

Abstract
Phthalates are commonly included as ingredients in personal care products such as cosmetics, shampoos and perfumes. Diethyl phthalate (DEP) has been found to be anti-androgenic and linked with adverse reproductive effects on males, but effects on females are poorly understood. We designed an integrative and translational study to experimentally examine the effects of DEP exposure at a human-equivalent dose on the mammary transcriptome in rats and to subsequently examine the DEP gene signature in breast tissues (both pre-malignant and tumor) from a population study. In Sprague-Dawley rats treated orally with DEP from birth to adulthood, we identified a signature panel of 107 genes predominantly down-regulated by DEP exposure. Univariate analysis of this 107 DEP gene signature in pre-malignant breast tissues revealed that six genes (P4HA1, MPZL3, TMC4, PLEKHA6, CA8, AREG) were inversely associated with monoethyl phthalate (MEP; the urinary metabolite of DEP) concentration (p < 0.05) among postmenopausal women; all six genes loaded on to one of seven factors identified by factor analysis. Transcription factor enrichment analysis revealed that genes in this factor were enriched for androgen receptor binding sites. These six genes were also significantly down-regulated in pre-malignant adjacent tissues compared to the corresponding tumor tissues in pair-wise analyses (p < 0.05). Results from our translational study indicate that low level exposure to diethyl phthalate results in measurable genomic changes in breast tissue with implications in breast carcinogenesis.
Subject terms: Breast cancer, Breast cancer
Introduction
Phthalates are a family of chemicals with diverse properties that are added to a wide variety of consumer products and used in industrial and medical applications1. One of the congeners, diethyl phthalate (DEP), is commonly used as a plasticizer and solvent in personal care products such as cosmetics, shampoos and perfumes and in pharmaceutical products1. The United States (U.S.) Food and Drug Administration found DEP to be the most common phthalate in personal care products2, a major source of exposure to DEP3. The urinary metabolite of DEP, monoethyl phthalate (MEP), is detectable in U.S. population samples at levels that are often an order of magnitude higher than other phthalates such as di(2-ethylhexyl) phthalate and dibutyl phthalate4. Due to non-covalent bonding to its parent materials, DEP is leached easily into the environment resulting in widespread human exposure1.
DEP has been shown to have anti-androgenic effects in males, including shorter anogenital distance5 and increased sperm DNA damage6,7. MEP has been detected in breast milk, suggesting lactational exposure, and was associated with lowered testosterone in infant males8. Links between phthalate exposures and female developmental and reproductive and cancer outcomes, however, have been less well studied, despite their potential as endocrine disruptors. Reduced fecundity with MEP exposure was reported in a Danish cohort9. An epidemiologic study in Mexican women showed an association between MEP exposure and increased breast cancer risk10. High serum level of MEP in postmenopausal women was associated with elevated breast density11, a marker for breast cancer risk12. We recently reported results from the Long Island Breast Cancer Study Project (LIBCSP) in which we did not observe any significant associations between urinary level of MEP and breast cancer risk, while inverse associations were observed for mono(3-carboxypropyl) phthalate (MCPP) and monocarboxyoctyl phthalate (MCOP), metabolites of anti-androgenic phthalates13.
Breast cancer is considered a hormone-driven disease14. Given the hormone-disrupting properties of DEP and its widespread exposure through the use of personal care and consumer products, there is an urgent need to systematically study whether DEP exposures may impact breast cancer development. In rats, fetal and prepubertal exposures to high doses of butyl benzyl phthalate led to changes in gene expression profiles of mammary glands15,16. Less is known about the effects of DEP exposure particularly at doses that are relevant to human exposure.
The aim of our present study is to employ an animal model to experimentally identify a gene signature of DEP exposure at a human-equivalent dose and to subsequently translate these findings to humans. We exposed female Sprague-Dawley (SD) rats to DEP orally from birth to adulthood at a dose that was previously shown to produce a urinary metabolite (MEP) level similar to that of the U.S. population17. We identified a DEP gene signature in normal developing mammary glands in SD rats, and subsequently examined this signature in pre-malignant and breast cancer tissues from a subsample of women who participated in a population-based study. The overall goal of the study is to examine whether human level exposure to DEP induces measurable transcriptomic changes in target tissues, thus shedding light on the possible causal relationship between phthalate exposure and breast cancer.
Materials and Methods
Test compound
Diethyl phthalate (DEP) (CAS # 84-66-2, lot # STBB0862V, 99% purity) was supplied in plastic containers (Sigma Aldrich, Italy). Olive oil, supplied in glass bottles (Montalbano Agricola Alimentare Toscana, Florence, lot # 111275, Italy), was used as the vehicle to prepare the dosing solution. The experimental oral dose was 0.1735 mg/Kg/day, which represented 1/10,000 the no observed adverse effect levels (NOAEL) of DEP18–20. DEP was stored in the dark at room temperature (20 °C). The solutions were prepared weekly on the basis of mean body weight of each group and were continuously stirred before and during the treatment. To minimize external contamination, olive oil and DEP were stored in glass containers and administered using 5 mL glass syringes. Biological samples were collected in polypropylene vials. Chemical analyses and stability testing have been described previously21.
Animal studies
Animal studies were carried out at Cesare Maltoni Cancer Research Centre/Ramazzini Institute (CMCRC/RI) (Bentivoglio, Italy) in accordance with the rules of Italian law for Animal Welfare (Decreto Legislativo 26, 2014), following the principles of Good Laboratory Practices and Standard Operating Procedures of the CMCRC/RI facility, which include authorization by the ethical committee. At Mount Sinai, the study was approved by the Institutional Animal Care and Use Committee (IACUC). The experiment used female Sprague-Dawley (SD) rats which belong to the colony that has been used for over 40 years in the laboratory of the CMCRC/RI. There were no siblings in each treatment group and they were randomized so as to have minimal differences in body weight among them (standard deviation <10% of the average). Animals were housed in makrolon cages (41 × 25 × 15 cm) at two or three per cage, with a stainless steel wire top and a shallow layer of white wood shavings as bedding (Giuseppe Bordignon supplier, Treviso, Italy). All animals were kept in a single room at 23 ± 3 °C and 40–60% relative humidity. Lighting was artificial and the light/dark cycles were approximately 12 hours each. All animals were given the same standard “Corticella” pellet diet (Piccioni Laboratory, Milan, Italy). Feed and tap water were available ad libitum and were both periodically analyzed to exclude biological and chemical contamination (mycotoxins, pesticides, arsenic, lead, mercury, selenium).
There were four experimental groups: parous and age-matched nulliparous, treated with DEP or vehicle, olive oil. There were five animals per experimental group. Animals were weighed weekly to determine treatment dose. F0 generation corresponds to breeders of the experimental animals (F1). F1 animals were treated daily from postnatal day (PND) one to PND 28 through milk of their dams (F0), which were gavaged with DEP or vehicle. After weaning at PND 28, F1 animals were treated with DEP or vehicle by gavage three times a week until PND 180 and sacrificed at PND 181. Parous rats were mated (outbred) at PND 97 and treatment was continued through pregnancy, delivery of pups (F2) and lactation. At the time of sacrifice of parous rats on PND 181, F2 pups had completed weaning. Animals were euthanized via carbon dioxide inhalation and necropsy was immediately performed for collection of mammary tissues. Animals were sacrificed on the same day and at random during the estrous cycle.
Transcriptome profiling in rats
The fifth left and right caudal mammary glands were collected, pulverized in liquid nitrogen and total RNA was extracted using Maxwell 16 LEV simplyRNA Blood kit (Promega, WI) or by Direct-zol RNA MiniPrep kit (Zymo Research, CA). RNA concentration was determined using NanoDrop (Thermo Scientific, MA) and RNA quality was assessed using a 2100 Bioanalyzer (Agilent Technologies, CA); samples with RNA Integrity Number ≥ 7 were used for microarrays. Transcriptomes were profiled by GeneChip Rat Gene 2.0 ST arrays (Affymetrix, CA) at the Yale Center for Genome Analysis (Yale School of Medicine, CT) as described previously22. Quality control of .CEL files and pre-processing by robust multiarray average (RMA) method were done using expression console software (Affymetrix, CA). Batch effects due to RNA extraction method were removed using ComBat package23 in RStudio (R 3.0.2). We applied a signal intensity filter to retain only those probesets with high and stable expression (signal value >30th percentile in at least one experimental group). A variance-based filter was used to retain the top 50% of probesets with high interquartile range resulting in a final dataset containing 7,532 genes. Microarray data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE95554 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE95554)
Study population
We utilized resources from the Long Island Breast Cancer Study Project (LIBCSP), which includes a population-based sample of women newly diagnosed with first primary breast cancer who participated in the main study interview within two to three months of diagnosis24. Approval of the Institutional Review Board (Program for the Protection of Human Subjects) was obtained by all participating institutions and informed consent was obtained from all study participants. Study participants included 1,508 women diagnosed with first primary in situ or invasive breast cancer between 1996 and 1997 who resided in Nassau and Suffolk counties on Long Island, NY. At the time of the in-person baseline interview, 93% of participants donated 25 mL spot urine samples, which were shipped overnight on ice, processed, and banked at −20 °C. Archived tumor tissue of the first primary breast cancer was obtained from the diagnosing hospitals25. Demographic, reproductive and lifestyle characteristics of women with available pre-malignant adjacent tissue and urinary MEP concentrations (n = 294), are provided in Table 1.
Table 1.
Participant characteristics.
| n | Subset | Full |
|---|---|---|
| 294 | 892 | |
| Age at diagnosis: mean (SD) | 57.9 (12.7) | 59.3 (12.8) |
| Stage = invasive (%) | 262 (89.1) | 768 (86.1) |
| Race = white (%) | 274 (93.2) | 835 (93.6) |
| Menopausal status = postmenopausal (%) | 189 (65.6) | 605 (67.8) |
| Parity = 1 or more (%) | 254 (86.4) | 769 (86.2) |
| Lactated (%) | 96 (32.7) | 292 (32.7) |
| Age at menarche: mean (SD) | 12.6 (1.6) | 12.5 (1.6) |
| Age at first birth: mean (SD) | 25.7 (4.8) | 25.7 (4.9) |
| Oral contraceptive use (%) | 141 (48.0) | 377 (42.3) |
| Hormone replacement therapy use (%) | 82 (28.0) | 246 (27.6) |
| Current or past smoker (%) | 165 (56.1) | 485 (54.3) |
| Alcohol (%) | 180 (61.2) | 556 (62.3) |
| BMI: mean (SD) | 26.9 (6.1) | 26.9 (5.8) |
| Education: | ||
| ≤High School | 142 (48.5) | 443 (49.8) |
| College | 112 (38.2) | 316 (35.5) |
| Post-college | 39 (13.3) | 131 (14.7) |
| Income: | ||
| ≤$24,999 | 50 (19.5) | 175 (22.2) |
| $25,000-$49,999 | 90 (35) | 237 (30.2) |
| ≥$50,000 | 117 (45.5) | 374 (47.6) |
All LIBCSP women with urinary MEP measurements (n = 892) compared with subset with both urinary MEP measurement and DEP gene signature measurement in pre-malignant adjacent tissues (n = 294).
Tumor tissue was excised prior to treatment initiation. Formalin-fixed, paraffin-embedded (FFPE) tumor sections were histopathologically reviewed by a trained pathologist and the cancer tissue was separated using manual microdissection for 745 women, and histologically normal pre-malignant adjacent tissue was retrieved for 586 of them. Following RNA extraction and quality control, samples were cross-referenced with DEP exposure information to obtain the final number of samples used in this study: 294 pre-malignant adjacent tissues, of which 243 had a paired tumor tissue.
Monoethyl phthalate (MEP) was measured in spot urine samples donated by 892 women with breast cancer using standard laboratory techniques by staff at the Centers for Disease Control and Prevention. Briefly, metabolites were deconjugated enzymatically, matrix removal and analyte enrichment were accomplished by solid phase extraction, and instrumental analysis was done with high performance liquid chromatography–tandem mass spectrometry using isotope dilution quantification as described before26.
Gene expression profiling in LIBCSP
Genes found to be differentially expressed by DEP exposure in rat mammary tissues were converted to orthologous human IDs using the Gene Annotator tool in the rat genome database27, resulting in a panel of 107 genes. These genes were included as part of a custom-designed code-set (NanoString Technologies, WA) to determine expression of the DEP gene signature in breast tumor and pre-malignant adjacent tissues of the LIBCSP. Total RNA was extracted from FFPE breast tumor and adjacent tissues using the Qiagen miRNeasy FFPE kits (Qiagen, MD). RNA concentration and quality were determined using NanoDrop (Thermo Scientific, MA). Only those samples exceeding concentration of 10 ng/µL were used. 100 ng RNA was incubated with reporter and capture probes overnight at 65 °C. Following hybridization, unbound probes were removed, and the purified complexes were aligned and immobilized on imaging cartridges using an nCounter Prep station. Code count detection was carried out by scanning cartridges in an nCounter Digital Analyzer to determine gene expression levels. Raw nCounter code counts were first normalized against the geometric mean of spike-in control probes to minimize the impact of sample preparation and detection. For background determination, counts that fell below two standard deviations of the mean of negative control probes were deemed unexpressed and assigned a value equivalent to the background threshold divided by the square root of two. Finally, data were normalized against the geometric mean of 6 housekeeping genes with high and stable expression: CLTC, GAPDH, GUSB, HPRT1, PGK1 and TUBB. The final dataset was log2 transformed.
Statistical methods
Differential gene expression analysis in rats
Differential gene expression analysis between DEP and control in rats was performed using linear models for microarray data (limma) package28. To obtain a robust DEP gene signature, we bootstrapped samples by choosing four out of five samples in treatment groups and in control groups resulting in 25 rounds of differential gene expression analysis each for parous and for nulliparous groups. A lenient false discovery rate (FDR) of 25% using Benjamini-Hochberg (BH) correction29 and a fold change of ≥1.5 fold was used. For functional enrichment analysis of differentially expressed genes we used gene ontology (GO)30 via EnrichR31. Fisher’s exact test32 was used to assess significance of enrichment at a FDR of 5% by BH method.
Signature gene analysis in the LIBCSP
Gene expression data of pre-malignant adjacent tissues as well as urinary MEP concentration were available for 294 breast cancer cases. Examination of the association between urinary MEP concentration and expression of each gene in the DEP gene signature was carried out using generalized linear models33 before and after stratifying by menopausal status. Natural log transformed urinary MEP concentration was quartiled, and the median of each quartile was considered as the predictor. To reduce the dimensionality of the dataset, we performed factor analysis to group highly correlated genes into latent factors using the psych package34 with a correlation cutoff of 0.3 to load a gene into a factor. We subsequently carried out an examination of the association between quartiles of urinary MEP concentration with the latent factors, such as oral contraceptive use, hormone replacement therapy, lactation, smoking status, religion etc. Differential gene expression analysis between pre-malignant adjacent and corresponding tumors in this study (n = 243) as well as in the breast cancer TCGA microarray data (n = 61 tumors and adjacent samples) filtered to include only the DEP gene signature, was carried out using Wilcoxon signed rank test35. To gain insight about upstream molecular regulators of genes contained within factors, we performed enrichment for transcription factors using ‘ChEA 2016’ via Enrichr31 which uses Fisher’s exact test32. Multiple comparisons were adjusted using BH-based FDR of 5%.
A full analysis of the association between phthalate exposure and breast cancer incidence in the LIBCSP cases and population-based controls has been reported previously13.
Ethics approval and consent to participate
Animal studies were performed following the principles of Good Laboratory Practices and Standard Operating Procedures of the CMCRC/RI facility, which include authorization by the ethical committee; the study was approved by the Institutional Animal Care and Use Committee (IACUC) of Mount Sinai. For the Long Island Breast Cancer Study Project (LIBCSP), Institutional Review Board approval was obtained by all participating institutions and informed consent was obtained from all study participants; study was approved by the Institutional Review Board of Mount Sinai (Program for the Protection of Human Subjects). All experiments were performed in accordance with relevant guidelines and regulations.
Results
Identifying a DEP gene signature in rats
Our final dataset of ~7,500 genes was obtained after filtering the mammary microarray data to retain only those genes with both high expression and variation. We then performed differential gene expression analysis between DEP-treated rats and controls to identify a phthalate gene signature. To identify a robust signature, we bootstrapped samples in parous and nulliparous rats separately given the potential difference in mammary transcriptome. This resulted in 339 and 43 differentially expressed genes by DEP treatment in the parous and nulliparous rat groups, respectively. For the parous rat group, we ranked genes by their difference in median expression between treatment and control and selected the top 100 genes. Finally, we identified the human orthologs of the rat genes resulting in 91 orthologous human genes in the parous group (‘DEP parous gene signature’) and 34 orthologous human genes in the nulliparous group (‘DEP nulliparous gene signature’) (Supplementary Table 1). These gene signatures separated DEP-treated from the control groups (Fig. 1A,B). All genes in both parous and nulliparous signatures were down-regulated by DEP exposure, except for Rps16 which was up-regulated in the nulliparous rat group. Eighteen genes overlapped between the parous and nulliparous signature, resulting in a final gene panel of 107 genes (‘DEP gene signature’) (Fig. 1C). Pathway enrichment analysis of these 107 genes revealed enrichment of gene ontology (GO) terms ‘mammary gland alveolus development’ (TPH1, AREG, PRLR), ‘response to hypoxia’ (MUC1, ACAA2, CLDN3, TGFB3, ALDOC, ANGPTL4, CD24, TLR2), ‘response to steroid hormone’ (FOXA1, SLC34A2, AR, KRT19, TGFB3, PTGDS, CD24, AREG, TLR2, ABCG2) and ‘cellular amine metabolic process’ (SLC44A3, TPH1, SLC44A4, AADAT, PLA2G2A, MBOAT1) (FDR < 0.05).
Figure 1.

DEP gene signatures in adult (A) parous and (B) age-matched nulliparous Sprague-Dawley rats identified by limma analysis of bootstrapped samples (FDR < 0.25, fold change ≥1.5). (C) Overlap of parous and nulliparous gene signatures. A full list of gene names in each group is provided in Supplemental Information (Table S1).
DEP gene signature in humans
Among the LIBCSP case cohort, we have 294 subjects with both gene profile and urinary MEP measurement. We compared the full group of cases with urinary MEP information (n = 892) with the subset (n = 294) used in our investigation and found no major differences in clinical, demographic and reproductive factors (Table 1), nor did we detect any difference in MEP distribution between the two groups (Fig. 2).
Figure 2.
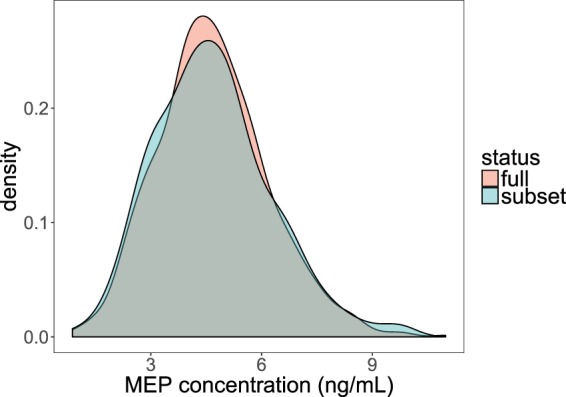
Comparative density distributions of monoethyl phthalate (MEP) concentrations between all samples with urinary MEP concentration (n = 892) and subset of samples with both MEP concentration and DEP gene signature measurement in pre-malignant adjacent breast tissues (n = 294) among LIBCSP case women.
We examined the expression of the 107 DEP gene signature in pre-malignant adjacent breast tissues and found that 85 out of the 107 genes were consistently expressed in over 75% of the 294 tissues examined. Univariate analyses were used to test the association between the expression of these 85 genes with quartiles of urinary MEP using generalized linear models. Overall, four genes were significantly associated with MEP (AREG, TMEM50B, CA8, C4ORF19) regardless of menopausal status. Given the different risk factor profiles associated with pre- and postmenopausal breast cancer36–38 we stratified our analysis by menopausal status and found that nine and three genes were significantly associated with MEP in the pre-malignant adjacent breast tissue among postmenopausal (n = 189) and premenopausal (n = 99) women, respectively (p < 0.05, Table 2, Fig. 3A). In postmenopausal women, six genes (AREG, TMC4, MPZL3, P4HA1, CA8, PLEKHA6) had decreasing expression with increasing levels of MEP, which was consistent with results from rats where we observed reduced gene expression levels with DEP exposure. Given the limited sample size in premenopausal women, we carried out subsequent analysis in postmenopausal women only. To reduce multiple comparisons and to take advantage of the strong correlation structure among genes, a dimensionality reduction technique, i.e. factor analysis, was carried out (Fig. 4). Factor analysis revealed seven loading factors among the genes in the DEP gene signature (Table 3), of which one of the factors, factor #2, was inversely associated with urinary MEP (p < 0.05, Fig. 3B). The six genes that were identified to be inversely associated with urinary MEP in univariate analysis loaded onto factor #2. We performed transcription factor enrichment analysis of all 20 genes that loaded into factor #2 to identify putative upstream molecules that may regulate these genes. Results revealed enrichment for several transcription factor binding sites including SOX2, AR, GATA1, FOXA1 and TBX20 among these genes (p < 0.05, Table 4). Genes contributing to enrichment of androgen receptor (AR) included SLC44A3, P4HA1, CXADR, AADAT, GALNT3 and ABCG2. We then restricted our analysis to AR + cases, which we defined as AR expression above the lower whisker of Tukey’s boxplot39 (n = 167) and found six more genes to be inversely associated with urinary MEP (Table 2).
Table 2.
Association between DEP gene signature and MEP concentration in pre-malignant adjacent breast tissues (p < 0.05) among LIBCSP case women.
| All (n = 294) | P-value | Beta | Postmenopausal (n = 189) | P-value | Beta | Postmenopausal with high AR expression (n = 167) | P-value | Beta | Premenopausal (n = 99) | P-value | Beta |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AREG | 0.014 | −0.14 | AREG | 0 | −0.22 | AREG | 0 | −0.244 | HMGCS2 | 0.006 | 0.41 |
| TMEM50B | 0.026 | 0.07 | TMC4 | 0.008 | −0.23 | SLC5A3 | 0.002 | 0.124 | CLDN8 | 0.023 | 0.26 |
| CA8 | 0.03 | −0.11 | TMEM50B | 0.008 | 0.11 | SPINT1 | 0.008 | −0.117 | MUC1 | 0.039 | 0.22 |
| C4ORF19 | 0.045 | 0.13 | MPZL3 | 0.013 | −0.17 | NAT8L | 0.01 | 0.262 | |||
| P4HA1 | 0.014 | −0.18 | TSPAN1 | 0.011 | −0.284 | ||||||
| CA8 | 0.015 | −0.16 | P4HA1 | 0.015 | −0.186 | ||||||
| SLC5A3 | 0.034 | 0.09 | TACSTD2 | 0.015 | −0.137 | ||||||
| PLEKHA6 | 0.039 | −0.15 | TMC4 | 0.016 | −0.22 | ||||||
| NAT8L | 0.045 | 0.20 | CA8 | 0.021 | −0.16 | ||||||
| SLC44A4 | 0.022 | −0.238 | |||||||||
| FXYD3 | 0.023 | −0.125 | |||||||||
| MPZL3 | 0.032 | −0.153 | |||||||||
| KRT18 | 0.036 | −0.098 | |||||||||
| PLEKHA6 | 0.037 | −0.161 | |||||||||
| TMEM50B | 0.039 | 0.075 |
Figure 3.

(A) Association between DEP gene signature and MEP concentration in pre-malignant adjacent breast tissues among postmenopausal women (n = 189; p < 0.05). (B) Association between factor 2 and MEP concentration (p < 0.05). Points on graph depict mean +/− standard error.
Figure 4.

Correlation structure of DEP gene signature in pre-malignant adjacent breast tissues among LIBCSP postmenopausal women, where genes are ordered by factors as indicated by numbers on top of figure.
Table 3.
Factor analysis of DEP gene signature in pre-malignant adjacent breast tissues among postmenopausal women revealed 7 latent factors.
| Factor | Genes loading onto factor |
|---|---|
| Factor 1 | FOXA1,AR,KRT19,TMEM50B,CBLC,TMEM30B,KRT18,PRSS8,WFDC2,ST6GALNAC2,MUC1,NAT8L,TGFB3,FAM134B,ICA1,HMGCS2,TFAP2B |
| Factor 2 | P4HA1,RNF180,MPZL3,TMC4,TMEM47,PRLR,PLEKHA6,ACAA2,TOM1L1,GALNT3, CA8,AADAT,AP1M2,MBOAT1,SLC44A3,CXADR,TNFRSF21,TLR2,AREG,ABCG2 |
| Factor 3 | RPS16,FXYD3,CD59,XBP1,SPINT2,IRX3,QSOX1,PTGDS,SLC5A3,TACSTD2,SH3YL1,CRABP2,MAL2 |
| Factor 4 | LCN2,SERPINB5,ANXA8L1,PLEKHB1,SLC34A2,PROM1,C4ORF19,CLDN8,SPINT1 |
| Factor 5 | SECTM1,CD14,PLIN2,CPVL,CD24,ALDOC,ANGPTL4,PPAP2C,ABHD14B |
| Factor 6 | AQP5,FOLR1,EPCAM,KCNK1,PDZK1IP1,CLDN3,RAB25,ESRP1,FRK,SLC12A2 |
| Factor 7 | TSPAN1,SLC44A4,F13A1 |
| No factor assignment | ASPN,CMTM8,PLA2G2A,TMEM184A |
Genes in bold are those that were significant at p < 0.05 from univariate analysis (Fig. 3), many of which load onto factor 2.
Table 4.
Transcription factor enrichment analysis of genes in factor #2 using ChIP-X Enrichment Analysis (ChEA) database via EnrichR.
| Transcription Factor | Contexta | Species | P-valueb | Adjusted P-value |
|---|---|---|---|---|
| SOX2 | SW620 | Human | 0.000 | 0.187 |
| AR | LNCAP | Human | 0.003 | 0.340 |
| SOX2 | STOMACH | Mouse | 0.011 | 0.519 |
| FOXA1 | ENDOMETRIOID-ADENOCARCINOMA | Human | 0.020 | 0.564 |
| GATA1 | MEL | Mouse | 0.031 | 0.623 |
| AR | VCAP | Human | 0.036 | 0.623 |
| TBX20 | HEART | Mouse | 0.043 | 0.623 |
| TBX20 | HEART | Mouse | 0.043 | 0.623 |
| GATA1 | MEL86 | Mouse | 0.043 | 0.623 |
| FOXA1 | PDAC | Human | 0.043 | 0.623 |
aRefers to cell-line or tissue in which the experiment was done.
bThe p-value is computed from a Fisher’s exact test (FET), a proportion test that assumes a binomial distribution and independence for probability of any gene belonging to any gene-set.
The top 5 transcription factors that appeared at least twice with a p-value < 0.05 are shown. ChEA is a gene-set enrichment analysis tool that tests if query gene-sets are enriched with genes that are putative targets of transcription factors, utilizing a gene-set library of transcription factors and their putative target genes curated from published ChIP-chip, ChIP-seq, and similar experiments (Lachmann et al. 2010).
We compared expression levels of 107 DEP signature genes in adjacent and their tumor counterparts (n = 243 pairs), and found that 33 genes were down-regulated and 34 genes were up-regulated in adjacent compared to corresponding tumor tissues at FDR < 0.05 (Fig. 5). In particular, all six genes found to be inversely associated with urinary MEP in adjacent tissues among postmenopausal women were also down-regulated in adjacent tissues compared to paired tumors. Results on four of these genes (P4HA1, TMC4, MPZL3, PLEKHA6) were also validated in the TCGA dataset (FDR < 0.05); however, AREG and CA8 showed the opposite trend.
Figure 5.
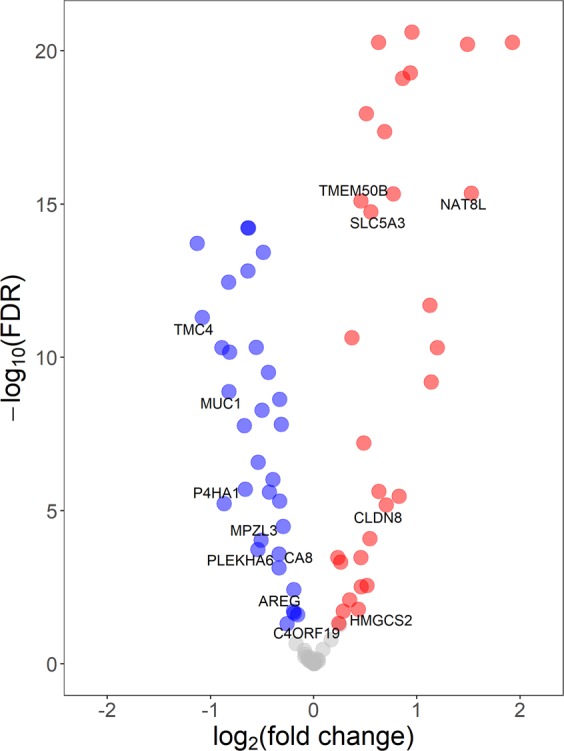
Genes significantly differentially expressed between paired tumor and adjacent breast tissues among LIBCSP postmenopausal women (FDR < 0.05). Blue indicates down-regulation and red indicates up-regulation in pre-malignant compared to tumor, respectively. Labels indicate genes found to be associated with MEP concentration (see Table 2).
Discussion
In the present study, we identified a DEP-induced gene signature using a rodent model and translated it to a population-based sample of women newly diagnosed with primary breast cancer, who had available pre-malignant adjacent and tumor tissue. To our knowledge, this is the first study of its kind to translate animal data to humans in an appropriate target tissue using a DEP dose relevant to human exposure. Rats have been widely used to study breast cancer since the pre-malignant stages of mammary cancer in rats closely recapitulate the human disease40.
We first identified a panel of 107 genes modified by DEP exposure in rats treated chronically from birth to adulthood using a human-level exposure that has previously been shown to result in urinary MEP concentrations within the range reported for the U.S. population17. By bootstrapping samples to overcome the limitation of small sample size for each experimental group (n = 5), we were able to show that even at a low, human level exposure, measurable changes in the transcriptome were detectable. The 107 modified genes in adult rats were predominantly down-regulated by DEP exposure and were enriched for biological processes such as ‘mammary gland alveolus development’. Interestingly, overexpression of at least eight genes among the DEP gene signature has been previously reported in breast cancer tissues; these include the genes FOXA141, PRLR42, TPH143, AREG44, AR45, XBP146, TGFB347 and HMGCS248. Coordinated down-regulated expression of these genes by DEP exposure in our study seemed to suggest an inverse relationship between DEP and breast cancer in animal models. While we could not obtain such information in our animal study (all rats were sacrificed by PND 181, which is too early for spontaneous tumor formation to occur), the suggestive inverse association in our population study13 seems to corroborate this postulation.
We next examined the DEP gene signature in pathologically non-malignant tissues because we hypothesized that gross genomic abnormalities in tumor tissues would mask the likely subtle effects of low-dose environmental chemical exposures. In particular we identified by univariate analysis, a subset of six out of 107 genes to be negatively associated with urinary MEP in pre-malignant adjacent tissues of postmenopausal women. The down-regulated expression of these six genes with urinary MEP in humans was consistent with direction of expression change observed in rats by DEP exposure. These six genes have diverse functions. P4HA1 encodes a component of prolyl 4-hydroxylase, a key enzyme in collagen synthesis; it was shown to be an activator of the HIF-1 pathway in breast cancer and biomarker of poor prognosis in various types of cancers including melanomas49, pancreatic cancer50 and head and neck squamous cell carcinomas51. MPZL3 (Myelin protein zero like 3) is involved in mediating cell adhesion but little is known about its possible role in cancer pathways. TMC4 encodes a transmembrane channel protein52; while its expression was upregulated in tumor-versus-adjacent tissue in our dataset as well as TCGA, other studies reported its downregulation in high-metastatic breast cancer cell lines compared to their low metastatic counterparts53 as well as breast-cancer cells compared to normal breast epithelium54, and its increased expression in tumors was associated with better survival of breast cancer patients55. PLEKHA6 (Pleckstrin Homology Domain Containing 6) has been shown to be differentially methylated in head and neck cancer56; its high expression was shown to be a positive prognostic factor in lung57 and breast55,58 cancers. CA8 (Carbonic Anhydrase 8) encodes an inhibitor of inositol trisphosphate inhibitors which regulate intracellular calcium release fundamental to many cellular processes including mitochondrial energy production and cell fate59. AREG (Amphiregulin) is a member of the epidermal growth factor family that is expressed in the mammary gland during development and has been shown to promote the growth of normal epithelial cells while inhibiting the growth of aggressive carcinoma cell lines52.
The strong correlation structure underlying genes in adjacent tissues enabled us to use factor analysis to derive seven factors, clusters of highly correlated genes, of which one factor, factor 2, showed independent negative association with urinary MEP. Interestingly, all six genes identified by univariate analysis loaded into factor #2, indicating similar expression patterns of these genes. Furthermore, we showed that the six genes that were negatively associated with urinary MEP were also coordinately down-regulated in adjacent compared to paired tumor tissue, and four out of six of these genes were also down-regulated in adjacent compared to paired tumors of the TCGA dataset. Collectively, our results from humans and animals seem to suggest an inverse relationship between DEP exposure and breast cancer development. However, we acknowledge the cross-sectional nature of our study, where urinary MEP concentrations were obtained only at one time-point after breast cancer diagnosis, and may not necessarily reflect lifetime exposure to DEP or whether the exposure occurred prior to diagnosis of breast cancer. Additionally, there is no control (cancer-free) population in the study. Hence, whether phthalate-induced transcriptome changes modify breast cancer susceptibility needs to be further investigated to clarify the etiologic mechanism of phthalates in breast carcinogenesis. Design of the current study does not allow us to claim the direct link between DEP exposure and breast cancer risk. We also have to acknowledge a seeming discrepancy of our results with the studies of López-Carrillo et al.10 (higher urine levels of MEP associated with increased BC risk) and Sprague et al.11 (elevated serum levels of MEP associated with increased breast density) for which we currently don’t have an explanation.
To gain insight about upstream molecular regulators of genes associated with urinary MEP in postmenopausal tumor-adjacent breast tissues, we performed transcription factor enrichment analysis of the 20 genes belonging to factor 2 and observed enrichment for AR binding sites among these genes. The anti-androgenic activity of phthalates has been extensively reported, which include endpoints such as shorter anogenital distance and reduced testosterone levels5,60–62. While the mechanism of anti-androgenic activity of phthalates has been shown to be mediated by inhibition of testosterone synthesis leading to Leydig cell dysfunction62,63, several phthalates have been shown to be able to bind to AR weakly suggesting that phthalates may elicit their effects through both receptor- and non-receptor-mediated pathways64. Even though in our study AR was among the signature panel of genes disrupted by DEP in rats, its expression was not significantly altered with urinary MEP exposure in human tissues. However, we noted that among postmenopausal women with high AR expression, six more genes were significantly associated with urinary MEP, suggesting that AR could play a role in the association between phthalate exposure and gene expression.
Our results indicated stronger associations of DEP signature genes among adjacent tissues of postmenopausal women, and not premenopausal women; part of this could be attributed to the smaller sample size among premenopausal (n = 99) compared to postmenopausal (n = 189) women. Premenopausal breast cancers tend to be more aggressive36, be associated with poorer survival37 and have distinct molecular profiles compared to postmenopausal breast cancers38. There was no major difference in distribution of MEP concentrations between premenopausal and postmenopausal women in our dataset. Whether there are different environmental etiologies of premenopausal compared to postmenopausal breast cancers remains to be elucidated.
We found a larger number of genes modified among parous rats (339 genes) compared to nulliparous rats (43 genes) suggesting that parity may play a role in modifying the effects of phthalate exposure on the mammary gland transcriptome. The parous rats in our study completed pregnancy and lactation and were sacrificed 35 days after the end of lactation. Post-lactational involution occurs within a few days of end of lactation whereby apoptosis of secretory structures remodels the gland to its pre-pregnant ductal architecture resembling its virgin counterpart65. That we did not observe significant gene expression changes between parous and nulliparous rats suggests that the parous rats in our study had finished involution. Parity is known to reduce the risk of developing breast cancer66; however how environmental factors interact with parity to influence cancer susceptibility is unknown. Our results suggesting that phthalate exposure had a more profound effect on the transcriptome of parous compared to nulliparous rats warrants further investigation, for example, by examining rates of spontaneous mammary tumor formation among exposed and control groups later in life.
We acknowledge some limitations of our study. First is the translation of findings from rats to humans. While rats have been a useful model system to study breast cancer, important species-specific differences exist, including an accelerated lifespan leading to condensed mammary developmental timing in rats, six pairs of mammary glands in rats compared to one in humans67 as well as species-specific differences in response to environmental chemicals68. Nevertheless, Sprague-Dawley rats have been shown to be one of the most physiologically relevant and genetically defined animal models for studying human sporadic breast cancer69,70. For example, they share similar age-equivalent distribution of mammary tumors as in human population; mammary carcinomas from this animal model also share many morphological and molecular features, including estrogen-dependence, chromosomal instability, aneuploidy, and deregulation of cell cycle with breast cancer in human populations71. Additionally, we did not have a technical possibility to run additional assays such as in situ hybridization experiments or immunohistochemical staining to confirm the expression changes in the caudal mammary glands. Second is the translation of findings from normal mammary glands in rats to cancer-adjacent breast tissues in humans. Extratumoral benign-appearing tissues lying adjacent to breast tumors have been shown to harbor genomic alterations such as somatic loss of heterozygosity, methylation changes and shortened telomeres, and in some cases may have been infiltrated with tumor cells72 – the so-called “field effect” – making it harder to untangle the subtle effects of low-dose environmental chemical exposures on transcriptome pattern. Finally, the route of exposure used in animal studies involved oral gavage, while exposures to many environmental chemicals, including phthalates have been shown to occur via multiple routes other than oral, such as dermal or by inhalation62. However, we attempted to resolve this issue by calibrating the exposure dose to achieve similar urinary biomarker concentration in rats and humans.
Conclusion
Using an integrative and translational approach, we identified a DEP gene signature in an animal model which was partially validated in a human population. Functional relevance of the gene signature indicated by in silico pathway analysis further support the potential causal role of phthalate in breast carcinogenesis.
Supplementary information
Acknowledgements
The authors gratefully acknowledge the technical support of Dr. Antonia M. Calafat at National Center for Environmental Health, Center for Disease Control and Prevention for analyzing urinary metabolites of phthalates for this study and provide her critical review of the manuscript. This work was supported by the grants from the National Institutes of Health (U01 ES019459, UO1 CA/ES66572, P30ES009089 and K01 ES12645).
Author contributions
J.C., S.L.T., M.D.G., and F.M. designed the study; F.M., L.F., S.P., and F.B. carried out the animal experiments; K.G. performed RNA extraction and microarray profiling of rat samples; H.H., G.G., R.M.S. coordinated the transaction and provided histopathological review of LIBCSP samples; K.G. and V.N.A. performed gene expression profiling of LIBCSP samples; K.G. and V.N.A. analyzed the data; H.P. provided expertise on phthalate exposure and breast cancer survival. All authors contributed to writing and reviewing the manuscript.
Data availability
Microarray data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE95554.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kalpana Gopalakrishnan and Vasily N. Aushev.
Supplementary information
is available for this paper at 10.1038/s41598-020-63904-w.
References
- 1.National Research Council. Phthalates and Cumulative Risk Assessment: The Tasks Ahead., 10.17226/12528 (2008). [PubMed]
- 2.Witorsch RJ, Thomas JA. Personal care products and endocrine disruption: A critical review of the literature. Crit. Rev. Toxicol. 2010;40(Suppl 3):1–30. doi: 10.3109/10408444.2010.515563. [DOI] [PubMed] [Google Scholar]
- 3.Philippat C, Bennett D, Calafat AM, Picciotto IH. Exposure to select phthalates and phenols through use of personal care products among Californian adults and their children. Environ. Res. 2015;140:369–376. doi: 10.1016/j.envres.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CDC. CDC - National Report on Human Exposure to Environmental Chemicals - NER. https://www.cdc.gov/exposurereport/index.html (2017).
- 5.Swan SH, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ. Health Perspect. 2005;113:1056–1061. doi: 10.1289/ehp.8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duty SM, et al. The relationship between environmental exposures to phthalates and DNA damage in human sperm using the neutral comet assay. Environ. Health Perspect. 2003;111:1164–1169. doi: 10.1289/ehp.5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hauser R, et al. DNA damage in human sperm is related to urinary levels of phthalate monoester and oxidative metabolites. Hum. Reprod. Oxf. Engl. 2007;22:688–695. doi: 10.1093/humrep/del428. [DOI] [PubMed] [Google Scholar]
- 8.Main KM, et al. Human breast milk contamination with phthalates and alterations of endogenous reproductive hormones in infants three months of age. Environ. Health Perspect. 2006;114:270–276. doi: 10.1289/ehp.8075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomsen AML, et al. Female exposure to phthalates and time to pregnancy: a first pregnancy planner study. Hum. Reprod. Oxf. Engl. 2017;32:232–238. doi: 10.1093/humrep/dew291. [DOI] [PubMed] [Google Scholar]
- 10.López-Carrillo L, et al. Exposure to phthalates and breast cancer risk in northern Mexico. Environ. Health Perspect. 2010;118:539–544. doi: 10.1289/ehp.0901091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sprague BL, et al. Circulating serum xenoestrogens and mammographic breast density. Breast Cancer Res. BCR. 2013;15:R45. doi: 10.1186/bcr3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyd NF, et al. Mammographic density and the risk and detection of breast cancer. N. Engl. J. Med. 2007;356:227–236. doi: 10.1056/NEJMoa062790. [DOI] [PubMed] [Google Scholar]
- 13.Parada H, et al. Urinary Phthalate Metabolite Concentrations and Breast Cancer Incidence and Survival following Breast Cancer: The Long Island Breast Cancer Study Project. Environ. Health Perspect. 2018;126:047013. doi: 10.1289/EHP2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer. 2012;12:381–385. doi: 10.1038/nrc3263. [DOI] [PubMed] [Google Scholar]
- 15.Moral R, et al. In utero exposure to butyl benzyl phthalate induces modifications in the morphology and the gene expression profile of the mammary gland: an experimental study in rats. Environ. Health Glob. Access Sci. Source. 2011;10:5. doi: 10.1186/1476-069X-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moral R, et al. The plasticizer butyl benzyl phthalate induces genomic changes in rat mammary gland after neonatal/prepubertal exposure. BMC Genomics. 2007;8:453. doi: 10.1186/1471-2164-8-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teitelbaum SL, et al. Paired Serum and Urine Concentrations of Biomarkers of Diethyl Phthalate, Methyl Paraben, and Triclosan in Rats. Environ. Health Perspect. 2016;124:39–45. doi: 10.1289/ehp.1409586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brown D, Butterworth KR, Gaunt IF, Grasso P, Gangolli SD. Short-term oral toxicity study of diethyl phthalate in the rat. Food Cosmet. Toxicol. 1978;16:415–422. doi: 10.1016/S0015-6264(78)80258-2. [DOI] [PubMed] [Google Scholar]
- 19.Moody DE, Reddy JK. Hepatic peroxisome (microbody) proliferation in rats fed plasticizers and related compounds. Toxicol. Appl. Pharmacol. 1978;45:497–504. doi: 10.1016/0041-008X(78)90111-4. [DOI] [PubMed] [Google Scholar]
- 20.Oishi S, Hiraga K. Testicular atrophy induced by phthalic acid esters: effect on testosterone and zinc concentrations. Toxicol. Appl. Pharmacol. 1980;53:35–41. doi: 10.1016/0041-008X(80)90378-6. [DOI] [PubMed] [Google Scholar]
- 21.Manservisi F, et al. Effect of maternal exposure to endocrine disrupting chemicals on reproduction and mammary gland development in female Sprague-Dawley rats. Reprod. Toxicol. Elmsford N. 2015;54:110–119. doi: 10.1016/j.reprotox.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gopalakrishnan K, et al. Changes in mammary histology and transcriptome profiles by low-dose exposure to environmental phenols at critical windows of development. Environ. Res. 2017;152:233–243. doi: 10.1016/j.envres.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinforma. Oxf. Engl. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gammon MD, et al. The Long Island Breast Cancer Study Project: description of a multi-institutional collaboration to identify environmental risk factors for breast cancer. Breast Cancer Res. Treat. 2002;74:235–254. doi: 10.1023/A:1016387020854. [DOI] [PubMed] [Google Scholar]
- 25.Mordukhovich I, et al. Associations between polycyclic aromatic hydrocarbon-related exposures and p53 mutations in breast tumors. Environ. Health Perspect. 2010;118:511–518. doi: 10.1289/ehp.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato K, Silva MJ, Needham LL, Calafat AM. Determination of 16 phthalate metabolites in urine using automated sample preparation and on-line preconcentration/high-performance liquid chromatography/tandem mass spectrometry. Anal. Chem. 2005;77:2985–2991. doi: 10.1021/ac0481248. [DOI] [PubMed] [Google Scholar]
- 27.Shimoyama M, et al. The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 2015;43:D743–750. doi: 10.1093/nar/gku1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple. Testing. J. R. Stat. Soc. Ser. B Methodol. 1995;57:289–300. [Google Scholar]
- 30.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fisher RA. On the Interpretation of χ2 from Contingency Tables, and the Calculation of P. J. R. Stat. Soc. 1922;85:87–94. doi: 10.2307/2340521. [DOI] [Google Scholar]
- 33.Nelder JA, Wedderburn RWM. Generalized Linear Models. J. R. Stat. Soc. Ser. Gen. 1972;135:370–384. doi: 10.2307/2344614. [DOI] [Google Scholar]
- 34.Revelle, W. R. psych: Procedures for Personality and Psychological Research. Northwest. Univ. Evanst. Ill. USA (2017).
- 35.Wilcoxon F. Individual Comparisons by Ranking Methods. Biom. Bull. 1945;1:80–83. doi: 10.2307/3001968. [DOI] [Google Scholar]
- 36.Assi HA, et al. Epidemiology and prognosis of breast cancer in young women. J. Thorac. Dis. 2013;5:S2–S8. doi: 10.3978/j.issn.2072-1439.2013.05.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fredholm H, et al. Breast cancer in young women: poor survival despite intensive treatment. Plos One. 2009;4:e7695. doi: 10.1371/journal.pone.0007695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao S, et al. The molecular landscape of premenopausal breast cancer. Breast Cancer Res. BCR. 2015;17:104. doi: 10.1186/s13058-015-0618-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tukey, J. W. Exploratory Data Analysis. (Addison-Wesley Publishing Company, 1977).
- 40.Russo J, et al. Comparative study of human and rat mammary tumorigenesis. Lab. Investig. J. Tech. Methods Pathol. 1990;62:244–278. [PubMed] [Google Scholar]
- 41.Bernardo GM, et al. FOXA1 represses the molecular phenotype of basal breast cancer cells. Oncogene. 2013;32:554–563. doi: 10.1038/onc.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swaminathan G, Varghese B, Fuchs SY. Regulation of prolactin receptor levels and activity in breast cancer. J. Mammary Gland Biol. Neoplasia. 2008;13:81–91. doi: 10.1007/s10911-008-9068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pai VP, Marshall AM, Hernandez LL, Buckley AR, Horseman ND. Altered serotonin physiology in human breast cancers favors paradoxical growth and cell survival. Breast Cancer Res. BCR. 2009;11:R81. doi: 10.1186/bcr2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baillo A, Giroux C, Ethier SP. Knock-down of amphiregulin inhibits cellular invasion in inflammatory breast cancer. J. Cell. Physiol. 2011;226:2691–2701. doi: 10.1002/jcp.22620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cochrane DR, et al. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. BCR. 2014;16:R7. doi: 10.1186/bcr3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen X, et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature. 2014;508:103–107. doi: 10.1038/nature13119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghellal A, et al. Prognostic significance of TGF beta 1 and TGF beta 3 in human breast carcinoma. Anticancer Res. 2000;20:4413–4418. [PubMed] [Google Scholar]
- 48.Gromov P, et al. FABP7 and HMGCS2 are novel protein markers for apocrine differentiation categorizing apocrine carcinoma of the breast. PloS One. 2014;9:e112024. doi: 10.1371/journal.pone.0112024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eriksson, J. et al. Prolyl 4-hydroxylase subunit alpha 1 (P4HA1) is a biomarker of poor prognosis in primary melanomas and its depletion inhibits melanoma cell invasion and disrupts tumor blood vessel walls. Mol. Oncol., 10.1002/1878-0261.12649 (2020). [DOI] [PMC free article] [PubMed]
- 50.Cao XP, Cao Y, Li WJ, Zhang HH, Zhu ZM. P4HA1/HIF1α feedback loop drives the glycolytic and malignant phenotypes of pancreatic cancer. Biochem. Biophys. Res. Commun. 2019;516:606–612. doi: 10.1016/j.bbrc.2019.06.096. [DOI] [PubMed] [Google Scholar]
- 51.Li, Q. et al. High P4HA1 expression is an independent prognostic factor for poor overall survival and recurrent-free survival in head and neck squamous cell carcinoma. J. Clin. Lab. Anal. e23107, 10.1002/jcla.23107 (2019). [DOI] [PMC free article] [PubMed]
- 52.Rebhan M, Chalifa-Caspi V, Prilusky J, Lancet D. GeneCards: a novel functional genomics compendium with automated data mining and query reformulation support. Bioinforma. Oxf. Engl. 1998;14:656–664. doi: 10.1093/bioinformatics/14.8.656. [DOI] [PubMed] [Google Scholar]
- 53.Pandit TS, et al. Lymphatic metastasis of breast cancer cells is associated with differential gene expression profiles that predict cancer stem cell-like properties and the ability to survive, establish and grow in a foreign environment. Int. J. Oncol. 2009;35:297–308. [PubMed] [Google Scholar]
- 54.Liu R, et al. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- 55.Aushev VN, et al. Tumor expression of environmental chemical-responsive genes and breast cancer mortality. Endocr. Relat. Cancer. 2019;26:843–851. doi: 10.1530/ERC-19-0357. [DOI] [PubMed] [Google Scholar]
- 56.Langevin SM, et al. Peripheral blood DNA methylation profiles are indicative of head and neck squamous cell carcinoma: an epigenome-wide association study. Epigenetics. 2012;7:291–299. doi: 10.4161/epi.7.3.19134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Relli V, Trerotola M, Guerra E, Alberti S. Distinct lung cancer subtypes associate to distinct drivers of tumor progression. Oncotarget. 2018;9:35528–35540. doi: 10.18632/oncotarget.26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Möllerström E, et al. High-resolution genomic profiling to predict 10-year overall survival in node-negative breast cancer. Cancer Genet. Cytogenet. 2010;198:79–89. doi: 10.1016/j.cancergencyto.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Zhuang GZ, et al. Carbonic anhydrase-8 regulates inflammatory pain by inhibiting the ITPR1-cytosolic free calcium pathway. PloS One. 2015;10:e0118273. doi: 10.1371/journal.pone.0118273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kwack SJ, Kim KB, Kim HS, Lee BM. Comparative toxicological evaluation of phthalate diesters and metabolites in Sprague-Dawley male rats for risk assessment. J. Toxicol. Environ. Health A. 2009;72:1446–1454. doi: 10.1080/15287390903212923. [DOI] [PubMed] [Google Scholar]
- 61.Lee BM, Koo HJ. Hershberger assay for antiandrogenic effects of phthalates. J. Toxicol. Environ. Health A. 2007;70:1365–1370. doi: 10.1080/15287390701432285. [DOI] [PubMed] [Google Scholar]
- 62.Swan SH. Environmental phthalate exposure in relation to reproductive outcomes and other health endpoints in humans. Environ. Res. 2008;108:177–184. doi: 10.1016/j.envres.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gray, L. E. et al. Adverse effects of environmental antiandrogens and androgens on reproductive development in mammals. Int. J. Androl. 29, 96–104; discussion 105–108 (2006). [DOI] [PubMed]
- 64.Fang H, et al. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem. Res. Toxicol. 2003;16:1338–1358. doi: 10.1021/tx030011g. [DOI] [PubMed] [Google Scholar]
- 65.Watson CJ. Key stages in mammary gland development - Involution: apoptosis and tissue remodelling that convert the mammary gland from milk factory to a quiescent organ. Breast Cancer Res. 2006;8:203. doi: 10.1186/bcr1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Russo J, Moral R, Balogh GA, Mailo D, Russo IH. The protective role of pregnancy in breast cancer. Breast Cancer Res. 2005;7:131–142. doi: 10.1186/bcr1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neville, M. C. & Daniel, C. W. The Mammary Gland: Development, Regulation, and Function. (Springer, 1987).
- 68.Habert R, et al. Concerns about the widespread use of rodent models for human risk assessments of endocrine disruptors. Reprod. Camb. Engl. 2014;147:R119–129. doi: 10.1530/REP-13-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maltoni C, Minardi F, Pinto C, Belpoggi F, Bua L. Results of three life-span experimental carcinogenicity and anticarcinogenicity studies on tamoxifen in rats. Ann. N. Y. Acad. Sci. 1997;837:469–512. doi: 10.1111/j.1749-6632.1997.tb56895.x. [DOI] [PubMed] [Google Scholar]
- 70.Maltoni C, Minardi F, Pinto C, Soffritt M, Mobiglia A. Long term bioassays on the toxic, carcinogenic and tumour chemopreventive effects (particularly on mammary carcinomas) of Tamoxifen on Sprague-Dawley rats: ongoing studies and early results. Acta Oncol. 1990;11:289–305. [Google Scholar]
- 71.Maltoni, C., Minardi, F. & Soffritt, M. Chemoprevention of experimental mammary cancer by tamoxifen. in Progress and Perspectives in Chemoprevention of Cancer. (eds. De Palo, G. & Sporn, M. & Veronesi, U.) 23–45 (1992).
- 72.Troester MA, et al. DNA defects, epigenetics, and gene expression in cancer-adjacent breast: a study from The Cancer Genome Atlas. NPJ Breast Cancer. 2016;2:16007. doi: 10.1038/npjbcancer.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number GSE95554.