

Abstract
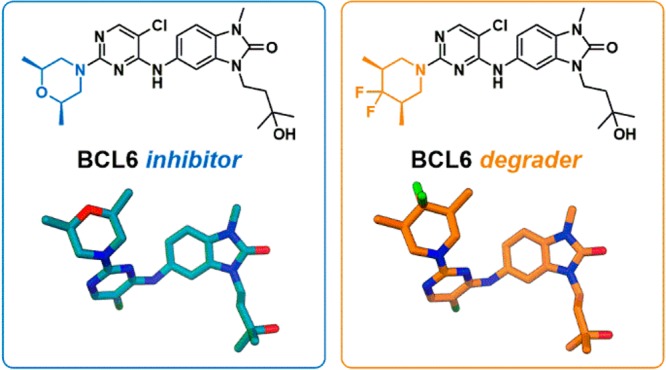
Deregulation of the transcriptional repressor BCL6 enables tumorigenesis of germinal center B-cells, and hence BCL6 has been proposed as a therapeutic target for the treatment of diffuse large B-cell lymphoma (DLBCL). Herein we report the discovery of a series of benzimidazolone inhibitors of the protein–protein interaction between BCL6 and its co-repressors. A subset of these inhibitors were found to cause rapid degradation of BCL6, and optimization of pharmacokinetic properties led to the discovery of 5-((5-chloro-2-((3R,5S)-4,4-difluoro-3,5-dimethylpiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (CCT369260), which reduces BCL6 levels in a lymphoma xenograft mouse model following oral dosing.
Introduction
BCL6 (B-cell lymphoma 6 protein) is a transcriptional repressor that plays a key role in the formation and maintenance of germinal centers during the process of antibody affinity maturation.1−3 By binding to DNA via its zinc fingers and recruiting one of its co-repressors (NCoR, SMRT, or BCOR) to its dimeric BTB domain, BCL6 represses genes involved in cell cycle control, cell death, differentiation, and the DNA damage response. This action enables B-cells in the germinal center to proliferate rapidly, evade growth checkpoint controls, and tolerate high levels of DNA damage. These features are required for the process of somatic hypermutation of antibodies but are also among the hallmarks of cancer;4,5 hence deregulation of BCL6 can lead to lymphomagenesis. Most B-cell lymphomas arise from germinal center B-cells, which are dependent on continued expression of BCL6 for survival.1 Recruitment of co-repressors is essential for the repressive and oncogenic functions of BCL6, and disruption of this interaction is sufficient to inhibit lymphoma cell growth.6,7 We sought to discover compounds that disrupt the protein–protein interaction (PPI) between the BTB domain of BCL6 and its co-repressors to alleviate BCL6-mediated gene repression, inhibit lymphoma cell growth, and identify new treatments for BCL6-driven lymphomas. Following on from earlier publications that identified small molecule or peptidomimetic compounds with moderate potency against BCL6,8−10 the recent publication of high-potency small molecules has established the ligandability of the BCL6 BTB domain dimer.11−13 Proteolysis targeting chimeras (PROTAC)14 and non-PROTAC degrader compounds that trigger the proteosomal degradation of BCL615 have also been reported. However, to date, no compound has been reported to be suitable for use as an in vivo probe to investigate the effect of degradation of BCL6. In this study we report the discovery not only of compounds that can potently inhibit BCL6 function by disrupting the PPI with its co-repressors but also of a subset of molecules that trigger the rapid degradation of BCL6. Further optimization of this series led to compound 1 (CCT369260), which shows degradation of tumoral BCL6 in vivo following oral dosing in a lymphoma xenograft mouse model.
Results
Chemistry: Synthesis of Compounds
Initial hit compounds 2 and 3 and follow-up compound 4 were obtained from commercial vendors, while the cyclopropyl compound 5 and benzimidazolone 6 (CCT365386) were prepared by single step nucleophilic aromatic substitution (SNAr) reaction from available building blocks. Compounds with alkyl substituents in the benzimidazolone-N3 position could be synthesized by stepwise alkylation of nitro-compound 7, reduction, and SNAr reaction, as exemplified in the preparation of 11a (Scheme 1). In order to facilitate the optimization of this position (as shown in Table 2), we developed conditions to allow the addition of this group as the final step (Scheme 1, steps d and e), using intermediate 9. The use of alkyl halides or epoxides as electrophiles under these conditions gave primarily the desired regioisomer, although in most cases minor products corresponding to alkylation at the 4-pyridylamine were also observed in the reaction mixture and required HPLC purification to separate.
Scheme 1. Final Step Diversification of Benzimidazolone N3 Position.

Reagents and conditions: (a) 2-ethyloxirane, cesium carbonate, DMF, 120 °C, 1 h; (b) sodium dithionite, ethanol/DMSO, rt to 90 °C; (c) 2,4-dichloropyridine-3-carbonitrile, DIPEA, DMA, 120 °C, 30–45 min; (d) (2R)- or (2S)-ethyloxirane, cesium carbonate, DMF, 120 °C, 1 h, then 140 °C, 1 h; (e) alkyl bromide or tosylate, cesium carbonate, DMF, 60 °C, 3 days or 120–140 °C, 1 h.
Table 2. Structure-Activity Relationships of the Benzimidazolone-N3 Position (Labeled as R3 Group).
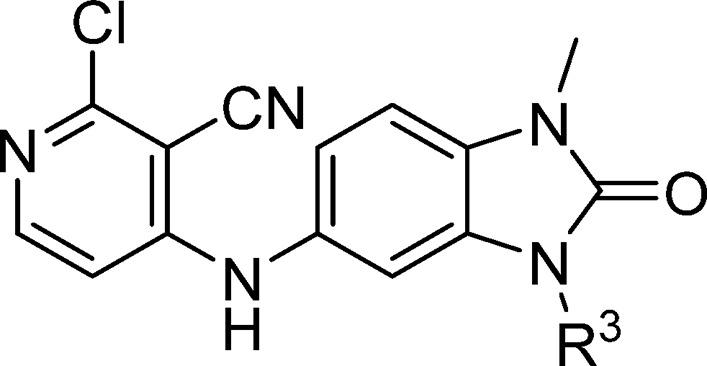

Data represent the geometric mean of at least three replicates. See Supporting Information Table S2 for full statistics.
Kinetic solubility measured by NMR in HEPES buffer (containing 5% DMSO) at pH 8. Where this was <30 μM, solubility by HPLC in PBS buffer and 1% DMSO at pH 7.4 was also measured and is shown in parentheses.
In order to vary the substituent of the 5-amino group on the benzimidazolone (labeled as R4 in Table 3), it was necessary first to scale up the synthesis of the 1,3-disubstituted 5-aminobenzimidazol-2-one. Cyclization of commercially available nitrodianiline 12 using disuccinimidylcarbonate proceeded in high yield and was followed by alkylation with tosylate 14a, then hydrogenation to reduce the nitro group and form aniline 16 (Scheme 2). These steps were readily amenable to scale up, and multigram quantities were produced without the need for column chromatography.
Table 3. Structure–Activity Relationships of the R4-Group-Substituted Pyridine and Pyrimidines.
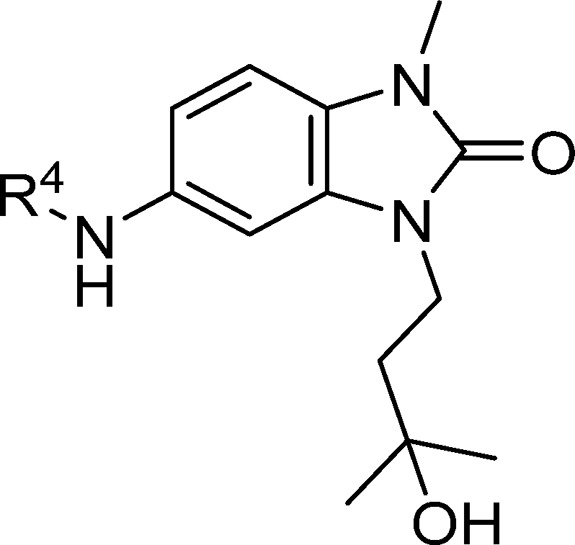

Data represent the geometric mean of at least three replicates. See Supporting Information Table S2 for full statistics.
Kinetic solubility measured by NMR in HEPES buffer (containing 5% DMSO) at pH 8. Where this was <30 μM, solubility by HPLC in PBS buffer and 1% DMSO at pH 7.4 was measured and is shown in parentheses.
Scheme 2. Synthesis of Key Intermediate 16.

Reagents and conditions: (a) disuccinimidylcarbonate, MeCN, rt, 18 h, up to 11g scale, 86–99% yield; (b) cesium carbonate, MeCN, reflux, 4 h, 12 g scale, 92% yield; (c) TsCl, NEt3, DCM, 0 °C to rt, up to 40g scale, 60–72% yield; (d) 10% Pd/C, 1 atm H2, ethanol, 60 °C, 3 h, 6 g scale, quant.
Aromatic nucleophilic substitution or (for less reactive pyridine substituents) palladium coupling reactions were used to prepare the set of compounds shown in Table 3 (Scheme 3).
Scheme 3. Synthesis of Compounds from Table 3, Investigating Different Substitution Patterns on Pyrimidine or Pyridine Rings.

Reagents and conditions: (a) heteroaryl chloride, DIPEA, NMP or DMF, 80–120 °C, 30 min; (b) 4-bromo- or iodopyridine, Xantphos, Pd2(dba)3, cesium carbonate, NMP/toluene, 140 °C, 1 h.
In order to explore substitution at the pyrimidine-2-position (Tables 4 and 5), dichloropyrimidine 18 was prepared on multigram scale. Final compounds were prepared from this, again via SNAr or metal-catalyzed reactions (Scheme 4).
Table 4. Structure–Activity Relationships of the Pyrimidine and Pyridine 2-Position (Labeled as R3 Group).


Data represent the geometric mean of at least three replicates. See Supporting Information Table S2 for full statistics.
Data represent the geometric mean of at least two replicates except where superscript 1 is indicated. n.d. = not done.
Kinetic solubility measured by NMR in HEPES buffer and 5% DMSO at pH 8, and/or where data are shown in brackets, by HPLC in PBS buffer and 1% DMSO at pH 7.4. #These compounds showed <10% degradation of BCL6 in single concentration experiments at 10 μM, n = 2.
Table 5. Modifications to the Pyrimidine 2-Substituent.
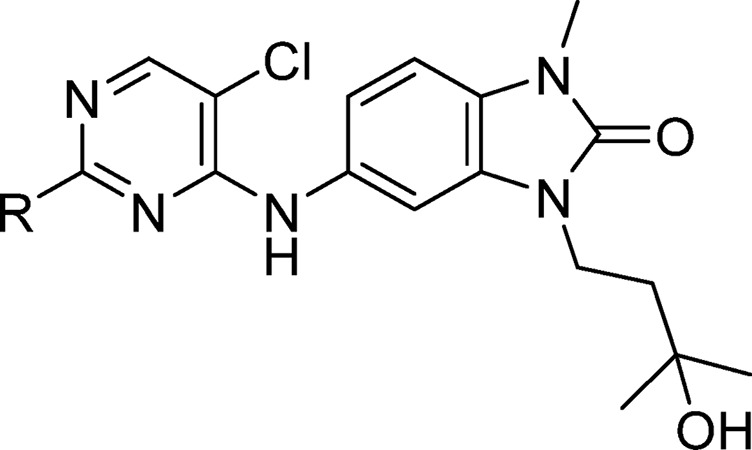

Data represent the geometric mean of at least three replicates. See Supporting Information Table S2 for full statistics.
Kinetic solubility measured by NMR in HEPES buffer and 5% DMSO at pH 8, and/or where data are shown in brackets, by HPLC in PBS buffer and 1% DMSO at pH 7.4. #These compounds showed <25% degradation of BCL6 in single concentration experiments at 10 μM. n.d. = not done.
Scheme 4. Synthesis of 2-Substituted Pyrimidine Compounds from Tables 4 and 5, Starting from Common Intermediate 18.

Reagents and conditions: (a) dimethylamine HCl, cesium carbonate, NMP, 180 °C, 1 h; (b) cyclic amine, DIPEA, NMP, 140–180 °C, 1–2 h; (c) pyrazole, cesium carbonate, NMP, 170 °C, 1 h; (d) 2,4-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiazole, sodium carbonate, PdCl2(PPh3)2, 1,4-dioxane/water, 130 °C, 30 min; (e) 1-methyl-2-(tributylstannyl)-1H-imidazole, PdCl2(PPh3)2, 1,4-dioxane, 90 °C, 18 h; (f) bicyclic amine, DIPEA, NMP, 140 °C, 2–8 h.
The reduced reactivity of the pyridine core in comparison to the pyrimidine meant that metal-catalyzed reactions on bromo- or iodopyridines or SNAr reactions using 2-fluoropyridines were used to prepare the pyridyl examples in Table 4 (Scheme 5).
Scheme 5. Synthesis of 2,4,5-Substituted Pyridine Derivatives.

Reagents and conditions: (a) 16, DIPEA, NMP, 180 °C, 1 h; (b) 3,5-dimethylpyrazole, Xantphos, cesium carbonate. Pd2(dba)3, DMF/toluene, 140 °C, 1 h; (c) DIPEA, THF, 100 °C, 16 h; (d) 16, Xantphos, Pd2(dba)3, cesium carbonate, DMF/toluene, 80 °C, 1 h.
Hit Discovery
A detailed description of our hit discovery campaign for BCL6 will be presented elsewhere.16 Briefly, we identified hit compounds 2 and 3 (Table 1) from an in-house high-throughput screen using a fluorescence polarization (FP) assay, based on the displacement of a fluorescently labeled peptide derived from the BCOR co-repressor.7 In common with other hits from this screen, these compounds showed weak potency (IC50 ∼100 μM). Initial attempts to obtain ligand-bound X-ray structures were hindered by solubility which was lower than their biochemical assay activity (Table 1). To address this, we made analogues with reduced lipophilicity and fewer aromatic rings. Compounds 4 and 5 demonstrated comparable potency to initial hits and improved solubility (Table 1), enabling X-ray structure determination.17 Like the BCL6 co-repressors SMRT and BCOR,7 compounds were found to bind at the dimer interface of the BTB domain of BCL6 (Figure 1A and Figure 1B). Two interaction features are common to both 4 and 5: a hydrogen bond from an NH to the backbone carbonyl of Met51 and the intercalation of their cyanopyridine moieties into a cleft between Tyr58 and Asn21 on the protein surface, creating a critical hydrophobic interaction between the cyanopyridine and the side chain of Tyr58. Compound 4 forms additional interactions via the oxygen atom of its benzimidazolone core to the backbone N–H of Glu115 and to the backbone N–H of His116 via a mediating water molecule. The cyclopropyl group of 5 makes only weak hydrophobic interactions with the backbone of residues 53–55 (in blue in Figure 1, also observed for the benzimidazole of 4), suggesting that the chlorocyanopyridine of 5 provides much of the binding affinity of this compound. We therefore hypothesized that combining the benzimidazolone core with the chlorocyanopyridine of 5 would lead to an increase in potency. Gratifyingly, the resulting compound 6 (CCT365386) showed a significant improvement in binding affinity, down to ∼10 μM (Table 1).
Table 1. Potency and Solubility Data for Hit and Early Lead Compounds.

Data represent the geometric mean. See Supporting Information Table S1 for full statistics. Potency was later confirmed in the TR-FRET assay described in the manuscript; see Supporting Information Table S2.
Kinetic solubility measured by NMR in HEPES buffer (containing 5% DMSO) at pH 8. LE calculated as 1.4 × pIC50/HAC.
Figure 1.

Structure-guided merging of 5 (A, PDB code 6TOG) and 4 (B, PDB code 6TOF) yielded the more potent 6 (CCT365386) (C, PDB code 6TOH). The surface of the BCL6 dimer is shown as a grey transparent surface. The two individual monomers are highlighted in gray and cyan ribbons except for residues 53–55 which are indicated in blue. Key protein residues are shown in line representation. The compounds are shown as orange ball and sticks, a selected water molecule is shown as a red sphere, and H-bonds are shown as yellow dashed lines. In panel C the orange arrow indicates an exit vector towards a large accessible pocket close to our compounds. Additional images containing overlays of 4 and 5 with 6 are shown in the Supporting Information Figure S1.
The X-ray structure of 6 bound to BCL6 confirmed the hypothesized binding mode, combining the features of 4 and 5 (Figure 1C). Low microsomal clearance and high PAMPA permeability, combined with good ligand efficiency for a protein–protein interaction inhibitor hit compound (LE = 0.32 based on the FP assay) further established 6 as a promising hit compound for optimization. Despite these good properties, the suboptimal biochemical activity and solubility meant that we did not observe significant displacement of co-repressor SMRT from BCL6 in the NanoBRET cellular target engagement assay (IC50 > 30 μM).16
Initial Hit Optimization
Having discovered an attractive starting point, our next objective was to improve the biochemical potency. To avoid being constrained by the tight binding limit of the FP assay, we developed new TR-FRET assay conditions with reduced protein concentration (from 3 μM used in the FP assay to 1 nM), allowing us to measure compound activities in the nM range.16 From the X-ray structure of 6, we identified two main areas for optimization: first, growing from the benzimidazolone-N3 position into a relatively large cavity, indicated by the arrow in Figure 1C, and second, by further optimization of the interactions between the pyridine and the pocket formed by Tyr58.
Our aim was to gain potency by extending from the N3 position and filling the pocket while ensuring that hydrogen bond donor or acceptor groups in the binding site were appropriately satisfied. In our design, we sought to exploit a hydrophobic patch close to Val18 (Figure 1C) with a n-butyl chain. This chain would pass close to Asn21, so we incorporated an alcohol (donor) or nitrile (acceptor) group to satisfy this interaction, which we hypothesized would also address the moderate solubility. However, the resulting compounds 11a and 11b showed no improvement in potency (Table 2). The X-ray structure of 11a shows both enantiomers bound to BCL6 with similar occupancies (0.46 and 0.54 for enantiomers (S) and (R), respectively, Figure 2). We hypothesized that the weak (3.25 Å) hydrogen bond between the hydroxyl of the (S) enantiomer of 11a and the side chain of Asn21 is not sufficient to outweigh the desolvation penalty18,19 and is hence not contributing positively to binding. This was supported by testing of single enantiomers 11c and 11d and des-hydroxy 11e, which all have comparable potency.
Figure 2.

Binding mode of enantiomers of 11a (PDB code 6TOO). Panel A and panel B show the binding mode of the respective (S)- and (R)-enantiomers of 11a. Panel C shows the (S)-enantiomer and highlights the four water molecules targeted to gain potency. In all panels the surface of the BCL6 dimer is shown as a gray transparent surface, with the two individual monomers highlighted as ribbons and colored in gray and cyan, respectively. Selected residues are shown in line representation. Enantiomers of 11a are shown as orange ball and sticks, selected water molecules as a red spheres, and H-bonds as yellow dashed lines.
We sought instead to interact indirectly with BCL6 via the network of water molecules (Figure 2C) which were observed in the 11a-bound crystal structure, by changing the position of the hydroxyl group from the C-2 to C-3 carbon of the butyl chain. Compound 11f showed a 3-fold improvement in potency (Table 2), and a crystal structure of 11f bound to BCL6 (Figure 3A) confirmed the expected binding mode with interactions to three water molecules. The addition of a methyl group to form the achiral tertiary alcohol 17a provided a further improvement in potency, consistent with its increase in lipophilicity, while maintaining the binding mode to BCL6 (Figure 3B).
Figure 3.

Binding mode of 11f and 17a. Panel A and Panel B show the respective BCL6 binding modes of 11f (PDB code 6TOI) and 17a (PDB code 6TOJ). Compounds are shown as orange ball and sticks, selected water molecules as red spheres, and H-bonds as yellow dashed lines. The surface of BCL6 dimer is shown as a gray transparent surface. The two individual monomers are displayed as ribbons and colored in gray and cyan, respectively. Key residues are shown as lines.
17a represented our first submicromolar compound. Further characterization in the NanoBRET assay demonstrated that 17a is able to disrupt the protein–protein interaction between full-length co-repressor and BCL6 in cells, albeit with a ∼10-fold drop-off in activity (NanoBRET IC50 = 8.8 μM, compared with TR-FRET IC50 = 0.86 μM). To gain further binding affinity and hence improve cellular potency, we explored modifications to the pyridine ring (Table 3). The 3-nitrile group points into a small lipophilic pocket, does not appear to be making polar interactions, and could be replaced by a chloro atom (17b). We also assessed whether the potentially reactive 2-chloropyridine in 17b could be removed and found that resulting compound 17c maintains activity and improves solubility.
The crystal structure of BCL6 bound to 17a shows the pyridine ring making hydrophobic contacts with the electron rich π-system of Tyr58, and the pyridine nitrogen engaging in a water-mediated H-bond with Arg28 (Figure 3B). We hypothesized that a pyrimidine ring could maintain both these features, and indeed 17d and 17e showed comparable activity to their pyridine equivalents.
Further functionalization of the pyrimidine or pyridine ring was explored in order to gain potency. The pyridine 2-position offers only limited scope for further substitution; the 2-chloro group in 17a (Figure 3) is largely filling the available space in this region. Substitution in the pyridine 5-position could give rise to a steric clash with the phenyl ring of the benzimidazolone, preventing the molecule from adopting the binding conformation observed crystallographically: for example, addition of a chlorine at this position in 17f leads to a 37-fold drop in activity compared to parent 17c. The remaining position (the 2-position on the pyrimidine) was hence selected for further exploration. Initial examples 17g, 18, and 19 showed that substitution was tolerated, and a set of 2-heterocyclyl- and heteroarylpyrimidines was therefore prepared to probe this region (Table 4). A variety of groups including aliphatic amines and both carbon- or nitrogen-linked heteroaryl groups were well tolerated in this position, consistent with the space available in this solvent-exposed region of the binding site, with hydrophilic groups such as morpholine 20a showing 4-fold better potency than the more hydrophobic piperidine 20b.
We attempted to increase potency further by increasing steric bulk to more completely fill this area of the pocket and derive additional hydrophobic surface contacts. Addition of one or two methyl groups to pyrazole (compare 23a, 23b, 23c, and 23d), piperidine (compare 20b, 24a, and 24b) and morpholine (compare 20a and 25a) subseries did indeed lead to an increase in potency, as measured in the TR-FRET assay. For the most potent examples, pyridine matched-pairs were prepared but were less potent than their pyrimidine counterparts (compare 23d and 26a, or 24b and 26b, or 25a and 26c, Table 4), in contrast to the matched-pairs for simpler analogues (compare 17a and 17d, and 17b and 17e; shown in Table 3). This may be due to the different conformational preferences of the added substituent: the additional steric clash resulting from the pyridyl CH compared to the pyrimidine N is likely to lead to a more twisted minimum energy conformation. Compounds with submicromolar activity in TR-FRET were profiled in the cellular NanoBRET assay and showed inhibition in the low micromolar range. Further substitution on the morpholine moiety led to 25b, which was our first compound to show both sub-100 nM activity in the TR-FRET assay and submicromolar activity in the cellular NanoBRET assay.
X-ray structures obtained for 23d and 25b provided possible explanations for the observed increases in potency. The pyrazole group of 23d forms new interactions: a possible cation−π stacking interaction with Arg24, and a hydrogen bond from one of the pyrazole nitrogen atoms to Arg28, which adopts an alternative conformation (Figure 4C and Figure 4D). In contrast, no new polar interactions were observed for 25b (Figure 4A and Figure 4B), which adopts the same binding mode as parent 17a. We hypothesize that the observed potency enhancement results from the improved hydrophobic contacts with Tyr58 and the displacement of water molecules in this region (Figure 4A and Figure 4B).
Figure 4.

Binding mode of 25b and 23d. Panels A and B show two different views of the binding mode of 25b (PDB code 6TON). Panels C and D show two views of the binding mode of 23d (PDB code 6TOK). The panels show the changed conformation of Arg28 and the new interaction with one of the compound’s pyrazole nitrogen atoms. In all panels, the surface of the BCL6 BTB dimer is shown as a gray transparent surface, with the two individual monomers displayed as ribbons and colored in gray and cyan. Key residues are shown as lines. Compounds are shown as orange ball and sticks, selected water molecules as red spheres, and H-bonds as yellow dashed lines.
Discovery of BCL6 Degraders
In contrast to the morpholine analogues 25a and 25b, we were surprised to see no cellular activity for dimethylpiperidine 24b (CCT368682) in the NanoBRET assay, despite submicromolar potency in the TR-FRET assay, and good passive permeability (PAMPA Papp is high [36 × 10–6 cm/s]). A large drop-off from TR-FRET to NanoBRET potencies was also observed for monomethylpiperidine 24a. Upon detailed examination of the NanoBRET data for 24b, we observed a dose-dependent reduction in total luminescence, suggesting either cellular toxicity or depletion of one of the assay components, for example, BCL6 degradation, as has been previously reported for another series of inhibitors.15 To investigate this further, we treated SU-DHL-4 and OCI-Ly1 cells with 24b; Western analysis of cell lysates demonstrated a concentration-dependent reduction in BCL6 protein levels, consistent with compound-mediated BCL6 degradation (Figure 5). We note also an apparent elevation in concentration of BCL6 at compound concentrations below the binding IC50 which is discussed further below.
Figure 5.

Treatment of DLBCL cell lines with 24b (CCT368682) leads to degradation of BCL6. SU-DHL-4 or OCI-Ly1 cells were treated with 24b at the indicated concentration for 4 h at 37 °C. Cells were collected, lysed, and examined by Western blot for BCL6 and GAPDH protein levels. D: DMSO control.
Degradation of BCL6 represents an attractive alternative to inhibition. As the protein is removed rather than just inhibited, activity at substoichiometric concentrations of compound is possible as the small molecule degrader may act catalytically. This removes the need for complete continuous occupancy of BCL6, as the loss of function would be maintained until the target is regenerated.20 We decided to focus on optimization of this novel BCL6 degrader, aiming to identify compounds with suitable physicochemical and pharmacokinetic properties to investigate in vivo degradation of BCL6 in a tumor xenograft model.
To support optimization, we developed an immunofluorescence assay to quantify BCL6 degradation in SU-DHL-4 cells. DC50 values from this assay are shown in Table 4. Initial SAR appeared tight, with relatively small changes in chemical structure resulting in the abolition of degradation activity (Table 4): only the monomethyl- and dimethylpiperidines 24a, 24b, and 26b were identified as degraders, with DC50 values similar to or below their IC50 values in the TR-FRET assay. No degradation was observed for pyrazole and morpholine analogues.
Optimization of DMPK Properties
Piperidine degraders 24a and 24b were subject to rapid metabolism in mouse microsomes, which we expected would limit our ability to achieve sufficient exposure to demonstrate degradation in vivo. We noted that morpholine 25a (Table 5) had lower microsomal clearance, leading us to hypothesize that metabolism of 24a and 24b may be occurring on the more lipophilic piperidine group. We aimed to reduce microsomal clearance and hence improve the likelihood of sufficient exposure21 first by replacing hydrogen atoms with fluorine to block the potential metabolic sites and second by lowering lipophilicity.
Trifluorination of the piperidine 3-methyl group showed modest improvement in microsomal stability (27a), and a more substantial reduction in clearance was achieved by the introduction of a difluoro group in the 4-position of the piperidine. The addition of these fluorine atoms also provided an improvement in both degradation (DC50) and biochemical inhibition (IC50), for monomethyl (Table 5, compare 24a and 27c) and dimethyl (compare 24b and 1) piperidine analogues.
In the previously reported series of BCL6 degraders, only hydrophobic groups were shown to cause degradation.15 We therefore designed analogues with lower lipophilicity to investigate whether degradation could be retained with more hydrophilic groups in this region and as an alternative approach to lowering metabolic clearance. We chose to modify the 3-position of the piperidine, as we had already shown that compounds with hydrophilic groups in the 4-position (morpholine 25a and piperazine 25c) were not capable of inducing BCL6 degradation. Hydroxymethyl 27d induced incomplete degradation, with the response plateauing at ∼60%. Addition of the 4,4-difluoro moiety was found again to improve both metabolic stability and the ability of the molecule to trigger BCL6 degradation, with 27e (CCT369900) giving potent and full (>85%) degradation while maintaining excellent aqueous solubility. With 27e we had thus identified an alternative and, importantly, less lipophilic group that induced degradation.
Further modifications demonstrated that the structural requirements for induction of degradation are quite specific: methylation of the OH reduced degradation (27f); alternative hydrophilic groups such as nitrile 27g in this position did not enable degradation; other discrete changes in structure such as moving the trifluoromethyl group (27h) and constraining the ring by forming bicyclic groups (28a, 28b) also led to a loss of degradation activity.
To confirm that degradation was not cell line specific, we developed an MSD (Meso Scale Discovery) assay to measure BCL6 levels. Compound 1 (CCT369260), along with analogues 27b and 27e, shows full (>85%) degradation of BCL6 in OCI-Ly1 and Karpas 422 cells, at DC50 values comparable to those observed in the SU-DHL-4 immunofluorescence assay (Table 6). With degradation activity confirmed, we investigated whether this translated into antiproliferative activity; examination of degrader compounds in a 14-day proliferation assay revealed growth inhibition in both SU-DHL-4 and OCI-Ly1 cell lines. Inhibitor 25b was less effective, suggesting that depletion of BCL6 gives a stronger effect on proliferation than inhibition alone, consistent with previous findings.15 Degraders showed a 5- to 40-fold reduction in activity in the BCL6 low-expressing22 OCI-Ly3 cell line compared to OCI-Ly1 (Table 6). Degrader 1 was further profiled in a panel of BCL6-negative cell lines and showed no antiproliferative activty (Supporting Information Figure S2). These findings suggest that the observed antiproliferative activity is driven by on-target effects.
Table 6. Antiproliferative Activity of Key Compounds.

Data represent the geometric mean of at least three replicates. See Supporting Information Table S2 for full statistics.
Data represent the geometric mean of at least two replicates except where superscript i is indicated. #This compound showed <10% degradation of BCL6 in a single concentration experiment at 10 μM, n = 2.
In Vivo Profiling of 1 (CCT369260)
On the basis of its acceptable microsomal clearance and robust effects in degradation and proliferation assays, compound 1 was selected for further profiling. A pharmacokinetic study was carried out in female Balb/C mice, dosing at 1 mg/kg iv (n = 3) and 5 mg/kg po (n = 3). All mice appeared normal after dosing and 24 h after dose. Compound 1 demonstrated moderate clearance (CL 20 mL min–1 kg–1) with mean oral bioavailability of 54%. Protein binding measurements using equilibrium dialysis showed the compound is highly bound (0.07% free in SCID mouse plasma, n = 6). To enable comparison of free concentrations in vivo with free levels in the OCI-Ly1 DC50 MSD assay, the free fraction in the DC50 assay medium was also measured (2.2% fraction unbound in IMDM medium, n = 3). This was used to estimate that a free concentration of greater than 1.1 nM (based on the OCI-Ly1 DC50) is required to elicit degradation in vivo, corresponding to a total plasma concentration of 1.5 μM or a total blood concentration of 2.8 μM (SCID mouse blood to plasma ratio of 1.9). PK studies were carried out at three doses (5, 15, and 50 mg/kg po, Figure 6) to determine if sufficient compound exposure could be achieved in the SCID mouse strain that would be used for xenograft studies. Exposure increased with dose between 5 and 15 mg/kg; however, no improvement was obtained by a further increase to 50 mg/kg. This is likely due to compound solubility; the compound was dosed as a clear solution in saline containing 10% DMSO and 5% Tween 20 at the lower doses but was not completely soluble and hence was dosed as a suspension in the same vehicle at the 50 mg/kg dose. The mean total blood concentration at the 15 mg/kg dose was above the desired concentration for approximately 10 h, confirming that this compound was suitable for progression to a PK/PD study at this dose to determine if, and at what concentration, BCL6 depletion could be achieved.
Figure 6.

(Left) Total SCID mouse blood concentrations of 1 (CCT369260) after po dosing at 5 (blue triangles), 15 (red circles), and 50 (green squares) mg/kg vs predicted active concentration (dashed line). (Right) Data for individual animals showing cmax, tmax, and total AUC10h in SCID mouse at 5, 15, and 50 mg/kg doses. All experiments were carried out according to the U.K. guidelines for animal experimentation.
A PK/PD study was carried out in mice using a single dose of 1 at 15 mg/kg po in an OCI-Ly1 DLBCL xenograft model. A clear decrease in levels of BCL6 in the tumor was observed up to 10 h after dosing with compound 1 (Figure 7 and Supporting Information Figure S3), with a maximal effect at ∼4 h. No visible adverse events were observed in this study. Examination of the relationship between compound 1 and BCL6 levels (Figure 7 and Supporting Information Figure S4) shows that, consistent with predictions, free concentrations of >1 nM degrader 1 lead to degradation of BCL6. Less substantial degradation is observed at 0.5 h; this delayed onset may be due to the time required for the compound to distribute into tumor. After compound levels drop below the calculated free DC50 at the 10 h time point, BCL6 levels remain low but rebound by 12 and 16 h, presumably due to the time required for BCL6 resynthesis. This study demonstrates that degradation of BCL6 in tumors following oral dosing with a small molecule is a feasible approach.
Figure 7.
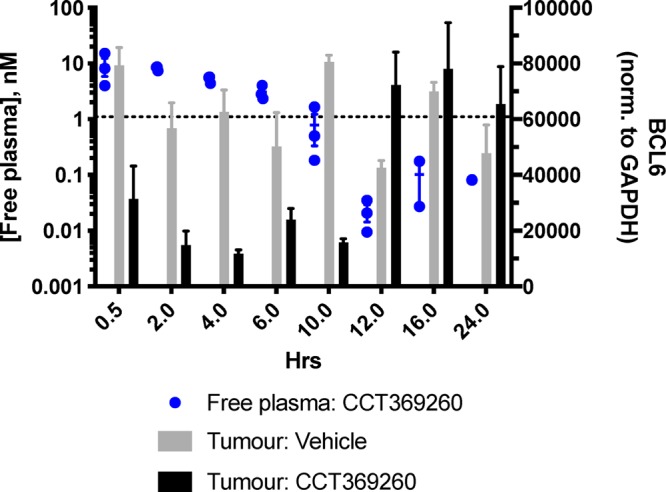
PK/PD study with 1 (CCT369260) at 15 mg/kg po. Tumour xenografts were prepared by subcutaneous injection of 1 × 107 OCI-Ly1 cells in female SCID mice, with dosing of compound commencing 20 days after injection, to mice with xenografts between 0.5 and 0.8 cm3, as described in more detail in the Supporting Information. Sampling took place at 0.5, 2, 4, 6, 10, 12, 16 and 24 h postdose. All experiments were carried out according to the U.K. guidelines for animal experimentation. BCL6 levels in tumor were quantified using capillary electrophoresis and normalized to a GAPDH loading control and are shown as gray (vehicle-treated) or black (compound treated) bars. Free compound levels at 0.5–24 h are shown (blue dots); dotted line indicates free DC50.
Discussion and Conclusions
We describe the discovery of a series of benzimidazolone inhibitors of BCL6, including 1 (CCT369260), an orally bioavailable degrader of BCL6. Weakly active (100 μM) hits derived from high throughput screening were merged to give benzimidazolone lead compound 6 (CCT365386). By filling a hydrophobic pocket and forming hydrogen bonds to water molecules, we were able to both improve binding affinity and incorporate hydrophilic groups to improve solubility. Further potency improvements were achieved through additional hydrophobic surface contacts in a solvent-exposed region of the pocket, resulting in sub-100 nM inhibitors. A subset of these compounds, including 24b (CCT368682), were shown to cause rapid degradation of BCL6, initially by Western blot and by an immunofluorescence assay in SU-DHL-4, and later confirmed in different cell lines using a MSD assay.
We noted that our initial Western blot data for 24b showed an apparent increase in BCL6 levels at low concentrations. A similar effect is also visible in previously presented work on BCL6 degraders,15 but analysis of curves from MSD experiments shows that this effect is not observed in all experiments and, when observed, corresponds to a < 25% increase. It is noteworthy that we and others15 also observe an increase in BCL6 levels when cells are treated with nondegrading inhibitors of BCL6. This suggests that binding may trigger a feedback mechanism leading to increased expression of BCL6.
Further optimization of initially identified degrader 24b enabled us to identify compounds with reduced metabolic clearance including 27e (CCT369900) and 1 (CCT369260). CCT369260 showed sub-100 nM activity in a degradation assay in SU-DHL-4 cells, and robust antiproliferative activity, in common with previously identified degraders15 but in contrast to BCL6-targetting PROTACs.14
Similar to previously reported degraders,15 we found that degradation mediated by our compounds is proteasome-dependent. It is noteworthy that we observe no significant difference in binding mode (as observed by X-ray crystallography) between 1 (CCT369260) and closely related nondegrader 25a (Figure 8), suggesting that the inhibitor or degrader behavior of a compound may depend on the nature of the substituents in this region of the molecule rather than on a distinct binding mode.
Figure 8.

Binding modes of the BCL6 degrader 1 (CCT369260) (A) and inhibitor 25a (B). Crystal structures of the BCL6 BTB domain bound to the BCL6 degrader 1 (CCT369260) (A, PDB code 6TOM) and inhibitor 25a (B, PDB code 6TOL) show a nearly identical binding mode. In both panels, the surface of the BCL6 dimer is shown as a gray transparent surface, with the two individual monomers displayed in ribbons and colored in gray and cyan. Selected residues are shown as lines. Compounds are shown as orange ball and sticks, selected water molecules as red spheres, and H-bonds as yellow dashed lines.
One possible hypothesis for how degradation is induced is via formation of a ternary complex of the degrader, BCL6, and an E3 ligase. In this context, we note that the degrader compounds including CCT369260 are largely buried in the BTB domain, with a relatively small area remaining solvent-exposed and available to bind to another component (for example, an E3 ligase) and that in contrast to bivalent PROTAC-type degraders, no hook effect is observed with increased concentration.23−25 These data suggest that the degrader molecule alone does not bind potently to an E3 ligase in the absence of BCL6. One related potential mechanism by which a small molecule could promote proteosomal degradation is through “hydrophobic tagging”; the degrader binds to the target protein and presents as a hydrophobic patch on the protein surface, mimicking a partially unfolded protein state, which is then targeted for proteosomal degradation.23,24,26 Should this be the case for our BCL6 degraders, then the incorporation of hydrophilic groups in the solvent-exposed region would not be expected to be tolerated, and a broad range of lipophilic groups could be expected to trigger degradation. However, we show first that certain hydrophilic groups are tolerated with degradation retained and second that the structural requirements for degradation are quite specific. These data together support an alternative hypothesis: that the combination of the protein surface and the bound degrader surface generates a “neo-substrate” or “neo-degron” that can be recognized by an E3 ubiquitin ligase or other effector of proteasomal degradation. This is similar conceptually to the mechanism of action of IMiDs which bind strongly to an E3 ligase and generate a “neo-enzyme” surface which modifies the substrate specificity of the ligase, although in our case it is the substrate surface, rather than that of the E3 ligase, that we hypothesize to be modified.
CCT369260 was progressed into PK studies, and we showed that the levels of compound needed to mediate degradation could be achieved in vivo following oral dosing, enabling us to demonstrate depletion of BCL6 levels in tumors following oral dosing in a mouse xenograft model. Ongoing work includes further optimization of CCT369260 and the use of our BCL6 degraders as in vitro and in vivo chemical tools to pharmacologically validate BCL6 as a target for the treatment of hematological cancer.
Experimental Section
All in vivo experiments were carried out according to the U.K. guidelines for animal experimentation. Cell lines were supplied by the German Collection of Microorganisms and Cell Cultures (DSMZ). Cell lines were authenticated by STR profiling and were routinely screened for Mycoplasma, using an in-house PCR-based assay.
General Synthetic Information
All anhydrous solvents and reagents were obtained from commercial suppliers and used without further purification. Evaporation of solvent was carried out using a rotary evaporator under reduced pressure at a bath temperature of up to 60 °C. Flash column chromatography was carried out using a Biotage purification system using SNAP KP-Sil cartridges or on reverse-phase mode using SNAP Ultra C18 cartridges. Semipreparative separations were carried out using an Agilent 1200 series preparative HPLC instrument over a 15 min gradient elution. Microwave-assisted reactions were carried out using a Biotage Initiator microwave system. Final compounds were purified to ≥95% purity. NMR data were collected on a Bruker Avance 500 spectrometer equipped with a 5 mm BBO/QNP probe or on a Bruker Avance Neo 600 spectrometer equipped with a 5 mm TCI CryoProbe. NMR data are presented in the form of chemical shift δ (multiplicity, coupling constants, integration) for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS), referenced to the internal deuterated solvent. HRMS was assessed using an Agilent 1200 series HPLC instrument and diode array detector coupled to a 6120 time-of-flight mass spectrometer with dual multimode APCI/ESI source or on a Waters Acquity UHPLC and diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source.
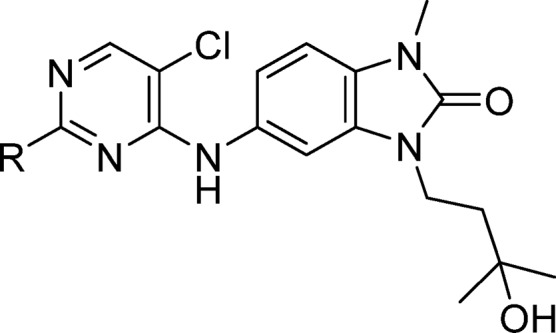
Preparation of Compounds. 5-((5-Chloro-2-((3R,5S)-4,4-difluoro-3,5-dimethylpiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (1, CCT369260)
To a mixture of 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (2.0 g, 5.0 mmol) were added (3R,5S)-4,4-difluoro-3,5-dimethylpiperidine hydrochloride (1.03 g, 5.6 mmol), NMP (20.2 mL), and DIPEA (2.64 mL, 15.1 mmol). The resulting mixture was heated to 140 °C for 2 h, cooled to room temperature, and then poured into ice–water. The resulting precipitate was collected and washed thoroughly with water to remove residual NMP, then further washed with ethanol to give a brown solid. Ethanol filtrate was concentrated and washed with water, then ethanol to give a second crop of solid. The two solid crops were combined and suspended in water. Ethanol was added until the brown color was removed from the solid, which was collected by filtration, washed again with water, and dried under vacuum to give compound 1 (1.97 g, 3.87 mmol, 77%) as an off-white solid. HRMS (ESI +ve): found 509.2269, expected 509.2243 for C24H32ClF2N6O2 [M + H]+. δH (600 MHz, CDCl3) 7.98 (s, 1H), 7.32 (d, J = 2.0 Hz, 1H), 7.22 (dd, J = 8.3, 2.0 Hz, 1H), 6.99 (s, 1H), 6.94 (d, J = 8.3 Hz, 1H), 4.60 (br d, J = 12.7 Hz, 2H), 4.04 (t, J = 7.4 Hz, 2H), 3.43 (s, 3H), 2.73 (t, J = 12.7 Hz, 2H), 2.32 (s, 1H), 2.02–1.90 (m, 2H), 1.89 (t, J = 7.4 Hz, 2H), 1.29 (s, 6H), 1.05 (d, J = 6.7 Hz, 6H).
2-Chloro-4-((cyclopropylmethyl)amino)nicotinonitrile (5)
A suspension of cyclopropylmethanamine (17 μL, 0.2 mmol), triethylamine (25 μL, 0.18 mmol), and 2,4-dichloronicotinonitrile (30 mg, 0.17 mmol) in DMA (0.35 mL) was stirred at rt for 18 h. The reaction was quenched with brine and extracted with EtOAc. The combined organics were washed with water and brine, dried, and concentrated, then purified by flash column chromatography (0–15% EtOAc in cyclohexane) to give 5 (26 mg, 0.13 mmol, 72%). δH (500 MHz, CDCl3) 8.09 (d, J = 6.1 Hz, 1H), 6.48 (d, J = 6.1 Hz, 1H), 5.31 (br s, 1H), 3.16–3.10 (m, 2H), 1.19–1.07 (m, 1H), 0.74–0.64 (m, 2H), 0.36–0.30 (m, 2H).
2-Chloro-4-((1,3-dimethyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (6, CCT365386)
A mixture of 5-amino-1,3-dimethyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (30 mg, 0.17 mmol), 2,4-dichloronicotinonitrile (30 mg, 0.17 mmol), and DIPEA (0.032 mL, 0.18 mmol) in DMA (0.4 mL) was heated in the microwave to 120 °C for 30 min, then added to a 1:1 mixture of methanol:water. The resulting precipitate was collected, washed sequentially with water, methanol, diethyl ether and dried to give 6 (32.3 mg, 0.10 mmol, 61%) as a brown solid. HRMS (ESI +ve): found 314.0810, expected 314.0803 for C15H13ClN5O+ [M + H]+. δH (500 MHz, DMSO-d6) 9.50 (s, 1H), 8.01 (d, J = 6.2 Hz, 1H), 7.21 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 2.0 Hz, 1H), 7.00 (dd, J = 8.2, 2.0 Hz, 1H), 6.67 (d, J = 6.2 Hz, 1H), 3.36 (s, 3H), 3.32 (s, 3H).
1-Methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (7)
To a mixture of N1-methyl-4-nitrobenzene-1,2-diamine (10.5 g, 62.8 mmol) in acetonitrile (150 mL) cooled to 0 °C was added portionwise disuccinimidyl carbonate (21.7 g, 84.7 mmol) over 20 min. The mixture was stirred at rt for 18 h and then poured into ice–water, forming a precipitate. The solid was collected by suction filtration and washed sequentially with water, DCM, and diethyl ether to give 7 (11.12 g) as an orange-red solid. HRMS (ESI +ve): found 194.0572, expected 194.0560 for C8H8N3O3+ [M + H]+. δH (500 MHz, DMSO-d6) 11.43 (s, 1H), 8.03 (dd, J = 8.7, 2.3 Hz, 1H), 7.76 (d, J = 2.3 Hz, 1H), 7.31 (d, J = 8.7 Hz, 1H), 3.36 (s, 3H).
2-Chloro-4-((1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (9)
To a mixture of 5-amino-1-methyl-1H-benzo[d]imidazol-2(3H)-one (750 mg, 4.6 mmol) and 2,4-dichloropyridine-3-carbonitrile (760 mg, 4.4 mmol) under argon was added DMA (10 mL) followed by DIPEA (0.90 mL, 5.19 mmol). The reaction mixture was heated at 120 °C under microwave irradiation for 45 min then allowed to cool to rt and added dropwise to a stirring mixture of methanol:water (1:1; 120 mL). The resulting precipitate was filtered, washed with water (2 × 25 mL) and diethyl ether (2 × 30 mL), affording 2-chloro-4-[(1-methyl-2-oxo-3H-benzimidazol-5-yl)amino]pyridine-3-carbonitrile (1.30 g, 99%, 4.3 mmol) as a beige solid. δH (500 MHz, DMSO-d6) 10.95 (br s, 1H), 9.39 (br s, 1H), 7.99 (d, J = 6.2 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 6.95 (dd, J = 8.3, 1.9 Hz, 1H), 6.90 (d, J = 1.9 Hz, 1H), 6.65 (d, J = 6.2 Hz, 1H), 3.29 (s, 3H).
3-(2-Hydroxybutyl)-1-methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (10)
To a mixture of 1-methyl-5-nitro-1H-benzo[d]imidazol-2(3H)-one (240 mg, 1.242 mmol) and cesium carbonate (485 mg, 1.489 mmol) in DMF (3 mL) was added 2-ethyloxirane (0.119 mL, 1.367 mmol), and the resulting mixture was heated in the microwave to 120 °C for 1 h, then added to water (10 mL). The resulting mixture was extracted with DCM, and combined organics were dried over sodium sulfate, filtered, and evaporated onto silica gel for purification by flash column chromatography (10 g silica, 40–60% ethyl acetate in cyclohexane). Fractions were combined and evaporated to give the title compound (200 mg, 60%) as a yellow-orange oil which solidified on standing. δH (500 MHz, DMSO-d6) 8.13 (d, J = 2.2 Hz, 1H), 8.05 (dd, J = 8.7, 2.3 Hz, 1H), 7.36 (d, J = 8.8 Hz, 1H), 4.90 (d, J = 5.4 Hz, 1H), 3.88 (dd, J = 14.2, 4.2 Hz, 1H), 3.82 (dd, J = 14.2, 7.4 Hz, 1H), 3.70 (m, 1H), 3.41 (s, 3H), 1.49 (m, 1H), 1.37 (m, 1H), 0.92 (t, J = 7.4 Hz, 3H).
2-Chloro-4-((3-(2-hydroxybutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11a)
To a suspension of 3-(2-hydroxybutyl)-1-methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (10) (34 mg, 0.13 mmol) in ethanol (1.5 mL) and DMSO (0.5 mL) was added sodium dithionite (116 mg, 0.67 mmol). The resulting mixture was heated to 90 °C for 5 h, then stirred at room temperature for 2 days, then heated again to 90 °C for 2 h. Additional sodium dithionite (116 mg, 0.67 mmol) was added, and the mixture was heated to 90 °C for 2 h. Water (1 mL) was added, and the resulting cloudy suspension was stirred at rt for 3 days, then diluted with water until all was in solution. The pH was adjusted to 10 using 2 M sodium carbonate, and the resulting mixture was extracted with DCM (×3), dried by passing through a phase separator, and evaporated under reduced pressure. The resulting material was diluted with DMA (0.5 mL), and then 2,4-dichloronicotinonitrile (15 mg, 0.087 mmol) and DIPEA (25 μL, 0.143 mmol) were added. The resulting mixture was heated in the microwave to 120 °C for 30 min, then partitioned between DCM and water. The aqueous layer was acidified to pH 7 using 10% citric acid, then extracted with DCM (×3). Combined organics were evaporated and purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 60:40 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min. The resulting yellow oil was triturated with diethyl ether to give compound 11a (4.2 mg, 0.011 mmol, 8.5%) as pale yellow solid. HRMS (ESI +ve): found 372.1230, expected 372.1222 for C18H19ClN5O2+ [M + H]+. δH (500 MHz, CD3OD) 7.94 (d, J = 6.2 Hz, 1H), 7.22 (d, J = 8.2 Hz, 1H) overlapping with 7.22 (d, J = 1.9 Hz, 1H), 7.07 (dd, J = 8.4, 1.9 Hz, 1H), 6.70 (d, J = 6.3 Hz, 1H), 3.98–3.89 (m, 1H), 3.89–3.80 (m, 2H), 3.47 (s, 3H), 1.67–1.55 (m, 1H), 1.48 (dp, J = 14.6, 7.3 Hz, 1H), 1.03 (t, J = 7.4 Hz, 3H).
2-Chloro-4-((3-(2-cyanobutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11b)
To a mixture of cesium carbonate (70 mg, 0.21 mmol) and 2-chloro-4-((1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (9) (25 mg, 0.08 mmol) in DMF (0.50 mL) was added 2-(bromomethyl)butanenitrile (25 mg, 0.15 mmol). The resulting mixture was heated to 60 °C for 3 days, then partitioned between water and DCM, and the aqueous layer acidified to pH 5 using a 10% citric acid solution. Layers were separated, the aqueous phase was extracted with further DCM, and combined organics were evaporated under reduced pressure and then purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 45:55 to 30:70 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 11b as an off-white solid. HRMS (ESI +ve): found 381.1209, expected 381.1225 for C19H18ClN6O+ [M + H]+. δH (500 MHz, CDCl3) 8.04 (d, J = 6.1 Hz, 1H), 7.08 (d, J = 1.9 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.02 (dd, J = 8.2, 1.9 Hz, 1H), 6.96 (s, 1H), 6.71 (d, J = 6.1 Hz, 1H), 4.11 (dd, J = 14.4, 5.7 Hz, 1H), 4.04 (dd, J = 14.5, 8.8 Hz, 1H), 3.48 (s, 3H), 3.11 (tt, J = 9.1, 5.4 Hz, 1H), 1.89–1.69 (m, 2H), 1.19 (t, J = 7.4 Hz, 3H).
(S)-2-Chloro-4-((3-(2-hydroxybutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11c)
To a mixture of 2-chloro-4-((1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (9) (20 mg, 0.067 mmol) and cesium carbonate (23 mg, 0.07 mmol) in DMF (0.50 mL) was added (2S)-2-ethyloxirane (7 μL, 0.08 mmol), and the resulting mixture was heated in the microwave to 120 °C for 1 h. Two further batches of (2S)-2-ethyloxirane (7 μL, 0.08 mmol) were added, with microwave heating to 140 °C for 30 min after each. The mixture was partitioned between water and DCM, and the aqueous layer was acidified to pH 5 using a 10% citric acid solution. Layers were separated, the aqueous phase was extracted with further DCM, and combined organics were evaporated under reduced pressure and then purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 50:50 to 35:65 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 11c (7.5 mg, 0.020 mmol, 30%) as an off white solid. HRMS (ESI +ve): found 372.1218, expected 372.1222 for C18H19ClN5O2+ [M + H]+. δH (500 MHz, CD3OD) identical to racemate 11a.
(R)-2-Chloro-4-((3-(2-hydroxybutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11d)
Prepared by the same method as its enantiomer 11c, starting from (2R)-2-ethyloxirane. HRMS (ESI +ve): found 372.1218, expected 372.1222 for C18H19ClN5O2+ [M + H]+. δH (500 MHz, CD3OD) identical to racemate 11a.
4-((3-Butyl-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)-2-chloronicotinonitrile (11e)
To 2-chloro-4-((1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (9) (25 mg, 0.084 mmol) and cesium carbonate (33 mg, 0.10 mmol) in DMF (0.85 mL) was added 1-bromobutane (9.5 μL, 0.089 mmol), and the resulting mixture was stirred at 140 °C under microwave irradiation for 1 h. Water (10 mL) was added, and the aqueous mixture was acidified with 1 M HCl. The aqueous mixture was extracted with EtOAc (5 × 10 mL). The organic extracts were combined, dried (Na2SO4), and concentrated in vacuo, then purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 40:60 to 20:80 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 11e (12.3 mg, 41%, 0.035 mmol) as an off-white solid. HRMS (ESI +ve): found 356.1263, expected 356.1273 for C18H19ClN5O+ [M + H]+. δH (500 MHz, CDCl3) 8.03 (d, J = 6.0 Hz, 1H), 7.02 (d, J = 8.1 Hz, 1H), 6.98 (dd, J = 8.1, 1.7 Hz, 1H), 6.93 (br s, 1H), 6.87 (d, J = 1.7 Hz, 1H), 6.60 (d, J = 6.0 Hz, 1H), 3.88 (t, J = 7.3 Hz, 2H), 3.46 (s, 3H), 1.77–1.68 (m, 2H), 1.45–1.35 (m, 2H), 0.97 (t, J = 7.3 Hz, 3H).
(R)-2-Chloro-4-((3-(3-hydroxybutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11f)
To 2-chloro-4-((1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (9) (40 mg, 0.13 mmol) and cesium carbonate (60 mg, 0.18 mmol) in DMF (0.6 mL) was added [(3R)-3-hydroxybutyl] 4-methylbenzenesulfonate (14b) (55 mg, 0.23 mmol), and the resulting mixture was heated in the microwave to 120 °C for 1 h. The mixture was partitioned between water and DCM, and the aqueous layer was acidified to pH 5 using a 10% citric acid solution. Layers were separated, the aqueous phase was extracted with further DCM, and combined organics were evaporated under reduced pressure and then purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 50:50 to 35:65 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 11f (21.5 mg, 0.057 mmol, 42%). HRMS (ESI +ve): found 372.1228, expected 372.1222 for C18H19ClN5O2+ [M + H]+. δH (500 MHz, CDCl3) 8.05 (d, J = 6.2 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.03 (dd, J = 2.0, 8.2 Hz, 1H), 6.97 (d, J = 2.0 Hz, 1H), 6.94 (s, 1H), 6.63 (d, J = 6.2 Hz, 1H), 4.25 (ddd, J = 15.0, 11.0, 4.6 Hz, 1H), 3.88 (dt, J = 15.0, 5.5, 3.8 Hz, 1H), 3.72 (dqd, J = 10.2, 6.2, 2.8 Hz, 1H), 3.49 (s, 3H), 1.88 (dddd, J = 14.1, 11, 5.5, 2.8 Hz, 1H), 1.71 (dddd, J = 14.1, 10.2, 4.6, 3.8 Hz, 1H), 1.22 (d, J = 6.2 Hz, 3H).
(S)-2-Chloro-4-((3-(3-hydroxybutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (11g)
Prepared by the same method as its enantiomer 11f. HRMS (ESI +ve): found 372.1219, expected 372.1222 for C18H19ClN5O2+ [M + H]+. δH (500 MHz, DMSO-d6) 9.48 (s, 1H), 8.00 (d, J = 6.0 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 7.17 (d, J = 1.9 Hz, 1H), 7.00 (dd, J = 1.9, 8.2 Hz, 1H), 6.68 (d, J = 6.0 Hz, 1H), 4.58 (d, J = 4.7 Hz, 1H), 3.94–3.79 (m, 2H), 3.65–3.58 (m, 1H), 3.34 (s, 3H), 1.72–1.58 (m, 2H), 1.08 (d, J = 6.3 Hz, 3H).
3-Hydroxy-3-methylbutyl 4-methylbenzenesulfonate (14a)
To a solution of 3-methylbutane-1,3-diol (27 g, 0.26 mol) and triethylamine (40 mL, 0.29 mol) in DCM (250 mL) at 0 °C was added dropwise 4-methylbenzenesulfonyl chloride (50 g, 84 mmol) in 200 mL of DCM over 1 h. The resulting mixture was stirred at 0 °C for 4 h, then water was added (200 mL) and stirred for 45 min. Adjustment to pH 10 was made by addition of 2 M NaOH and separated. Organic phase was washed sequentially with 200 mL of half-saturated sodium bicarbonate solution (×2), water, 1 M HCl, brine, then dried over magnesium sulfate, filtered, and concentrated to give a yellow oil. This was dissolved in DCM/cyclohexane and purified by flash column chromatography (0–40% ethyl acetate in cyclohexane) to give the title compound (39.9 g) as a colorless oil. δH (500 MHz, CDCl3) 7.85–7.78 (m, 2H), 7.40–7.34 (m, 2H), 4.23 (t, J = 6.8 Hz, 2H), 2.47 (s, 3H), 1.88 (t, J = 6.8 Hz, 2H), 1.23 (s, 6H).
[(3R)-3-Hydroxybutyl] 4-methylbenzenesulfonate (14b)
A solution of (3R)-butane-1,3-diol (0.86 mL, 9.5 mmol) in dry dichloromethane (10 mL) under a nitrogen atmosphere was cooled in a salt–ice bath (bath temp −12 °C). Triethylamine (2.25 mL, 16.1 mmol) was added followed by a solution of 4-methylbenzenesulfonyl chloride (2 g, 10.5 mmol) in dry dichloromethane (6 mL) over 10 min. The resulting mixture was allowed to warm slowly to room temperature and stirred for 20 h, then diluted with DCM, washed with 10% citric acid, sat. sodium bicarbonate solution, brine, dried over sodium sulfate, filtered, and evaporated under reduced pressure to give a clear oil. This was purified by flash column chromatography (50 g silica, 10–30% ethyl acetate in cyclohexane) to give 14b (1.65 g, 6.75 mmol, 71%) as a clear oil. δH (500 MHz, CDCl3) 7.82 (br d, J = 8.3 Hz, 2H), 7.37 (br d, J = 7.9 Hz, 2H), 4.26 (ddd, J = 10.0, 8.7, 5.0 Hz, 1H), 4.13 (dt, J = 10.0, 5.5 Hz, 1H), 3.96 (dqd, J = 9.7, 6.2, 3.5 Hz, 1H), 2.47 (s, 3H), 1.85 (dddd, J = 14.5, 8.7, 5.8, 3.6 Hz, 1H), 1.71 (ddt, J = 14.2, 8.9, 5.0 Hz, 1H), 1.21 (d, J = 6.3 Hz, 3H).
3-(3-Hydroxy-3-methylbutyl)-1-methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (15)
A mixture of 1-methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (7, 9.05 g, 46.9 mmol), cesium carbonate (30.5 g, 93.7 mmol), and 3-hydroxy-3-methylbutyl 4-methylbenzenesulfonate (14a, 16.4 g, 63.5 mmol) in acetonitrile (234 mL) was heated to reflux for 4 h. The mixture was concentrated under reduced pressure. The residue was then diluted with water (500 mL) and stirred for 30 min, forming a precipitate, which was filtered and washed thoroughly with water. The collected solid was dried under vacuum to give 15 (12 g, 43 mmol, 92%) as a red-brown solid. δH (500 MHz, CDCl3) 8.13 (dd, J = 8.7, 2.0 Hz, 1H), 7.95 (d, J = 2.0 Hz, 1H), 7.04 (d, J = 8.7 Hz, 1H), 4.13 (m, 2H), 3.50 (s, 3H), 1.93 (m, 2H), 1.34 (s, 6H).
5-Amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16)
Palladium on activated charcoal (10% Pd, 240 mg) was added to a solution of 3-(3-hydroxy-3-methylbutyl)-1-methyl-5-nitro-1,3-dihydro-2H-benzo[d]imidazol-2-one (15) (6.3 g, 22.6 mmol) in ethanol (113 mL), which was then stirred at 60 °C under an atmosphere of hydrogen for 3 h. The mixture was filtered through a pad of Celite and washed through with ethanol. The resulting solution was then concentrated under reduced pressure to give compound 16 (5.85 g) as a pale pink solid. δH (500 MHz, CD3OD) 6.90 (d, J = 8.1 Hz, 1H), 6.63 (d, J = 1.9 Hz, 1H), 6.58 (dd, J = 8.1, 1.9 Hz, 1H), 3.95 (m, 2H), 3.36 (s, 3H), 1.84 (m, 2H), 1.29 (s, 6H).
2-Chloro-4-((3-(3-hydroxy-3-methylbutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)nicotinonitrile (17a)
A mixture of 2,4-dichloropyridine-3-carbonitrile (11 mg, 0.064 mmol), 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16) (15 mg, 0.06 mmol), and DIPEA (15 μL, 0.086 mmol) in NMP (0.5 mL) was heated in the microwave to 120 °C for 30 min and then purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 45:55 to 20:80 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 17a (11.2 mg, 0.027 mmol, 46%) as a white solid. HRMS (ESI +ve): found 386.1388, expected 386.1378 for C19H21ClN5O2+ [M + H]+. δH (500 MHz, DMSO-d6) 9.46 (s, 1H), 8.00 (d, J = 6.1 Hz, 1H), 7.20 (d, J = 8.3 Hz, 1H), 7.11 (d, J = 1.9 Hz, 1H), 7.00 (dd, J = 8.3, 2.0 Hz, 1H), 6.68 (d, J = 6.2 Hz, 1H), 4.44 (s, 1H), 3.92–3.85 (m, 2H), 3.34 (s, 3H), 1.73–1.66 (m, 2H), 1.15 (s, 6H).
5-((2,3-Dichloropyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (17b)
A mixture of 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16) (20 mg, 0.08 mmol), Xantphos (12 mg, 0.02 mmol), 2,3-dichloro-4-iodopyridine (30 mg, 0.11 mmol), Pd2(dba)3 (2.3 mg, 0.004 mmol), and cesium carbonate (67 mg, 0.21 mmol) in NMP:toluene (1:1 v/v, 1.8 mL) was degassed and heated in the microwave under a nitrogen atmosphere at 140 °C for 1 h. Purification was by reverse-phase flash chromatography (Biotage Ultra C18 12g, 30–90% methanol in water (0.1% formic acid modifier)) to give 17b (7 mg, 0.018 mmol, 22%). HRMS (ESI +ve): found 395.1038, expected 395.1036 for C18H21Cl2N4O2+ [M + H]+. δH (600 MHz, CDCl3) 7.90 (d, J = 5.6 Hz, 1H), 7.03 (d, J = 8.2 Hz, 1H), 7.01 (dd, J = 8.2, 1.8 Hz, 1H), 6.95 (d, J = 1.8 Hz, 1H), 6.68 (s, 1H), 6.64 (d, J = 5.7 Hz, 1H), 4.09–4.03 (m, 2H), 3.46 (s, 3H), 1.93–1.87 (m, 2H), 1.32 (s, 6H).
5-((3-Chloropyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (17c)
Method as for 17b, using 4-bromo-3-chloropyridine. Purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 17c (21 mg, 69%, 0.055 mmol) as the formic acid salt. HRMS (ESI +ve): found 361.1422, expected 361.1426 for C18H22ClN4O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.30 (s, 1H), 8.16 (s, 1H), 8.06 (d, J = 5.7 Hz, 1H), 7.18–7.13 (m, 2H), 7.06 (dd, J = 8.1, 2.0 Hz, 1H), 6.84 (d, J = 5.7 Hz, 1H), 4.03 (t, J = 8.2 Hz, 2H), 3.41 (s, 3H), 1.85 (t, J = 8.2 Hz, 2H), 1.25 (s, 6H).
4-Chloro-6-((3-(3-hydroxy-3-methylbutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)pyrimidine-5-carbonitrile (17d)
Method as for 17a, using 4,6-dichloropyrimidine-5-carbonitrile and heating at 80 °C for 30 min. 17d (8.2 mg, 0.02 mmol, 33%) obtained as an off white solid. HRMS (ESI +ve): found 387.1340, expected 387.1331 for C18H20ClN6O2+ [M + H]+ . δH (500 MHz, DMSO-d6) 10.25 (br s, 1H), 8.49 (s, 1H), 7.23 (s, 1H), 7.14 (s, 2H), 4.45 (br s, 1H), 3.93–3.81 (m, 2H), 3.33 (s, 3H), 1.75–1.64 (m, 2H), 1.16 (s, 6H).
5-((5,6-Dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (17e)
A mixture of 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16) (30 mg, 0.12 mmol), DIPEA (30 uL, 0.17 mmol), and 4,5,6-trichloropyrimidine (26 mg, 0.14 mmol) in DMF (0.9 mL) was heated in the microwave to 120 °C for 30 min. The mixture was partitioned between water and DCM, and the aqueous layer was acidified to pH 4 using a 10% citric acid solution. Layers were separated, the aqueous phase was extracted with further DCM, and combined organics were evaporated under reduced pressure and then purified by reverse phase flash chromatography (Biotage 12g SNAP Ultra C18, 20–100% methanol in water, 0.1% formic acid modifier) and then further triturated with diethyl ether to give compound 17e as a beige solid (41 mg, 0.10 mmol, 86%). HRMS (ESI +ve): found 396.0997, expected 396.0989 for C17H20Cl2N5O2+ [M + H]+. δH (500 MHz, DMSO-d6) 9.46 (s, 1H), 8.26 (s, 1H), 7.30 (d, J = 2.0 Hz, 1H), 7.21 (dd, J = 8.4, 2.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 4.45 (s, 1H), 3.91–3.84 (m, 2H), 3.33 (s, 3H, obscured by solvent), 1.74–1.66 (m, 2H), 1.16 (s, 6H).
5-((3,5-Dichloropyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (17f)
Method as for 17b, using 4-bromo-3,5-dichloropyridine. Obtained compound 17f (8 mg, 0.02 mmol, 25%). HRMS (ESI +ve): found 395.1027, expected 395.1036 for C18H21Cl2N4O2+ [M + H]+. δH (600 MHz, CDCl3) 8.34 (s, 2H), 6.92 (d, J = 8.2 Hz, 1H), 6.78 (dd, J = 8.2, 2 Hz, 1H), 6.76 (d, J = 2.0 Hz, 1H), 6.58 (s, 1H), 4.04 (t, J = 7.4 Hz, 2H), 3.45 (s, 3H), 1.89 (t, J = 7.4 Hz, 2H), 1.30 (s, 6H).
5-((5-Chloro-2-(methylthio)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (17g)
Method as for 17e, using 4,5-dichloro-2-methylsulfanylpyrimidine. Obtained compound 17g as white solid (27 mg, 0.063 mmol, 78%). HRMS (ESI +ve): found 408.1208, expected 408.1255 for C18H23ClN5O2S+ [M + H]+ ]+. δH (500 MHz, DMSO-d6) 8.43 (br s, 1H), 8.24 (s, 1H), 7.41 (d, J = 1.6 Hz, 1H), 7.27 (dd, J = 8.4, 1.6 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 3.87 (m, 2H), 3.32 (s, 3H), 2.38 (s, 3H), 1.72 (m, 2H), 1.17 (s, 6H).
5-((2,5-Dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18)
A mixture of 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16) (1.61 g, 6.47 mmol), 2,4,5-trichloropyrimidine (0.88 mL, 7.69 mmol), and cesium carbonate (4.21 g, 12.93 mmol) in DMF (15 mL) was heated in the microwave to 120 °C for 30 min. The resulting mixture was diluted with water, acidified to pH 5 by addition of 10% citric acid, and extracted with DCM. The combined organics were evaporated under reduced pressure, and the resulting sticky solid was dissolved in a minimum volume of ethyl acetate and precipitated by addition of diethyl ether. The resulting solid was collected by filtration and washed with diethyl ether and dried under reduced pressure, giving the title product (1.96 g, 72%, 4.69 mmol) as a solid. δH (500 MHz, DMSO-d6) 9.57 (s, 1H), 8.34 (s, 1H), 7.35 (d, J = 1.9 Hz, 1H), 7.19 (dd, J = 8.4, 1.9 Hz, 1H), 7.15 (d, J = 8.4 Hz, 1H), 4.44 (s, 1H), 3.92–3.86 (m, 2H), 3.33 (s, 3H), 1.76–1.69 (m, 2H), 1.17 (s, 6H).
5-((5-Chloro-2-(dimethylamino)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (19)
A mixture of 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (20 mg, 0.05 mmol), dimethylamine hydrochloride (37 mg, 0.45 mmol), cesium carbonate (164 mg, 0.50 mmol) in NMP (0.5 mL) was heated in the microwave to 180 °C for 1 h. Mixture was partitioned between DCM and water, aqueous layer was extracted with DCM, and combined organics were washed with Brine. Organic layers were combined and evaporated, and the resulting NMP solution was purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 19 (8 mg, 0.0188 mmol, 37%). HRMS (ESI +ve): found 405.1790, expected 405.1800 for C19H26ClN6O2+. δH (500 MHz, acetone-d6) 7.96 (s, 1H), overlapping with 7.98–7.94 (0.6H, partly exchanged NH), 7.73 (d, J = 1.7 Hz, 1H), 7.35 (dd, J = 8.1, 1.7 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 4.02 (t, J = 7.9 Hz, 2H), 3.38 (s, 3H), 3.13 (s, 6H), 1.85 (t, J = 7.9 Hz, 2H), 1.25 (s, 6H).
5-((5-Chloro-2-morpholinopyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (20a)
A mixture of morpholine (0.031 mL, 0.25 mmol), 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18, 20 mg, 0.05 mmol), and DIPEA (0.04 mL, 0.25 mmol) in NMP (1 mL) was heated in the microwave to 180 °C for 1 h, then partitioned between DCM and water. The aqueous layer was extracted with DCM, and combined organics were washed with brine. The organic layers were combined and evaporated, and the resulting material (containing residual NMP) was purified by preparative HPLC (ACE 5 C18-PFP column (5 μm, 250 mm × 21.2 mm), 15 min gradient elution from 40:60 to 25:75 water:methanol (both modified with 0.1% formic acid) at a flow rate of 20 mL/min) to give 20a (18 mg, 0.038 mmol, 76%). HRMS (ESI +ve): found 447.1897, expected 447.1906 for C21H28ClN6O3+ [M + H]+. δH (500 MHz, CD3OD) 7.92 (s, 1H), 7.47 (d, J = 1.7 Hz, 1H), 7.31 (dd, J = 8.4, 1.7 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 4.00 (m, 2H), 3.70–3.67 (m, 4H), 3.66–3.62 (m, 4H), 3.41 (s, 3H), 1.84 (m, 2H), 1.28 (s, 6H).
5-((5-Chloro-2-(piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (20b)
Method as for 20a, using piperidine and heating to 170 °C for 1 h. Obtained compound 20b (19 mg, 0.04 mmol, 80%) following HPLC. HRMS (ESI +ve): found 445.2111, expected 445.2113 for C22H30ClN6O2+ [M + H]+. δH (500 MHz, acetone-d6) 7.97 (br, partly exchanged NH, 0.3H), 7.96 (s, 1H), 7.61 (d, J = 1.8 Hz, 1H), 7.31 (dd, J = 8.7, 1.3 Hz, 1H), 7.05 (d, J = 8.7 Hz, 1H), 4.02 (m, 2H), 3.73 (br t, J 4.8 Hz, 4H), 3.38 (s, 3H), 1.86 (m, 2H), 1.67–1.62 (m, 2H), 1.59–1.52 (m, 4H), 1.25 (s, 6H).
5-((5-Chloro-2-(2,4-dimethylthiazol-5-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (21)
A mixture of 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (20 mg, 0.05 mmol), 2,4-dimethyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiazole (12.07 mg, 0.05 mmol), sodium carbonate (10.7 mg, 0.10 mmol), and bis(triphenylphosphine)palladium(II) chloride (1.8 mg, 0.0025 mmol) in 1,4-dioxane (0.40 mL) and water (0.40 mL) was heated in the microwave at 130 °C for 30 min. The mixture was partitioned between DCM and water and pH adjusted to 5 using 10% citric acid before separation and extraction with further DCM. The organic layers were combined and evaporated, and the resulting solution was purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 21 (2 mg, 0.0042 mmol, 8%) as a white solid. HRMS (ESI +ve): found 473.1501, expected 473.1521 for C22H26ClN6O2S+ [M + H]+. δH (500 MHz, acetone-d6) 8.50 (br, 1H), 8.38 (s, 1H), 7.54 (d, J = 1.8 Hz, 1H), 7.34 (dd, J = 8.3, 1.8 Hz, 1H), 7.13 (d, J = 8.3 Hz, 1H), 4.07 (t, J = 7.9 Hz, 2H), 3.41 (s, 3H), 2.63 (s, 3H), 2.60 (s, 3H), 1.89 (t, J = 7.9 Hz, 2H), 1.24 (s, 6H).
5-((5-Chloro-2-(1-methyl-1H-imidazol-2-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (22)
A mixture of 1-methyl-2-(tributylstannyl)-1H-imidazole (21 mg, 0.06 mmol), 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (20 mg, 0.05 mmol), and bis(triphenylphosphine)palladium(II) chloride (3.5 mg, 0.005 mmol) in 1,4-dioxane (1 mL) was heated for 18 h at 90 °C. The mixture was partitioned between DCM and water and pH adjusted to 5 using 10% citric acid before separation and extraction with further DCM. The organic layers were combined and evaporated, and the resulting solution was purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column, 15 min gradient from 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 22 (3 mg, 0.0068 mmol, 13%) as the formic acid salt. HRMS (ESI +ve): found 442.1740, expected 442.1753 for C21H25ClN7O2+ [M + H]+. δH (500 MHz,, acetone-d6) 8.50 (br, 1H), 8.37 (s, 1H), 8.12 (s, 1H), 7.74 (br s, 1H), 7.64 (br s, 1H), 7.55 (d, J = 2.0 Hz, 1H), 7.35 (dd, J = 8.6, 2.0 Hz, 1H), 7.14 (d, J = 8.6 Hz, 1H), 4.04 (t, J = 8.3 Hz, 2H), 3.91 (s, 3H), 3.40 (s, 3H), 1.88 (t, J = 8.3 Hz, 2H), 1.24 (s, 6H).
5-((5-Chloro-2-(1H-pyrazol-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (23a)
Method as for 19, using 1H-pyrazole, heating in the microwave to 170 °C for 1 h. Purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 60:40 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 23a. HRMS (ESI +ve): found 428.1552, expected 428.1596 for C20H23ClN7O2+ [M + H]+. δH (500 MHz, DMSO-d6) 9.42 (br, 1H), 8.45 (s, 1H), 8.38 (d, J = 2.2 Hz, 1H), 7.79 (s, 1H), 7.63 (d, J = 2.2 Hz, 1H), 7.42 (dd, J = 8.4, 2.2 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 6.52 (s, 1H), 3.91 (t, J = 7.6 Hz, 2H), 3.34 (s, 3H), 1.75 (t, J = 7.6 Hz, 2H), 1.14 (s, 6H).
5-((5-Chloro-2-(3-methyl-1H-pyrazol-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (23b) and 5-((5-Chloro-2-(5-methyl-1H-pyrazol-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (23c)
A mixture of 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (25 mg, 0.063 mmol), 3-methylpyrazole (0.05 mL, 0.62 mmol), and cesium carbonate (100 mg, 0.31 mmol) in NMP (0.5 mL) was heated in the microwave to 170 °C for 1 h. The mixture was partitioned between DCM and water and pH adjusted to 5 using 10% citric acid before separation and extraction with further DCM. The organic layers were combined and evaporated, and the resulting solution was purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 45:55 to 20:80 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min. Two products were obtained: the major, later eluting regioisomer was assigned as compound 23b (18 mg, 0.039 mmol, 61%). HRMS (ESI +ve): found 442.1755, expected 442.1753 for C21H25ClN7O2+ [M + H]+. δH (500 MHz, CDCl3) 8.34 (s, 1H), 8.27 (d, J = 2.6 Hz, 1H), 7.63 (d, J = 2.0 Hz, 1H), 7.32 (s, 1H, NH), 7.18 (dd, J = 8.3, 2.0 Hz, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.23 (d, J = 2.6 Hz, 1H), 4.15–4.08 (m, 2H), 3.47 (s, 3H), 2.40 (s, 3H), 1.94 (t, J = 7.3 Hz, 2H), 1.30 (s, 6H). The minor, earlier eluting regioisomer was assigned as compound 23c (2.5 mg, 0.005 mmol, 8.5%). HRMS (ESI +ve): found 442.1756, expected 442.1753 for C21H25ClN7O2+ [M + H]+. δH (500 MHz,, CDCl3) 8.37 (s, 1H), 7.74 (d, J = 2.0 Hz, 1H), 7.63 (m, 1H), 7.29 (s, 1H, NH), 7.08 (dd, J = 8.3, 2.0 Hz, 1H), 6.98 (d, J = 8.3 Hz, 1H), 6.16 (s, 1H), 4.13–4.06 (m, 2H), 3.45 (s, 3H), 2.46 (s, 3H), 1.97–1.91 (m, 2H), 1.29 (s, 6H).
5-((5-Chloro-2-(3,5-dimethyl-1H-pyrazol-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (23d)
Method as for 23b, using 3,5-dimethyl-1H-pyrazole. Purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min, then further purified by flash column chromatography (12 g KP-SIL, 0–6% methanol in DCM) to give compound 23d. HRMS (ESI +ve): found 456.1928, expected 456.1909 for C22H27ClN7O2 [M + H]+. δH (500 MHz, CDCl3) 8.37 (s, 1H), 7.44 (d, J = 1.9 Hz, 1H), 7.22 (s, 1H), 7.13 (dd, J = 1.9, 8.2 Hz, 1H), 6.98 (d, J = 8.2 Hz, 1H), 5.95 (s, 1H), 4.08 (t, J = 7.3 Hz, 2H), 3.45 (s, 3H), 2.33 (s, 3H), 2.31 (s, 3H), 1.89 (t, J = 7.4 Hz, 2H), 1.35–1.22 (m, 6H).
5-((5-Chloro-2-(3-methylpiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (24a)
Method as for 20a, using 3-methylpiperidine and heating to 170 °C for 1 h. Obtained 24a (18 mg, 0.037 mmol, 74%) as the formic acid salt after HPLC purification. HRMS (ESI +ve): found 459.2239, expected 459.2270 for C23H32ClN6O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.14 (s, 1H, formic acid), 7.98 (br, 0.8H, partly exchanged NH), 7.95 (s, 1H), 7.57 (d, J = 2.0 Hz, 1H), 7.35 (dd, J = 8.8, 2.0 Hz, 1H), 7.06 (d, J = 8.8 Hz, 1H), 4.59–4.50 (m, 2H), 4.01 (m, 2H), 3.37 (s, 3H), 2.83 (td, J = 12.5, 2.9 Hz, 1H), 2.51 (dd, J = 12.9, 10.6 Hz, 1H), 1.86 (m, 2H), 1.80 (dm, J = 12.9 Hz, 1H),1.69 (dp, J = 13.2, 3.4 Hz, 1H), 1.57 (m, 1H), 1.46 (qt, J = 13.2, 12.5, 12, 4 Hz, 1H), 1.25 (s, 6H), 1.16 (app qd, J = 12.1, 3.8 Hz, 1H), 0.90 (d, J = 6.1 Hz, 2H).
5-((5-Chloro-2-((3S,5R)-3,5-dimethylpiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (24b)
Method as for 20a using (3R,5S)-3,5-dimethylpiperidine. Obtained 24b (11 mg, 0.022 mmol, 88%) as the formic acid salt. HRMS (ESI +ve): found 473.2392, expected 473.2426 for C24H34ClN6O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.31 (br s, 0.6H, partly exchanged NH), 8.13 (s, 1H), 7.96 (s, 1H), 7.51 (d, J = 2.0 Hz, 1H), 7.39 (dd, J = 8.4, 2.0 Hz, 1H), 7.09 (d, J = 8.4 Hz, 1H), 4.66 (br d, J = 12.6 Hz, 2H), 4.04 (m, 2H), 3.39 (s, 3H), 2.33 (dd, J = 12.6, 11.7 Hz, 2H), 1.86 (m, 2H), 1.81 (m, 1H), 1.59 (m, 2H), 1.26 (s, 6H), 0.90 (d, J = 6.5 Hz, 6H), 0.82 (m, 1H).
5-((5-Chloro-2-((2R,6S)-2,6-dimethylmorpholino)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (25a)
Method as for 20a, using (2R,6S)-2,6-dimethylmorpholine and heating to 170 °C for 1 h. Obtained compound 25a (16 mg, 0.032 mmol, 63%) after HPLC purification. HRMS (ESI +ve): found 475.2169, expected 475.2219 for C23H32ClN6O3 [M + H]+. δH (500 MHz, acetone-d6) 8.07 (br, 1H), 7.98 (s, 1H), 7.57 (d, J = 1.8 Hz, 1H), 7.31 (dd, J = 8.7, 1.8 Hz, 1H), 7.07 (d, J = 8.7 Hz, 1H), 4.46 (dd, J = 13.3, 2.3 Hz, 2H), 4.03 (t, J = 8.0 Hz, 2H), 3.55 (dqd, J = 10.5, 6.2, 2.3 Hz, 2H), 3.37 (s, 3H), 2.48 (dd, J = 13.3, 10.5 Hz, 2H), 1.86 (t, J = 8.0 Hz, 2H), 1.26 (s, 6H), 1.16 (d, J = 6.2 Hz, 6H).
5-((5-Chloro-2-(2,2,6,6-tetramethylmorpholino)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (25b)
A mixture of 5-((2,5-dichloropyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (18) (30 mg, 0.076 mmol), 2,2,6,6-tetramethylmorpholine (22 mg, 0.15 mmol), and DIPEA (40 μL, 0.23 mmol) in NMP (0.67 mL) was heated in the microwave to 140 °C for 2 h. Once cooled, the mixture was diluted with DMSO (0.5 mL) and then purified by reverse-phase chromatography eluting from 30% to 100% methanol in water (each containing 0.1% formic acid) to give compound 25b (32 mg, 0.064 mmol, 84%) as a pale brown solid. HRMS (ESI +ve): found 503.2521, expected 503.2532 for C25H36ClN6O3+ [M + H]+. δH (600 MHz, CDCl3) 7.97 (s, 1H), 7.36 (d, J = 2.0 Hz, 1H), 7.24 (dd, J = 8.4, 2.0 Hz, 1H), 7.00 (s, 1H), 6.93 (d, J = 8.4 Hz, 1H), 4.04 (t, J = 7.2 Hz, 2H), 3.58 (s, 4H), 3.42 (s, 3H), 1.89 (t, J = 7.2 Hz, 2H), 1.28 (s, 6H), 1.23 (s, 12H).
5-((5-Chloro-2-((3S,5R)-3,4,5-trimethylpiperazin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (25c)
Method as for 25b, using (2S,6R)-1,2,6-trimethylpiperazine and heating for 1 h at 140 °C. Purification by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 90:10 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min followed by further purification using an SCX-2 column gave compound 25c (2 mg, 0.004 mmol, 11%). HRMS (ESI +ve): found 488.2518, expected 488.2535 for C24H35ClN7O2+ [M + H]+. δH (600 MHz, CD3OD) 7.94 (s, 1H), 7.43 (d, J = 2.0 Hz, 1H), 7.37 (dd, J = 8.4, 2.0 Hz, 1H), 7.14 (d, J = 8.4 Hz, 1H), 4.40 (br d, J = 13.4 Hz, 2H), 4.04 (m, 2H), 3.45 (s, 3H), 2.70 (dd, J = 13.4, 11.0 Hz, 2H), 2.34 (s, 3H), 2.32–2.22 (m, 2H), 1.87 (m, 2H), 1.30 (s, 6H), 1.15 (d, J = 6.2 Hz, 6H).
5-((5-Chloro-2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (26a)
A mixture of 3,5-dimethyl-1H-pyrazole (3 mg, 0.03 mmol), Pd2(dba)3 (1.5 mg, 0.0017 mmol), Xantphos (4.1 mg, 0.007 mmol), cesium carbonate (14 mg, 0.04 mmol), and 5-((2-bromo-5-chloropyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (29, 9 mg, 0.02 mmol) in NMP:toluene (1:1 v/v, 0.8 mL) was heated in the microwave to 140 °C for 1 h. The resulting mixture was diluted with water, acidified to pH 5 by addition of 10% citric acid, then extracted with DCM (5 mL × 3). Organic layer was collected and passed through a Si-DMT palladium scavenging column, then evaporated under reduced pressure. Product was purified by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 40:60 to 25:75 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give 26a (4 mg, 0.0084 mmol, 41%). HRMS (ESI +ve): found 455.1943, expected 455.1957 for C23H28ClN6O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.19–8.14 (m, 1.6H, including partly exchanged NH), 7.39 (s, 1H), 7.19–7.16 (m, 2H), 7.09 (dd, J = 8.2, 2.0 Hz, 1H), 5.97 (s, 1H), 4.03 (t, J = 7.9 Hz, 2H), 3.42 (s, 3H), 2.57 (s, 3H), 2.10 (s, 3H), 1.87 (t, J = 7.9 Hz, 2H), 1.23 (6H, s).
5-((5-Chloro-2-((3S,5R)-3,5-dimethylpiperidin-1-yl)pyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (26b)
A mixture of 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16, 15 mg, 0.06 mmol), 5-chloro-2-((3S,5R)-3,5-dimethylpiperidin-1-yl)-4-iodopyridine 30a (25.3 mg, 0.072 mmol), cesium carbonate (157 mg, 0.48 mmol), Xantphos (20.9 mg, 0.036 mmol), and Pd2(dba)3 (5.5 mg, 0.006 mmol) in toluene:DMF (3:1 v/v, 0.8 mL) was heated in the microwave at 80 °C for 1 h. EtOAc and aq NH4Cl were added, layers were separated, and aqueous layer was extracted with further EtOAc. The organic layers were combined, dried over MgSO4, and concentrated under vacuum. Purification was by flash column chromatography (0–10% MeOH in DCM), followed by further purification by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 60:40 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min to give compound 26b (13 mg, 0.028 mmol, 46%) as formic acid salt. HRMS (ESI +ve): found 472.2482, expected 472.2474 for C25H35ClN5O2+ [M + H]+. δH (500 MHz, CD3OD) 8.27 (br, 1H), 7.84 (s, 1H), 7.22 (d, J = 8.3 Hz, 1H), 7.18 (d, J = 1.8 Hz, 1H), 7.10 (dd, J = 8.3, 1.8 Hz, 1H), 6.12 (s, 1H), 4.03 (m, 2H), 3.81 (dm, J = 13 Hz, 2H), 3.46 (s, 3H), 2.30 (dd, J = 13, 11.4 Hz, 2H), 1.86 (m, 2H), 1.80 (m, 1H), 1.62 (m, 2H), 1.28 (s, 6H), 0.87 (d, J = 6.6 Hz, 6H), 0.76 (dt, J = 12.9, 11.8 Hz, 1H).
5-((5-Chloro-2-((2R,6S)-2,6-dimethylmorpholino)pyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (26c)
Method as for 26b, using (2S,6R)-4-(5-chloro-4-iodo-2-pyridyl)-2,6-dimethylmorpholine 30b. Further purification by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 45:55 to 20:80 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min gave compound 26c (5 mg, 0.011 mmol, 26%). HRMS (ESI +ve): found 474.2246, expected 474.2266 for C24H33ClN5O3+ [M + H]+. δH (500 MHz,, CD3OD) 7.86 (s, 1H), 7.21 (d, J = 8.3 Hz, 1H), 7.16 (d, J = 1.6 Hz, 1H), 7.09 (dd, J = 8.3, 1.6 Hz, 1H), 6.13 (s, 1H), 4.03 (m, 2H), 3.73 (br dd, J = 12.3, 2.3 Hz, 2H), 3.63 (m, 2H), 3.46 (s, 3H), 2.34 (dd, J = 12.5, 10.6 Hz, 2H), 1.86 (m, 2H), 1.29 (s, 6H), 1.16 (d, J = 6.3 Hz, 6H).
5-((5-Chloro-2-(3-(trifluoromethyl)piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27a)
Method as for 20a, using 3-(trifluoromethyl)piperidine, with heating for 1 h at 170 °C. Obtained compound 27a (13 mg, 0.025 mmol, 50%) after purification by HPLC. HRMS (ESI +ve): found 513.1980, expected 513.1987 for C23H29ClF3N6O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.11 (br s, 1H), 8.00 (s, 1H), 7.49 (d, J = 1.6 Hz, 1H), 7.37 (dd, J = 8.4, 1.6 Hz, 1H), 7.05 (d, J = 8.4 Hz, 1H), 4.88 (br d, J = 12.5 Hz, 1H), 4.63 (br d, J = 13.3 Hz, 1H), 4.06–3.98 (m, 2H), 3.38 (s, 3H), 2.96–2.85 (m, 3H), 2.40 (m, 1H), 1.88–1.79 (m, 3H), 1.66–1.50 (m, 2H), 1.29 (s, 6H).
5-((5-Chloro-2-(4,4-difluoropiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27b)
Method as for 25b, using 4,4-difluoropiperidine, with heating for 1 h at 140 °C. Purification by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 90:10 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min gave compound 27b (11 mg, 0.023 mmol. 55%). HRMS (ESI +ve): found 503.1729, expected 503.1744 for C22H27ClF2N6NaO2+ [M + Na]+. δH (600 MHz, CD3OD) 7.94 (s, 1H), 7.44 (d, J = 1.9 Hz, 1H), 7.32 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.11–3.95 (m, 2H), 3.84 (br t, J = 5.9 Hz, 4H), 3.43 (s, 3H), 1.95 (tt, J = 13.8, 5.9 Hz, 4H), 1.89–1.79 (m, 2H), 1.29 (s, 6H).
5-((5-Chloro-2-(4,4-difluoro-3-methylpiperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27c)
Method as for 27b, using 4,4-difluoro-3-methylpiperidine. Obtained compound 27c (11 mg, 0.022 mmol, 52%) as hemiformic acid salt. HRMS (ESI +ve): found 495.2087, expected 495.2081 for C23H30ClF2N6O2+ [M + H]+. δH (600 MHz, CD3OD) 8.28 (s, 0.5H, formic), 7.94 (s, 1H), 7.42 (d, J = 1.9 Hz, 1H), 7.35 (dd, J = 8.4, 2.0 Hz, 1H), 7.13 (d, J = 8.4 Hz, 1H), 4.40 (br d, J = 13.6 Hz, 1H), 4.32 (br d, J = 13.4 Hz, 1H), 4.09–3.96 (m, 2H), 3.44 (s, 3H), 3.28 (ddd, J = 13.6, 11.4, 3.4 Hz, 1H)), 2.98 (dd, J = 13.4, 10.2 Hz, 1H), 2.15–1.95 (m, 2H), 1.87 (m, 2H) overlapping with 1.88–1.78 (m, 1H), 1.29 (s, 6H), 1.00 (d, J = 6.8 Hz, 3H).
5-((5-Chloro-2-(3-(hydroxymethyl)piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27d)
Method as for 27b, using piperidin-3-ylmethanol. Obtained 27d (18.5 mg, 0.039 mmol, 77%) as formic acid salt. HRMS (ESI +ve): found 475.2224, expected 475.2219 for C23H32ClN6O3+ [M + H]+. δH (600 MHz, CD3OD) 8.14 (br s, 1H), 7.89 (s, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.39 (dd, J = 8.4, 2.0 Hz, 1H), 7.10 (d, J = 8.4 Hz, 1H), 4.47 (dd, J = 13.1, 3.7 Hz, 1H), 4.32 (dt, J = 13.1, 4.2 Hz, 1H), 4.02 (m, 2H), 3.46 (dd, J = 10.9, 5.7 Hz, 1H), 3.42 (s, 3H), overlapping with 3.41 (dd, J = 10.9, 7.2 Hz, 1H), 3.01 (td, J = 13.1, 11.2, 3.1 Hz, 1H), 2.80 (dd, J = 13.1, 10.1 Hz, 1H), 1.89–1.80 (m, 3H), 1.74–1.66 (m, 2H), 1.49 (m, 1H), 1.34–1.26 (m, 1H) overlapping with 1.29 (s, 6H).
5-((5-Chloro-2-(4,4-difluoro-3-(hydroxymethyl)piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27e)
Method as for 27b, using (4,4-difluoro-3-piperidyl)methanol. Obtained 27e (13 mg, 0.025 mmol, 65%) as formic acid salt. HRMS (ESI +ve): found 511.2036, expected 511.2030 for C23H30ClF2N6O3+ [M + H]+. δH (600 MHz, CD3OD) 8.22 (s, 1H), 7.95 (s, 1H), 7.43 (d, J = 1.9 Hz, 1H), 7.41 (dd, J = 8.4, 1.9 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.50 (br d, J = 13.6 Hz, 1H), 4.26 (m, 1H), 4.03 (m, 2H), 3.88 (dd, J = 11.2, 4.1 Hz, 1H), 3.48 (dd, J = 11.2, 9.2 Hz, 1H), 3.44 (m, 1H) overlapping with 3.43 (s, 3H), 3.31 (dd, J = 13.6, 9.4 Hz, 1H), 2.13 (m, 1H), 2.01 (m, 1H), 1.96–1.86 (m, 1H) overlapping with 1.87 (m, 2H), 1.30 (s, 6H).
5-((5-Chloro-2-(4,4-difluoro-3-(methoxymethyl)piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27f)
Method as for 27b, using 4,4-difluoro-3-(methoxymethyl)piperidine. Obtained 27f (5 mg, 0.001 mmol, 22%) as formic acid salt. HRMS (ESI +ve): found 525.2171, expected 525.2187 for C24H32ClF2N6O3+ [M + H]+. δH (600 MHz, CD3OD) 8.22 (formic acid), 7.96 (s, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.36 (dd, J = 8.4, 1.9 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.56–4.46 (br d, J = 13.6 Hz, 1H), 4.39–4.29 (dm, J = 13.6 Hz, 1H), 4.05 (m, 2H), 3.66 (dd, J = 9.6, 3.6 Hz, 1H), 3.45 (s, 3H), 3.41–3.35 (m, 1H), 3.32–3.33 (m, 1H), 3.27–3.22 (m, 1H), 3.22 (s, 3H), 2.28–2.17 (m, 1H), 2.07–1.97 (m, 1H), 1.97–1.90 (m, 1H), 1.87 (dd, J = 9.1, 7.3 Hz, 2H), 1.30 (s, 6H).
1-(5-Chloro-4-((3-(3-hydroxy-3-methylbutyl)-1-methyl-2-oxo-2,3-dihydro-1H-benzo[d]imidazol-5-yl)amino)pyrimidin-2-yl)piperidine-3-carbonitrile (27g)
Method as for 27b, using piperidine-3-carbonitrile hydrochloride. Obtained 27g (20 mg, 0.04 mmol, 80%). HRMS (ESI +ve): found 470.2053, expected 470.2066 for C23H29ClN7O2+ [M + H]+. δH (600 MHz, CD3OD) 7.93 (d, J = 1.1 Hz, 1H), 7.45 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.0 Hz, 1H), 7.11 (dd, J = 8.4, 1.1 Hz, 1H), 4.06–3.99 (m, 2H), 3.93 (d, J = 5.2 Hz, 2H), 3.78–3.66 (m, 2H), 3.42 (s, 3H), 2.95–2.88 (m, 1H), 2.06–1.98 (m, 1H), 1.96–1.84 (m, 2H + 1H), 1.80–1.70 (m, 1H), 1.64–1.54 (m, 1H), 1.29 (s, 6H).
5-((5-Chloro-2-(4-(trifluoromethyl)piperidin-1-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (27h)
Method as for 27b, using 4-(trifluoromethyl)piperidine. Obtained 27h (18 mg, 0.035 mmol, 82%) as formic acid salt. HRMS (ESI +ve): found 535.1787, expected 535.1807 for C23H28ClF3N6NaO2+ [M + Na]+. δH (600 MHz, CD3OD) 8.21 (s, 1H), 7.92 (s, 1H), 7.46 (d, J = 1.9 Hz, 1H), 7.33 (dd, J = 8.4, 1.9 Hz, 1H), 7.11 (d, J = 8.4 Hz, 1H), 4.68 (dt, J = 13.5, 2.8 Hz, 2H), 4.01 (m, 2H), 3.43 (s, 3H), 2.85 (td, J = 13.5, 12.7, 2.6 Hz, 2H), 2.42 (m (tqt), 1H), 1.95–1.77 (m, 4H), 1.46 (qd, J = 12.6, 4.4 Hz, 2H), 1.29 (s, 6H).
5-((5-Chloro-2-(8,8-difluoro-3-azabicyclo[3.2.1]octan-3-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (28a)
Method as for 25b, using 8,8-difluoro-3-azabicyclo[3.2.1]octane hydrochloride. Purification by HPLC (ACE 5 C18-PFP 250 mm × 21.2 mm column; 15 min gradient of 90:10 to 0:100 water:methanol, 0.1% formic acid modifier) at a flow rate of 20 mL/min gave compound 28a (9.5 mg, 0.019 mmol, 45%) as hemiformic acid salt. HRMS (ESI +ve): found 507.2073, expected 507.2081 for C24H30F2N6O2Cl+ [M + H]+. δH (600 MHz, CD3OD) δ 8.19 (s, 0.5H, formic), 7.94 (s, 1H), 7.47 (d, J = 2.0 Hz, 1H), 7.33 (dd, J = 8.4, 2.0 Hz, 1H), 7.12 (d, J = 8.4 Hz, 1H), 4.41 (br d, J = 13.4 Hz, 2H), 4.02 (m, 2H), 3.43 (s, 3H), 3.23 (br d, J = 12.9 Hz, 2H), 2.28 (m, 2H), 1.90–1.83 (m, 4H), 1.65–1.53 (m, 2H), 1.29 (s, 6H).
5-((5-Chloro-2-(3,3-difluoro-8-azabicyclo[3.2.1]octan-8-yl)pyrimidin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (28b)
Method as for 27b, using 3,3-difluoro-8-azabicyclo[3.2.1]octane hydrochloride and heating in the microwave at 140 °C for 8 h. Obtained compound 28b (12.5 mg, 0.025 mmol, 59%) as a cream solid. HRMS (ESI +ve): found 507.2087, expected 507.2081 for C24H30ClF2N6O2+ [M + H]+. δH (600 MHz, DMF-d7) 8.84 (s, 1H), 8.14 (s, 1H), 7.66 (d, J = 1.9 Hz, 1H), 7.47 (dd, J = 8.4, 2.0 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 4.71 (br s, 2H), 4.52 (s, 1H), 4.04 (m, 2H), 3.42 (s, 3H), 2.33–2.12 (m, 4H), 2.09–1.93 (m, 4H), 1.86 (m, 2H), 1.26 (s, 6H).
5-((2-Bromo-5-chloropyridin-4-yl)amino)-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (29)
A mixture of 5-amino-3-(3-hydroxy-3-methylbutyl)-1-methyl-1,3-dihydro-2H-benzo[d]imidazol-2-one (16) (20 mg, 0.08 mmol), 2,4-dibromo-5-chloropyridine (25 mg, 0.092 mmol), and DIPEA (16 mg, 0.12 mmol) in NMP (0.5 mL) was heated in the microwave to 180 °C for 1 h, then diluted with water, acidified to pH 5 by addition of 10% citric acid, then extracted with DCM (5 mL × 3). Combined organics were evaporated under reduced pressure and purified by reverse phase column chromatography (12 g C18 column, 30–80% MeOH in water, both modified with 0.1% formic acid) to give 29 (9 mg, 0.019 mmol, 24%). HRMS (ESI +ve): found 439.0512, expected 439.0531 for C18H21BrClN4O2+ [M + H]+. δH (500 MHz, acetone-d6) 8.08 (s, 1H), 7.92 (br, 0.6H, partly exchanged NH), 7.17 (d, J = 8.4 Hz, 1H), 7.15 (m, 1H), 7.08 (dt, J = 8.4, 1.7 Hz, 1H), 6.83 (d, J = 4.3 Hz, 1H), 4.04 (m, 2H), 3.42 (s, 3H), 1.86 (m, 2H), 1.26 (s, 6H).
5-Chloro-2-((3S,5R)-3,5-dimethylpiperidin-1-yl)-4-iodopyridine (30a)
A solution of cis-3,5-dimethylpiperidine (0.23 g, 2.0 mmol), 5-chloro-2-fluoro-4-iodopyridine (0.53 g, 2.0 mmol), and DIPEA (0.53 mL, 3.1 mmol) in THF (8 mL) was heated in a sealed vial to 100 °C for 16 h. When cooled, water was added to the THF solution and extracted with EtOAc. The combined organic layers were washed with water twice and dried with sodium sulfate. Flash column chromatography (4% ethyl acetate in cyclohexane) gave 30a (488 mg, 1.39 mmol, 68%) as a white solid. δH (500 MHz, DMSO-d6) 8.08 (s, 1H), 7.40 (s, 1H), 4.36–4.10 (m, 2H), 2.25 (dd, J = 12.9, 11.4 Hz, 2H), 1.75 (dtd, J = 10.6, 3.6, 1.9 Hz, 1H), 1.62–1.46 (m, 2H), 0.88 (d, J = 6.6 Hz, 6H), 0.84–0.70 (m, 1H).
(2S,6R)-4-(5-Chloro-4-iodopyridin-2-yl)-2,6-dimethylmorpholine (30b)
Prepared as for 30a, using (2S,6R)-2,6-dimethylmorpholine, with heating for 3 days. Flash column chromatography (10% ethyl acetate in cyclohexane) gave 30b (54 mg, 0.15 mmol, 39%) as a white waxy solid. δH (500 MHz, DMSO-d6) 8.13 (s, 1H), 7.43 (s, 1H), 4.22–4.06 (m, 2H), 3.66–3.50 (m, 2H), 2.39 (dd, J = 12.8, 10.5 Hz, 2H), 1.14 (d, J = 6.1 Hz, 6H).
Acknowledgments
We thank Dr. Nora Cronin from the Division of Structural Biology at the Institute of Cancer Research and the staff of Diamond Light Source and the European Synchrotron Radiation Facility for their support during X-ray crystallography data collection. We also thank Meirion Richards and Dr. Amin Mirza of the Structural Chemistry team for their expertise and assistance with LC, NMR, and mass spectrometry, and Dr. Matthew Cheeseman for input concerning statistical analysis of data.
Glossary
Abbreviations Used
- APCI
atmospheric pressure chemical ionization
- BCL6
B-cell lymphoma 6 protein
- BCOR
BCL-6 co-repressor protein
- BRET
bioluminescence resonance energy transfer
- BTB
broad-complex, tramtrack, and bric a brac (domain), also known as POZ (poxvirus and zinc finger) domain
- CL
clearance
- CLint
intrinsic clearance
- cmax
maximum (peak) concentration achieved after a single dose
- cPr
cyclopropyl
- DC50
concentration of compound at which 50% of the maximally observed protein degradation (Dmax) is achieved
- DIPEA
N,N-diisopropylethylamine
- DLBCL
diffuse large B-cell lymphoma
- Dmax
maximal percentage degradation of protein achieved
- EtOAc
ethyl acetate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HAC
heavy atom count
- IMDM
Iscove’s modified Dulbecco’s medium
- IMiD
immunomodulatory imide drug, such as thalidomide and its analogues
- MeCN
acetonitrile
- MLM
mouse liver microsome
- MSD
meso scale discovery, an assay method similar to ELISA, using electrochemiluminescence as a detection technique
- NanoBRET
a BRET-based assay format commercialized by Promega that uses NanoLuc luciferase to generate the donor signal and a HaloTag ligand as the fluorescent energy acceptor
- NCoR
nuclear receptor co-repressor 1
- PFP
pentafluorophenyl
- PPI
protein–protein interaction
- PROTAC
proteolysis targeting chimera
- QToF
quadrupole time-of-flight
- SCID
severe combined immunodeficient (mouse model)
- SCX-2
SCX (strong cation exchange)-2, which is a propylsulfonic acid bonded sorbent
- Si-DMT
silica bound equivalent of 2,4,6-trimercaptotriazine
- SMRT
silencing mediator for retinoid or thyroid hormone receptor (also known as nuclear receptor co-repressor 2 or NCoR2)
- SNAr
nucleophilic aromatic substitution
- sol.
solubility
- STR
short tandem repeat (analysis)
- tmax
time after dosing at which the maximum (peak) concentration cmax is observed
- TR-FRET
time-resolved fluorescence energy transfer
- Xantphos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b02076.
Supplementary experimental details on protein synthesis and purification, assay conditions, PK and PD methods; analytical data for compound 1; tables listing statistics and replicate values and crystallographic data; figures showing binding modes, cell proliferation assay data, PD effects, and PK/PD relationship (PDF)
Molecular formula strings and some data (CSV)
Accession Codes
Atomic coordinates and structure factors for the crystal structures of BCL6 with compounds 1, 4, 5, 6, 11a, 11f, 17a, 23d, 25a, and 25b can be accessed using PDB codes 6TOM, 6TOF, 6TOG, 6TOH, 6TOO, 6TOI, 6TOJ, 6TOK, 6TOL, and 6TON respectively. Authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by Cancer Research UK (Grant C309/A11566), CRT Pioneer Fund and Sixth Element Capital, who we thank for their generous funding. We also acknowledge NHS funding to the NIHR Biomedical Research Centre.
The authors declare no competing financial interest.
Supplementary Material
References
- Hatzi K.; Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol. Med. 2014, 20 (6), 343–352. 10.1016/j.molmed.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K.; Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunological Reviews 2012, 247 (1), 172–183. 10.1111/j.1600-065X.2012.01112.x. [DOI] [PubMed] [Google Scholar]
- Jardin F.; Ruminy P.; Bastard C.; Tilly H. The BCL6 proto-oncogene: a leading role during germinal center development and lymphomagenesis. Pathol. Biol. 2007, 55 (1), 73–83. 10.1016/j.patbio.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. The Hallmarks of Cancer. Cell 2000, 100 (1), 57–70. 10.1016/S0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144 (5), 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Polo J. M.; Dell’Oso T.; Ranuncolo S. M.; Cerchietti L.; Beck D.; Da Silva G. F.; Prive G. G.; Licht J. D.; Melnick A. Specific peptide interference reveals BCL6 transcriptional and oncogenic mechanisms in B-cell lymphoma cells. Nat. Med. 2004, 10 (12), 1329–1335. 10.1038/nm1134. [DOI] [PubMed] [Google Scholar]
- Ghetu A. F.; Corcoran C. M.; Cerchietti L.; Bardwell V. J.; Melnick A.; Privé G. G. Structure of a BCOR corepressor peptide in complex with the BCL6 BTB domain dimer. Mol. Cell 2008, 29 (3), 384–391. 10.1016/j.molcel.2007.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti L. C.; Ghetu A. F.; Zhu X.; Da Silva G. F.; Zhong S.; Matthews M.; Bunting K. L.; Polo J. M.; Farès C.; Arrowsmith C. H.; Yang S. N.; Garcia M.; Coop A.; MacKerell A. D. Jr; Privé G. G.; Melnick A. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010, 17 (4), 400–411. 10.1016/j.ccr.2009.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerchietti L. C.; Yang S. N.; Shaknovich R.; Hatzi K.; Polo J. M.; Chadburn A.; Dowdy S. F.; Melnick A. A peptomimetic inhibitor of BCL6 with potent antilymphoma effects in vitro and in vivo. Blood 2009, 113 (15), 3397–3405. 10.1182/blood-2008-07-168773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas M. G.; Yu W.; Beguelin W.; Teater M. R.; Geng H.; Goldstein R. L.; Oswald E.; Hatzi K.; Yang S.-N.; Cohen J.; Shaknovich R.; Vanommeslaeghe K.; Cheng H.; Liang D.; Cho H. J.; Abbott J.; Tam W.; Du W.; Leonard J. P.; Elemento O.; Cerchietti L.; Cierpicki T.; Xue F.; MacKerell A. D. Jr.; Melnick A. M. Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J. Clin. Invest. 2016, 126 (9), 3351–3362. 10.1172/JCI85795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y.; Sakai N.; Sogabe S.; Ida K.; Oki H.; Sakamoto K.; Lane W.; Snell G.; Iida M.; Imaeda Y.; Sakamoto J.; Matsui J. Discovery of a B-cell lymphoma 6 protein–protein interaction inhibitor by a biophysics-driven fragment-based approach. J. Med. Chem. 2017, 60 (10), 4358–4368. 10.1021/acs.jmedchem.7b00313. [DOI] [PubMed] [Google Scholar]
- Yasui T.; Yamamoto T.; Sakai N.; Asano K.; Takai T.; Yoshitomi Y.; Davis M.; Takagi T.; Sakamoto K.; Sogabe S.; Kamada Y.; Lane W.; Snell G.; Iwata M.; Goto M.; Inooka H.; Sakamoto J.-i.; Nakada Y.; Imaeda Y. Discovery of a novel B-cell lymphoma 6 (BCL6)–corepressor interaction inhibitor by utilizing structure-based drug design. Bioorg. Med. Chem. 2017, 25 (17), 4876–4886. 10.1016/j.bmc.2017.07.037. [DOI] [PubMed] [Google Scholar]
- McCoull W.; Abrams R. D.; Anderson E.; Blades K.; Barton P.; Box M.; Burgess J.; Byth K.; Cao Q.; Chuaqui C.; Carbajo R. J.; Cheung T.; Code E.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gangl E.; Gao N.; Grist M.; Hargreaves D.; Howard M. R.; Hu J.; Kemmitt P. D.; Nelson J. E.; O’Connell N.; Prince D. B.; Raubo P.; Rawlins P. B.; Robb G. R.; Shi J.; Waring M. J.; Whittaker D.; Wylot M.; Zhu X. Discovery of pyrazolo[1,5-a]pyrimidine B-cell lymphoma 6 (BCL6) binders and optimization to high affinity macrocyclic inhibitors. J. Med. Chem. 2017, 60 (10), 4386–4402. 10.1021/acs.jmedchem.7b00359. [DOI] [PubMed] [Google Scholar]
- McCoull W.; Cheung T.; Anderson E.; Barton P.; Burgess J.; Byth K.; Cao Q.; Castaldi M. P.; Chen H.; Chiarparin E.; Carbajo R. J.; Code E.; Cowan S.; Davey P. R.; Ferguson A. D.; Fillery S.; Fuller N. O.; Gao N.; Hargreaves D.; Howard M. R.; Hu J.; Kawatkar A.; Kemmitt P. D.; Leo E.; Molina D. M.; O’Connell N.; Petteruti P.; Rasmusson T.; Raubo P.; Rawlins P. B.; Ricchiuto P.; Robb G. R.; Schenone M.; Waring M. J.; Zinda M.; Fawell S.; Wilson D. M. Development of a novel B-cell lymphoma 6 (BCL6) PROTAC to provide insight into small molecule targeting of BCL6. ACS Chem. Biol. 2018, 13 (11), 3131–3141. 10.1021/acschembio.8b00698. [DOI] [PubMed] [Google Scholar]
- Kerres N.; Steurer S.; Schlager S.; Bader G.; Berger H.; Caligiuri M.; Dank C.; Engen J. R.; Ettmayer P.; Fischerauer B.; Flotzinger G.; Gerlach D.; Gerstberger T.; Gmaschitz T.; Greb P.; Han B.; Heyes E.; Iacob R. E.; Kessler D.; Kölle H.; Lamarre L.; Lancia D. R.; Lucas S.; Mayer M.; Mayr K.; Mischerikow N.; Mück K.; Peinsipp C.; Petermann O.; Reiser U.; Rudolph D.; Rumpel K.; Salomon C.; Scharn D.; Schnitzer R.; Schrenk A.; Schweifer N.; Thompson D.; Traxler E.; Varecka R.; Voss T.; Weiss-Puxbaum A.; Winkler S.; Zheng X.; Zoephel A.; Kraut N.; McConnell D.; Pearson M.; Koegl M. Chemically induced degradation of the oncogenic transcription factor BCL6. Cell Rep. 2017, 20 (12), 2860–2875. 10.1016/j.celrep.2017.08.081. [DOI] [PubMed] [Google Scholar]
- Pierrat O. A.; Liu M.; Rodrigues M.J.; Collie G. W.; Le Bihan Y-V; Shetty K. N.; McAndrew P. C.; Richards M.; Stubbs M.; Rowlands M. G.; Yahya N.; Pickard L.; Shehu E.; Talbot R.; Bellenie B. R.; Davis O. A.; Varela A.; Cheung K-M. J.; Drouin L.; Innocenti P.; Woodward H.; Lloyd M. G.; Huckvale R.; Broccatelli F.; Carter M.; Galiwango D.; Hayes A.; Raynaud F. I.; Bryant C.; Whittaker S.; Hoelder S.; Burke R.; van Montfort R. L. M. Unpublished results.
- Hill A. P.; Young R. J. Getting physical in drug discovery: a contemporary perspective on solubility and hydrophobicity. Drug Discovery Today 2010, 15 (15–16), 648–655. 10.1016/j.drudis.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Bissantz C.; Kuhn B.; Stahl M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53 (14), 5061–5084. 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnarchuk M. S. Water, water, everywhere...It’s time to stop and think. Drug Discovery Today 2016, 21 (7), 1139–1146. 10.1016/j.drudis.2016.05.009. [DOI] [PubMed] [Google Scholar]
- Lai A. C.; Crews C. M. Induced protein degradation: an emerging drug discovery paradigm. Nat. Rev. Drug Discovery 2017, 16, 101. 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccatelli F.; Hop C. E. C. A.; Wright M. Strategies to optimize drug half-life in lead candidate identification. Expert Opin. Drug Discovery 2019, 14 (3), 221–230. 10.1080/17460441.2019.1569625. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Kuai Y.; Lei L.; Weng Y.; Berberich-Siebelt F.; Zhang X.; Wang J.; Zhou Y.; Jiang X.; Ren G.; Pan H.; Mao Z.; Zhou R. The feedback loop of LITAF and BCL6 is involved in regulating apoptosis in B cell non-Hodgkin’s-lymphoma. Oncotarget 2016, 7 (47), 77444–77456. 10.18632/oncotarget.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Dixit V. M. Drugging the undruggables: exploring the ubiquitin system for drug development. Cell Res. 2016, 26 (4), 484–498. 10.1038/cr.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins I.; Wang H.; Caldwell J. J.; Chopra R. Chemical approaches to targeted protein degradation through modulation of the ubiquitin–proteasome pathway. Biochem. J. 2017, 474 (7), 1127–1147. 10.1042/BCJ20160762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Mares A.; Smith I. E. D.; Ko E.; Campos S.; Miah A. H.; Mulholland K. E.; Routly N.; Buckley D. L.; Gustafson J. L.; Zinn N.; Grandi P.; Shimamura S.; Bergamini G.; Faelth-Savitski M.; Bantscheff M.; Cox C.; Gordon D. A.; Willard R. R.; Flanagan J. J.; Casillas L. N.; Votta B. J.; den Besten W.; Famm K.; Kruidenier L.; Carter P. S.; Harling J. D.; Churcher I.; Crews C. M. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11 (8), 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson J. L.; Neklesa T. K.; Cox C. S.; Roth A. G.; Buckley D. L.; Tae H. S.; Sundberg T. B.; Stagg D. B.; Hines J.; McDonnell D. P.; Norris J. D.; Crews C. M. Small-molecule-mediated degradation of the androgen receptor through hydrophobic tagging. Angew. Chem., Int. Ed. 2015, 54 (33), 9659–9662. 10.1002/anie.201503720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.