

Abstract
Congenital heart disease (CHD) survivors are at risk for neurodevelopmental disability (NDD), and recent studies identify genes associated with both disorders, suggesting that NDD in CHD survivors may be of genetic origin. Genes contributing to neurogenesis, dendritic development and synaptogenesis organize neural elements into networks known as the connectome. We hypothesized that NDD in CHD may be attributable to genes altering both neural connectivity and cardiac patterning. To assess the contribution of de novo variants (DNVs) in connectome genes, we annotated 229 published NDD genes for connectome status and analyzed data from 3,684 CHD subjects and 1,789 controls for connectome gene mutations. CHD cases had more protein truncating and deleterious missense DNVs among connectome genes compared to controls (OR = 5.08, 95%CI:2.81–9.20, Fisher’s exact test P = 6.30E-11). When removing three known syndromic CHD genes, the findings remained significant (OR = 3.69, 95%CI:2.02–6.73, Fisher’s exact test P = 1.06E-06). In CHD subjects, the top 12 NDD genes with damaging DNVs that met statistical significance after Bonferroni correction (PTPN11, CHD7, CHD4, KMT2A, NOTCH1, ADNP, SMAD2, KDM5B, NSD2, FOXP1, MED13L, DYRK1A; one-tailed binomial test P ≤ 4.08E-05) contributed to the connectome. These data suggest that NDD in CHD patients may be attributable to genes that alter both cardiac patterning and the connectome.
Subject terms: Genetics, Paediatric research
Introduction
Congenital heart disease (CHD) affects nearly 1% of all births in the United States each year1,2, and 20–30% of subjects with CHD have extra-cardiac abnormalities or neuro-developmental disability (NDD)3–6. Approximately 1/3rd of these infants will be diagnosed with a severe CHD, or those that require intervention within the first postnatal year, and the incidence of NDD increases with the severity of the CHD4. Thus, approximately 20% of children with a mild CHD will have a NDD, while more than 50% of children with severe CHD experience NDDs ranging from developmental delay and intellectual disability to autism spectrum disorder and attention-deficit/hyperactivity disorder (ADHD)7–15. Further, although neonates with CHD are at risk for hypoxemia in the pre- and postnatal periods5,16–22 and as many as 1/3rd of cases have a genetic syndrome associated with their CHD23–25, currently identified risk factors explain less than half of neurodevelopmental outcomes in infants with severe CHD5,26–31. Therefore, as genomic data for CHD is becoming more and more available, we sought to explore a genetic basis for NDD in patients with CHD.
“Neural connectivity is the intermediate between molecules and behavior”32, and NDDs ranging from developmental delay, intellectual disability and autism to ADHD, depression and schizophrenia have been attributed to alterations in the microstructural and functional networks of the developing brain, or the “connectome”32,33. When compared to typically developing controls, magnetic resonance imaging (MRI) studies in over 1/3rd of fetuses with isolated CHD show reduced brain volumes and delayed cerebral maturation34,35. Similarly, neural connectivity data demonstrate alterations that mediate behavior in children and adolescents with CHD36–38, suggesting that disruption of the connectome perturbs neurocognition in this vulnerable population. Since alterations in neural networks are present in neonates with CHD prior to surgery39, these data suggest that changes in neural connectivity could be secondary to mutations in the genome rather than solely due to hemodynamic factors, and recent genomic studies have identified candidate genes common to children with CHD and those with NDD, suggesting a genetic basis for CHD-associated NDD23,24,40,41. In these subjects, genetic substitutions affect genes involved in chromatin modification, morphogenesis and transcriptional regulation of neuronal tissues as well as in the heart. De novo variants (DNVs) in certain chromatin modifiers, important for fetal brain development42–45, account for 14/35 (40%) of variants of patients with both CHD and autism spectrum disorder40.
As the adult CHD population is significantly expanding and the need to understand the many causes of NDD in these patients is urgently increasing46, we tested the hypothesis that NDD in severe CHD patients, or those at highest risk for NDDs, may be attributable to genes that alter both neural connectivity and cardiac patterning. To assess the contribution of DNVs in connectome genes we annotated 229 published NDD genes for connectome status47 and analyzed genomic data from 3,684 subjects with CHD and 1,789 controls for connectome gene mutations40,48. Genes contributing to the connectome were defined as those necessary for the development, growth and maintenance of neural networks in the developing brain and included those subserving neurogenesis, axonal migration, dendritic development and/or synaptogenesis (Fig. 1)49–51. Our second aim was to test the hypothesis that DNVs in chromatin modifiers contributing to the connectome occur more commonly in subjects with CHD than in control subjects. We defined chromatin modifiers as genes that alter DNA or protein in chromatin by the covalent addition or removal of chemical groups52.
Figure 1.

Characterization of the 229 Neurodevelopmental Disorder (NDD) genes. Review of the literature demonstrated that 129 of the published 229 NDD genes contributed to the connectome, 13 were chromatin modifiers, and 30 were chromatin modifiers that contributed to the connectome. Fifty-seven were neither chromatin modifiers nor connectome genes.
Results
To assess the contribution of DNVs within CHD to the developing connectome, we studied 3,684 unique, previously published, proband/parent trios with CHD and exome sequencing data. The CHD trios consist of affected probands and their two unaffected parents. These included 1039 probands as part of the multi-center/multi-study cohort from the Deciphering Developmental Disorders Project (“DDD Plus Study”)48 and 2,645 probands from the Pediatric Cardiac Genomics Consortium (PCGC) study (see Supplementary Table S1 for comparison of the study cohorts)40. Three hundred twenty six probands in the DDD Plus Study were PCGC cases, and these have been omitted from the DDD Plus cohort to avoid subject duplication. Participants in these studies were selected for structural CHD (excluding prematurity-associated patent ductus arteriosus). Patients with known chromosome abnormalities were the only patients excluded from both cohorts. Controls included 1,789 previously analyzed families that included one offspring with autism, one unaffected sibling and unaffected parents; only data from the unaffected siblings and parents were analyzed as controls in this study40. Two hundred twenty nine previously published NDD genes were individually annotated with respect to connectome and chromatin modifier status by two independent reviewers (Supplementary Table S2)47. PubMed search terms included connectome, neural connectivity, neural network, neuron, neurogenesis, axon, growth cone, dendrite, synapse, synaptogenesis, oligodendroglia, myelinogenesis, chromatin modifier, chromatin and methylation, acetylation and/or ubiquitination. Connectome genes represent a subset of the NDD genes; of the 229 NDD genes (Fig. 1), 159 fulfilled our definition for contribution to the developing connectome, including 30 genes that are also chromatin modifiers.
Analysis of DNVs of NDD genes in CHD cases and controls
For all analyses, we first report data for the DDD Plus Study (N = 1039) and the PCGC study (N = 2645) independently followed by those for the combined CHD cohort (N = 3684).
As shown in Table 1, we identified the frequency of DNVs in the 229 NDD genes occurring in both the individual CHD populations and the combined CHD cohort, and compared to those found in the control group using Fisher’s exact tests. Results were summarized using odds ratios and 95% Confidence Intervals (95% CI). Single nucleotide variants and small indels were classified into distinct categories: protein truncating (PT, i.e., nonsense, frameshift, splice-site variants), missense variants (MIS; including small in-frame insertions or deletions), and synonymous variants. All missense variants predicted by MetaSVM as deleterious were classified as D-mis variants40. Protein damaging variants refer PT or D-mis DNVs.
Table 1.
De novo variants of 229 NDD genes identified from CHD individuals of DDD Plus Study and PCGC cohorts.
| Variant Category | DDD Plus Study-Sifrim 2016 (N = 1039) | PCGC-Jin 2017 (N = 2645) | Combined CHD (N = 3684) | Controls (N = 1789) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | |
| Total | 94 | 2.80 | 2.00, 3.93 | 1.50E-09 | 163 | 1.79 | 1.32, 2.44 | 1.52E-04 | 257 | 2.07 | 1.54, 2.77 | 2.96E-07 | 57 |
| PT | 44 | 10.48 | 3.76, 18.31 | 1.33E-12 | 62 | 5.51 | 2.52, 12.07 | 2.60E-07 | 106 | 6.87 | 3.19, 14.78 | 1.72E-10 | 7 |
| Missense | 44 | 1.90 | 1.02, 2.38 | 0.0054 | 75 | 1.21 | 0.82, 1.80 | 0.38 | 119 | 1.40 | 0.97, 2.02 | 0.079 | 38 |
| D-mis | 23 | 3.13 | 1.19, 4.72 | 0.0013 | 33 | 1.69 | 0.87, 3.28 | 0.12 | 56 | 2.08 | 1.11, 3.90 | 0.017 | 12 |
| PT or D-mis | 67 | 5.97 | 2.79, 7.67 | 4.81E-14 | 95 | 3.13 | 1.90, 5.14 | 7.10E-07 | 162 | 3.89 | 2.41, 6.28 | 7.80E-11 | 19 |
| Non D-mis | 21 | 1.31 | 0.68, 2.01 | 0.37 | 42 | 0.99 | 0.60, 1.62 | 1.0 | 63 | 1.08 | 0.68, 1.70 | 0.82 | 26 |
| SYN | 6 | 0.80 | 0.25, 1.61 | 0.81 | 26 | 1.33 | 0.67, 2.64 | 0.50 | 32 | 1.18 | 0.61, 2.30 | 0.74 | 12 |
Compared to the controls, although there was a higher frequency of DNVs in the 229 NDD genes in both the DDD Plus Study and the PCGC cohort (OR = 2.80, 95%CI:2.00–3.93, Fisher’s exact test P = 1.50E-09 and OR = 1.79, 95%CI:1.32–2.44, Fisher’s exact test P = 1.52E-04, respectively), this excess was largely attributable to DNVs in PT (OR = 10.48, 95%CI:3.76–18.31, Fisher’s exact test P = 1.33E-12 (DDD Plus); OR = 5.51, 95%CI:2.52–12.07, Fisher’s exact test P = 2.60E-07 (PCGC)) or protein damaging DNVs (OR = 5.97, 95%:2.79–7.67, Fisher’s exact test P = 4.81E-14 (DDD Plus); OR = 3.13, 95%CI:1.90–5.14, Fisher’s exact test P = 7.10E-07 (PCGC)), as shown in Table 1. In contrast, we did not see any significant changes in synonymous or non-D-mis DNVs in the NDD genes between the CHD cases and controls in either of the separate primary studies or the analysis with combined data. Among the 3684 CHD subjects, 162 were carriers of PT or D-mis DNVs in NDD genes, which accounted for 4.3% of individuals in this cohort, including 3 individuals with 2 PT or D-mis variants in two different genes. In addition, the enrichment of gene burden of PT DNVs in the NDD genes was observed when compared to the whole exomes in the DDD Plus or PCGC primary study cohorts (OR = 10.48, 95%CI:3.76–18.31 or OR = 5.51, 95%CI:2.52–12.07 vs OR = 1.89, 95%CI:1.49–2.39 or OR = 1.60, 95%CI:1.31–1.95, respectively; Table 1 and Supplementary Table S1). The excess of protein damaging DNVs were aggregated in genes contributing to both heart development and NDD in this analysis, further confirming previous data from the PCGC study40.
Analysis of DNVs of connectome genes in CHD cases and controls
Of the 229 NDD genes (Fig. 1), 159 fulfilled our definition for contribution to the developing connectome (i.e., genes contributing to neurogenesis, axonal migration, dendritic development, myelinogenesis and/or synaptogenesis). We next compared the DNVs in these 159 connectome genes occurring in the CHD population to those found in the control group, using the same statistical methodology as above, as shown in Table 2. We identified 134 PT or D-mis DNVs derived from 47 connectome genes. Similar to the NDD data above, there were more protein damaging variants in connectome genes in either the DDD Plus Study (OR = 8.14, 95%CI:4.35–15.2, Fisher’s exact test P = 1.38E-14) or the PCGC cohort (OR = 3.95, 95%CI:2.14–7.28, Fisher’s exact test P = 5.99E-07) compared to controls. The odd ratios became even more striking for DDD Plus Study (OR = 14.93, 95%CI:5.30–42.0) and PCGC Study (OR = 7.13, 95%CI:2.56–19.8), respectively, if only PT DNVs were included.
Table 2.
De novo variants of connectome genes identified from the two CHD cohorts.
| Variant Category | DDD Plus Study-Sifrim 2016 (N = 1039) | PCGC-Jin 2017 (N = 2645) | Combined CHD (N = 3684) | Controls (N = 1789) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | |
| Total | 83 | 3.27 | 2.25, 4.77 | 2.25E-10 | 123 | 1.78 | 1.25, 2.54 | 1.08E-03 | 206 | 2.18 | 1.56, 3.05 | 1.07E-06 | 43 |
| PT | 36 | 14.93 | 5.30, 42.0 | 1.06E-11 | 46 | 7.13 | 2.56, 19.8 | 2.41E-06 | 82 | 9.25 | 3.39, 25.3 | 2.13E-09 | 4 |
| Missense | 42 | 2.56 | 1.57, 4.18 | 1.42E-04 | 55 | 1.25 | 0.79, 1.99 | 0.36 | 97 | 1.61 | 1.04, 2.47 | 0.03 | 27 |
| D-mis | 22 | 4.49 | 1.99, 10.1 | 1.07E-04 | 30 | 2.31 | 1.05, 5.04 | 0.042 | 52 | 2.91 | 1.11, 3.90 | 2.79E-03 | 8 |
| PT or D-mis | 58 | 8.14 | 4.35, 15.2 | 1.38E-14 | 76 | 3.95 | 2.14, 7.28 | 5.99E-07 | 134 | 5.08 | 2.81, 9.20 | 6.30E-11 | 12 |
| Non D-mis | 20 | 1.71 | 0.91, 3.21 | 0.10 | 25 | 0.80 | 0.44, 1.46 | 0.53 | 45 | 1.05 | 0.61, 1.80 | 1.0 | 19 |
| SYN | 5 | 0.67 | 0.24, 1.90 | 0.62 | 22 | 1.12 | 0.54, 2.27 | 0.86 | 27 | 1.00 | 0.51, 1.97 | 1.0 | 12 |
Similarly when the cohorts were combined, CHD subjects had a higher frequency of protein damaging DNVs among connectome genes than control subjects (OR = 5.08, 95%CI:2.81–9.20, Fisher’s exact test P = 6.30E-11, in which the excess was largely contributed by PT DNVs (OR = 9.25, 95%CI:3.39–25.3, Fisher’s exact test P = 2.13E-09 Table 2). Cases also had a higher fraction of total DNVs contributing to the connectome when compared to control subjects (OR = 2.18, 95% CI:1.56–3.05, Fisher’s exact test P = 1.07E-06). In contrast, we did not see any significant differences for synonymous or non-Dmis DNVs in the connectome genes between the CHD cases and controls.
Analysis of Chromatin Modifier DNVs in NDD genes in CHD cases and controls
Chromatin modifiers have been reported to play a significant role in neural development, and we identified 60 DNVs from 18 NDD genes described as chromatin modifiers. As shown in Table 3, there were more PT DNVs in chromatin modifier genes in the DDD Plus Study (OR = 15.53, 95%CI:3.61–66.8, Fisher’s exact test P = 9.53E-07) or the PCGC cohort (OR = 7.71, 95%CI:1.82–32.6, Fisher’s exact test P = 5.10E-04), compared to controls. There were also more protein damaging DNVs in chromatin modifier genes in the DDD Plus Study (OR = 6.33, 95%CI:2.75–14.6, Fisher’s exact test P = 1.13E-06) and the PCGC subjects (OR = 2.91, 95%CI:1.28–6.58, Fisher’s exact test P = 8.04E-03), respectively.
Table 3.
De novo variants of chromatin modifier genes identified from the two CHD cohorts.
| Variant Category | DDD Plus Study-Sifrim 2016 (N = 1039) | PCGC-Jin 2017 (N = 2645) | Combined CHD (N = 3684) | Controls (N = 1789) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | OR | 95% CI | p-value | Allele Count | |
| Total | 32 | 3.28 | 2.79, 6.01 | 7.15E-05 | 49 | 1.89 | 1.07, 3.33 | 0.028 | 81 | 2.27 | 1.32, 3.89 | 0.0017 | 16 |
| PT | 19 | 15.53 | 3.61, 66.8 | 9.53E-07 | 25 | 7.71 | 1.82, 32.6 | 5.10E-04 | 44 | 9.85 | 2.38, 40.7 | 1.55E-05 | 2 |
| Missense | 12 | 1.94 | 0.84, 4.51 | 0.13 | 17 | 1.04 | 0.47, 2.28 | 1.0 | 29 | 1.29 | 0.63, 2.65 | 0.60 | 10 |
| D-mis | 8 | 2.59 | 0.84, 7.92 | 0.094 | 8 | 0.98 | 0.32, 3.00 | 1.0 | 16 | 1.42 | 0.52, 3.88 | 0.64 | 5 |
| PT or D-mis | 27 | 6.33 | 2.75, 14.6 | 1.13E-06 | 33 | 2.91 | 1.28, 6.58 | 8.04E-03 | 60 | 3.84 | 1.75, 8.42 | 1.44E-04 | 7 |
| Non D-mis | 4 | 1.29 | 0.34, 4.81 | 0.74 | 9 | 1.10 | 0.37, 3.29 | 1.0 | 13 | 1.15 | 0.41, 3.24 | 1.0 | 5 |
| SYN | 0 | 0.18 | 0.01, 3.32 | 0.30 | 7 | 1.07 | 0.31, 3.66 | 1.0 | 7 | 0.77 | 0.23, 2.65 | 0.74 | 4 |
Comparison of the number of DNVs in chromatin modifier genes occurring in the NDD genes in the combined cohort of CHD cases and controls demonstrated an excess of protein damaging DNVs (OR = 3.84, 95%CI: 1.75–8.42, Fisher’s exact test P = 1.44E-04) in CHD subjects with greater contribution from PT DNVs (OR = 9.85, 95%CI:2.38–40.7, Fisher’s exact test P = 1.55E-05; Table 3). Of note, we did not observe any significant differences between the CHD cases and controls with respect to synonymous or non-Dmis DNVs of chromatin modifier genes.
Identification of 12 NDD candidate genes with higher DNV burden in the CHD population
To identify a subset of NDD genes in which damaging DNVs are over-represented in the CHD cohorts, we implemented a one-tailed binomial test to quantify the enrichment of protein damaging DNVs in 229 NDD genes in only CHD cases from the DDD Plus study and PCGC study. This method calculated the exact probability of the observed data under a binomial distribution with each gene mutation rate as a specified probability parameter53,54. The observed top 38 enriched NDD genes with protein damaging DNVs more than expected (ranked by one-tailed binomial test p-value, P < 0.05) are shown in Table 4.
Table 4.
Top NDD Genes (adjusted by mutation rate, one-tailed binomial test p < 0.05) with de novo PT or D-mis variants from the two CHD cohorts.
| Gene | Chromatin Modifiers | Connectome | Mutation Rate | Size | DDD-Plus | PCGC | Observed | Expected | P-value |
|---|---|---|---|---|---|---|---|---|---|
| PTPN11 | − | + | 2.58E-05 | 1801 | 7 | 9 | 16 | 0.80 | 1.54E-34 |
| CHD7 | + | + | 1.19E-04 | 8909 | 3 | 14 | 17 | 3.96 | 7.56E-26 |
| CHD4 | + | + | 8.01E-05 | 5678 | 3 | 3 | 6 | 2.52 | 2.29E-08 |
| KMT2A | + | + | 1.48E-04 | 11810 | 5 | 2 | 7 | 5.25 | 3.49E-08 |
| NOTCH1 | − | + | 1.59E-04 | 7699 | 1 | 5 | 6 | 3.42 | 1.21E-06 |
| ADNP | − | + | 3.92E-05 | 3299 | 4 | 0 | 4 | 1.47 | 1.59E-06 |
| SMAD2 | − | + | 1.71E-05 | 1410 | 0 | 3 | 3 | 0.63 | 6.84E-06 |
| KDM5B | + | + | 6.13E-05 | 4662 | 1 | 3 | 4 | 2.07 | 9.19E-06 |
| NSD2 | + | + | 2.56E-05 | 4192 | 1 | 2 | 3 | 1.86 | 2.27E-05 |
| FOXP1 | − | + | 3.02E-05 | 2216 | 3 | 0 | 3 | 0.99 | 3.69E-05 |
| MED13L | − | + | 8.79E-05 | 6615 | 3 | 1 | 4 | 2.94 | 3.72E-05 |
| DYRK1A | − | + | 3.12E-05 | 2387 | 3 | 0 | 3 | 1.06 | 4.08E-05 |
| POGZ | − | + | 5.66E-05 | 4179 | 0 | 3 | 3 | 1.86 | 2.35E-04 |
| PTEN | − | + | 1.40E-05 | 1326 | 0 | 2 | 2 | 0.59 | 3.97E-04 |
| KAT6A | + | + | 8.14E-05 | 5995 | 2 | 1 | 3 | 2.66 | 6.73E-04 |
| ARID1B | + | + | 9.02E-05 | 6412 | 2 | 1 | 3 | 2.85 | 9.03E-04 |
| SMAD4 | − | + | 2.14E-05 | 1670 | 1 | 1 | 2 | 0.74 | 9.28E-04 |
| EP300 | + | + | 9.32E-05 | 7214 | 2 | 1 | 3 | 3.21 | 9.92E-04 |
| WAC | + | − | 2.42E-05 | 1958 | 1 | 1 | 2 | 0.87 | 1.18E-03 |
| FRYL | − | − | 1.07E-04 | 9100 | 1 | 2 | 3 | 4.04 | 1.45E-03 |
| NAA15 | − | + | 2.84E-05 | 2621 | 0 | 2 | 2 | 1.17 | 1.61E-03 |
| DDX3X | − | + | 2.90E-05 | 2006 | 1 | 1 | 2 | 0.89 | 1.68E-03 |
| CUL3 | − | − | 2.98E-05 | 2323 | 1 | 1 | 2 | 1.03 | 1.77E-03 |
| CTNNB1 | − | + | 2.99E-05 | 2332 | 0 | 2 | 2 | 1.04 | 1.78E-03 |
| ANKRD11 | + | + | 1.59E-04 | 7964 | 3 | 0 | 3 | 3.54 | 4.44E-03 |
| LZTR1 | + | − | 4.81E-05 | 2544 | 0 | 2 | 2 | 1.13 | 4.50E-03 |
| SETD5 | + | − | 5.43E-05 | 4329 | 1 | 1 | 2 | 1.92 | 5.69E-03 |
| ASH1L | + | + | 1.07E-04 | 8868 | 1 | 1 | 2 | 3.94 | 2.05E-02 |
| NAT8L | − | + | 1.06E-05 | 912 | 0 | 1 | 1 | 0.41 | 2.13E-02 |
| TECTA | − | − | 1.10E-04 | 6255 | 1 | 1 | 2 | 2.78 | 2.18E-02 |
| ITPR1 | − | − | 1.18E-04 | 8337 | 2 | 0 | 2 | 3.71 | 2.45E-02 |
| NTM | − | + | 1.77E-05 | 1176 | 0 | 1 | 1 | 0.52 | 3.55E-02 |
| PSMD12 | − | − | 1.82E-05 | 1382 | 0 | 1 | 1 | 0.61 | 3.64E-02 |
| PCOLCE | − | − | 2.19E-05 | 1359 | 0 | 1 | 1 | 0.60 | 4.37E-02 |
| EP400 | + | − | 1.63E-04 | 9360 | 1 | 1 | 2 | 4.16 | 4.45E-02 |
| ANK3 | − | + | 1.67E-04 | 13243 | 0 | 2 | 2 | 5.89 | 4.63E-02 |
| CCSER1 | − | − | 2.35E-05 | 2705 | 0 | 1 | 1 | 1.20 | 4.68E-02 |
| KMT2C | + | + | 1.74E-04 | 14569 | 0 | 2 | 2 | 6.48 | 4.97E-02 |
Six genes (PTPN11, CHD7, CHD4, KMT2A, NOTCH1, ADNP) reached genome wide significance (P ≤ 2.5E-06, Bonferroni correction), and the top two genes were PTPN11 (one-tailed binomial test P ≤ 1.54E-34) and CHD7 (one-tailed binomial test P ≤ 7.56E-26), which are associated with Noonan syndrome (OMIM:163950) and CHARGE syndrome (OMIM:214800). The pathogenic missense variants encoding the same p.Asn308 residue of PTPN11 ([hg19], chr12:112915523 A > G, p.N308D; chr12:112915524 A > G, p.N308S; or chr12:112915524 A > C, p.N308T) were the most recurrent amino acid substitutions observed in 7 out of 16 individuals, whereas in CHD7, all were unique singleton variants - with 76.5% (13/17) as PT DNVs. These observations are consistent with the most likely pathogenic mechanisms underlying these two genetic conditions55–57.
Review of the top twelve NDD genes (PTPN11, CHD7, CHD4, KMT2A, NOTCH1, ADNP, SMAD2, KDM5B, NSD2, FOXP1, MED13L and DYRK1A) with higher burden for the protein damaging DNVs (one-tailed binomial test P ≤ 2.18E-04, statistical significance after Bonferroni correction) showed that all 12 contributed to the connectome, and 5 of them belonged to chromatin modifiers. This enrichment could also be observed in the gene numbers for the two categories (connectome Fisher’s exact test P = 0.02, and chromatin modifiers Fisher’s exact test P = 0.04). Finally, as shown in Table 5, review of the available literature showed that 11 of these genes contribute to neurogenesis; 6 play a role in dendrite formation, and 6 contribute to synaptogenesis. As shown in Fig. 2 there was a sizeable overlap among the roles of these genes, while ADNP58–61, DYRKIA62,63, NOTCH164–66 and PTPN1167–70 are reported to contribute to neurogenesis, dendrite formation and synaptogenesis.
Table 5.
Reported functions of the top 12 NDD genes (one-tailed binomial test p-value cutoff <2.18E-04, statistical significance after Bonferroni correction).
| Gene | Chromatin Modifier | Cerebellum | Connectome | Neurogenesis | Dendrites | Synapse | Syndrome |
|---|---|---|---|---|---|---|---|
| ADNP | − | − | + | + | + | + | Helsmoortel-van der AA |
| CHD4 | + | + | + | − | + | + | Sifrim-Hitz-Weiss Syndrome |
| CHD7 | + | + | + | + | − | − | CHARGE Syndrome |
| DYRK1A | − | − | + | + | + | + | Down Syndrome |
| FOXP1 | − | + | + | + | − | − | FOXP1-related ID Syndrome |
| KDM5B | + | − | + | + | − | − | ID syndrome |
| KMT2A | + | − | + | + | - | + | Wiedemann-Steiner Syndrome |
| MED13L | − | - | + | + | − | − | ID, corpus callosum changes |
| NOTCH1 | - | + | + | + | + | + | Adams Oliver Syndrome |
| NSD2 | + | − | + | + | − | − | Wolf Hirschorn Syndrome |
| PTPN11 | − | + | + | + | + | + | Noonan Syndrome |
| SMAD2 | − | + | + | + | + | − | ID, dysmorphic features, FTT |
Figure 2.
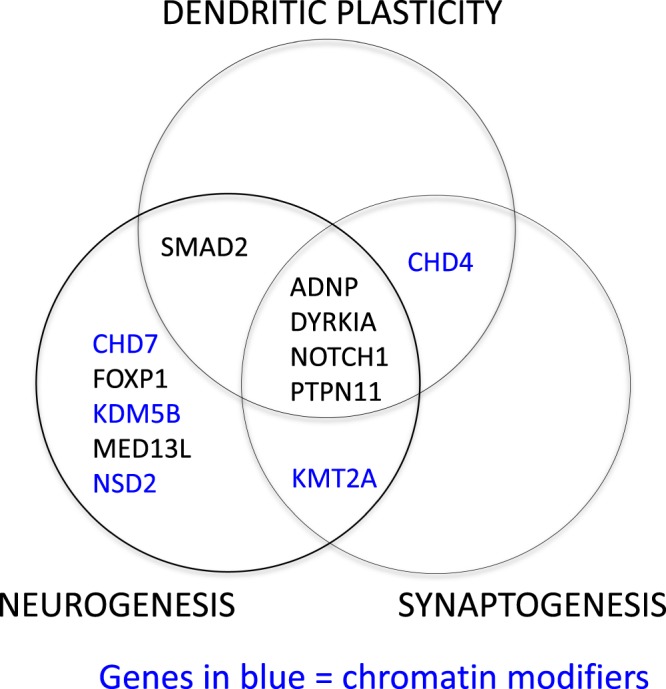
Functions of the top 12 over-represented genes with damaging DNVs. Review of the published literature for the identified top 12 enriched genes achieving statistical significance after correction for multiple comparisons revealed that 11 contributed to neurogenesis and one, CHD4, contributed to dendritic plasticity and synaptogenesis. Of the 11 contributing to neurogenesis, four genes, ADNP, DYRKIA, NOTCH1 and PTPN11, also contributed to dendritic plasticity and synaptogenesis. One, SMAD2, also subserved dendritic plasticity, and a single gene, KMT2A contributed to both neurogenesis and synaptogenesis. Five of the top genes were chromatin modifiers.
Discussion
Converging data suggest that genes that contribute to structural heart development also contribute to the connectome, and we identified 12 candidate genes supporting this hypothesis. These candidates, including ADNP, CHD4, CHD7, DYRK1A, FOXP1, KDM5B, KMT2A, MED13L, NOTCH1, PTPN11, SMAD2 and NSD2, contribute to a broad array of both well-established and more recently identified NDD syndromes. Eleven out of 12 sub-serve neurogenesis; 5 are chromatin modifiers and 2 are members of the NOTCH pathway (Table 5). Finally, the contribution of the cerebellum to cognition and behavior has recently been reported71,72, and 6 of the twelve identified genes are known to contribute to cerebellar development.
Three of these genes are associated with well-known pediatric syndromes.
Mutations in the RAS-MAPK pathway are associated with NDD, while Noonan syndrome is the most common clinical RASopathy. In preclinical models, PTPN11, the gene responsible for 50% of cases of Noonan Syndrome, regulates neurogenesis and is required for neuronal process extension67,68. In addition, PTPN11 differentially regulates expression of post-synaptic receptors and contributes to synaptic homeostasis, while variants in this gene alter surface expression of both AMPA and NMDA receptors during development69,70.
DYRK1A contributes to cognitive disability in Down syndrome. While trisomies were omitted from our -analysis, we noted 4 subjects with de novo DYRK1A variants. Three had PT DNVs, and a fourth was noted to have a deleterious missense DNV. Haplo-insufficiency of DYRK1A results in ID and CHD73, and preclinical studies demonstrate decreased striatal dopamine levels, reduced number of dopamine neurons in the substantia nigra pars compacta and altered behavioral responses to dopaminergic agents, suggesting that haplo-insufficiency of DYRK1A alters the connectome62. In contrast, over-expression of DYRK1A increases the population of GABA inter-neurons and alters the excitatory or inhibitory synapse balance in developing brain63.
Mutations in the ATP-dependent chromatin remodeler CHD7 are responsible for CHARGE syndrome (Coloboma, Heart defects, Atresia choanae, Retarded growth and development and Genital and Ear abnormalities). Variants in CHD7 have been described in subjects with both autism and ID, and many CHARGE patients show hypoplasia of the cerebellum. CHD7 is present in both neuronal precursors and stem cells, with genetic inactivation of CHD7 in cerebellar granule neuron progenitors leading to cerebellar hypoplasia in mice due to impaired granule neuron differentiation, apoptosis and abnormal Purkinje cell migration50.
Two additional candidates, CHD4 and SMAD2, also play important roles in cerebellar development, CHD and intellectual disability74.
In preclinical studies, knock-out of Chd4 impairs dendritic pruning in developing cerebellar granule neurons and impedes the establishment of granule neuron parallel fiber or Purkinje cell synapses in the rodent cerebellar cortex45,75,76. Recent publications describe an autosomal dominant ID syndrome attributable to variants in CHD4; affected patients also show cardiac, skeletal and urogenital malformations48,74.
Loss of function SMAD2 variants cause a wide spectrum of autosomal dominant aortic and arterial aneurysmal disease, and a recent report describes two patients with these variants who have complex CHD and NDD. Preclinical studies show delayed migration and maturation of granule cells and retardation of dendritic arborization of Purkinje cells, suggesting that Smad2 plays a key role in cerebellar connectivity76,77.
Language disability is common among children with CHD, and subjects with FOXP1, MED13L, NOTCH1 and ADNP variants show ID and specific language impairment, with or without ASD64,78,79.
FOXP1 is expressed in neural stem cells, and modulation of FOXP1 expression influences neuronal differentiation. In a preclinical model of cortical development, FOXP1-knockdown in utero reduced both neural stem cell differentiation and migration. Furthermore, FOXP1 repressed expression of Notch pathway genes, resulting in inhibition of Notch signaling80.
NOTCH1, responsible for Adams Oliver Syndrome, is required for neuronal differentiation, dendrite development and synaptic plasticity in developing brain64–66.
Similarly, subjects with haplo-insufficiently of MED13L show ID and severe speech delay; congenital heart defects are found in 20–50% of patients79,81. In preclinical studies, haploinsufficiency of MED13L shows defects in both neuronal migration and differentiation82,83.
ADNP variants are reported in children with autism and ID who carry a diagnosis of Helsmoortel-Van der Aa syndrome84. In preclinical studies, ADNP deficiency decreases neurogenesis, reduces dendritic spine density, impairs neurite outgrowth and alters synaptic gene expression58–61.
Finally, histone lysine methyltransferases (KMTs) and demethylases (KDMs) are posited to regulate gene regulation, while variants causing haplo-insufficiency of KMTs and KDMs are common in patients with NDD85.
Dominant DNVs in KMT2A have been reported in individuals with Wiedemann-Steiner syndrome, a developmental disorder with ID and cardiac anomalies; KMT2A peaks in expression in human fetal brains and is reported to be essential for both neurogenesis and synaptic plasticity86–88.
Haploinsufficiency of NSD2, a histone lysine methyltransferase, is associated with all known cases of Wolf-Hirschhorn Syndrome89. While little is known about the neurobiology of NSD2 variants, suppression of the functional homolog of NSD2 in zebrafish affects early embryogenesis, including incomplete neuron formation and endbrain or cerebellar volume changes, which are also observed in Wolf-Hirschhorn patients and Nsd2-deficient mice89–91.
KDM5B, a histone lysine demethylase, negatively regulates neurogenesis, represses Reln expression in neural stem cells in the adult subventricular zone and has been reported to cause ID92. Of note, a recent study reports a single patient with a KDM5B variant, ID and an atrial septal defect85.
In addition to their recognized NDDs, infants and children with CHD are at high risk for abnormal MRI studies of the brain, and recent data suggest a correlation of behavior with alterations in the connectome. To better explore these findings, prior studies have addressed either the impact of targeted CHD variants on brain development and neurodevelopmental outcome93, or identified genes that are both highly expressed in the developing heart and contribute to NDD or brain development. However, none have provided analyses of large cohorts with MRI measures of neural connectivity. In addition, the contribution of DNVs in brain or connectome development associated with fetal MRI studies, prior to hypoxemia, are largely lacking94.
Genetic testing is an important component of the evaluation for neonates with CHD as it may both impact strategies for clinical care and provide long-term outcome information. Diagnostic genetic variants are detected in 11.1% of fetuses with cardiac anomalies95. In addition, the incidence of extra-cardiac malformations patients with CHD has been reported to range from 10–26%96, and those CHD patients with extra-cardiac malformations are more likely to harbor pathogenic DNVs than those with only CHD48,97. In addition, significant developmental delay defined as a cognitive, language or motor score <70 has been reported to occur in almost 75% of children with a known genetic syndrome, compared to 33% of children with single-ventricle non-syndromic CHD and 20% of those with bi-ventricular non-syndromic CHD98. These data demonstrate that while syndromic cases have a higher incidence of developmental delay, children with non-syndromic CHD are also affected by this lifelong disability. The goal of our study is to suggest that these disabilities are not simply due to hypoxia but may indeed have a genetic origin.
The strengths of this study include the analysis of multiple previously published large CHD data sets and the independently reported NDD genes. The weaknesses include the lack of MR connectome imaging and limited phenotyping data for the study subjects. In addition, paternal age, which is a risk factor for DNVs, was not available for our analysis99,100. Although all the variants we examined are de novo variants and all control subjects were reported to have unremarkable clinical presentations, our controls were first-degree unaffected siblings of subjects with ASD. Finally, although we excluded subjects with aneuploidies, genes contributing to Noonan Syndrome, CHARGE Syndrome and Down Syndrome are among our significant candidates, suggesting a possible bias to syndromic intellectual disability. Nonetheless, after excluding these subjects from our analyses, our major findings persisted (Supplementary Table S3).
As the growing population of children with CHD becomes adolescents and these adolescents transition to adult cardiology care, the impact of NDD on this population does not wane28,101,102. Recent data demonstrate a significant prevalence and impact of neurocognitive deficits among adults with CHD; adult CHD subjects have lower academic levels and higher unemployment rates compared to reference populations. Furthermore, adult CHD patients are more likely than typical young adults to suffer from depression. [For review of adult CHD neurocognitive or behavioral deficits, please see103]. Establishing the determinants of neuro-behavior in those with CHD will permit both prognostication and targets for intervention, and future work should link genes contributing to cardiac development to the functional connectome.
Methods
To test the hypothesis that variants contributing to NDD are more common in subjects with CHD than in controls, we performed a secondary analysis of primary data from previously published cohorts of subjects with CHD and genomic data from those with NDD. High confidence DNVs (pp_dnm ≥ 0.9) from Sifrim et al. were included48, and DNVs from Jin et al. were qualified using the previously published filtering criteria followed by examination using in silico visualization40. We compared the frequency of DNVs in the NDD genes occurring in the CHD population to those found in the control group using Fisher’s exact tests. Results were summarized using odds ratios and 95% Confidence Intervals (95%CI)104. To test for over-representation of a gene set among cases, a one-tailed binomial test was conducted by comparing the observed number of variants to the expected count as previously reported40. Bonferroni correction was applied for multiple comparisons105. Analyses were performed using Excel, Microsoft Office 365, and online software MEDCALC (https://www.medcalc.org/calc/odds_ratio.php), Fisher’s Exact test (https://www.langsrud.com/stat/fisher.htm).
Subject population
CHD subjects with genomic data were ascertained from recent publications. Evaluation of available subjects from the multi-center/multi-study cohort reported by Deciphering Developmental Disorders Project (“DDD Plus Study”, N = 1039 cases)48 and a recent publication from the Pediatric Cardiac Genome Consortium (PCGC) study40 (N = 2645 cases) yielded 3684 unique cases of structural CHD (excluding prematurity-associated patent ductus arteriosus). Controls included 1,789 previously analyzed unaffected siblings of autism probands and their unaffected parents40.
Analysis of the overall de novo variants (DNVs) identified from whole exomes in the DDD Plus Study, PCGC Study and controls (Supplementary Table S1) demonstrated that the mean DNVs per individual are 1.09, 1.13, and 1.02 respectively. Compared to control subjects, both DDD Plus subjects and PCGC subjects had a higher prevalence of protein truncating (PT) variants (OR = 1.89, 95%CI:1.49–2.39, Fisher’s exact test P = 1.53E-07, DDD Plus Study; and OR = 1.60, 95%CI: 1.31–1.95, Fisher’s exact test P = 2.25E-06, PCGC Study). In addition, DDD Plus and PCGC subjects had similar prevalence of both de novo missense and synonymous variants when compared to controls, suggesting similarity of the data sets.
Target genes
The 229 target NDD disease-risk genes (ASD, DD and ID) were selected based on recently published sequencing studies47,106,107. Genes contributing to the connectome were defined as those necessary for the development, growth and maintenance of neural networks in developing brain49–51, while chromatin modifiers were defined as variants that alter the assembly and compaction of chromatin52. Each of the 229 candidate genes was individually annotated using PubMed search terms including connectome, neural connectivity, neuron, neurogenesis, axon, growth cone, dendrite, synapse, synaptogenesis, oligodendroglia, myelinogenesis, chromatin modifier, chromatin and methylation, acetylation and/or ubiquitination (Supplementary Table 2). Functions and murine phenotypes of these genes (Mouse Genome Informatics, http://www.informatics.jax.org/) are also shown in Supplementary Table 2. Gene assignments are effective 6–30–2018, and a diagram demonstrating the inter-relationship of NDD, connectome and chromatin modifier genes for the 229 target genes is shown in Fig. 1.
Ethical approval and informed consent
The Yale University IRB does not require approval for meta-analyses involving de-identified data.
Supplementary information
Acknowledgements
We thank the patients and their families; they are the inspiration for this study. This work is supported by NIH R01HD081379 (MKK), NIH UM1 HL098162 (MB), NIH R01 HL125885 (MB) and NIH P50HD093078 (LRM). MKK is a Mallinckrodt Fellow. This publication was made possible by CTSA Grant Number UL1 TR000142 from the National Center for Advancing Translational Science (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Author contributions
W.J., M.B., M.K.K. and L.R.M. designed research; D.S. and L.R.M. performed literature reviews; W.J. and V.S. performed analyses; W.J., D.F., J.C., V.S., D.S., M.B., M.K.K. and L.R.M. reviewed data; W.J., D.F., J.C., V.S., D.S., M.B., M.K.K. and L.R.M. wrote manuscript.
Data availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Martina Brueckner, Mustafa K Khokha and Laura R Ment.
Supplementary information
is available for this paper at 10.1038/s41598-020-63928-2.
References
- 1.Egbe A, Lee S, Ho D, Uppu S, Srivastava S. Prevalence of congenital anomalies in newborns with congenital heart disease diagnosis. Ann. Pediatr. Cardiol. 2014;7:86–91. doi: 10.4103/0974-2069.132474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van der Linde D, et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 3.Laraja K, et al. Neurodevelopmental Outcome in Children after Fetal Cardiac Intervention for Aortic Stenosis with Evolving Hypoplastic Left Heart Syndrome. J. Pediatr. 2017;184:130–136 e134. doi: 10.1016/j.jpeds.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marino BS, et al. Neurodevelopmental outcomes in children with congenital heart disease: evaluation and management: a scientific statement from the American Heart Association. Circulation. 2012;126:1143–1172. doi: 10.1161/CIR.0b013e318265ee8a. [DOI] [PubMed] [Google Scholar]
- 5.Mebius, M. J., Kooi, E. M. W., Bilardo, C. M. & Bos, A. F. Brain Injury and Neurodevelopmental Outcome in Congenital Heart Disease: A Systematic Review. Pediatrics140, 10.1542/peds.2016-4055 (2017). [DOI] [PubMed]
- 6.Naef N, et al. Neurodevelopmental Profiles of Children with Congenital Heart Disease at School Age. J. Pediatr. 2017;188:75–81. doi: 10.1016/j.jpeds.2017.05.073. [DOI] [PubMed] [Google Scholar]
- 7.Sananes R, et al. Neurodevelopmental outcomes after open heart operations before 3 months of age. Ann. Thorac. Surg. 2012;93:1577–1583. doi: 10.1016/j.athoracsur.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Brosig CL, et al. Preschool Neurodevelopmental Outcomes in Children with Congenital Heart Disease. J. Pediatr. 2017;183:80–86 e81. doi: 10.1016/j.jpeds.2016.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latal B. Neurodevelopmental Outcomes of the Child with Congenital Heart Disease. Clin. Perinatol. 2016;43:173–185. doi: 10.1016/j.clp.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer CJ, et al. Academic achievement and satisfaction in adolescents with CHD. Cardiol. Young. 2016;26:257–262. doi: 10.1017/S1047951115000074. [DOI] [PubMed] [Google Scholar]
- 11.Sarrechia I, et al. Neurodevelopment and behavior after transcatheter versus surgical closure of secundum type atrial septal defect. J. Pediatr. 2015;166:31–38. doi: 10.1016/j.jpeds.2014.08.039. [DOI] [PubMed] [Google Scholar]
- 12.Sarrechia I, et al. Neurocognitive development and behaviour in school-aged children after surgery for univentricular or biventricular congenital heart disease. Eur. J. Cardiothorac. Surg. 2016;49:167–174. doi: 10.1093/ejcts/ezv029. [DOI] [PubMed] [Google Scholar]
- 13.Sarrechia I, et al. Neurodevelopmental outcome after surgery for acyanotic congenital heart disease. Res. Dev. Disabil. 2015;45-46:58–68. doi: 10.1016/j.ridd.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Holland JE, et al. Psychiatric Disorders and Function in Adolescents with Tetralogy of Fallot. J. Pediatr. 2017;187:165–173. doi: 10.1016/j.jpeds.2017.04.048. [DOI] [PubMed] [Google Scholar]
- 15.DeMaso, D. R. et al. Psychiatric Disorders in Adolescents With Single Ventricle Congenital Heart Disease. Pediatrics139, 10.1542/peds.2016-2241 (2017). [DOI] [PMC free article] [PubMed]
- 16.Kelly CJ, et al. Abnormal Microstructural Development of the Cerebral Cortex in Neonates With Congenital Heart Disease Is Associated With Impaired Cerebral Oxygen Delivery. J. Am. Heart Assoc. 2019;8:e009893. doi: 10.1161/JAHA.118.009893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Licht DJ, et al. Preoperative cerebral blood flow is diminished in neonates with severe congenital heart defects. J. Thorac. Cardiovasc. Surg. 2004;128:841–849. doi: 10.1016/j.jtcvs.2004.07.022. [DOI] [PubMed] [Google Scholar]
- 18.Petit CJ, et al. Preoperative brain injury in transposition of the great arteries is associated with oxygenation and time to surgery, not balloon atrial septostomy. Circulation. 2009;119:709–716. doi: 10.1161/CIRCULATIONAHA.107.760819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg C, Gembruch O, Gembruch U, Geipel A. Doppler indices of the middle cerebral artery in fetuses with cardiac defects theoretically associated with impaired cerebral oxygen delivery in utero: is there a brain-sparing effect? Ultrasound Obstet. Gynecol. 2009;34:666–672. doi: 10.1002/uog.7474. [DOI] [PubMed] [Google Scholar]
- 20.Limperopoulos C. Disorders of the fetal circulation and the fetal brain. Clin. Perinatol. 2009;36:561–577. doi: 10.1016/j.clp.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Claessens NHP, Kelly CJ, Counsell SJ, Benders M. Neuroimaging, cardiovascular physiology, and functional outcomes in infants with congenital heart disease. Dev. Med. Child. Neurol. 2017;59:894–902. doi: 10.1111/dmcn.13461. [DOI] [PubMed] [Google Scholar]
- 22.Mebius MJ, et al. Cerebral oxygen saturation during the first 72h after birth in infants diagnosed prenatally with congenital heart disease. Early Hum. Dev. 2016;103:199–203. doi: 10.1016/j.earlhumdev.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Zaidi S, Brueckner M. Genetics and Genomics of Congenital Heart Disease. Circ. Res. 2017;120:923–940. doi: 10.1161/CIRCRESAHA.116.309140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Homsy J, et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262–1266. doi: 10.1126/science.aac9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blue GM, et al. Advances in the Genetics of Congenital Heart Disease: A Clinician’s Guide. J. Am. Coll. Cardiol. 2017;69:859–870. doi: 10.1016/j.jacc.2016.11.060. [DOI] [PubMed] [Google Scholar]
- 26.Gaynor JW, et al. Neurodevelopmental outcomes after cardiac surgery in infancy. Pediatrics. 2015;135:816–825. doi: 10.1542/peds.2014-3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wernovsky G, Licht DJ. Neurodevelopmental Outcomes in Children With Congenital Heart Disease-What Can We Impact? Pediatr. Crit. Care Med. 2016;17:S232–242. doi: 10.1097/PCC.0000000000000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW. Brain in Congenital Heart Disease Across the Lifespan: The Cumulative Burden of Injury. Circulation. 2016;133:1951–1962. doi: 10.1161/CIRCULATIONAHA.115.019881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Russell, M. W., Chung, W. K., Kaltman, J. R. & Miller, T. A. Advances in the Understanding of the Genetic Determinants of Congenital Heart Disease and Their Impact on Clinical Outcomes. J Am Heart Assoc7, 10.1161/JAHA.117.006906 (2018). [DOI] [PMC free article] [PubMed]
- 30.Pierpont ME, et al. Genetic Basis for Congenital Heart Disease: Revisited: A Scientific Statement From the American Heart Association. Circulation. 2018;138:e653–e711. doi: 10.1161/CIR.0000000000000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blue GM, et al. The promises and challenges of exome sequencing in familial, non-syndromic congenital heart disease. Int. J. Cardiol. 2017;230:155–163. doi: 10.1016/j.ijcard.2016.12.024. [DOI] [PubMed] [Google Scholar]
- 32.Sporns O. Structure and function of complex brain networks. Dialogues Clin. Neurosci. 2013;15:247–262. doi: 10.31887/DCNS.2013.15.3/osporns. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Di Martino A, et al. Unraveling the miswired connectome: A developmental perspective. Neuron. 2014;83(6):1335–53. doi: 10.1016/j.neuron.2014.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mlczoch E, et al. Structural congenital brain disease in congenital heart disease: results from a fetal MRI program. Eur. J. Paediatr. Neurol. 2013;17:153–160. doi: 10.1016/j.ejpn.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Schellen, C. et al. Fetal MRI detects early alterations of brain development in Tetralogy of Fallot. Am J Obstet Gynecol, 10.1016/j.ajog.2015.05.046 (2015). [DOI] [PubMed]
- 36.Panigrahy A, et al. Relationship of white matter network topology and cognitive outcome in adolescents with d-transposition of the great arteries. Neuroimage Clin. 2015;7:438–448. doi: 10.1016/j.nicl.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watson CG, Stopp C, Newburger JW, Rivkin MJ. Graph theory analysis of cortical thickness networks in adolescents with d-transposition of the great arteries. Brain Behav. 2018;8:e00834. doi: 10.1002/brb3.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson CG, et al. Altered White Matter Microstructure Correlates with IQ and Processing Speed in Children and Adolescents Post-Fontan. J. Pediatr. 2018;200:140–149 e144. doi: 10.1016/j.jpeds.2018.04.022. [DOI] [PubMed] [Google Scholar]
- 39.De Asis-Cruz J, Donofrio MT, Vezina G, Limperopoulos C. Aberrant brain functional connectivity in newborns with congenital heart disease before cardiac surgery. Neuroimage Clin. 2018;17:31–42. doi: 10.1016/j.nicl.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jin SC, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blue GM, et al. Genetic burden and associations with adverse neurodevelopment in neonates with congenital heart disease. Am. Heart J. 2018;201:33–39. doi: 10.1016/j.ahj.2018.03.021. [DOI] [PubMed] [Google Scholar]
- 42.Tyssowski K, Kishi Y, Gotoh Y. Chromatin regulators of neural development. Neuroscience. 2014;264:4–16. doi: 10.1016/j.neuroscience.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Iwase S, et al. Epigenetic Etiology of Intellectual Disability. J. Neurosci. 2017;37:10773–10782. doi: 10.1523/JNEUROSCI.1840-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Valnegri P, et al. RNF8/UBC13 ubiquitin signaling suppresses synapse formation in the mammalian brain. Nat. Commun. 2017;8:1271. doi: 10.1038/s41467-017-01333-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 2016;353:300–305. doi: 10.1126/science.aad4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan, A. & Gurvitz, M. Epidemiology of ACHD: What Has Changed and What Is Changing? Prog Cardiovasc Dis, 10.1016/j.pcad.2018.08.004 (2018). [DOI] [PubMed]
- 47.Stessman HA, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 2017;49:515–526. doi: 10.1038/ng.3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sifrim A, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 2016;48:1060–1065. doi: 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silver DL. Genomic divergence and brain evolution: How regulatory DNA influences development of the cerebral cortex. Bioessays. 2016;38:162–171. doi: 10.1002/bies.201500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng W, et al. Chd7 is indispensable for mammalian brain development through activation of a neuronal differentiation programme. Nat. Commun. 2017;8:14758. doi: 10.1038/ncomms14758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jakovcevski M, Akbarian S. Epigenetic mechanisms in neurological disease. Nat. Med. 2012;18:1194–1204. doi: 10.1038/nm.2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat. Rev. Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conover, W. J. Practical nonparametric statistics. 97–104 (John Wiley & Sons, 1971).
- 54.Hollander, M. & Wolfe, D. A. Nonparametric statistical inference. (John Wiley & Sons, 1973).
- 55.Zentner GE, et al. CHD7 functions in the nucleolus as a positive regulator of ribosomal RNA biogenesis. Hum. Mol. Genet. 2010;19:3491–3501. doi: 10.1093/hmg/ddq265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tartaglia M, et al. PTPN11 mutations in Noonan syndrome: molecular spectrum, genotype-phenotype correlation, and phenotypic heterogeneity. Am. J. Hum. Genet. 2002;70:1555–1563. doi: 10.1086/340847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tartaglia M, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001;29:465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 58.Hacohen-Kleiman G, et al. Activity-dependent neuroprotective protein deficiency models synaptic and developmental phenotypes of autism-like syndrome. J. Clin. Invest. 2018;128:4956–4969. doi: 10.1172/JCI98199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amram N, et al. Sexual divergence in microtubule function: the novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol. Psychiatry. 2016;21:1467–1476. doi: 10.1038/mp.2015.208. [DOI] [PubMed] [Google Scholar]
- 60.Mandel S, Gozes I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem. 2007;282:34448–34456. doi: 10.1074/jbc.M704756200. [DOI] [PubMed] [Google Scholar]
- 61.Gozes I. Microtubules (tau) as an emerging therapeutic target: NAP (davunetide) Curr. Pharm. Des. 2011;17:3413–3417. doi: 10.2174/138161211798072553. [DOI] [PubMed] [Google Scholar]
- 62.Martinez de Lagran M, et al. Dopaminergic deficiency in mice with reduced levels of the dual-specificity tyrosine-phosphorylated and regulated kinase 1A, Dyrk1A(+/-) Genes. Brain Behav. 2007;6:569–578θ. doi: 10.1111/j.1601-183X.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- 63.Souchet B, et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis. 2014;69:65–75. doi: 10.1016/j.nbd.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 64.Ding XF, Gao X, Ding XC, Fan M, Chen J. Postnatal dysregulation of Notch signal disrupts dendrite development of adult-born neurons in the hippocampus and contributes to memory impairment. Sci. Rep. 2016;6:25780. doi: 10.1038/srep25780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dahlhaus M, et al. Notch1 signaling in pyramidal neurons regulates synaptic connectivity and experience-dependent modifications of acuity in the visual cortex. J. Neurosci. 2008;28:10794–10802. doi: 10.1523/JNEUROSCI.1348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lutolf S, Radtke F, Aguet M, Suter U, Taylor V. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development. 2002;129:373–385. doi: 10.1242/dev.129.2.373. [DOI] [PubMed] [Google Scholar]
- 67.Zhu Y, et al. Loss of Shp2 within radial glia is associated with cerebral cortical dysplasia, glial defects of cerebellum and impaired sensorymotor development in newborn mice. Mol. Med. Rep. 2018;17:3170–3177. doi: 10.3892/mmr.2017.8236. [DOI] [PubMed] [Google Scholar]
- 68.Rosario M, Franke R, Bednarski C, Birchmeier W. The neurite outgrowth multiadaptor RhoGAP, NOMA-GAP, regulates neurite extension through SHP2 and Cdc42. J. Cell Biol. 2007;178:503–516. doi: 10.1083/jcb.200609146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oh JY, Rhee S, Silva AJ, Lee YS, Kim HK. Noonan syndrome-associated SHP2 mutation differentially modulates the expression of postsynaptic receptors according to developmental maturation. Neurosci. Lett. 2017;649:41–47. doi: 10.1016/j.neulet.2017.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang B, Lu W. Src homology 2 domain-containing phosphotyrosine phosphatase 2 (Shp2) controls surface GluA1 protein in synaptic homeostasis. J. Biol. Chem. 2017;292:15481–15488. doi: 10.1074/jbc.M117.775239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schmahmann, J. D. The cerebellum and cognition. Neurosci Lett, 10.1016/j.neulet.2018.07.005 (2018). [DOI] [PubMed]
- 72.Wang SS, Kloth AD, Badura A. The cerebellum, sensitive periods, and autism. Neuron. 2014;83:518–532. doi: 10.1016/j.neuron.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luco SM, et al. Case report of novel DYRK1A mutations in 2 individuals with syndromic intellectual disability and a review of the literature. BMC Med. Genet. 2016;17:15. doi: 10.1186/s12881-016-0276-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Weiss K, et al. De Novo Mutations in CHD4, an ATP-Dependent Chromatin Remodeler Gene, Cause an Intellectual Disability Syndrome with Distinctive Dysmorphisms. Am. J. Hum. Genet. 2016;99:934–941. doi: 10.1016/j.ajhg.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yamada T, et al. Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron. 2014;83:122–134. doi: 10.1016/j.neuron.2014.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang L, et al. Smad2 protein disruption in the central nervous system leads to aberrant cerebellar development and early postnatal ataxia in mice. J. Biol. Chem. 2011;286:18766–18774. doi: 10.1074/jbc.M111.223271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Granadillo, J. L. et al. Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum Mutat, 10.1002/humu.23627 (2018). [DOI] [PubMed]
- 78.Meerschaut I, et al. FOXP1-related intellectual disability syndrome: a recognisable entity. J. Med. Genet. 2017;54:613–623. doi: 10.1136/jmedgenet-2017-104579. [DOI] [PubMed] [Google Scholar]
- 79.Asadollahi R, et al. Genotype-phenotype evaluation of MED13L defects in the light of a novel truncating and a recurrent missense mutation. Eur. J. Med. Genet. 2017;60:451–464. doi: 10.1016/j.ejmg.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 80.Braccioli L, et al. FOXP1 Promotes Embryonic Neural Stem Cell Differentiation by Repressing Jagged1 Expression. Stem Cell Rep. 2017;9:1530–1545. doi: 10.1016/j.stemcr.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jimenez-Romero S, Carrasco-Salas P, Benitez-Burraco A. Language and Cognitive Impairment Associated with a Novel p.Cys63Arg Change in the MED13L Transcriptional Regulator. Mol. Syndromol. 2018;9:83–91. doi: 10.1159/000485638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Utami KH, et al. Impaired development of neural-crest cell-derived organs and intellectual disability caused by MED13L haploinsufficiency. Hum. Mutat. 2014;35:1311–1320. doi: 10.1002/humu.22636. [DOI] [PubMed] [Google Scholar]
- 83.Snijders Blok, L. et al. De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum Genet, 10.1007/s00439-018-1887-y (2018). [DOI] [PMC free article] [PubMed]
- 84.Van Dijck, A. et al. Clinical Presentation of a Complex Neurodevelopmental Disorder Caused by Mutations in ADNP. Biol Psychiatry, 10.1016/j.biopsych.2018.02.1173 (2018). [DOI] [PMC free article] [PubMed]
- 85.Faundes V, et al. Histone Lysine Methylases and Demethylases in the Landscape of Human Developmental Disorders. Am. J. Hum. Genet. 2018;102:175–187. doi: 10.1016/j.ajhg.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vallianatos CN, Iwase S. Disrupted intricacy of histone H3K4 methylation in neurodevelopmental disorders. Epigenomics. 2015;7:503–519. doi: 10.2217/epi.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Miyake N, et al. Delineation of clinical features in Wiedemann-Steiner syndrome caused by KMT2A mutations. Clin. Genet. 2016;89:115–119. doi: 10.1111/cge.12586. [DOI] [PubMed] [Google Scholar]
- 88.Jakovcevski M, et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci. 2015;35:5097–5108. doi: 10.1523/JNEUROSCI.3004-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Righini A, et al. Brain magnetic resonance imaging in Wolf-Hirschhorn syndrome. Neuropediatrics. 2007;38:25–28. doi: 10.1055/s-2007-981685. [DOI] [PubMed] [Google Scholar]
- 90.Yamada-Okabe T, et al. Functional characterization of the zebrafish WHSC1-related gene, a homolog of human NSD2. Biochem. Biophys. Res. Commun. 2010;402:335–339. doi: 10.1016/j.bbrc.2010.10.027. [DOI] [PubMed] [Google Scholar]
- 91.Naf D, et al. Mouse models for the Wolf-Hirschhorn deletion syndrome. Hum. Mol. Genet. 2001;10:91–98. doi: 10.1093/hmg/10.2.91. [DOI] [PubMed] [Google Scholar]
- 92.Zhou Q, et al. Inhibition of the histone demethylase Kdm5b promotes neurogenesis and derepresses Reln (reelin) in neural stem cells from the adult subventricular zone of mice. Mol. Biol. Cell. 2016;27:627–639. doi: 10.1091/mbc.E15-07-0513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson, E. M. et al. PTPN11 Gain-of-Function Mutations Affect the Developing Human Brain, Memory, and Attention. Cereb Cortex, 10.1093/cercor/bhy158 (2018). [DOI] [PMC free article] [PubMed]
- 94.Hu P, et al. Clinical application of targeted next-generation sequencing in fetuses with congenital heart defect. Ultrasound Obstet. Gynecol. 2018;52:205–211. doi: 10.1002/uog.19042. [DOI] [PubMed] [Google Scholar]
- 95.Lord J, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–757. doi: 10.1016/S0140-6736(18)31940-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stoll C, Dott B, Alembik Y, Roth MP. Associated noncardiac congenital anomalies among cases with congenital heart defects. Eur. J. Med. Genet. 2015;58:75–85. doi: 10.1016/j.ejmg.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 97.Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mussatto KA, et al. Risk and prevalence of developmental delay in young children with congenital heart disease. Pediatrics. 2014;133:e570–577. doi: 10.1542/peds.2013-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Girard SL, et al. Paternal Age Explains a Major Portion of De Novo Germline Mutation Rate Variability in Healthy Individuals. PLoS One. 2016;11:e0164212. doi: 10.1371/journal.pone.0164212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kong A, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gilboa SM, et al. Congenital Heart Defects in the United States: Estimating the Magnitude of the Affected Population in 2010. Circulation. 2016;134:101–109. doi: 10.1161/CIRCULATIONAHA.115.019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Marelli AJ, et al. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–756. doi: 10.1161/CIRCULATIONAHA.113.008396. [DOI] [PubMed] [Google Scholar]
- 103.Cohen, S. & Earing, M. G. Neurocognitive Impairment and its Long-Term Impact on Adults with Congenital Heart Disease. Prog Cardiovasc Dis, 10.1016/j.pcad.2018.08.002 (2018). [DOI] [PubMed]
- 104.Altman, D. G. Practical statistics for medical research., (Chapman and Hall, 1991).
- 105.Bonferroni, C. E. Teoria statistica delle classi e calcolo delle probabilita. (PUbblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze, 1936).
- 106.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on request.