Jolla, CA), anti-phospho-c-Src (Biosource International Inc. Camarillo, CA).
In vivo studies.
Five-week old female athymic nude mice (Hsd: Athymic Nude-nu from Charles River laboratories, Lecco, Italy) were fed ad libitum and kept in optimal hygienic conditions in a 12 hr/12 hr light/dark cycle. Upon arrival, animals were kept in the animal’s facility for one week before starting the experiments. Mice were anesthetized with ketamine (Imalgene 1000, Merial Italia SpA., Milan, Italy) and xylazine (Bayer AG, Leverkusen, Germany), subjected to laparotomy, and injected with HTLA-230 or SY5Y cells (1 × 106 cells in 20 μL of medium without serum) into the capsule of the left adrenal gland, as previously described24. Treatment started 1 or 7 days after tumour cell injection (day 0). Animals (8–10 each in the Dasatinib or vehicle only groups) were treated daily by oral gavage either with Dasatinib (30mg/kg/day or 60mg/Kg/day depending on the schedule) or with vehicle (80 mM citric acid, pH 3.1) (control groups). At the end of drug or vehicle-only treatment (40 days after tumour cell injection), animals were sacrificed by CO2 inhalation, tumour masses collected and volumes calculated according to the following formula: (length x width x height x π / 6). A small aliquot was immediately frozen in liquid nitrogen for biochemical analyses, whilst the remaining tissue was formalin-fixed for histological and immunohistochemical analyses. In vivo studies were approved by the Animal Care and Use Committee of ENEA, Rome, and all animal care was in accordance with the European guidelines.
Histology and immunohistochemistry.
Paraffin-embedded tissues were cut in 7 μm sections and processed for hematoxilin-eosin staining according to standard techniques. For immunohistochemistry, 4 μm sections were cut and Ki-67 antigen was retrieved by microwave exposure (3 min, 790 W). Slides were then treated with 1% H2O2 for 10 min to inhibit endogenous peroxidase activity. Immunohistochemical analysis to detect Ki-67 was carried out using a specific antibody (Novocastra Laboratories Ltd, Newcastle, UK) diluted 1:1000 according to the manufacturer’s specifications. Secondary anti-rabbit antibody HRP-conjugated (DAKO Cytomation Inc., Carpinteria, CA) was diluted 1:100. Control reactions were carried out by substituting the Ki-67 specific antibody with normal rabbit serum. Staining was carried out using DAB substrate (DAKO).
Statistical analysis.
Student’s t test was applied to evaluate significance of difference in tumour volumes between treated and control groups using GraphPad Prism 4 for Windows (GraphPad Software Inc., San Diego, CA). Results are presented as mean ± standard error of the mean (SEM). The level of significance was set at p < 0.05.
Results
Dasatinib is effective in reducing in vitro viability and anchorage-independent growth of human NB cells.
We tested the effect of Dasatinib on the viability of ten human NB cell lines (ACN, GI-CA-N, KCNR, RN-GA, SH-EP, HTLA-230, LAN-5, SK-N-AS, SK-N-BE2c, SY5Y). NB cells were treated with increasing drug concentrations (10, 100 and 1000 nM) and MTT assay was carried out after 48 hours. Dasatinib reduced the viability of all NB cell lines tested, although to different extents (Fig. 1 A). IC50 was calculated, by linear regression analysis, only in the cell lines in which at least one of the tested concentrations was able to reduce cell viability below 50%. IC50 values were in the range of sub-micromolar concentrations in 7/10 cell lines (Fig. 1 A).
Figure 1.
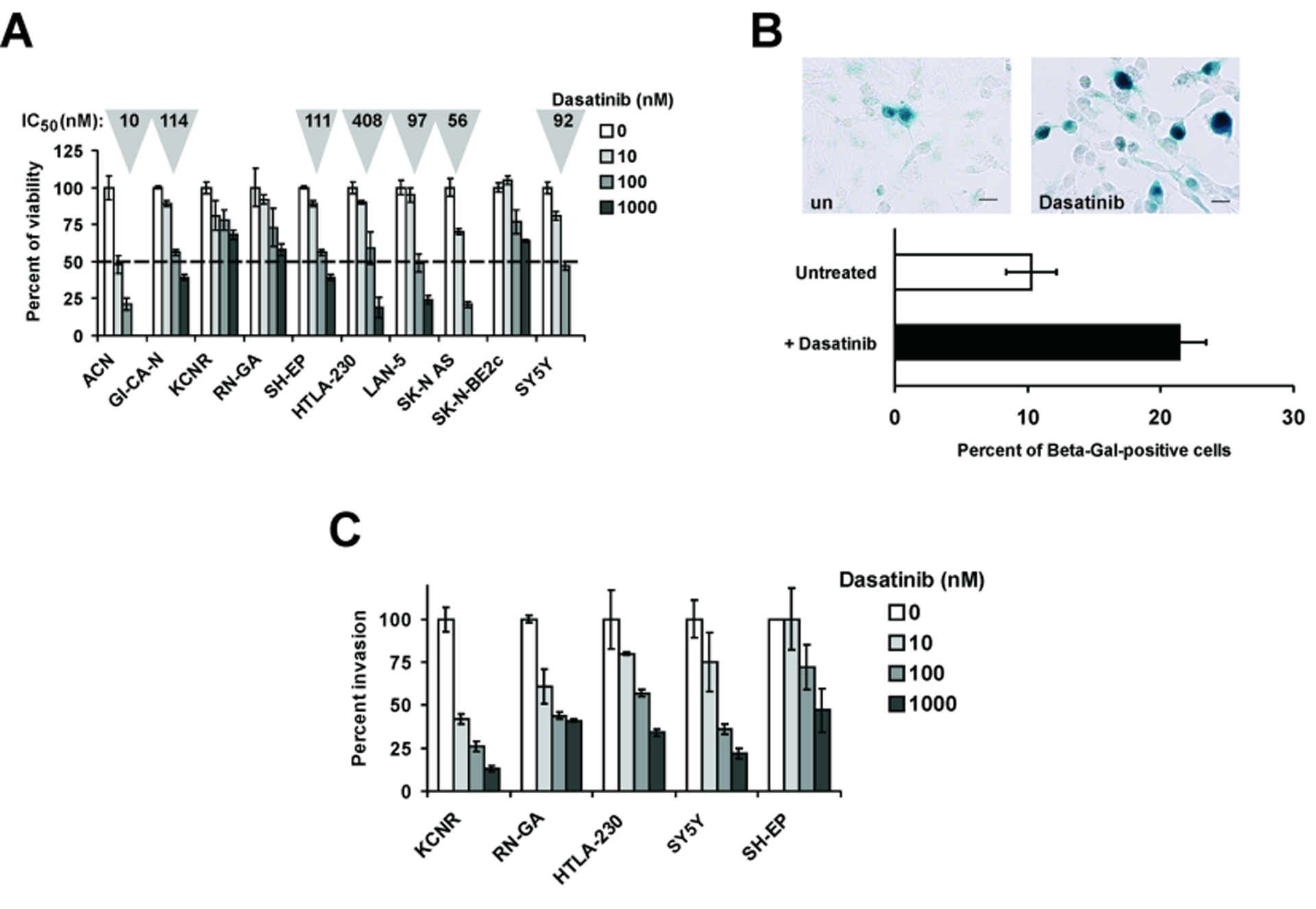
A: Cell viability assays were carried out by exposing the indicated NB cell lines to increasing concentrations of Dasatinib (10, 100 and 1000 nM) for 48 hours. IC50 values (in grey triangles on the top of each column) were calculated by regression analysis in the cell lines where at least one drug concentration caused a 50 % decrease in cell viability. Triplicate assays were performed for each drug concentration. Values ± SE are given. B: top panels show beta-galactosidase staining of untreated (un) and Dasatinib-treated SH-EP cells. Percentages of beta-galactosidase positive cells counted in randomly selected microscopic fields (at least 1000 elements) are reported in the bottom graph. C: invasion assays were carried out in vitro in Matrigel® - coated chambers (see Materials and Methods) using the indicated NB cell lines treated with increasing concentration of Dasatinib (10, 100 and 1000 nM) for 20 hours. Percent invasion was arbitrarily set to 100 in untreated cells. Experiments were carried out in triplicate. Values ± SE are given.
Next, we evaluated the activity of Dasatinib in reducing anchorage-independent growth, a well established hallmark of tumour aggressiveness. For this analysis, we selected 3 Dasatinib-sensitive (SY5Y, SH-EP and HTLA-230) and 2 partially resistant (RN-GA and KCNR) cell lines. Cells were seeded in semi-solid (soft agar) medium containing Dasatinib (10, 100 and 1000 nM) and cell clones were allowed to growth for 11–13 days. Cultures were stained and clones (aggregates of >20 cells) were counted. Dasatinib-treated cells were markedly less clonogenic than their untreated counterparts (Table 1). Interestingly, anchorage-independent growth was strongly inhibited also in the cell lines RN-GA and KCNR that were partially resistant to Dasatinib in the cell viability assays (see Fig. 1 A). In the semi-solid agar assays, the SY5Y and SH-EP cell lines were not informative because did not form clones of more than 20 cells.
Table 1.
Semi-solid agar assay in NB cell lines treated with Dasatinib
| Cell line | Dasatinib concentration (nM) | Aggregates >20 cells (clones) | Aggregates ≤20 cells (single cells) | Clonogenic inhibition (%) | p* |
|---|---|---|---|---|---|
| RN-GA | 0 | 83 | 458 | 0.0 | |
| 10 | 36 | 438 | 50.5 | >0.001 | |
| 100 | 25 | 518 | 70.0 | >0.001 | |
| 1000 | 3 | 531 | 96.3 | >0.001 | |
| KCNR | 0 | 119 | 304 | 0.0 | |
| 10 | 64 | 313 | 39.7 | >0.001 | |
| 100 | 32 | 373 | 71.9 | >0.001 | |
| 1000 | 27 | 494 | 81.6 | >0.001 | |
| HTLA-230 | 0 | 19 | 389 | 0.0 | |
| 10 | 8 | 500 | 66.2 | >0.001 | |
| 100 | 6 | 458 | 72.2 | >0.001 | |
| 1000 | 4 | 488 | 82.5 | >0.001 | |
Note: The assay was carried out by counting > 300 total objects (aggregates of >20 cells or aggregates ≤20 cells) in 10 randomly selected microscopic fields (see also Materials and Methods).
Statistical significance was determined by χ2 test.
Different cell fates after Dasatinib in vitro treatment.
In some cell lines, Dasatinib treatment induced morphological changes consistent with the induction of senescence or apoptosis. To evaluate whether drug treatment could induce senescence, NB cells were treated for 48 h with Dasatinib at a concentration 20% above the calculated IC50 and stained for expression of beta-galactosidase, a well known marker of cellular senescence25. Beta-galactosidase-positive cells were significantly increased only in Dasatinib-treated SH-EP cells compared to untreated cells (Fig. 1 B), whereas no significant increase in beta-galactosidase positivity was detected in the other cell lines tested (not shown). Next, cell cycle distribution and sub G1 DNA content were determined by flow cytometry in RN-GA, KCNR, HTLA-230, SY5Y and SH-EP cells after Dasatinib treatment (at concentrations 20% above the calculated IC50 for each cell line, for 1, 2 and 3 days) (Table 2). Massive subG1 increase was detected in RN-GA (58.4 %) after 2 days of treatment, and in HTLA-230 (56.9 %) after 3 days. At this time point (3 days), the viability of HTLA-230 cells as assessed by trypan blue staining was less than 1%. In the other tested cell lines, the increase in the sub G1 fraction was more modest (SY5Y and SH-EP) or not detectable (KCNR). SY5Y cells underwent a G1 accumulation and simultaneous decrease of S and G2/M phases (Table 2). Caspase 3/7 activity assay (data not shown) carried out after 48 hours of Dasatinib treatment was consistent with the data obtained by the flow cytometric analysis. Of interest, Dasatinib treatment in SH-EP cells induced apoptosis determined by subG1 accumulation (Table 2), caspase 3/7 increase (data not shown), and senescence (beta-GAL staining) (Fig. 1 B). Altogether, these experiments suggest that Dasatinib treatment exerts inhibitory effects inducing distinct, cell line-specific outcomes.
Table 2.
Cell cycle distribution and sub G1 DNA content after Dasatinib treatment.
| Cell line | Treatment | Days | Percent | |||
|---|---|---|---|---|---|---|
| Sub G1 | G1 | S | G2/M | |||
| HTLA-230 | Untreated | 1.6 | 35.9 | 24.1 | 38.4 | |
| Dasatinib (490 nM)* | 1 | 3.0 | 40.2 | 16.4 | 40.4 | |
| 2 | 6.4 | 36.8 | 18.6 | 38.2 | ||
| 3 | 56.9 | 24.1 | 9.4 | 9.6 | ||
| RN-GA | Untreated | 0.6 | 41.3 | 33.0 | 25.1 | |
| Dasatinib (1000 nM)** | 1 | 5.1 | 67.4 | 16.2 | 11.3 | |
| 2 | 58.4 | 32.7 | 4.9 | 4.0 | ||
| 3 | 51.6 | 43.2 | 2.5 | 2.7 | ||
| SH-EP | Untreated | 7.8 | 66.5 | 14.0 | 11.7 | |
| Dasatinib (133 nM)* | 1 | 7.6 | 69.8 | 13.2 | 9.4 | |
| 2 | 6.9 | 69.1 | 13.5 | 10.5 | ||
| 3 | 13.4 | 70.8 | 9.7 | 6.1 | ||
| SY5Y | Untreated | 11.5 | 66.9 | 7.2 | 14.4 | |
| Dasatinib (110 nM)* | 1 | 10.0 | 81.3 | 3.1 | 5.5 | |
| 2 | 13.1 | 79.4 | 1.7 | 6.1 | ||
| 3 | 23.4 | 67.0 | 2.9 | 6.7 | ||
| KCNR | Untreated | 1.0 | 63.1 | 12.4 | 23.4 | |
| Dasatinib (1000 nM)** | 1 | 7.6 | 54.2 | 13.3 | 25.0 | |
| 2 | 6.8 | 51.6 | 19.4 | 21.2 | ||
| 3 | 4.9 | 55.8 | 18.9 | 20.4 | ||
Dasatinib concentration equal to calculated IC50 + 20 %.
Maximum Dasatinib concentration used in this study.
Dasatinib reduces in vitro invasion of NB cells.
Many NB cell lines are able to invade through extra-cellular matrix (ECM) barriers26. The ability of Dasatinib to inhibit in vitro invasion of NB cells was tested by using Matrigel-coated invasion chambers. Cells were seeded in the upper chamber in medium supplemented with 5% FCS. In the lower chamber, medium supplemented with 20% FCS was used as chemo-attractant. After 18 hours, the experiment was terminated and cells migrated in the lower chamber were stained and counted. Dasatinib used at increasing concentrations (10, 100 and 1000 nM) inhibited invasion of all cell lines tested, albeit with different efficiencies (Fig. 1 C).
In vitro effects of Dasatinib on the Akt and Erk1/2 pathways.
The effect of Dasatinib treatment on Akt and Erk1/2 pathways was evaluated by western blot analysis by use of phospho-specific antibodies. HTLA-230 (MYCN amplified27) and SY5Y (MYCN non-amplified20) cells were selected for this analysis since they are also suitable for the in vivo studies described below. The primary targets of Dasatinib are Src-family tyrosine kinases such as Abl proteins and c-Src19, but Dasatinib also interacts with several other tyrosine and serine/threonine kinases and other non-kinase proteins28. Exponentially growing cells were treated at a concentration 20% higher than the calculated IC50 for 2 hours. Proteins were extracted and the effects on Akt and Erk activation tested by immunoblotting with anti-phospho Akt and Erk1/2. In HTLA-230 cells, Dasatinib treatment markedly inhibited Erk1/2 phosphorylation and had a more modest effect on Akt phosphorylation while the total amount of each protein remained constant (Fig. 2 A and B). Dasatinib treatment had similar effects on HTLA-230 cells stimulated to proliferate after a 16-h serum starvation (Fig. 2 A). In contrast, SY5Y cells did not show a significant decrease of Akt and Erk activation when treated (2 hours) at a concentration (110 nM) 20% higher than the calculated IC50 (not shown). By increasing Dasatinib concentration 5-fold (550 nM) and treating the cells for 2 or 4 hours, we obtained a weak down-regulation of Akt and Erk1/2 activation (Fig. 2 C and D). We also assessed expression and phosphorylation status of Src and the c-Kit receptor that is expressed in some NB cells and tumours29,30. Src phosphorylation was reduced by Dasatinib in HTLA-230 and SY5Y cell lines (Fig. 2E). c-Kit was detectable in HTLA-230 and its phosphorylation was weakly decreased by Dasatinib (Fig. 2 E). By contrast, SY5Y cells did not show detectable levels of c-Kit by immunoblotting (Fig. 2 E). The partial effect on Src phosphorylation and the low expression of c-Kit make unlikely that Dasatinib acts on HTLA-230 and SY5Y exclusively through these targets.
Figure 2.
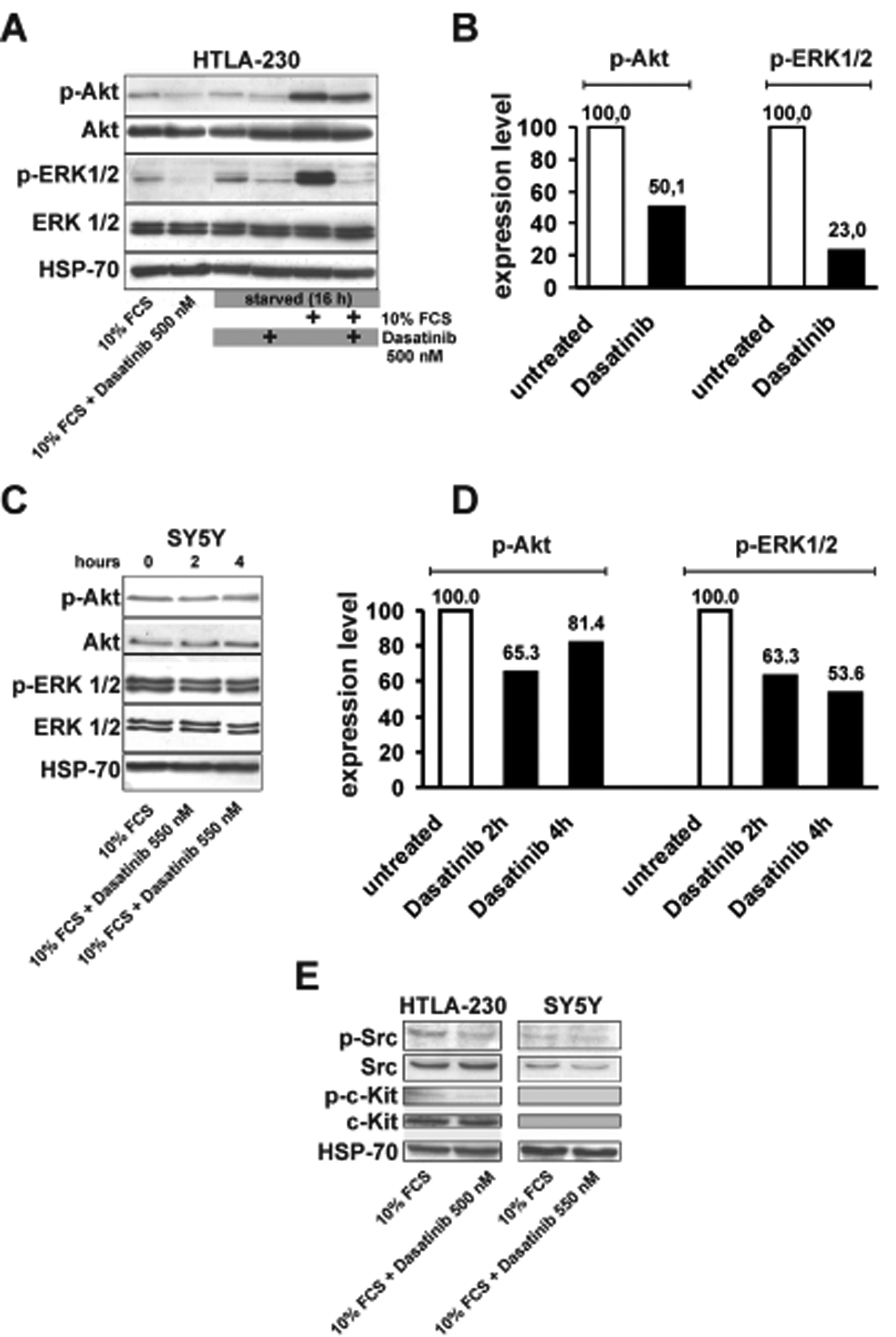
A and C: western blot analyses for phospho-Akt (p-Akt), Akt, phospho-Erk1/2 (p-Erk1/2) and Erk1/2 detection in Dasatinib-treated HTLA-230 (500 nM) and SY5Y (550 nM) cells. In HTLA-230 cells experiments were carried out either by adding Dasatinib to complete medium (lanes on the left) or by starvation in the absence of serum (20 hours), followed by treatment with Dasatinib (2 hours) and serum stimulation (1 hour).SY5Y cells in complete medium were treated with Dasatinib (550 nM) for 2 or 4 hours. B and D: Densitometric analyses of phospho-Akt and phospho-Erk1/2 in untreated and Dasatinib-treated HTLA-230 and SY5Y cells from blots in panels A and C. Densitometric values were normalized for the levels of total Akt and Erk1/2. Expression levels in untreated cells were arbitrarily set to 100. D: detection of phospho-c-Src (p-c-Src), c-Src, phospho-c-Kit (p-c-Kit) and c-Kit in untreated or Dasatinib-treated (2 hours) HTLA-230 and SY5Y cells. HSP-70 expression was utilized to normalize the amount of protein extracts loaded in each lane in A, C and in E.
In vivo activity of Dasatinib.
Immuno-deficient mice (nu/nu, Crl:CD1-Foxn1) were orthotopically transplanted in the left adrenal gland with HTLA-230 or SY5Y cells (1 × 106 cells/animal). Animals injected with HTLA-230 cells were treated with 30mg/Kg/day of Dasatinib starting 7 days after injection. Animals injected with SY5Y cells were treated according to three different schedules: i) 30mg/Kg/day of Dasatinib for 40 consecutive days starting 7 days after injection; ii) 60mg/Kg/day for 40 consecutive days starting 7 days after injection; iii) 60mg/Kg/day for 40 consecutive days, starting 1 day after injection. Each treatment and control group included 8–10 animals. All experiments were completed 40 days after transplantation. Treatments were carried out by gavage in a volume of 0.2 mL. Control groups were treated with the same volume of vehicle (0.2 mL of 80 mM citric acid pH 3.1). During treatment, the weight of the animals was measured twice a week. At the end of treatment, animals were sacrificed and necropsy was carried out. Tumours were removed and their volume was measured. Metastases or micro-metastatic foci were not detected in liver, spleen, intestine, kidneys and lungs in any group of animals. Tumours and the above-mentioned organs were fixed and processed for histological and immunohistochemical analyses.
In animals injected with HTLA-230 cells, although Dasatinib treatment did not suppress completely tumour growth, we observed a significant reduction in mass volume (Student’s t test, p= 0. 047), compared to control animals (Fig. 3 A). Throughout the length of the treatment, no sign of distress was observed, consistent with lack of weight loss in treated groups compared with controls (Fig. 3 C). Microscopic examination did not reveal significant changes in tumour vascularization or presence of apoptotic figures in any group of animals (Fig. 4 A and B), although tumour cellularity in Dasatinib-treated tumours (Fig. 4 B) was decreased compared to controls (Fig. 4 A). Evaluation of the active form of caspase 3 revealed that there were no changes in tumours from untreated or Dasatinib-treated mice (data not shown). On the contrary, immunohistochemical detection of the proliferation-specific Ki67 antigen revealed a significant decrease in the proliferative rate in Dasatinib-treated tumours (Fig. 4 D) compared to their controls (Fig. 4 C). Results and statistics of this analysis are summarized in Table 3.
Figure 3.
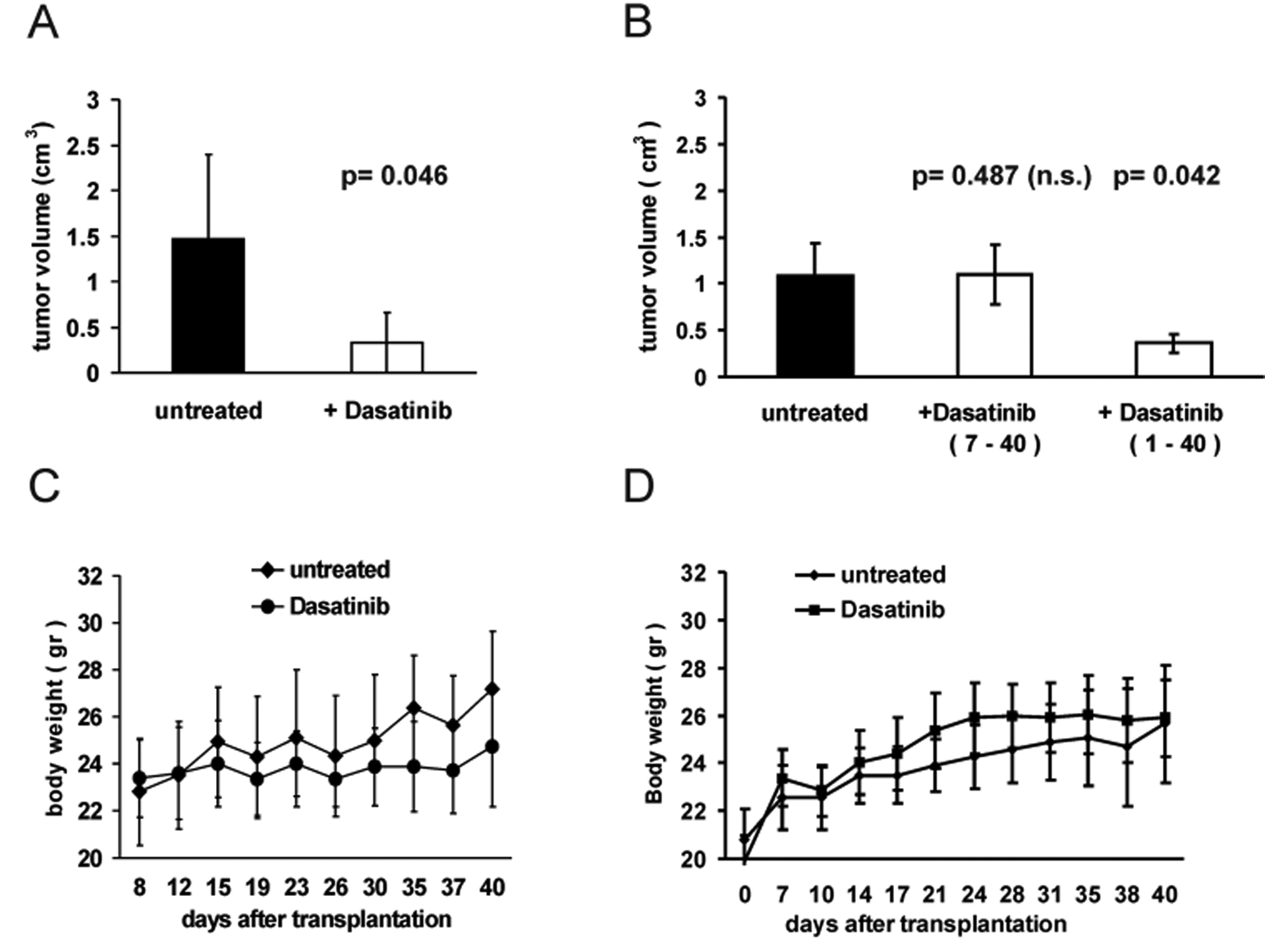
A: HTLA-230 (A) and SY5Y (B) cells were orthotopically injected in nude mice (see text). Forty days after injection, animals were sacrificed and mean tumour volume (bars ± SE) was calculated in untreated and in Dasatinib-treated groups. p was determined by Student’s t test. Dasatinib treatment was given at the dose of 30mg/Kg/day starting 7 days after orthotopic injection of HTLA-230 cells (A) or at 60mg/Kg/day starting either 1 or 7 days after injection of SY5Y cells (B). C and D: Body weight was periodically measured until the last day of treatment (day 40). The graph in D shows the comparison of body weight of untreated and Dasatinib-treated animals (60mg/Kg/day from day 1 to day 40). In all groups, body weight differences between untreated and Dasatinib-treated animals throughout the experiments were not significant.
Figure 4.
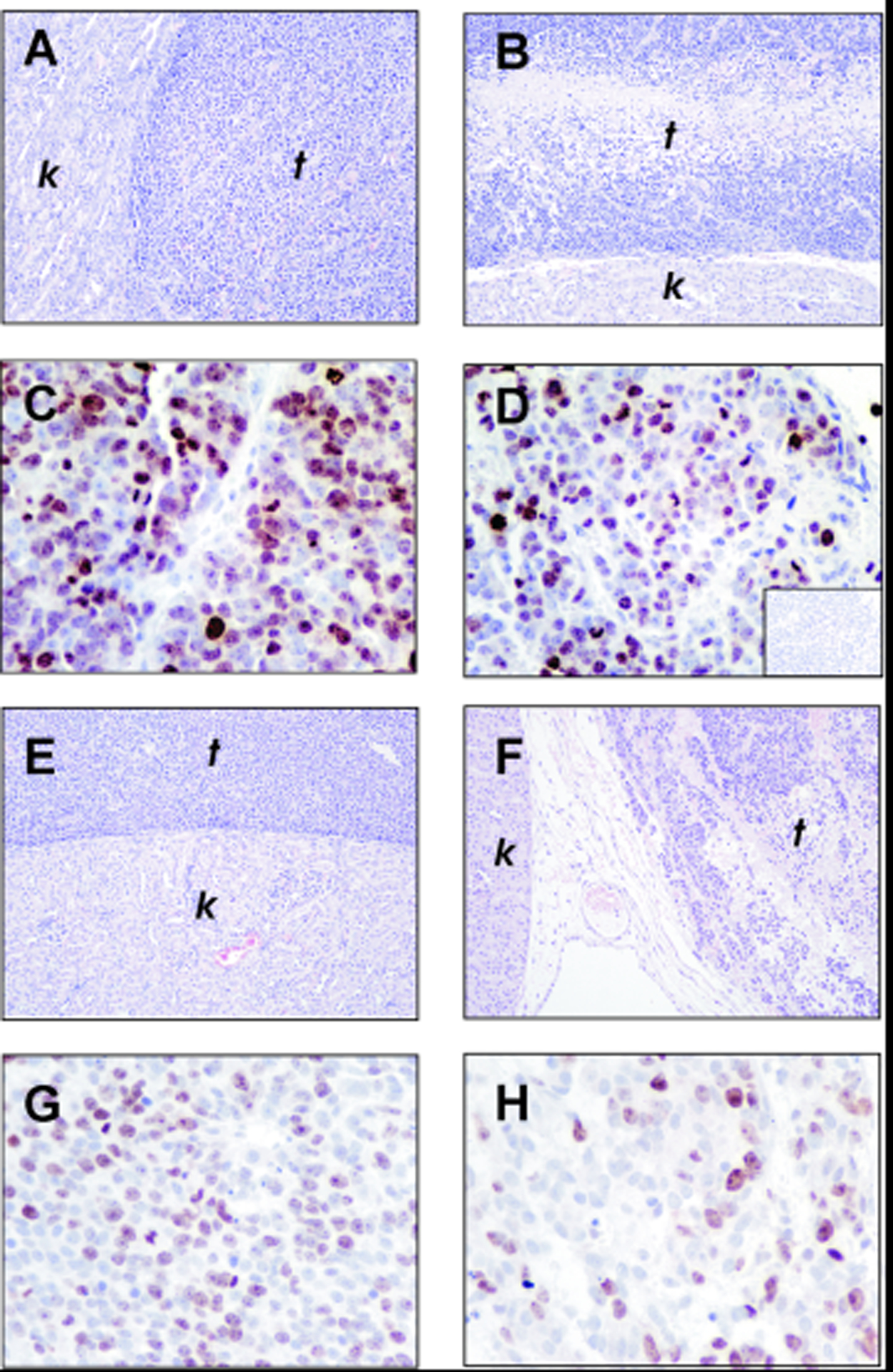
Histological sections of untreated (A and E) and Dasatinib-treated (B and F) tumours from animals orthotopically injected with HTLA-230 (A and B) and SY5Y (E and F) cells; t: tumour; k: kidney. Immunohistochemical staining for Ki-67 expression in untreated (C and G) and Dasatinib-treated (D and H) tumours from animals injected with HTLA-230 (C and D) or SY5Y cells (G and H). Inset in D shows the negative control in which staining with primary anti-Ki-67 antibody is replaced by staining with normal serum.
Table 3.
Ki-67 analysis in tumours from HTLA-230 and SY5Y transplants.
| Cell line | Treatment | Total cells | Ki-67+ cells | % Ki-67+ cells | p* |
|---|---|---|---|---|---|
| HTLA-230 | Untreated | 3528 | 2757 | 78.1 | <0.0001 |
| Dasatinib | 3427 | 2291 | 66.8 | ||
| SY5Y | Untreated | 4033 | 2282 | 56.6 | <0.0001 |
| Dasatinib | 5099 | 1732 | 34.0 |
Fisher’s exact test. Dasatinib treatments were: 30 mg/Kg/day from day 7 to day 40 for HTLA-230 and 60 mg/Kg/day from day 1 to day 40 for SY5Y.
Results in animals injected with SY5Y cells varied depending on the treatment’s schedule. Animals treated with 30/mg/Kg/day did not show any significant reduction in tumour volume compared to controls (not shown). Fig. 3 B shows that tumour volume in animals which received a dose of 60mg/Kg/day starting 7 days after injection was not statistically different from controls (Student’s t test, p= 0.487). In contrast, animals treated with 60mg/Kg/day starting 1 day after tumour cell injection showed a significant decrease in tumour volume compared to controls (Student’s t test, p= 0. 042) (Fig. 3 B). In the Dasatinib-treated tumours, we observed a marked decrease in cellularity (Fig. 4 F) and immunoreactivity of the proliferation-specific Ki67 antigen (Fig. 4 H and G and Table 3), similarly to what we observed in HTLA-230 derived tumours. Of interest, also at the dose of 60mg/Kg/day utilized in this group of animals we observed neither signs of distress nor a significant reduction in body weight compared to the controls (Fig. 3 D).
To gain a deeper understanding of the Dasatinib’s effect(s) at the molecular level, protein extracts were prepared from HTLA-230- and SY5Y-derived tumours and assessed for activation of signal transduction pathways and expression of pro-apoptotic proteins. Consistent with the in vitro data (see Figure 2 A and B), we detected a striking down-regulation of phospho-Erk1/2 and a more modest decrease of phospho-Akt in Dasatinib-treated HTLA-230-derived tumours compared to their controls (Fig. 5 A and B). In addition, a slight increase in Bax and Bad expression was only detected in Dasatinib-treated tumours (Fig. 5 A).
Figure 5.
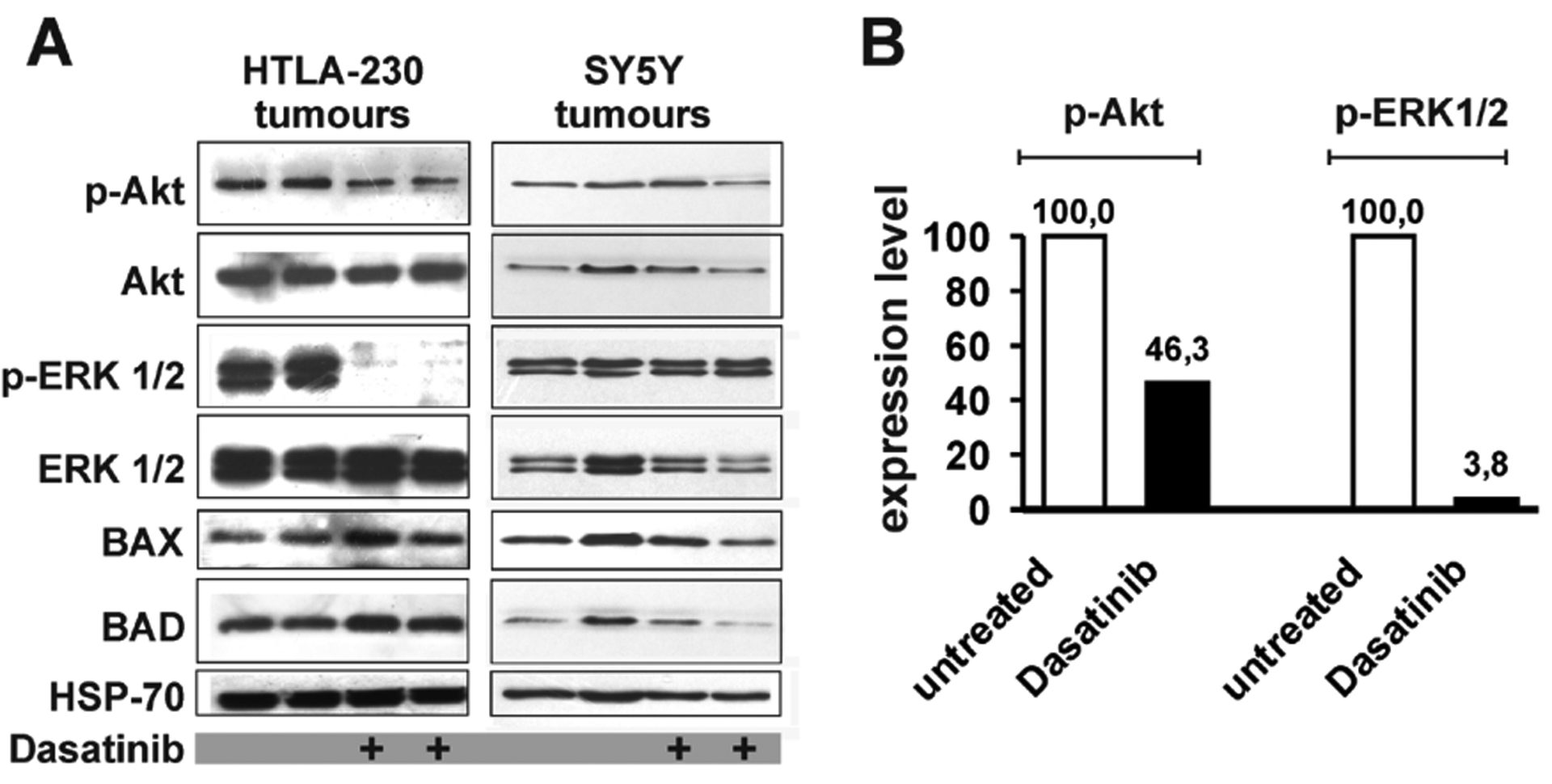
A: western blot analyses to detect phospho-Akt (p-Akt), Akt, phospho-Erk1/2 (p-Erk1/2) and Erk1/2, BAX and BAD in cell extracts from tumours of untreated and Dasatinib-treated animals transplanted with HTLA-230 (blots on the left) or with SY5Y cells (blots on the right). Protein amount loaded in each lane was normalized by HSP-70 levels. B: Densitometric analysis of phospho-Akt and phospho-Erk1/2 in untreated and Dasatinib-treated animals injected with HTLA-230 cells. Each densitometric value represents the mean of 2 untreated and 2 Dasatinib-treated samples from blot in panel A. Values were normalized for the levels of total Akt and Erk1/2. Expression levels in untreated samples were arbitrarily set to 100.
In protein extracts of SY5Y-derived tumours prepared from mice treated with 60mg/Kg/day starting 1 day after injection, we did not observe a consistent variation of the activity of Akt and Erk1/2 or of the levels of Bax and Bad (Fig. 5 A). These findings, together with the weak down-regulation of Akt and Erk1/2 activity observed in vitro, suggest that Dasatinib likely acts on other unidentified targets in SY5Y cells.
Discussion
We have demonstrated here the efficacy of Dasatinib used at submicromolar concentrations in reducing cell viability, anchorage-independent growth and invasion in vitro in a panel human NB cell lines. Of interest, the cellular outcome induced by Dasatinib treatment in sensitive cells was rather heterogeneous. In the SH-EP cell line, induction of senescence, detected by beta-galactosidase expression and G1 accumulation, was accompanied by apoptosis analyzed by caspase 3/7 activation and increase of subG1 DNA content. In contrast, other Dasatinib-sensitive cell lines (HTLA-230, RN-GA, SY5Y) did not show any sign of senescence but underwent massive apoptosis with different kinetics (Table 2). Moreover, the pro-apoptotic activity induced by Dasatinib in our study contrasts with the findings described in a previous report31, but this difference may be due to the use of different NB cell lines in the two studies. In this regard, the heterogeneous behaviour of Dasatinib-treated tumour cells may depend on cell line-specific targets. A recent paper has described 38 different proteins that physically interact with Dasatinib28. Unexpectedly, although Dasatinib was originally described as a specific inhibitor of Src-family kinases, it also interacts with several Ser/Threo kinases and proteins without kinase activity, some of which may be functionally altered. In addition, the target profile of Dasatinib also depends on the cell type28. By testing some cellular pathways that are important for cell proliferation and survival, we found that Erk1/2 and Akt activation was potently inhibited by Dasatinib in HTLA-230, the cell line subsequently used for the in vivo experiments. Conceivably, this finding may be due to upstream inhibition of c-Kit and c-Src that occurs in this cell line, although we cannot exclude that the effect is also due to inhibition of other relevant targets of Dasatinib that could be identified by a proteomic/transcriptomic approach. We obtained different results with SY5Y cells in which Dasatinib was ineffective in inducing a detectable inactivation of Akt and Erk1/2 unless used at a concentration (550 nM) more than five times higher than the calculated IC50. In addition, c-Src activation was only partially inhibited in SY5Y suggesting that, in these cells, the inhibition of other, yet unknown, targets is responsible for the sensitivity to Dasatinib.
Regardless of the cell type-specific processes triggered by Dasatinib, the inhibitory effects on anchorage-independent growth and on in vitro invasion were demonstrated in the same range of concentrations used in the cell viability assays. This analysis expands previous data on Dasatinib activity in vitro31,32. Interestingly, the inhibition of anchorage-independent growth and in vitro invasion did not faithfully parallel the inhibition of cell viability in the same cell line. RN-GA cells that were partially resistant to concentrations of Dasatinib as high as 1000 nM, showed a >50% inhibition of clonogenic activity in semi-solid agar at 10 nM and >50% reduction of invasion at 100 nM. The latter finding may represent an interesting observation for future clinical use of this drug in NB, although further work is needed to verify whether the inhibition of invasion is also achievable in vivo.
In order to evaluate the efficacy of Dasatinib in vivo we used a murine orthotopic model because: a) NB cells injected directly in the adrenal gland are embedded in a microenvironment that closely mimics that of the human disease24; and b) drug uptake is more similar to that of spontaneous NB which frequently occurs in the adrenals1 compared to subcutaneous transplants. HTLA-230 and SY5Y cells were chosen for the in vivo study since they derive from aggressive NB and have been successfully used in pseudometastatic and orthotopic models24,27. Drug treatment in the orthotopic model utilizing HTLA-230 cells produced a significant reduction of tumour burden. Nevertheless, it should be stressed that Dasatinib activity in vivo appears weaker than in vitro. HTLA-230 tumours were reduced in size and cellularity, with proliferation being also significantly inhibited, but complete tumour eradication was not achieved. This partial effectiveness may depend on suboptimal drug concentration at the tumour site. However, we treated animals with a dose (30 mg/Kg/day) higher than that utilized in CML pre-clinical models19 and in other solid tumours33. In addition, HTLA-230 tumours were vascularized and Dasatinib treatment did not induce any significant change, at least as assessed by microscopic observation. Using SY5Y cells in vivo in the orthotopic model, we failed to demonstrate Dasatinib antitumour activity at 30mg/Kg/day and at 60mg/Kg/day when starting treatment 7 days after transplantation. We could detect a significant decrease in tumour volume at a dose of 60mg/Kg/day only by starting treatment 1 day after tumour cell injection. This suggests that Dasatinib is effective in reducing growth of SY5Y in vivo only when the number of tumour cells is still low. Nevertheless, it should be stressed that SY5Y cells were among the most Dasatinib-sensitive cell lines in vitro (IC50= 92 nM). Somehow in agreement with these observations, it has been reported that the efficacy of cytotoxic drugs can be different when tumour cells are grown in vitro in tri-dimensional cultures (spheroids) compared to bi-dimensional ones34. This behaviour is unlikely to be due to a less efficient diffusion of a low molecular weight drug like Dasatinib (MW 506) into tri-dimensional structures. Instead, inter-cellular signals that promote drug-resistance of tumour cells may be activated in spheroids34 similarly to what can occur in the in vivo environment. An additional possible reason for the partial efficacy of Dasatinib in vivo may rest in a different supply of growth factors and nutrients. The adrenal gland is the most favourable milieu for NB growth, being the most frequent site of occurrence of primary NB1. In addition, occurrence of NB in adrenals represents a well known marker of aggressiveness35. Thus, it is conceivable that some survival factors that help NB cells to resist Dasatinib treatment are available in the adrenal microenvironment, but not in vitro. Taken together, these data emphasize the critical importance of assessing the efficacy of novel drugs with demonstrated in vitro anti-tumour activity in in vivo settings mimicking as much as possible the individual microenvironment of spontaneous human cancer.
Despite the partial efficacy, Dasatinib induced in vivo changes in ERK1/2 and Akt activation similar to those detected in vitro in animals orthotopically injected with HTLA-230. Of interest, protein kinases upstream of Erk1/2 in the MAP kinase pathway are among those shown to interact physically with Dasatinib28. Thus, the marked efficacy of Dasatinib in reducing Erk1/2 activation in vitro and in vivo might be due to a direct effect on some component(s) of this pathway in HTLA-230 cells. In contrast, we could not detect a clear inhibitory effect of Dasatinib on Akt and Erk1/2 activation in tumours of animals injected with SY5Y cells. This observation is consistent with the minor inhibitory effect of Dasatinib on Akt and Erk1/2 activity in SY5Y cells in vitro.
A rational combination of Dasatinib with existing cytotoxic drugs may achieve greater effectiveness in NB growth inhibition in vivo. In this regard, it has been recently demonstrated that low doses of Imatinib mesylate (0.5–2.5 μM) that have limited activity when used alone, appear to act synergistically with cytotoxic drugs already utilized in therapy36.
In summary, our data demonstrate that Dasatinib is active on NB cells in vitro and in vivo. The orthotopic model, used in our study for the first time to test Dasatinib effectiveness in NB, highlights the importance of choosing appropriate in vivo models to evaluate the efficacy of novel compounds of potential therapeutic interest. Based on the findings of this study, further investigations utilizing Dasatinib in combination with existing cytotoxic drugs may lead to the development of more effective and less toxic therapies of NB.
Acknowledgements
This work was partially supported by grants from “Fondazione Italiana per la Lotta al Neuroblastoma” (G.R.), Italian Ministry of Health (C.D), “Io…domani - Associazione per la Lotta contro i Tumori Infantili” (ALTI) (C.D.), and NCI RO1 CA95111 (B.C). R.V. and F.S. are fellows of “Fondazione Italiana per la Lotta al Neuroblastoma”.
Abbreviations:
- NB
neuroblastoma
- Erk
Extracellular signal-Regulated Kinase
- CML
Chronic Myelogenous Leukemia
- FCS
Fetal Calf Serum
Footnotes
Statements
Advanced neuroblastoma (stage 4) still remains an unsolved clinical problem. Tyrosine kinase inhibitors such as Dasatinib may represent an effective and less toxic alternative for future therapeutic intervention.
References
- 1.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003;3:203–16. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet 2007;369:2106–20. [DOI] [PubMed] [Google Scholar]
- 3.Spitz R, Hero B, Simon T, Berthold F. Loss in chromosome 11q identifies tumors with increased risk for metastatic relapses in localized and 4S neuroblastoma. Clin Cancer Res 2006;12:3368–73. [DOI] [PubMed] [Google Scholar]
- 4.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, Faldum A, Hero B, Iehara T, Machin D, Mosseri V, Simon T et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 2009;27:289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sano H, Bonadio J, Gerbing RB, London WB, Matthay KK, Lukens JN, Shimada H. International neuroblastoma pathology classification adds independent prognostic information beyond the prognostic contribution of age. Eur J Cancer 2006;42:1113–9. [DOI] [PubMed] [Google Scholar]
- 6.Brodeur GM, Seeger RC. Gene amplification in human neuroblastomas: basic mechanisms and clinical implications. Cancer Genet Cytogenet 1986;19:101–11. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki T, Bogenmann E, Shimada H, Stram D, Seeger RC. Lack of high-affinity nerve growth factor receptors in aggressive neuroblastomas. J Natl Cancer Inst 1993;85:377–84. [DOI] [PubMed] [Google Scholar]
- 8.Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol 1994;14:759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosse YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008;455:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008;455:971–4. [DOI] [PubMed] [Google Scholar]
- 11.George RE, Sanda T, Hanna M, Frohling S, Luther W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008;455:975–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Bernardi B, Nicolas B, Boni L, Indolfi P, Carli M, Cordero Di Montezemolo L, Donfrancesco A, Pession A, Provenzi M, di CA, Rizzo A, Tonini GP et al. Disseminated neuroblastoma in children older than one year at diagnosis: comparable results with three consecutive high-dose protocols adopted by the Italian Co-Operative Group for Neuroblastoma. J Clin Oncol 2003;21:1592–601. [DOI] [PubMed] [Google Scholar]
- 13.Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med 2005;353:172–87. [DOI] [PubMed] [Google Scholar]
- 14.Vitali R, Cesi V, Nicotra MR, McDowell HP, Donfrancesco A, Mannarino O, Natali PG, Raschella G, Dominici C. c-Kit is preferentially expressed in MYCN-amplified neuroblastoma and its effect on cell proliferation is inhibited in vitro by STI-571. Int J Cancer 2003;106:147–52. [DOI] [PubMed] [Google Scholar]
- 15.Meco D, Riccardi A, Servidei T, Brueggen J, Gessi M, Riccardi R, Dominici C. Antitumor activity of imatinib mesylate in neuroblastoma xenografts. Cancer Lett 2005;228:211–9. [DOI] [PubMed] [Google Scholar]
- 16.Beppu K, Jaboine J, Merchant MS, Mackall CL, Thiele CJ. Effect of imatinib mesylate on neuroblastoma tumorigenesis and vascular endothelial growth factor expression. J Natl Cancer Inst 2004;96:46–55. [DOI] [PubMed] [Google Scholar]
- 17.Rossler J, Zambrzycka I, Lagodny J, Kontny U, Niemeyer CM. Effect of STI-571 (imatinib mesylate) in combination with retinoic acid and gamma-irradiation on viability of neuroblastoma cells. Biochem Biophys Res Commun 2006;342:1405–12. [DOI] [PubMed] [Google Scholar]
- 18.Te Kronnie G, Timeus F, Rinaldi A, Crescenzio N, Spinelli M, Rosolen A, Ricotti E, Basso G. Imatinib mesylate (STI571) interference with growth of neuroectodermal tumour cell lines does not critically involve c-Kit inhibition. Int J Mol Med 2004;14:373–82. [PubMed] [Google Scholar]
- 19.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 2004;305:399–401. [DOI] [PubMed] [Google Scholar]
- 20.Lovat PE, Di SF, Corazzari M, Fazi B, Donnorso RP, Pearson AD, Hall AG, Redfern CP, Piacentini M. Gangliosides link the acidic sphingomyelinase-mediated induction of ceramide to 12-lipoxygenase-dependent apoptosis of neuroblastoma in response to fenretinide. J Natl Cancer Inst 2004;96:1288–99. [DOI] [PubMed] [Google Scholar]
- 21.Sieuwerts AM, Klijn JG, Peters HA, Foekens JA. The MTT tetrazolium salt assay scrutinized: how to use this assay reliably to measure metabolic activity of cell cultures in vitro for the assessment of growth characteristics, IC50-values and cell survival. Eur J Clin Chem Clin Biochem 1995;33:813–23. [DOI] [PubMed] [Google Scholar]
- 22.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O,. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92:9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raschella G, Tanno B, Bonetto F, Negroni A, Claudio PP, Baldi A, Amendola R, Calabretta B, Giordano A, Paggi MG. The RB-related gene Rb2/p130 in neuroblastoma differentiation and in B-myb promoter down-regulation. Cell Death Differ 1998;5:401–7. [DOI] [PubMed] [Google Scholar]
- 24.Brignole C, Marimpietri D, Pastorino F, Nico B, Di PD, Cioni M, Piccardi F, Cilli M, Pezzolo A, Corrias MV, Pistoia V, Ribatti D et al. Effect of bortezomib on human neuroblastoma cell growth, apoptosis, and angiogenesis. J Natl Cancer Inst 2006;98:1142–57. [DOI] [PubMed] [Google Scholar]
- 25.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436:720–4. [DOI] [PubMed] [Google Scholar]
- 26.Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschella G. Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo. Clin Cancer Res 2006;12:6772–80. [DOI] [PubMed] [Google Scholar]
- 27.Bogenmann E A metastatic neuroblastoma model in SCID mice. Int J Cancer 1996;67:379–85. [DOI] [PubMed] [Google Scholar]
- 28.Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007;110:4055–63. [DOI] [PubMed] [Google Scholar]
- 29.Matsunaga T, Takahashi H, Ohnuma N, Tanabe M, Yoshida H, Iwai J, Shirasawa H, Simizu B. Expression of N-myc and c-src protooncogenes correlating to the undifferentiated phenotype and prognosis of primary neuroblastomas. Cancer Res 1991;51:3148–52. [PubMed] [Google Scholar]
- 30.Uccini S, Mannarino O, McDowell HP, Pauser U, Vitali R, Natali PG, Altavista P, Andreano T, Coco S, Boldrini R, Bosco S, Clerico A et al. Clinical and molecular evidence for c-kit receptor as a therapeutic target in neuroblastic tumors. Clin Cancer Res 2005;11:380–9. [PubMed] [Google Scholar]
- 31.Timeus F, Crescenzio N, Fandi A, Doria A, Foglia L, Cordero Di Montezemolo L. In vitro antiproliferative and antimigratory activity of dasatinib in neuroblastoma and Ewing sarcoma cell lines. Oncol Rep 2008;19:353–9. [PubMed] [Google Scholar]
- 32.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Lock RB, Tajbakhsh M, Reynolds CP, Maris JM, Keir ST, Billups CA, Smith MA. Initial testing of dasatinib by the pediatric preclinical testing program. Pediatr Blood Cancer 2008;50:1198–206. [DOI] [PubMed] [Google Scholar]
- 33.Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z, Gallick GE. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res 2008;68:3323–33. [DOI] [PubMed] [Google Scholar]
- 34.Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007;99:1441–54. [DOI] [PubMed] [Google Scholar]
- 35.Brodeur GM and Maris JM, Principles and Practice of Pediatric Oncology, In: P. &. Pizzo PD (ed.) , Principles and Practice of Pediatric Oncology , pp. 895–937, (2002). [Google Scholar]
- 36.Palmberg E, Johnsen JI, Paulsson J, Gleissman H, Wickstrom M, Edgren M, Ostman A, Kogner P, Lindskog M. Metronomic scheduling of imatinib abrogates clonogenicity of neuroblastoma cells and enhances their susceptibility to selected chemotherapeutic drugs in vitro and in vivo. Int J Cancer 2009;124:1227–34. [DOI] [PubMed] [Google Scholar]