

Abstract
Objective:
There are no effective systemic therapies for adenoid cystic cancer (ACC) and lack of tumor lines and mouse models have hindered drug development. We aim to develop MYB-activated models for testing new therapeutic agents.
Materials and Methods:
We studied new ACC patient-derived xenograft (PDX) models and generated a matched cell line from one patient. In addition, we generated a genetically-engineered MYB-NFIB mouse model (GEMM) that was crossed with Ink4a+/−/Arf+/− mice to study tumor spectrum and obtain tumor lines. Using human and murine ACC-like tumor lines, we analyzed MYB expression by RNA-Seq and immunoblot and tested efficacy of new MYB inhibitors.
Results:
We detected MYB-NFIB transcripts in both UFH1 and UFH2 PDX and observed tumor inhibition by MYB depletion using shRNA in vivo. We observed rapid loss of MYB expression when we cultured UFH1 in vitro, but were able to generate a UFH2 tumor cell line that retained MYB expression for 6 months. RNA-Seq expression detected an ACC-like mRNA signature in PDX samples and we confirmed an identical KMT2A/MLL variant in UFH2 PDX, matched cell line, and primary biopsy. Although the predominant phenotype of the MYB-NFIB GEMM was B-cell leukemia, we also generated a MYB-activated ACC-like mammary tumor cell line. We observed tumor inhibition using a novel MYB peptidomimetic in both human and murine tumor models.
Conclusions:
We generated and studied new murine and human MYB-activated tumor samples and detected growth inhibition with MYB peptidomimetics. These data provide tools to define treatment strategies for patients with advanced MYB-activated ACC.
Keywords: adenoid cystic cancer, MYB gene, peptidomimetic inhibitor, B-cell leukemia
Introduction
Management of malignant salivary gland tumors is a challenge for clinicians due to lack of effective therapy for patients with unresectable disease and also due to the large number of distinct clinico-histologic subtypes that must be studied separately as unique biologic entities. Adenoid cystic cancer (ACC) is the most common malignant tumor arising from major or minor salivary glands and can also present as primary tumors within breast, lung, skin, and other tissues (1–7). The identification of a recurrent MYB-NFIB fusion oncogene in most ACC cases provided a new tool for diagnosis and insight into a molecular pathogenesis for this malignancy. This chromosomal rearrangement typically truncates carboxy-terminal MYB domains while fusing the terminal NFIB exon encoding a long 3’ untranslated region (8). The detection of MYBL1 fusions in cases of MYB fusion–negative ACC confirmed importance of MYB/MYBL1 activation as the signature oncogene driving pathogenesis of ACC (9–12). Further, there is evidence suggesting a role for the translocated NFIB locus to redirect a 3’ super-enhancer to generate positive feedback activation sustaining deregulated MYB expression (13).
An obstacle to study the role of MYB activation in ACC and to develop effective MYB inhibitor compounds as therapeutic agents is the lack of validated human or mouse tumor cell lines. Although ACC PDX models have been studied, these are cumbersome for drug discovery projects (14–16). Until recently, there were no validated MYB-activated tumor cells adapted for in vitro growth to allow drug development screening and, currently there is only one report of a human cell line with MYB activation (17).
Methods and material:
Peptidomimetics.
The MYBMIM peptidomimetic and its TG3 inactive analogue were produced by solid phase synthesis and validated by mass spectrometry as previously published (24). Peptidomimetics were dissolved in PBS as previously established (24).
Cell viability analysis.
5x103 cells were seeded in 96-well plates overnight and treated with indicated compounds for 48 hours prior to addition of 40 μL 2 mg/mL MTT reagent (Promega) in PBS for 4 hours, and analyzed using SpectraMax M5 microplate reader (Molecular Devices, Sunnyvale, CA).
Cell line generation and culture conditions.
Patient tumor biopsies were minced and incubated with HBSS digestion solution (Thermo Fisher Scientific Inc), HEPES (Mediatech, Inc), CaCl2, FBS, DNase I(Sigma Aldrich) and collagenase A(Sigma Aldrich), at 37°C for 60 min. Cell suspensions were obtained by passing mixture through 40-μm sieve (Fisher Scientific, Pittsburgh, PA). Washed cells were cultured in high-glucose Dulbecco’s modification of Eagle medium (DMEM; Invitrogen), supplemented with Penicillin-Streptomycin (Sigma-Aldrich), 10% Fetal Bovine Serum (Invitrogen).
H&E and Immunohistochemistry.
The tissue specimens were fixed with 10% formalin, dehydrated using ethanol, and paraffin-embedded sections obtained. Images were taken by Leica DM6000B fluorescence microscope.
Generation of MYB-NFIB transgenic mice.
MYB exons 1-14 was fused with NFIB exon 12 and subcloned into Lox-stop-Lox conditional pCBR-based plasmid (Promega) and confirmed by nucleotide sequencing. Transgenic MYB-NFIB injection was performed using FVB mice. MMTV-Cre FVB mice and INK4/ARF knockout B6 mice were crossed with MYB-NFIB fusion mice to generate MYB-NFIB/MMTV-Cre and MYB-NFIB/MMTV-CRE/Ink4a+/−Arf+/− mice. Primer sequences used to identify the MYB-NFIB fusion; forward primer pCBR-BN-376F: 5′-CAGATGTGCAGTGCCAGCACCGATG-3′; reverse primer pCBR-BN-1268R: 5′-TGGCGAGGCGCTTTCTTCAGGTAGG-3′.
Flow cytometric analysis.
Peripheral blood (PB) was collected from indicated mice genotypes and subjected to an automated blood count (Element HT5® Hematology Analyzer, HESK, Colorado, USA). After lysing RBCs, total mononuclear cells were stained with fluorochrome-conjugated antibodies including B220 (PE-Cy7), IgM (APC-Cy7), CD3 (FITC), CD4 (PE), CD8 (Pacific Blue), CD11b (PE-Cy7) and Gr.1 (APC) for 45 minutes at 4°C. Cells were washed with PBS and acquired on a LSR Fortessa flow cytometer (BD Biosciences). All data were analyzed by FlowJo-V10 software (BD Biosciences).
Lentivirus production and cell transduction.
3 x 106 293T cells were seeded in 10-cm dishes overnight and transfected with pLKO.1 vector as recommended (Qiagen, Germantown, MD). X-tremeGENE™ HP DNA Transfection Reagent (Sigma Aldrich, St. Louis, MO) was used for transfection and viral supernatants were used to infect target cells in presence of 8 μg/mL polybrene (Sigma Aldrich, St. Louis, MO). shRNA clones TRCN0000040058, TRCN0000040059, TRCN0000040060 from TRC-Hs1.0 (Human) (Horizon Discovery Group Co) were used as indicated.
Xenograft Tumor Studies.
2x10^6 PDX tumor single cells transduced with shRNA vector control or MYB-shRNA lentivirus were mixed with 200 μl Matrigel (BD Biosciences, Franklin Lakes, NJ) and subcutaneously injected in 6-week-old NOD.CB17-Prkdcscid/NcrCrl (NOD/SCID) (Jackson Laboratory, Bar Harbor, ME). Control and treatment groups were matched by sex and age. Tumors were measured every other week with a dial caliper, and tumor volumes were calculated using the formula: tumor volume= (width2 × length)/2.
MYB dual color breakapart fluorescence in situ hybridization (FISH)
Four-micron paraffin-embedded sections were analyzed with MYB breakapart probe (ZytoVision, Bremerhaven, Germany). Using a Vysis VP2000™ slide pretreatment instrument and reagents, slides were mounted with probe solution, denatured at 80°C and hybridized for 24 hours at 37°C. (Abbott, Abbott Park, Illinois). Slides were washed, counterstained with DAPI and analyzed with a Leica DM6000B microscope and Ariol Software v4.0 (LeicaBiosystems, Wetzlar, Germany).
RNAseq Data Processing and Analysis.
Sequencing reads were mapped to the human transcriptome (hg 19) using bowtie2 (38), and gene expression levels were summarized into TPM values from uniquely mapped reads using RSEM (39). Clustered heatmaps of gene expression were generated with R package ‘heatmap3’ (40) based on a set of previously defined gene signature that was highly expressed in ACC tumors (21). We then applied single-sample gene get variation analysis (ssGSVA) (41) to determine the molecular phenotypes of each sample using log2(TPM+1) data. We obtained GSVA scores for hallmark gene sets or canonic pathways (MSiDB hallmark H set and canonic pathway C2 set) (42) for each sample. Comparison of GSVA scores was conducted using R package ‘limma’ (43). MYB-NFIB gene fusion detection was performed using Star-Fusion (44).
Statistical Methods
Statistical analyses of experimental data were carried out using Prism Graphpad 6.0. The two-sided t test was used to analyze data from cell viability and xenograft assays. Data were presented as mean and standard deviation (±SD). The log-rank test was used to determine the P value in the survival difference between MMTV-Cre/MYB-NFIB and MMTV-Cre/MYB-NFIB/Ink4a+/−/Arf+/− groups in Kaplan-Meier survival analysis. All statistical tests were two-sided and P values were considered statistically significant when not greater than .05.
Ethics statement.
IACUC guidelines and regulations followed U.S. National Institute of Health guidelines, U.S. Public Health Service policy, and U.S. Food and Drug Administration regulations to minimize stress and pain on research animals.
Results:
Characterization of human ACC tumor models
To develop new ACC tumor models, we obtained informed consent to collect excess ACC tumor from two patients undergoing medically indicated procedures using an IRB-approved protocol. Although we are able to maintain UFH1 ACC as a PDX model (UFH1PDX) by passaging through immunodeficient SCID mice, we were unable to adapt tumor cells to in vitro growth. This observation has been noted in previous reports where ACC tumor samples can only be studied in vitro as short-term cultures (8). Interestingly, while UFH1 tumor cells could be maintained by passaged through SCID mice, these tumor cells remained viable without proliferation in cell culture in vitro for > 6 months with no passaging and infrequent changing of media (data not shown). We tested MYB mRNA and protein expression from UFH1 cells immediately after excision from a PDX xenograft and plating onto plastic tissue culture dishes (5-hour time period to allow for attachment) and compared this to MYB expression after 24 hours in cell culture. We observed loss of MYB mRNA by quantitative reverse transcription polymerase chain reaction (RT-PCR) and protein expression within 24 hours that correlated with loss of tumor cell growth in vitro (Supplemental Figure 1a). The mechanism for this MYB inactivation is unknown and we were unable to rescue this by supplementing with conditioned media, ROCK inhibitor, or murine feeder layers (36). We studied a second tumor sample from a palliative debulking procedure in a patient with locally recurrent and metastatic parotid gland ACC. The patient required multiple resections and post-operative photon radiation therapy and salvage palliative proton beam radiation therapy for unresectable recurrent tumor over several years. The patient presented with recurrent tumor (Fig. 1a) and lung metastases that progressed despite dual nivolumab/ipilumumab therapy. A biopsy sample of the recurrent tumor showed poorly differentiated ACC with elevated nuclear MYB expression (Fig. 1b). A complex MYB-NFIB rearrangement with 3’ MYB duplication was detected by hybrid-capture technology on an earlier tumor biopsy (Foundation Medicine, Cambridge, MA). Tumor cells from the biopsy were cultured in vitro and the UFH2 cell line has been maintained using high-glucose DMEM, supplemented with 1% Penicillin-Streptomycin antibiotic and 10% FBS with a doubling time of 36 hours (Fig. 1c). UFH2 identity was matched to the patient using CLIA-certified NGS assay sequencing performed on genomic DNA extracted from UFH2 cell line with detection of both ATRX E1433*frameshift and MLL/KTM2A S1325N variants detected in earlier tumor surgical sample (data not shown). To test for MYB expression, we performed protein immunoblot analysis using a control ACCX22 PDX (gift from Christopher Moskaluk, University of Virginia), the new UFH1 and UFH2 PDX models, the matched UFH2 cell line (in vitro passage 9 and late passage 34), and a control human mucoepidermoid tumor cell line (H3118). We observed elevated MYB expression in the PDX ACC samples and the matched UFH2 cell line (passage 9) but absence of MYB at passage 34 (Fig. 1d). Multiple MYB bands seen on protein immunoblot have been attributed to alternate splicing or protein processing (10) and the non-ACC salivary gland tumor (H3318) showed absent MYB expression. The early and late passage UFH2 tumor cell line were tumorigenic in SCID mice and the early passage excised xenograft showed poorly differentiated, solid histology on H&E staining similar to the primary tumor surgical biopsy (Fig. 1e). In summary, tumor cells obtained from UFH1 and UFH2 ACC patients exhibited distinct growth properties in vitro, perhaps suggesting that the ability to generate the UFH2 line was associated with the undifferentiated histology. Despite this difference in growth pattern, we confirmed MYB rearrangements by fluorescent in situ hybridization using break-apart probes in sections from both UFH1 and UFH2 PDX (data not shown).
Figure 1.
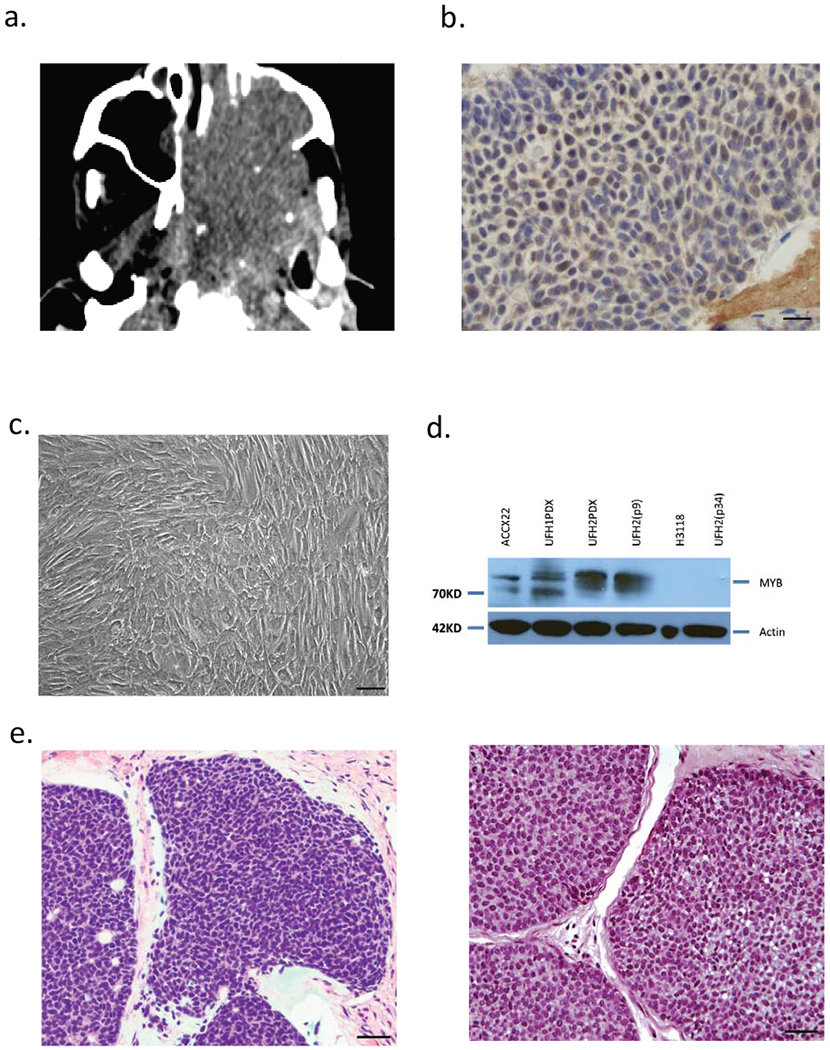
Establishment of human ACC PDX and cell line (UFH2). (a) CT image of unresectable ACC requiring palliative debulking surgery. (b) Nuclear anti-MYB immunohistochemical staining in tumor biopsy. (c) UFH2 tumor cells in vitro at confluence. (d) MYB protein immunoblot (Anti-Myb, abcam, #ab45150) using ACCX22 (positive control), H3118 mucoepidermoid (negative control) and UFH1 PDX and UFH2 PDX and cell line at indicated passage number. (e) H&E staining primary ACC tumor biopsy section (left) and excised matched UFH2 PDX xenograft section (right).
RNA-Seq analysis of new ACC PDX models
We also extracted RNA from UFH1 PDX and UFH2 PDX and from the late passage 34 UFH2 cell line for RNA-Seq analysis. We confirmed detection of distinct MYB-NFIB fusion transcripts in both UFH1 PDX (MYB exon14 fused to either NFIB exon10 or exon11) and UFH2 PDX (MYB exonl4 fused to either NIFIB exon11 or exon12) (Fig. 2a) but not in the late passage UFH2 tumor cell line. We compared RNA-Seq expression pattern of UFH1 and UFH2 with gene expression signature previously identified in a microarray study of 12 primary ACC tumors with matched adjacent normal tissues (21). While UFH1 and UFH2 were collected from recurrent metastatic disease compared to the microarray gene signature primarily collected from primary ACC cases, we noted clustering of UFH1, UFH2 PDX tumor samples, as well as the late passage UFH2 cell line, with similar expression patterns within the ACC signature (Fig 2b).
Figure 2.
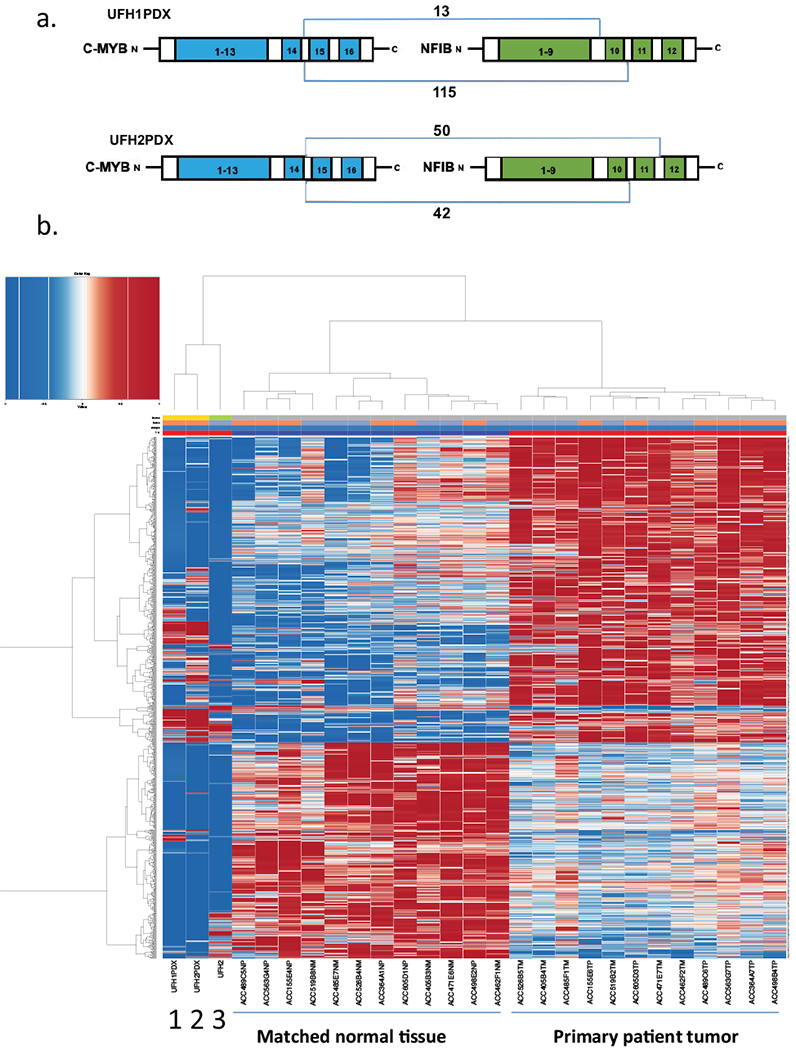
MYB-NFIB fusion transcripts and gene expression pattern in ACC PDXs and ACC cell line. (a) Schematic of distinct MYB fusion transcripts detected in either UFH1PDX or UFH2PDX. (b) hierarchical clustering of mRNA expression by RNA-Seq using ACC UF PDX tumors and ACC cell lines compared with previously defined mRNA signature (21) from 12 ACC patient tumors and matched normal samples as indicated. Lane 1 (UFH1 PDX), Lane 2 (UFH2 PDX), Lane 3 (UFH2 cell line passage 34).
ACC mouse model development
To further study ACC biology and investigate new drug development, we also generated a genetically-engineered mouse model (GEMM) with a conditional LoxP-Stop-LoxP MYB-NFIB transgene (Fig. 3a). MYB-NFIB mice were crossed with MMTV-Cre mice to obtain bi-allelic mice that express MYB-NFIB in secretory epithelium of virgin and lactating mammary gland, the salivary gland, seminal vesicle, skin, erythrocytes, B cells and T cells (Fig. 3b) (18). To test Cre activation of MYB-NFIB transgene, HEK293 cells were transfected with the MYB-NFIB pCBR-based transgene construct alone or co-transfected with Adeno-Cre. We confirmed by immunoblotting that the fusion transgene was activated only in presence of Adeno-Cre (Fig. 3c). To verify fusion transgene expression in mice crossed with MMTV-Cre, we performed RT-PCR and immunoblot on samples collected from murine tissues. Although, we detected expression of the transgene in whole salivary gland tissues by RT-PCR (Fig. 3d), we did not observe MYB expression by protein immunoblot in extracts from whole normal submandibular glands (n=9 mice) (data not shown). In addition, we did not detect epithelial salivary gland tumors in the GEMM (n=42 mice), however there was shorter overall survival compared to historical controls (37) due to development of a B-cell neoplasm in majority of mice (see below).
Figure 3.
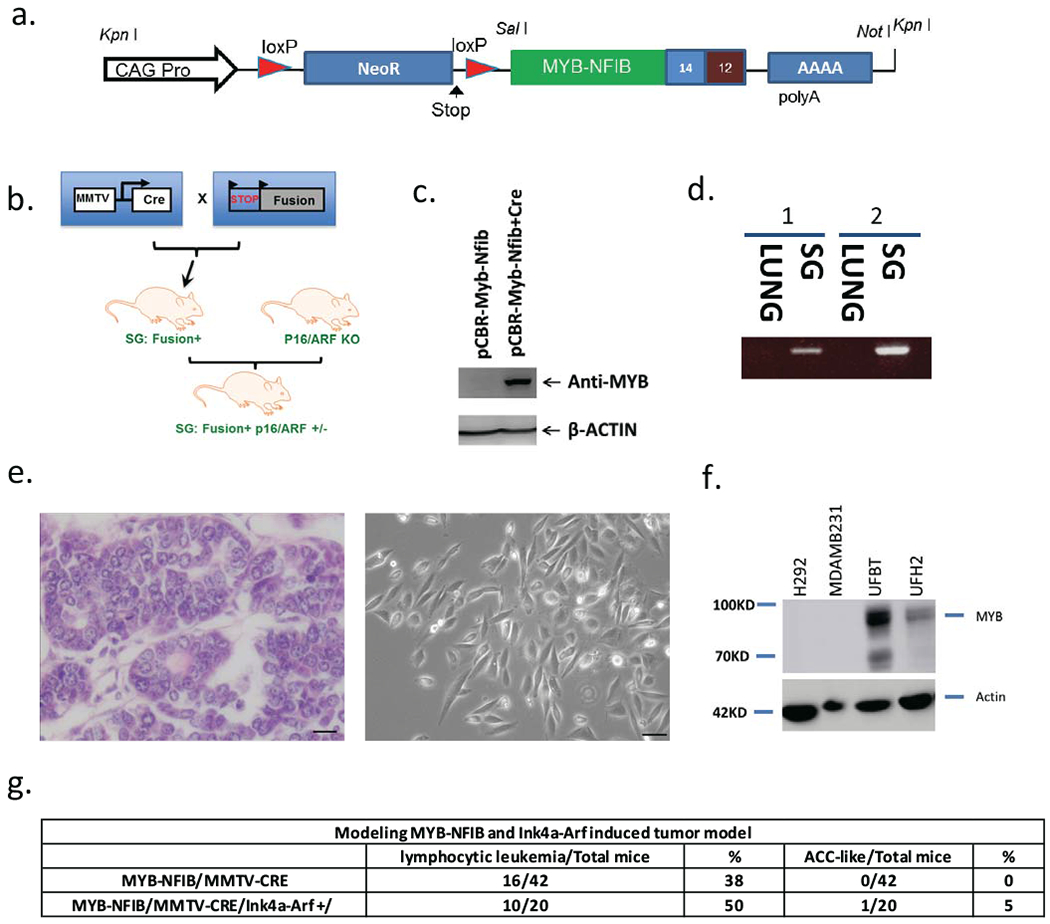
Conditional MYB-NFIB transgene model. (a) Schematic lox-stop-lox design pCBR-based transgene driven by ubiquitously expressed CAG promoter. (b) Breeding strategy for MYB-NFIB/MMTV-Cre mice and mice crossed to Ink4a/ARF+/−. (c) CRE-dependent expression of MYB-NFIB transgene verified by immunoblot analysis using 293 cells transfected construct alone or co transfected with adeno-Cre. (d) RT-PCR analysis of RNA extracted from MYB-NFIB transgene-positive normal salivary gland (SG) or lung. (e) H&E staining of resected mammary tumor from MYB-NFIB/p16/ARF+/− animal (left) and photomicrograph image of subconfluent mammary tumor line (UFBT) (right). (f) MYB immunoblot of H292 mucoepidermoid tumor cell line and MDAMB231 triple-negative breast tumor line (negative controls) and mammary UFBT and UFH2 tumor cell lines. (g) Summary of predominant tumor phenotypes.
MYB-activated mammary breast tumor generated in transgenic mice
Since the p16 pathway is reported to undergo inactivation in a subset of salivary gland cancers including ACC (20, 21,22), we also crossed MYB-NFIB/MMTV-CRE transgenic mice with Ink4a+/−/Arf+/− mice and generated tri-allelic mice that expressed heterozygous Ink4a+/−/Arf+/− alleles. Within MYB-NFIB/MMTV/Ink4a+/−/Arf+/− cohort (n=20 mice) we detected a large mammary tumor with cribriform ACC-like adenocarcinoma histology at 6 months of age (Fig. 3e). We generated a murine tumor cell line from this breast tumor (UFBT) and observed elevated steady-state MYB protein expression (Fig 3f). RT-PCR and nucleotide sequencing confirmed expression of the human MYB-NFIB transgene (data not shown) suggesting this tumor arose as a MYB-dependent event. Although, we did not detect salivary gland tumors in the MYB-NFIB/Ink4a/Arf+/− mice (Fig. 3g), we again observed predominantly B cell leukemia phenotype. While this work was in progress, we were aware of another report studying a similar MMTV-CRE driven MYB-NFIB GEMM crossed with mutant p53 that did not generate salivary gland tumors (32).
These observations may be explained by negative selection of MYB expression in murine salivary gland tissues via unknown mechanism or due to shortened survival for MYB-NFIB transgene mice that exhibited a predominant phenotype of leukocytosis, thrombocytosis, and splenomegaly (Supplemental Fig. 1b, 1c). To define the hematopoietic phenotype, we performed flow cytometry on peripheral blood (PB) cells of control MMTV-Cre mice and compared to both MYB-NFIB and MYB-NFIB/Ink4a(+/−)/Arf(+/−) mice. We analyzed the B220+ cell population and identified a statistically significant expanded B-cell population with an increase in immature B220+IgM+ B-cells in MMTV-Cre/MYB-NFIB transgene mice compared to MMTV-Cre controls. In contrast, we found a decreased proportion of myeloid CD11b+Gr.1+ and Gr.1− cells and a decreased CD3+CD4+CD8− and CD3+CD8+CD4− T lymphocyte populations in PB of MMTV-CRE/MYB-NFIB transgene positive mice compared to MMTV-Cre control mice. In addition, we observed inhibition of other hematopoietic lineages in MMTV-Cre/MYB-NFIB mice (Supplemental Fig. 1d). Histologic analysis also showed disrupted splenic architecture and infiltration of clusters of clonal lymphocytes in spleen, liver, murine femur bone marrow sections, and also within salivary glands (Supplemental Fig. 2a). For all parameters, we observed a more aggressive B-cell leukemia phenotype in MMTV-Cre/MYB-NFIB/Ink4a(+/−)/Arf(+/−) mice compared with MYB-NFIB/MMTV-Cre or MMTV-Cre alone, including higher leukocyte and lower platelet counts (Supplemental Fig. 2b).
MYB depletion inhibits ACC cell viability in vivo and in vitro
To study if MYB depletion affects growth proliferation of the new ACC UFH1 and UFH2 samples, we obtained three different LKO.1 lentiviral MYB shRNA clones and tested the efficiency of each constructs using a MYB-activated hematopoietic leukemic cell line, K562. Using immunoblot analysis we validated that transient expression of shRNA clones 59 and 60 depletes MYB expression (Fig. 4a). Therefore, we used lentiviral clones 59 and 60 for transient transfection into the UFH2 and UFBG cell lines. We observed a statistically significant decrease in cell viability/metabolism by MTT assay (Fig. 4b). We also tested the effect of MYB inhibition in vivo using MYB shRNA or control vectors on PDX xenograft growth from tumor cells obtained from UFH1PDX or ACCX22. MYB depletion significantly inhibited the tumor volume of xenograft tumors (Fig. 4c) suggesting that sustained MYB expression is required for efficient tumor growth of these cell lines.
Figure 4.
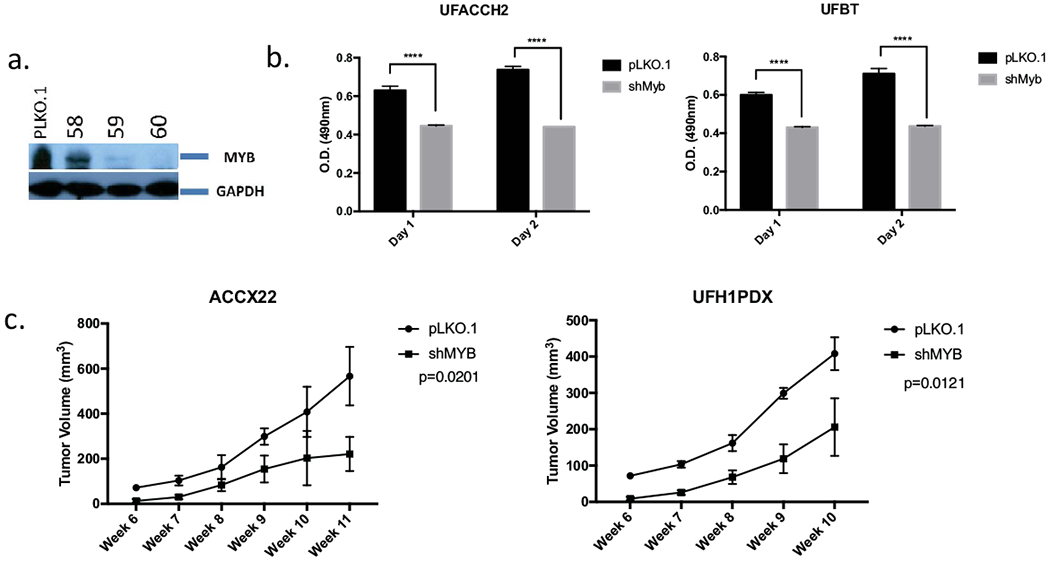
MYB inhibitors suppress ACC viability in vitro and in vivo. (a) Immunoblot analysis testing efficiency of MYB depletion with PLKO.1 control and three different lentiviral MYB shRNAi clones. (b) Viability of UFBT and UFH2 cell lines after transduced with control or MYB shRNA lentiviruses (error bars represent standard deviation of UFBT (n=3 experiments) or UFH2 (n=5 experiments). (c) 2x10^6 ACCX22 (left) or UFH1PDX (right) cells were treated with shMYB or control and injected subcutaneously to dorsal flanks of NOD/SCID mice. ACCX22 PDX (left; n=3 mice control and n=3 mice shMYB), UFH1PDX (right; n=3 mice control and n=6 mice shMYB). Error bars represent median and standard deviations.
Novel MYB inhibitors suppress MYB-activated ACC cell lines viability
Recent studies have proposed that Celastrol, a natural low-molecular-weight compound, can inhibit MYB function through disruption of its interaction with the KIX domain of p300/CBP, a transcription co-activator of MYB protein (22). To test the efficacy of Celastrol on suppressing tumor growth/metabolism of the human UFH2 early passage cell line and the murine ACC-like mammary tumor cell line (UFBT), we performed MTT assay after incubation with increasing concentrations of either the control DMSO solvent or Celastrol. We observed that Celastrol inhibits viability at 1.25 μM (Fig. 5a) that was similar to the effective Celastrol dose in MYB-activated leukemia cells (23). However, we detected a similar reduction in cell viability following exposure of Celastrol to tumor lines that were not associated with MYB activation (3t3 and AsPC1) suggesting a more general cytotoxicity of Celastrol (Fig. 5b). As an alternate strategy to test for MYB-specific cytotoxicity, we tested the effect of a peptidomimetic inhibitor (MYBMIM) that was recently developed using a novel structure-guided molecular design to target and interfere with the assembly of the MYB:CBP/P300 co-transcriptional protein complex (24). To test the effect of MYBMIM on our new human and murine MYB-activated tumor cell lines (UFH2 early passage tumor cell line and murine mammary UFBT tumor cell line), we incubated tumor cells with varying concentrations of either MYBMIM and performed MTT assay for growth viability/metabolism. To control for non-specific peptidomimetic effects, we also treated cells with the inactive MYBMIM analogue termed TG3, which lacks 3 key amino acid sidechains that are responsible for specific binding to CBP/P300 (24). We observed a dose-dependent tumor cell viability inhibition in both MYB-activated tumor cells by MYBMIM but not TG3 (Fig. 5c). To test for MYB-specific toxicity or growth inhibition of the MYB peptides, we incubated MYBMIM or TG3 with 3T3, Panc-1 and AsPC1 tumor cell lines that are not associated with MYB activation. We did not detect cell viability inhibition on these MYB-negative tumor cell lines using MYBMIM (Fig. 5d). This suggests that MYBMIM may have limited toxicity on normal tissues and therefore represents a promising probe for the development of selective MYB inhibitors for ACC patients and tumors with MYB activation.
Figure 5.
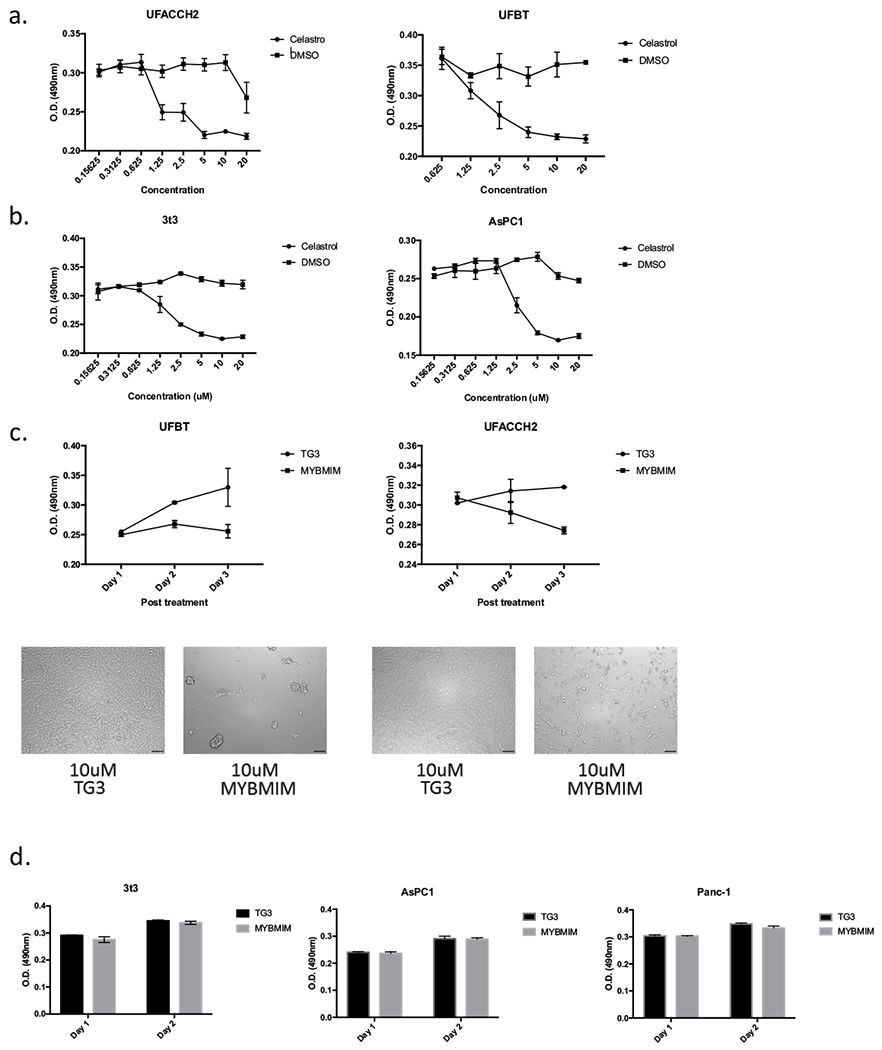
MYB inhibitors suppress ACC cell lines viability in vivo. (a) Viability of early passage human UFACCH2 or murine ACC-like breast tumor UFBT cell lines after exposure to Celastrol at indicated concentrations or DMSO control for 48 hrs. Error bars represent standard deviations of four biological replicates. (b) Viability of control tumor cell lines that are not associated with MYB activation (3T3 and AsPC1) after 48 hrs exposure to Celastrol. (c) Viability (top panel) of UFBT (left) or UFH2 (right) cells, treated with 10 μM of inactivated TG3 analogue peptide or 10 μM MYBMIM. Error bars represent standard deviations of three biological replicates. Photomicrographs (bottom panel) after incubation with indicated peptide mimic inhibitors (UFBT, left and UFH2, right). (d) Viability of non-MYB-activated control tumor cells (3T3, AsPC1 and Panc-1) after exposure to 10 μM control TG3 or MYBMIM for 48 hrs. Error bars represent standard deviations of four biological replicates.
Discussion:
It has been 40 years since the first report describing isolation and characterization of a defective avian tumor virus encoding the MYB oncogene (25). Over the following decades, the MYB gene has been identified as a key transcriptional regulator essential for the normal maturation for all hematopoietic elements (26–29). However, defining the role of MYB in tumorigenesis has been challenging due to the inability to develop functional oncogene assays using non-hematopoietic cells or tissues and, until recently, the lack of recurrent structural somatic mutations that provide further support for a ‘driver oncogene’ role in human cancer. The identification of recurrent chromosomal translocations resulting in either MYB or MYBL1 fusion transcripts in the majority of human adenoid cystic cancer (30) provides the strongest support for MYB activation as an etiologic event in carcinogenesis that also including the important subset of breast, colon, and hematopoietic tumors associated with MYB overexpression. Despite the direct evidence for structural MYB rearrangements, it has been difficult to generate validated human ACC tumor cell lines (31) and there are no activated MYB mouse models that recapitulate ACC salivary gland tumors. These obstacles have limited the ability to study MYB biology and develop new therapeutic agents. For example, we observed elevated steady-state MYB protein expression in extracts from excised human ACC xenografts but rapid loss of MYB protein expression within 5 hours of plating cell culture in vitro limits the ability to study MYB and to develop therapeutic strategies (Supplemental Fig. 2c). The mechanism for MYB down-regulation following plating tumor cells on plastic dishes in vitro is unexplained but may be linked with the difficulty to adapt xenograft ACC tumors to cultured tumor cell lines in vitro. We also generated and studied a new MYB-NFIB GEMM under MMTV-Cre regulation. As recently reported using a similar conditional MYB-fusion transgene strategy (32) we did not observe epithelial salivary gland tumors in either MYB-NFIB alone or in mice crossed with Ink4+/−/Arf+/− null allele gene. While this might be partly explained by shortened survival due to the predominant phenotype of lethal B-cell leukemia in majority of mice, MMTV may be a suboptimal CRE promoter as we were unable to detect steady-state MYB-NFIB expression in murine salivary gland tissues while the human transgene was expressed in murine leukemia tumor cells. Similar to the prior report (32), we observed mammary tumors and were able to successfully generate a murine breast cell line associated with constitutive MYB transgene activation that allowed us to pursue drug testing for growth inhibition.
There are 7 candidate MYB inhibitors that have been recently proposed: celastrol and naphthoquinone (23,33), crizotinib (MET/ALK inhibitor) and linstinib (dual IGF1/insulin receptor inhibitor (34), MYBMIM, a novel MYB peptidomimetic (24), and the related semisynthetic epipodophyllotoxins chemotherapy agents, teniposide and etoposide (35). We selected for testing Celastrol, a natural product from Tripterygium wilfordii, and MYBMIM, a novel MYB peptidomimetic. In contrast to several of the other proposed pharmacological agents which had different known specificities and appeared to targeted MYB indirectly, Celastrol and MYBMIM offered two different pharmacologic strategies to interfere with the ability of MYB to achieve a functional protein complex in cancer cells (22, 23, 24). We have now observed specific effects of MYBMIM to preferentially suppress the growth and survival MYB-activated tumor cell lines validating this strategy for further pharmacologic development. Recently, there is a report of another human ACC tumor cell line (17) and the activated tumor cell lines reported here will offer new opportunities for drug candidate screening and development.
Supplementary Material
Supplemental Figure 1.
(a) Rapid loss of MYB mRNA expression by qRT-PCR (left) and loss of MYB protein by immunoblot (right) after plating UFH1 xenograft into cell culture in vitro. (b) Tumor phenotype of MYB-NFIB/Ink4a/ARF mice (n=20 mice; summary of tumor phenotype of MYB-NFIB alone mice n=42 shown in Figure 3). (c) Kaplan-Meier overall survival analysis of MYB-NFIB/MMTV-Cre (n=42) and MYB-NFIB/MMTC-Cre/Ink4a/ARF+/− (n=20) mice (p=0.0299). The log-rank test was used to determine the P value in the survival difference between MMTV-Cre/MYB-NFIB and MMTV-Cre/MYB-NFIB/ Ink4a+/−/Arf+/− groups in Kaplan-Meier survival analysis. (d) Flow cytometry analysis of peripheral blood from MMTV-Cre control (n=3), MMTV-Cre/MYB-NFIB (n=3), and MMTV-Cre/MYB-NFIB/Ink4a+/− mouse (n=3) with B-cell and T cell specific markers.
Supplemental Figure 2
(a) Splenomegaly disrupted splenic architecture and tissue infiltration of leukemia cells in spleen, liver, salivary gland, and bone marrow of indicated MYB-NFIB mice. (b) Complete blood count showing median and standard deviation of WBC, platelets, and hemoglobin levels of control MMTV-Cre (n=3), MMTV-Cre/MYB-NFIB (n=3), and MMTV-Cre/MYB-NFIB/Ink4a/Arf+/− mice (n=3).
We have isolated and studied two new ACC patient-derived xenograft (PDX) models and developed one matched ACC tumor cell line. We also generated and analyzed the tumor phenotype of a conditional MYB-NFIB transgene genetically engineered mouse model (GEMM). We did not detect salivary gland tumors with this GEMM or with MYB-NFIB mice crossed to Ink4a+/−/Arf+/− mouse model. However, we were able to isolate a murine mammary carcinoma associated with elevated MYB-NFIB transgene expression and ACC-like histology. We observed reduced survival of mice due to immature B-cell leukemia consistent with a role for MYB deregulation in blocking hematopoietic cell lineage differentiation. We have focused on characterizing these new MYB-activated human and murine cell lines to test candidate MYB inhibitors. These data will provide tools to study the biology of ACC and help develop new therapeutic strategies for both ACC and other common adult malignancies associated with MYB activation.
Highlights.
We studied a new ACC PDX sample that express MYB-NFIB with a matched cell line
We studied a MYB-NFIB mouse model and generated an ACC-like mammary tumor cell line
We show MYB-specific inhibition in ACC tumor lines using a peptidomimic inhibitor
Acknowledgements
We thank patients who consented to allow studies on adenoid cystic cancer. We are grateful to Jeffrey and Marnie Kaufman and for support through the Adenoid Cystic Carcinoma Research Foundation (AACRF). We thank Christopher Moskaluk (University of Virginia) for the ACCX22 PDX sample. We thank Jinping Lai (UF Department of Pathology) for assistance with IHC slide imaging and Wei Nei (UF Department of Molecular Genetics and Microbiology) for advice on analyzing samples from the GEMM mouse model. This work was supported by the Gatorade Trust through funds distributed by the University of Florida, Department of Medicine (FK), and by the Damon Runyon-Richard Lumsden Foundation Clinical Investigator and St. Baldrick’s Foundation Arceci Innovation Awards, and the NCI R01 CA204396 and P30 CA008748 (AK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ramos A, Pozo CD, Chinchurreta A, García F, Lorenzo M, Gismero S. Adenoid cystic carcinoma of the lacrimal sac: Case report. Arq Bras Oftalmol. 2016;79(5):333–335. doi: 10.5935/0004-2749.20160095. [DOI] [PubMed] [Google Scholar]
- 2.Min R, Siyi L, Wenjun Y, et al. Salivary gland adenoid cystic carcinoma with cervical lymph node metastasis: A preliminary study of 62 cases. Int J Oral Maxillofac Surg. 2012;41(8):952–957. doi: 10.1016/j.ijom.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 3.Sun J, Wu S, Chen S, et al. Adjuvant radiation therapy and survival for adenoid cystic carcinoma of the breast. BREAST. 2017;31:214–218. doi: 10.1016/j.breast.2016.11.017. [DOI] [PubMed] [Google Scholar]
- 4.Jaiswara C, Dhiman NK, Singh AK, Sharma NK, Verma V, Pandey A. Adenoid cystic carcinoma of the floor of the mouth - A rare presentation. J Oral Biol Craniofac Res. 2016;6(Suppl 1):S65–S69. doi: 10.1016/j.jobcr.2015.09.007 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song JY. Adenoid cystic carcinoma of the sublingual gland: A case report. Imaging Sci Dent. 2016;46(4):291–296. doi: 10.5624/isd.2016.46.4.291 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He S, Li P, Zhong Q, et al. Clinicopathologic and prognostic factors in adenoid cystic carcinoma of head and neck minor salivary glands: A clinical analysis of 130 cases. Am J Otolaryngol. 2016. doi:S0196-0709(16)30253-8 [pii]. [DOI] [PubMed] [Google Scholar]
- 7.Doggett S, Chino S, Lempert T, Federhart J. Percutaneous CT-fluoroscopic-guided radioisotope seed placement for the management of adenoid cystic carcinoma of the trachea. Brachytherapy. 2016. doi:S1538-4721(16)30605-5 [pii]. [DOI] [PubMed] [Google Scholar]
- 8.Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci U S A.2009;106(44):18740–18744. doi: 10.1073/pnas.0909114106 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013;45(6):602–612. doi: 10.1038/ng.2611 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brayer KJ, French CA, Kang FI, Ness SA. Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov.2016;6(2): 176–187. doi: 10.1158/2159-8290.CD-15-0859 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitani Y, Liu B, Rao PH, et al. Novel MYBL1 gene rearrangements with recurrent MYBL1-NFIB fusions in salivary adenoid cystic carcinomas lacking t(6;9) translocations. Clin Cancer Res. 2016;22(3):725–733. doi: 10.1158/1078-0432.CCR-15-2867-T [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreasen S, Tan Q, Agander TK, Steiner P, Bjørndal K, Høgdall E, Larsen SR, Erentaite D, Olsen CHv, Ulhøi BP, von Holstein SL, Wessel I, Heegaard S,Homøe P,. Adenoid cystic carcinomas of the salivary gland, lacrimal gland, and breast are morphologically and genetically similar but have distinct microRNA expression profiles. Modern pathology : an official journal of the United States and Canadian Academy of Pathology Inc. 2018. [DOI] [PubMed] [Google Scholar]
- 13.Drier Y, Cotton MJ, Williamson KE, et al. An oncogenic MYB feedback loop drives alternate cell fates in adenoid cystic carcinoma. Nat Genet. 2016;48(3):265–272. doi: 10.1038/ng.3502 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moskaluk CA, Baras AS, Mancuso SA, et al. Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91 (10):1480–1490. doi: 10.1038/labinvest.2011.105 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin T, Zhu L, Zhou B, et al. Establishment and characterization of a cell line from human adenoid cystic carcinoma of the lacrimal glands and a nude mouse transplantable model. Oncol Rep. 2015;33(6):2797–2806. doi: 10.3892/or.2015.3925 [doi]. [DOI] [PubMed] [Google Scholar]
- 16.Yarbrough WG, Panaccione A, Chang MT, Ivanov SV. Clinical and molecular insights into adenoid cystic carcinoma: Neural crest-like sternness as a target. Laryngoscope Investig Otolaryngol. 2016;1(4):60–77. doi: 10.1002/lio2.22 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warner KA, Oklejas AE, Pearson AT, et al. UM-HACC-2A: MYB-NFIB fusion-positive human adenoid cystic carcinoma cell line. Oral Oncol. 2018;87:21–28. doi: S1368-8375(18)30365-8 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wagner KU, Wall RJ, St-Onge L, et al. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25(21):4323–4330. doi: gka680 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maruya S, Kurotaki H, Shimoyama N, Kaimori M, Shinkawa H, Yagihashi S. Expression of p16 protein and hypermethylation status of its promoter gene in adenoid cystic carcinoma of the head and neck. ORL J Otorhinolaryngol Relat Spec. 2003;65(1):26–32. doi: 10.1159/000068658 [doi]. [DOI] [PubMed] [Google Scholar]
- 20.Li J, El-Naggar A, Mao L. Promoter methylation of p16INK4a, RASSF1A, and DAPK is frequent in salivary adenoid cystic carcinoma. Cancer. 2005;104(4):771–776. doi: 10.1002/cncr.21215 [doi]. [DOI] [PubMed] [Google Scholar]
- 21.Gao R, Cao C, Zhang M, et al. A unifying gene signature for adenoid cystic cancer identifies parallel MYB-dependent and MYB-independent therapeutic targets. Oncotarget. 2014;5(24):12528–12542. doi: 2985 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coulibaly Anna, Haas Astrid, Steinmann Simone, Jakobs Anke, Schmidt Thomas J., Klempnauer Karl-Heinz, Bunting Kevin D.,. The natural anti-tumor compound celastrol targets a myb-C/EBPβ-p300 transcriptional module implicated in myeloid gene expression. PLoS ONE PLOS ONE. 2018;13(2):e0190934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uttarkar S, Dasse E, Coulibaly A, et al. Targeting acute myeloid leukemia with a small molecule inhibitor of the Myb/p300 interaction. Blood. 2016;127(9):1173–1182. [DOI] [PubMed] [Google Scholar]
- 24.Ramaswamy Kavitha, Forbes Lauren, Minuesa Gerard, Gindin Tatyana, Brown Fiona, Kharas Michael G., Krivtsov Andrei V., Armstrong Scott A., Still Eric, de Stanchina Elisa, Knoechel Birgit, Koche Richard,Kentsis Alex,. Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat Commun Nature Communications. 2018;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roussel M, Saule S, Lagrou C, et al. Three new types of viral oncogene of cellular origin specific for haematopoietic cell transformation. Nature. 1979;281 (5731):452–455. [DOI] [PubMed] [Google Scholar]
- 26.Todokoro K, Watson RJ, Higo H, et al. Down-regulation of c-myb gene expression is a prerequisite for erythropoietin-induced erythroid differentiation. Proc Natl Acad Sci U S A. 1988;85(23):8900–8904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bianchi E, Bulgarelli J, Ruberti S, et al. MYB controls erythroid versus megakaryocyte lineage fate decision through the miR-486-3p-mediated downregulation of MAF. Cell Death Differ. 2015;22(12):1906–1921. doi: 10.1038/cdd.2015.30 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sala A, Watson R. B-myb protein in cellular proliferation, transcription control, and cancer: Latest developments. J Cell Physiol.1999;179(3):245–250. [DOI] [PubMed] [Google Scholar]
- 29.Sitzmann J, Noben-Trauth K, Kamano H, Klempnauer KH. Expression of B-myb during mouse embryogenesis. Oncogene. 1996;12(9):1889–1894. [PubMed] [Google Scholar]
- 30.Chahal M, Pleasance E, Grewal J, et al. Personalized oncogenomic analysis of metastatic adenoid cystic carcinoma: Using whole-genome sequencing to inform clinical decision-making. Cold Spring Harb Mol Case Stud. 2018;4(2): 10.1101/mcs.a002626 Print 2018 Apr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu o. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One. 2009;4(6):e6040. doi: 10.1371/journal.pone.0006040 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mikse OR, Tchaicha JH, Akbay EA, et al. The impact of the MYB-NFIB fusion proto-oncogene in vivo. Oncotarget. 2016;7(22):31681–31688. doi: 10.18632/oncotarget.9426 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Uttarkar S, Frampton J, Klempnauer KFI. Targeting the transcription factor myb by small-molecule inhibitors. Exp Hematol. 2017;47:31–35. [DOI] [PubMed] [Google Scholar]
- 34.Andersson MK, Afshari MK, Andren Y, Wick MJ, Stenman G. Targeting the oncogenic transcriptional regulator MYB in adenoid cystic carcinoma by inhibition of IGF1R/AKT signaling. J Natl Cancer Inst. 2017;109(9): 10.1093/jnci/djx017. [DOI] [PubMed] [Google Scholar]
- 35.Yusenko M, Jakobs A, Klempnauer KFI. A novel cell-based screening assay for small-molecule MYB inhibitors identifies podophyllotoxins teniposide and etoposide as inhibitors of MYB activity. Sci Rep. 2018;8(1): 13159-018-31620-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Ory V, Chapman S, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180(2):599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang P, Duda DG, Jain RK, Fukumura D. Histopathologic findings and establishment of novel tumor lines from spontaneous tumors in FVB/N mice. Comp Med. 2008;58(3):253–263. [PMC free article] [PubMed] [Google Scholar]
- 38.Langdon WB Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. BioData mining.8, 1, doi: 10.1186/s13040-014-0034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B & Dewey CN RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011; 12, 323, doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014; 512, 155–160, doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hanzelmann S, Castelo R & Guinney J GSVA: gene set variation analysis for microarray and RNA-seq data. BMC bioinformatics. 2013; 14, 7, doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bild AH et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006; 439, 353–357, doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 43.Ritchie ME et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015; 43,e47, doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brian JH et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. http://star-fusion.github.io 2017; doi: 10.1101/120295. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1.
(a) Rapid loss of MYB mRNA expression by qRT-PCR (left) and loss of MYB protein by immunoblot (right) after plating UFH1 xenograft into cell culture in vitro. (b) Tumor phenotype of MYB-NFIB/Ink4a/ARF mice (n=20 mice; summary of tumor phenotype of MYB-NFIB alone mice n=42 shown in Figure 3). (c) Kaplan-Meier overall survival analysis of MYB-NFIB/MMTV-Cre (n=42) and MYB-NFIB/MMTC-Cre/Ink4a/ARF+/− (n=20) mice (p=0.0299). The log-rank test was used to determine the P value in the survival difference between MMTV-Cre/MYB-NFIB and MMTV-Cre/MYB-NFIB/ Ink4a+/−/Arf+/− groups in Kaplan-Meier survival analysis. (d) Flow cytometry analysis of peripheral blood from MMTV-Cre control (n=3), MMTV-Cre/MYB-NFIB (n=3), and MMTV-Cre/MYB-NFIB/Ink4a+/− mouse (n=3) with B-cell and T cell specific markers.
Supplemental Figure 2
(a) Splenomegaly disrupted splenic architecture and tissue infiltration of leukemia cells in spleen, liver, salivary gland, and bone marrow of indicated MYB-NFIB mice. (b) Complete blood count showing median and standard deviation of WBC, platelets, and hemoglobin levels of control MMTV-Cre (n=3), MMTV-Cre/MYB-NFIB (n=3), and MMTV-Cre/MYB-NFIB/Ink4a/Arf+/− mice (n=3).