

Abstract
Parallel β-sheet-containing repeat proteins often display a structural motif in which conserved asparagines form a continuous ladder buried within the hydrophobic core. In such “asparagine ladders”, the asparagine side-chain amides form a repetitive pattern of hydrogen bonds with neighboring main chain NH and CO groups. Although asparagine ladders have been speculated to be important for stability, there is little experimental evidence to support such speculation. Here we test the contribution of a minimal asparagine ladder from the leucine-rich repeat protein pp32 to stability and investigate lattice rigidity and hydrogen bond character using solution NMR spectroscopy. Point substitutions of the two ladder asparagines of pp32 are strongly destabilizing and decrease the cooperativity of unfolding. The chemical shifts of the ladder side-chain HZ protons are shifted significantly downfield in the NMR spectrum and have low temperature coefficients, indicative of strong hydrogen bonding. In contrast, the HE protons are shifted upfield and have temperature coefficients close to zero, suggesting an asymmetry in hydrogen bond strength along the ladder. Ladder NH2 groups have weak 1H-15N cross-peak intensities; 1H-15N NOE and 15N CPMG experiments show this to be the result of high rigidity. Hydrogen exchange measurements demonstrate that the ladder NH2 groups exchange very slowly, with rates approaching the global exchange limit. Overall, these results show that the asparagine side-chains are held in a very rigid, non-dynamic structure, making a significant contribution to overall stability. In this regard, buried asparagine ladders can be considered “second backbones” within the cores of their elongated β-sheet host proteins.
Graphical Abstract
Introduction
Networks of polar interactions in protein interiors are common structural motifs that play important roles in stabilizing proteins 1–3. The unusual environments of these interior polar networks are likely to impart unusual energetic features on the networks, and have long been recognized as contributing to communication processes such as allostery 4. Determining the contributions of individual residues to folding and/or stabilizing the native states of proteins is not straightforward, as many studies can attest 5–7. An even greater challenge lies in identifying collective networks of residues whose interactions are key to protein cooperativity, allostery, and function. Because these networks are defined by sequence information at multiple positions, understanding interaction networks in proteins often requires a large amount of sequence information 8,9, accurate sequence alignments 10,11, sophisticated analyses 12–14, and creation and experimental characterization of large numbers of variants 15–17 compared to studies focused on single-site contributions.
Although it can be difficult to identify functionally relevant polar residues (specifically, those enhancing stability) and their interaction networks, there are some cases where key residues and interaction networks can be inferred using structural data and sequence conservation alone. One such example is the linear array of asparagine residues, often called an asparagine ladder, found in leucine-rich repeat (LRR) proteins. Asparagine ladders are highly conserved both in sequence and in structure, and are entirely buried in the hydrophobic interior of LRR proteins despite the polar character of their eponymous asparagine residue (Figure 1 and S1). Compared to other networks of interactions, the LRR asparagine ladder is an attractive target for experimental study since it is composed of the same residue (asparagine) at the same position within adjacent repeats, reflecting a simple symmetry across the network. Furthermore, the asparagine ladder is likely to be tightly coupled via hydrogen bonds (Figure 1A), providing a unique opportunity to study how a highly conserved network contributes to protein stability and cooperativity. A study of the asparagine ladder in LRR proteins may also provide a better understanding of asparagine/glutamine ladder architectures in other contexts, perhaps most notably in the amyloid protein aggregates responsible for neurodegenerative pathologies like Alzheimer’s (Aβ), Parkinson’s (α-synuclein), and Huntington’s (huntingtin) diseases 18
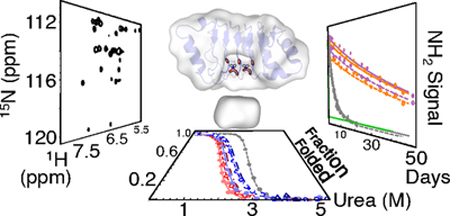
Figure 1. The structure and sequence of pp32 and its asparagine ladder.

(A) The hydrogen bond network of the side chain NHz groups of asparagines 74 and 98. Potential hydrogen bonds to backbone carbonyls identified in the crystal structure are shown as dashed lines. (B) Structure of pp32 (PDB ID: 4XOS) with substituted residues shown as sticks. Repeats are colored from blue (N-terminus) to red (C-terminus). Key residues in this study are colored as follows: T49, purple; N74 and N98, gray; L69, green; C123, orange; Y131 and D146, blue. (C) The canonical LRR motif and the pp32 sequence separated into caps and repeats. Residues are aligned based on the HMM logo in Figure S1; minor shifts in the alignment were introduced based on the three-dimensional structure of pp32. Positions corresponding to the LRR motif are highlighted gray.
In this study, we examine the role of the asparagine ladder in LRR folding and evaluate the behavior of its hydrogen bond network. Using the LRR protein pp32, we determined the thermodynamic contribution of the asparagine ladder to global stability and cooperativity using urea-induced unfolding monitored by circular dichroism. To explore the structural and dynamic features of the hydrogen bond network in pp32, we used NMR spectroscopy to measure chemical shifts, 15N dynamics of the backbone and asparagine side chains, temperature coefficients, and hydrogen exchange rates of asparagine ladder side chains compared to solvent-exposed asparagine side-chains. Our results show that the asparagine ladder plays an important role in stabilizing the LRR fold, and its disruption causes substantial changes to secondary structure and loss of stability. Furthermore, thermodynamic parameters indicate the asparagine ladder contributes to more cooperativity than the conserved leucine residues that define the LRR motif 19–21. NMR chemical shifts and temperature coefficients suggest that hydrogen bonds formed by NH2 groups of ladder asparagine side chains are stronger between repeats than they are within repeats. These data indicate the asparagine ladder acts like a second backbone, maintaining the LRR fold and supporting repeat-to-repeat interactions that promote global cooperativity.
Materials and Methods
Protein Cloning, Expression, and Purification.
Protein expression and purification were performed as described in 22. Mutated versions of the pp32 gene were generated using the Quikchange mutagenesis kit (Agilent Technologies). For 15N (and 13C) NMR samples, bacteria were grown in M9 minimal media supplemented with 15NH4Cl (and 13C glucose). Protein preparation was performed as described in 22. NMR samples were dialyzed into 20 mM NaPO4, 50 mM NaCl, 0.1 mM TCEP, pH 6.8 prior to data acquisition.
For side chain dynamics and E.COSY experiments, labeled protein was partially deuterated by unfolding in 50% 2H denaturant buffer (8 M Urea, 50 mM NaCl, 0.1 mM TCEP, 20 mM NaPO4 pH 6.8 (after adjustment for deuterium)) for at least 30 minutes to allow for complete exchange. 50% 2H refolding buffer (50 mM NaCl, 0.1 mM TCEP, 20 mM NaPO4 pH 6.8 (after adjustment for deuterium)) was then added to denatured samples to refold the protein and samples were concentrated using filtration columns (GE Healthcare). Residual denaturant was removed with a spin column 23.
Circular dichroism spectra and equilibrium unfolding.
All CD experiments were performed on an Aviv Model 400 CD spectropolarimeter using a computer-controlled Hamilton Microlab syringe titrator with samples in CD buffer (20 mM NaPO4, 150 mM NaCl, 0.1 mM TCEP, pH 7.8) at 20 °C. CD spectra were collected with 5 second signal averaging every nm from 280 to 200 nm; protein concentrations were 30 to 50 μM in a 0.1 cm path-length quartz cuvette. Equilibrium unfolding experiments were monitored at 220 nm using a 5-minute equilibration time and 30 second signal averaging; protein concentrations were 3–5 μM in a 1 cm path length quartz cuvette. Urea (VWR Life Sciences) used for denaturation studies was deionized using a mixedbed resin (BioRad) immediately prior to use; urea concentrations were determined using refractometry 24. Two-state analysis of equilibrium unfolding experiments was performed as described in 25. Errors in thermodynamic parameters from equilibrium unfolding experiments are from three independent experiments.
NMR spectroscopy.
Backbone resonances of wild-type and variant pp32 proteins were assigned as in 22 using a Bruker Avance II 600 MHz spectrometer equipped with a cryoprobe. Triple-resonance experiments (HNCA, HNCACB, CBCA(CO)NH, HNCO, 15N-edited 1H-1H NOESY) along with wild-type pp32 assignments 22 were used to assign variant proteins. Assignments were made using the CARA program 26.
Side-chain assignments were determined using NH2-filtered HSQC, CBCGCO 27, and CγO-coupled NH2-HSQC/HNCO E.COSY experiments 28–30. Side chain Cγ assignments were made using CBCGCO spectra in conjunction with Cβ assignments from backbone triple resonance experiments. Asparagine Cγ assignments were then linked to side chain HE/Z proton pairs with an NH2-filtered 1H-13C HSQC. Stereospecific assignments of asparagine HE and HZ resonances were determined from J-coupling values measured in E.COSY experiments (see Results) and were confirmed using proton-proton NOEs. All side chain assignments were made using Sparky 31. Wild-type (except for stereospecific assignments), Y131F/D146L (YD), and L69A side chain assignments were inferred by comparison with C123N, and T49L spectra.
Chemical shift perturbations (CSPs) were calculated as the weighted Euclidean distance between wild-type and variant chemical shifts (δH/NWT, δH/NVar) using the equation
| (1) |
where 0.14 is a weighting factor to normalize δN to δH 32.
15N transverse relaxation rates with attenuated exchange contributions (R2*) were determined using 2D constant-time (CT) CPMG experiments 33. For two-point relaxation dispersion experiments νcpmg was set to 200 Hz (νcpmg = 1/4 τcp, τcp = 1.25 ms) or 1 kHz (τcp = 250 μs) to observe relaxation with maximal and minimal effects of chemical exchange, respectively. R2* values were determined using νccpmg of 1 kHz to measure exchange-minimized relaxation of backbone and side-chain amides on the ps-ns timescale, with R2* values calculated using eq 1 from33.
1H-15N NOE experiments were performed using the pulse sequence in 34. The ratio between spectra with and without 1H saturation was used to determine 1H-15N NOE effects.
Temperature coefficients.
Chemical shift values used to determine temperature coefficients for wild-type and all peripheral variants were obtained from 1H, 15N-HSQC spectra using a Bruker Avance 600 MHz spectrometer equipped with a cryoprobe. Additional chemical shift data for the T49L variant was obtained from TROSY spectra of an 15N-labelled, 50% 2H T49L sample using a Varian 800 MHz spectrometer equipped with a room temperature triple-resonance probe. HSQC spectra were collected at 10, 15, 20, and 30 °C; TROSY spectra were collected at 5 °C intervals from 10 to 35 °C. Peak assignments for each variant were determined by referencing NH2 peaks from the 20 °C spectrum to the NH2 assignments of wild-type, C123N, and T49L pp32; peaks could then be tracked by overlaying all spectra from the temperature series. Chemical shifts for resolvable amides were fit to a linear model. Amides that were unresolved at any temperature were excluded from the analysis.
Hydrogen exchange of asparagine side-chain NH2 groups.
Hydrogen exchange rates from side-chain amides were measured using two types of experiments. To obtain an overall exchange rates for wild-type and variant pp32 constructs, we used NH2-filtered HSQC experiments. This approach has the advantage that it suppresses slow-exchanging resonances from amide NH groups. In this experiment, decrease in NH2 signal intensity results from exchange of either the HZ or the HE with a deuteron. As such, the apparent rate constant is the sum of the exchange rate constants for the HZ and HE protons. In addition, we used unfiltered 1H-15N HSQC spectra from the backbone hydrogen exchange studies of Dao et al [35] to resolve the exchange rates of the HE and HZ protons for wild-type pp32.
For the NH2-filtered exchange measurements, exchange was initiated as described in 21. A spin column was packed with 2 mL of pre-swollen G-25 fine Sephadex (GE Healthcare) and was washed three times with 2 mL water followed by three 2 mL washes with hydrogen exchange buffer (150 mM NaPO4, 50 mM NaCl, 0.1 mM TCEP, 100% D2O, pH 6.8 (after accounting for D2O)). Samples were applied to the equilibrated column, collected by centrifugation, and were immediately placed into a Bruker Avance 600 MHz spectrometer at 30 °C. NH2-filtered HSQC spectra were recorded every 30 minutes for up to 4 hours. After 4 hours, spectra were recorded at longer intervals (hours or days); samples were immersed in a 30 °C water bath in between spectra.
Exchange rates for NHD species were obtained from data collected in 21. Assignments were made by comparison to the wild-type NH2-filtered HSQC spectrum determined in the present study. Since there is substantial overlap between the NH2 and NHD peaks for both HZ and HE protons, peak heights were obtained by fitting cross peaks using a single two-dimensional lorentzian functions. The quality of fits was determined by visual inspection of 1D slices in the 1H and 15N dimensions; when fitting with a single lorentzian results in systematic residuals, a second lorentizan was included in the fit.
The decay of NH2-filtered NH2 peak heights during hydrogen exchange experiments was fitted with a single-exponential decay using the equation
| (2) |
where I(t) is the peak height at time t, A is the initial peak height (at t = 0) above the baseline B at t = 0, and kex is the rate constant for exchange. The build-up and subsequent decay of unfiltered asparagine 74 (NDE)HZ peak heights was fitted with the double exponential eq 3 below. The build-up of unfiltered asparagine 98 (NDE)HZ and (NDZ)HE peak heights were globally fitted with eqs 3 and 4,
| (3) |
| (4) |
where IZ(t) and IE(t) are the peak heights for the (NDE)HZ and (NDZ)HE isotopomers respectively, kex,HE and kex,HZ are the rate constants for hydrogen exchange of HE and HZ respectively and are shared globally during the fit, and AZ and AE are the initial peak heights of the (NHE)HZ and (NHZ)HE isotopomers above the baselines BZ and BE, respectively. Protection factors (PFs) were calculated using the formula
| (5) |
where ki is the intrinsic exchange rate constant for an asparagine side chain and kex is a fitted rate constant from eqs 2, 3, or 3 and 4. The ki value was determined from a linear fit of the temperature dependence of rate constants for asparagine side chain exchange from previous studies. 35–37. To account for potential sample degradation and spectrometer drift over the long exchange times of the wild-type and stabilizing variants (T49L and C123N), peak heights were normalized against non-exchangeable methyl peaks over the course of the experiment. For rapidly exchanging variants (L69A and YD, < 24 hours), normalization was not necessary.
For hydrogen exchange measurements on asparagine 74 in the L69A variant, we were only able to measure a few spectra before the NH2 signal intensity decayed to the baseline. Thus, we fixed the A parameter from eq 2 due to a lack of data in the decay portion of the curve. A was fixed at the average value from the other asparagine 74 hydrogen exchange experiments.
Results
The structure and sequence features of asparagine ladders in LRR proteins.
In the asparagine ladder motif of LRR proteins, the primary amide side chains of asparagines are completely buried within the nonpolar environment of the protein core. Owing to the high hydrogen bonding capacity of primary amides and their capacity to donate and accept hydrogen bonds equally (donating two hydrogen bonds from the NH2 group and accepting two hydrogen bonds to the carbonyl oxygen), these buried asparagine side chains are likely to form extended networks of hydrogen bonds. Crystal structures of LRR proteins show that this is accomplished through hydrogen bonds to the amide groups of peptide bonds (Figure 1A), wherein two peptide amide NH groups donate hydrogen bonds to each asparagine CγO group, and two peptide carbonyl oxygens accept hydrogen bonds from each asparagine NH2 group. Though other polar and charged protein side-chains (serine, threonine, histidine, arginine, aspartate and glutamate) have high hydrogen bonding capacities, there is a clear preference for asparagine at the ladder position of typical LRR repeats (84 percent of repeats, Figure S1). Only cysteine, serine, and threonine occur at the ladder position with frequencies greater than 1 percent (9.3, 1.5, and 1.4 percent), implying that the stereochemistry and hydrogen-bond patterning of the asparagine side-chain is well matched for the ladder environment.
Owing to the repetitive architecture of LRR proteins, analogous peptide groups from each repeat participate in hydrogen bonding to each buried asparagine. Three of these hydrogen bonds cross the interfaces between adjacent repeats. Specifically, the backbone NH donor groups to each asparagine CγOδ1 and one of the carbonyl acceptor groups to each asparagine NHZ are from the previous repeat (residue i-24 and i-22 for donors, residue i-27 for acceptor; Figure 1A). The fourth hydrogen bond is between the asparagine ladder NHE and the backbone CO of residue i-3 in the same repeat; in addition, this backbone CO also acts as the i-27 donor to the asparagine in the neighboring C-terminal repeat. Therefore, this network of backbone carbonyl acceptors forms a continuous hydrogen bond network (Oi-27...HZNiHE...Oi-3...HZNi+24HE) that spans the entire ladder. It seems likely that this network contributes to strong coupling of adjacent repeats.
Because the ladder asparagine is one of the most conserved residues in some LRR families (Figure S1), the length of these ladders can span large distances. For example, the asparagine ladder in YopM spans 14 continuous repeats 38. Although the length of this extended hydrogen bond network is impressive, a large protein like YopM would be a challenging target to apply high-resolution studies of hydrogen bonding, such as NMR spectroscopy. Thus, for the present study, we sought to identify a simple asparagine ladder in a relatively small LRR protein. The LRR domain from the human pp32 protein meets this criterion, with two ladder asparagines centered in a short array of LRRs (Figure 1B). Though internal asparagines in longer ladders may have different properties than the two-residue ladder in pp32, the shared structural features of all asparagine ladder residues (most notably, their hydrogen bonding patterns) suggests their salient properties are preserved in the short pp32 ladder 19,39. Furthermore, the central ladder position in pp32 also permits us to determine the effects of ladder extending substitutions in adjacent repeats. pp32 is also well-suited for the present studies because high-resolution structures have been determined 40,41 and its thermodynamic stability and folding kinetics have been well characterized21,22,42.
pp32 has five canonical LRRs flanked by N- and C-terminal caps (Figure 1B). As pp32 and its homologues are found predominantly in the animal taxon and have repeat lengths ranging from 21 to 26 residues, the LRRs of pp32are best represented by the “typical” LRR subfamily 19,43. The asparagine ladder in pp32 is composed of the side chains of asparagines 74 (repeat three) and 98 (repeat four). Unlike repeats three and four, residues at the analogous positions in repeats one and two are hydrophobic. The residue at the ladder position in repeat five is a cysteine, the second most common residue at the ladder position (Figure 1C, S1).
The effect of asparagine ladder substitutions on pp32 global stability.
To determine the importance of asparagines 74 and 98 to the stability and structural integrity of pp32, we generated a series of constructs in which asparagines 74 and/or 98 are substituted with alanine and leucine. With the exception of the N98A variant, far-UV CD spectra of these constructs differ significantly from the wild-type pp32 spectrum (Figure 2A), indicating that the secondary structure is perturbed by the loss of ladder asparagines. The increase in negative ellipticity from ~200 to 210 nm suggests that substitutions of asparagines 74 and 98 increase the amount of random coil present in pp32 for all variants except N98A. Nonetheless, all spectra retain a pronounced shoulder near 218nm, suggesting that some amount of the native β-sheet structure is preserved.
Figure 2. CD spectra and urea-induced unfolding of asparagine ladder and peripheral variants of pp32.

(A) Far-UV CD spectra of asparagine ladder variants. Alanine variants are colored red (N74A, solid line; N98A, short dashed line; N74A/N98A, long dashed line), leucine variants are colored blue (N74L, solid line; N98L, short dashed line; N74L/N98L, long dashed line), and wild-type pp32 is colored grey (dashed line). (B) Urea melts of asparagine ladder variants. Transitions were monitored by CD at 220 nm and were fitted using a two-state model (curves). Data and curves are transformed to fraction folded. Colors and line styles are as in (A). (C) Far-UV CD spectra of peripheral variants (T49L, purple; L69A, green; C123N, orange; YD, blue). (D) Urea melts of peripheral variants. Data were collected and transformed as in (B). Colors and line styles are as in (C). Raw titration data are shown in Figure S2. Conditions: 20 mM NaPO4, 150 mM NaCl, 0.1 mM TCEP, pH 7.8, 20 °C.
To determine how hydrophobic substitutions to the asparagine ladder impact folding stability and cooperativity, we collected urea-induced unfolding transitions using CD spectroscopy (Figure 2B, S2A). For all variants, substitution of ladder asparagines with hydrophobic residues is significantly destabilizing (Table 1, average ΔΔG°H2O = 4.3 kcal mol−1) and reduces the steepness of the unfolding transitions (Table 1, average decrease in m-value = 1 kcal mol−1 M−1). This decrease in the m-value suggests a decreased cooperativity in unfolding, though the reduced m-value may also result from partial disruption of structure in the absence of denaturant, consistent with the loss of secondary structure observed by far-UV CD. However, the N98A variant shows a significant decrease in cooperativity (Δm = 1.0 kcal mol−1 M−1) but has a far-UV spectrum that is the same as wild-type pp32 (Figure 1A), implying that the m-value is due to a genuine decrease in cooperativity rather than partial unfolding under native conditions.
Table 1.
Global stability for pp32 variants.
| Variant | ΔG°H2Oa | m-valuea |
|---|---|---|
| wild-typeb | −7.93 ± 0.18 | 2.86 ± 0.02 |
| S27N | −5.45 ± 0.18 | 2.66 ± 0.07 |
| T49L | −10.49 ± 0.33 | 2.67 ± 0.06 |
| T49V | −9.76 ± 0.42 | 2.76 ± 0.13 |
| V52N | −5.62 ± 0.4 | 2.77 ± 0.20 |
| T49L/V52Nc | −9.35 | 2.80 |
| C123N | −8.90 ± 0.23 | 2.87 ± 0.07 |
| L69Ab | −4.99 ± 0.13 | 3.02 ± 0.03 |
| YDb | −4.72 ± 0.14 | 2.69 ± 0.14 |
| N74A | −3.28 ± 0.22 | 1.98 ± 0.11 |
| N98A | −3.18 ± 0.14 | 1.85 ± 0.06 |
| N74A/N98A | −3.62 ± 0.25 | 1.94 ± 0.06 |
| N74L | −3.61 ± 0.03 | 1.94 ± 0.02 |
| N98L | −3.68 ± 0.2 | 1.59 ± 0.11 |
| N74L/N98L | −4.18 ± 0.35 | 1.83 ± 0.12 |
Global stabilities were determined from urea-induced unfolding transitions at 20 °C. Units for ΔG°H2O and m-values are kcal mol−1 and kcal mol−1 Murea−1. Uncertainties are standard deviations on the mean from at least three independent unfolding transitions.
Equilibrium unfolding data are from 21.
Only a single measurement was made so no error is reported.
This observation demonstrates that the asparagine ladder plays a role in promoting cooperative folding, in addition to stabilizing the LRR fold. It is noteworthy that substitution of both ladder residues with hydrophobic residues (for example, the doubly-substituted N74L/N98L variant) appears to be no more destabilizing than single substitution (the singly substituted N74L and N98L variants; Figure 2B, Table 1). Rather, comparison of unfolding transitions of single and double-substitutions suggests that replacement of the second (isolated) asparagine residue is modestly stabilizing, consistent with coupling of adjacent ladder asparagines through the hydrogen bonding network.
In addition to substitutions to asparagines 74 and 98 that disrupt the ladder, we attempted to extend the asparagine ladder of pp32 by introducing asparagine residues at equivalent ladder positions of repeats one, two, and five (serine 27, valine 52, and cysteine 123; Figure 1C). Asparagine substitutions in repeats one and two (S27N and V52N) were destabilizing (Figure S3), but substitution in repeat five (C123N) was stabilizing (Table 1). The C-terminal C123N variant has a far-UV CD spectrum similar to wild-type pp32 (Figure 2C). Although the increase in stability is significant, the magnitude of the ΔΔG°H2O value is less than that observed for hydrophobic substitutions of ladder asparagines 74 and 98 (Figure 2D, S2B and Table 1), suggesting that when in their native context, ladder asparagines are uniquely stabilizing.
The asymmetric effect of N- versus C-terminal extension on stability suggests local differences in context between repeats two and five. In attempting to determine the sequence origins of this variation, we identified a residue (threonine 49) in close proximity to valine 52 that differs from the strongly conserved leucine normally found at this position (Figure 1C). Substitution of threonine 49 with the consensus leucine is highly stabilizing (Figure 2D, ΔΔG°H2O = −2.6 kcal mol−1); moreover, the far-UV CD spectrum of the T49L variant is nearly identical to wild-type pp32. The isosteric hydrophobic substitution T49V is also stabilizing, although the stability increment (ΔΔG°H2O = −1.8 kcal mol−1) is not as large as that for T49L (Table 1), suggesting that the large increase in stability of the T49L variant results both from substitution of the hydroxyl group with a methyl, as well as improved hydrophobic packing of the leucine side chain 44. Although the T49L substitution significantly stabilizes the pp32 LRR domain, introduction of asparagine at position 52 is destabilizing in the T49L as well as the wild-type pp32 background (Table 1).
Assignments of backbone NH and asparagine side-chain NH2 resonances.
Owing to the unique structural features of asparagine ladders, which involve burial and extensive intramolecular hydrogen bonding, the side chain NH2 groups of ladder asparagines may be expected to have unique NMR signatures, including unusual chemical shifts and 1H chemical shift temperature coefficients, which are sensitive to hydrogen bonding, as well as unique relaxation and exchange properties, which are sensitive to dynamics. Moreover, these structural features are likely to be sensitive to nearby perturbations such as ladder extension and changes in local stability and chemical environment. To explore these structural features, we assigned backbone resonances for the stabilizing T49L and C123N variants, as well as the side chain asparagine NH2 resonances for wild type, T49L, and C123N pp32 variants.
The backbone of wild-type pp32 has been assigned previously using standard triple-resonance techniques 22. Like wild-type pp32, the T49L, and C123N variants both display well-dispersed 1H-15N HSQC spectra (Figure S4A), with most resonances overlaying well with wild-type resonances. To confirm the identities of overlapping resonances and assign the resonances that differ from wild-type pp32, we collected HNCACB, CBCACONH, and HNCO spectra for T49L and C123N. From these spectra we were able to assign backbone resonances of 146 (T49L) and 151 (C123N) out of 152 residues.
To assign asparagine side-chain NH2 resonances, we collected CBCGCO and NH2-filtered versions of the 1H-15N HSQC and HNCO spectra to connect backbone assignments to side-chain 15N nuclei. The NH2-filtered 1H-15N HSQC experiment enhances resolution of asparagine and glutamine side chains by filtering out backbone amide groups via exploitation of the proton multiplicity of NH2 versus NH groups (Figure 3A). Similarly, the NH2-filtered HNCO experiment selectively correlate asparagine and glutamine side chain NH2 protons with side chain CO nuclei. These spectra, in conjunction with CBCGCO spectra, were used to assign side chains starting with Cβ assignments from HNCACB and CBCACONH experiments. Using this approach, we assigned all 14 asparagines in the T49L variant, and the 15 asparagines in the C123N variant (Figure S4B). The asparagine side chain assignments from C123N and T49L were readily transferred to wild-type pp32, YD, and L69A.
Figure 3. Stereospecific NMR assignment of asparagine NH2 side chain protons.

(A) Wild-type pp32 NHz-filtered HSQC spectrum. Ladder side chain NH resonances are connected by dashed lines. For N98, only the upfield resonance is resolved, though the position of the downfield proton is known from HDX experiments where overlapping peaks exchange rapidly (Figure S7). (B) Schematic diagram of an E.COSY cross-peak pattern with a large positive J(A-X) coupling and a small positive J(B-X) coupling (brackets). Solid circles (black) represent observed resonances in which A/B nuclei are paired with the same X spin state (here, Xα) whereas dashed circles (grey) represent unobserved resonances in which A/B nuclei are paired with opposite X states (indicated by labels next to peaks). When J(A-X) and J(B-X) coupling constants have the same sign, the line connecting the observed resonances has a positive slope, as shown. J(A-X) and J(B-X) coupling constants of opposite sign have negative slope. (C) Selections from C123N CγO-coupled NH2-HSQC E.COSY spectra to measure small 2J(HE/Z-Cγ) values. 2J(HE-Cγ) is positive (1 to 5 Hz, same as (B)) whereas 2J(HZ-Cγ) is negative (−1 to −5 Hz), permitting the Hz and HE resonances to be assigned using the large positive 1J(Nδ2-Cγ) coupling constant. For the ladder asparagines 74 and 98, the upfield resonances are displaced with positive slope (as in B), identifying these resonances as originating from HE. The downfield resonances are displaced with negative slope, identifying these resonances as originating from Hz. For non-ladder asparagine residues such as N122 (right panel), this pattern is reversed. The full spectrum is shown in Figure S5. (D) Selections from T49L HNCO E.COSY to measure small 3J (HE/Z-Cβ) values. 3J(Hz-Cβ) is positive (5–10 Hz) whereas 3J(Hz-Cβ) is close to zero, permitting the Hz and HE resonances to be assigned using the large positive 1J(Cγ-Cβ) coupling constant. For the ladder asparagines, downfield resonances are displaced with a positive slope, identifying these resonances as originating from HZ, whereas upfield resonances have a vertical displacement, identifying these resonances as originating from HE. Again, for non-ladder asparagine side chains (e.g., N122, right panel), this pattern is reversed. The full spectrum shown in Figure S5.
NH2 groups produce two resonances in 1H-15N HSQC spectra. For each pair, resonances have identical 15N frequencies, but distinct 1H frequencies (Figure 3A), corresponding to the HZ and HE protons (Figure 1A). For surface exposed asparagines, the HZ proton is typically upfield of the HE proton, since this proton is closer to the shielding Oδ1 electrons 45. Because the unique structural environment of the ladder asparagines may produce significant chemical shift changes, we sought to directly assign the asparagine HE and HZ protons using E.COSY experiments 29,30.
In E.COSY experiments a large J(A-X) coupling constant is exploited to accurately resolve a small J(B-X) coupling constant in a system of mutually J-coupled nuclei, A-X-B. Characteristic E.COSY peak patterns arise in A-B correlated 2D spectra in which the X nuclei are not decoupled during the experiment, ensuring cross peaks are only observed between A and B nuclei with matched X states (e.g., AXα, BXα, but not AXβ, BXα, Figure 3B). We took advantage of relatively large negative 1J(Nδ2-Cγ) and large positive 1J(Cγ-Cβ) to measure the small 2J(HE/Z-Cγ) and 3J(HE/Z-Cβ) (Figure 3C and D respectively), allowing us to determine stereospecific assignments for asparagine HZ and HE protons from the sign of the small HE/Z-Cβ,γ coupling constants. The E.COSY experiments revealed that for asparagines 74 and 98, chemical shifts of HE and HZ protons are inverted with respect to the pattern expected for surface asparagine residues, with the HZ protons downfield relative to the HE protons (Figure 3C, D; Figure S5). These assignments are consistent with a number of NOEs between the asparagines 74 and 98 NH2 protons and protons that are nearby in the crystal structure of pp32 (data not shown). The inversion of the HE and HZ chemical shifts is unique to the asparagine ladder residues, as all of the 12 other asparagine side chains have HE chemical shifts downfield of their Hz chemical shifts, including asparagine 123 in C123N (Figure 4A). This inversion is unlikely to be due to local magnetic fields from nearby aromatics as the nearest aromatic group is more than 10Å from either asparagine ladder NH2 group.
Figure 4. Asparagine side chain proton chemical shifts and chemical shift perturbations (CSPs) of backbone amides in pp32 variants relative to WT pp32.

(A) HE and HZ chemical shifts for each asparagine side chain from wild-type pp32 and peripheral variants are shown as gray and red lines, respectively. Ladder asparagines 74 and 98 are outlined in black. (B) CSPs for T49L (purple) and C123N (orange) backbone amides. Black outlined circles denote perturbations ≥ 1 standard deviation (σCSP) from the mean (dashed vertical lines). Boxes indicate repeat and cap boundaries. Arrows indicate the location of each substitution.
Hydrogen bonding usually results in large downfield movements of proton chemical shifts.46 Measurement of the chemical shifts of asparagines 74 and 98 in wild-type pp32 provides a qualitative estimate of the strength of hydrogen bonding in these ladder residues. The inversion of Hz and HE proton chemical shifts results from movement of Hz resonances downfield by 0.6 to 1 ppm and HE resonances upfield by 0.4 to1.3 ppm (Figure 4A). This inversion suggests strong Hz hydrogen bonding with the i-2 7 backbone carbonyl group of the previous repeat for both asparagines and weak HE hydrogen bonding to the i-3 CO group. Note that the non-ladder asparagine 94 HE proton is also shifted quite far downfield in all sequence backgrounds, consistent with strong hydrogen bonding. In the high-resolution crystal structure of pp32 (PDB ID: 4XOS), the HE of asparagine 94 makes a 2.8 Å hydrogen bond with a side-chain carboxylate oxygen from aspartate 70.
Chemical shift sensitivities of backbone NH resonances to N- and C-terminal structural perturbation.
Asparagine ladders provide a direct network of hydrogen bonding from the N-terminus to the C-terminus of LRR proteins and may propagate local changes over large distances. To examine whether such propagation occurs in pp32, we examined the effects of two peripheral mutations (T49L and C123N) on the chemical shifts of nearby and distant residues. These variants were chosen because both are stabilizing and both maintain structured native states, based on CD and NMR spectroscopy, and because together they provide a comparison of perturbations from a potential ladder-extending variant (C123N) with a non-extending variant (T49L).
Comparison of backbone amide proton chemical shifts of the T49L and C123N variants to those of wild-type pp32 shows that the C123N substitution causes more distant 1H-15N chemical shift perturbations (CSPs) than the T49L variant (Figure 4B). In the C123N variant, backbone CSPs extend N-terminally from the site of the substitution. The largest backbone CSPs in C123N are in repeat four, adjacent to the substituted repeat. Though the backbone CSPs in repeat three are smaller than in repeat four, they are of the same magnitude as those in repeat five, the site of the C123N substitution. The ladder asparagines 98 and 74 have the second and third largest backbone CSPs respectively (Figure S6A). Propagation of structural changes from the C-terminal end of the asparagine ladder (98) to the N-terminal end (74) may be responsible for the large CSPs observed far from the substitution site. In contrast, CSPs of the T49L variant are more localized to the site of substitution and do not affect the backbone of either asparagine ladder residue (Figure S6B). Since both the T49L and C123N substitutions are stabilizing, the longer-range perturbations of the C123N substitution, especially at ladder residues 74 and 98, may reflect propagation of perturbations through the ladder.
Given the importance of side chain interactions in the asparagine ladder, CSPs resulting from T49L and C123N substitution were also determined for the HE/Z protons of all 14 asparagines (Figure S6C). In T49L, ladder side chain CSPs extend C-terminally, decreasing in magnitude with increasing distance from the substitution site (that is, N74 HZ >> N74 HE > N98 HZ; the chemical shift of the N98 HE is not significantly perturbed). These side-chain CSPs extend a full repeat further than backbone CSPs for T49L. In C123N, however, ladder side chain CSPs are significant only for HZ protons, even though asparagine 98 HE is closest to the substitution site. Thus, for both N- and C-terminal substitution (T49L and C123N), the HZ protons are more sensitive to perturbation than the HE protons, which may be related to their large downfield chemical shifts, and perhaps stronger hydrogen bonding, compared to the HE protons.
Dynamics of asparagine 74 and asparagine 98 side-chain NH2 groups.
One conspicuous feature of the NH2 cross peaks of the asparagine ladder residues is their low intensities compared to surface asparagine cross peaks (Figure 3A). This decreased intensity is seen in all peripheral variants, though interestingly, the NH2 resonances of potential ladder extending asparagine 123 are of high intensity, comparable to solvent-exposed asparagines. Reduced intensity could either result from large amplitude dynamics on the chemical shift timescale (μs-ms), or from rapid transverse relaxation (large R2 values) due to both the slow overall tumbling on the nanosecond timescale and a higher local concentration of hydrogen spin density, compared to surface asparagine NH2 groups. To resolve these two possibilities and to investigate the overall rigidity of the asparagine ladder, we measured 15N spin relaxation of the asparagine side-chain NH2 groups in pp32 using various NMR experiments that probe dynamics on different timescales.
Due to the low signal intensities of asparagine 74 and 98 side chain NH2s, conventional relaxation experiments suffered from low signal-to-noise. To increase signal intensity, we equilibrated 15N-labelled proteins in a 7 M urea solution containing 50% D2O. Under these conditions, all pp32 variants are unfolded, and undergo rapid hydrogen exchange with solvent. Upon refolding, exchangeable sites should be 50% deuterated on average. For the asparagine side-chain amides, this level of deuteration leads to an equal distribution of the four isotopomers (NH2, NHZDE, NHEDZ, and ND2). The singly-deuterated species have significantly reduced HZ-HE dipole-dipole relaxation compared to the NH2 species. Combining an NH-filtered pulse sequence with deuterium-decoupling to reduce scalar relaxation of the second kind suppresses signals from the NH2 species. Thus, the only cross peaks in this experiment are those of the singly protonated species (NHZDE and NHEDZ), which have higher signal-to-noise due to elimination of HZ-HE dipole-dipole relaxation. Thus, standard pulse sequences can be used to probe 15N dynamics (with minimal modifications such as 2H decoupling) while retaining high signal-to-noise.
To test whether the reduced signal intensities of asparagines 74 and 98 result from intermediate exchange on the μs-ms timescale we conducted “two-point” relaxation dispersion (RD) constant time (CT) CPMG experiments 33. In these experiments, two spectra are acquired with CPMG spin echoes applied with different spacings over a constant time period (T), typically 35 to 40 ms in duration. During this period, the NMR signal decays according to an apparent relaxation rate R2app = R2 + Rex(ν) where R2 is the contribution from ps-ns timescale dynamics and Rex(ν) is the ms-μs contribution, which depends on the time interval between the spin echo pulses. The peak intensity at the end of the CT period (T) is given by . In the first spectrum (A) the frequency of spin echoes is kept at the maximum value allowed by duty cycle considerations, minimizing the effects of exchange broadening on signal decay. If exchange contributions are completely quenched, R2app = R2. In reality, R2app = R2* ~ R2 where R2* is a best-case approximation to the ps-ns timescale dynamics contribution to the signal decay. Therefore, . In the second spectrum (B) the frequency of spin echoes is minimized, thereby maximizing signal decay from exchange broadenings. In this scenario, R2app ~ R2 + Rex where Rex is the ms-μs contribution to the signal decay. As a result, . From this it follows that cross-peak intensities with significant Rex contributions will have lower peak intensities in (B) than in (A). Typically, I(B)/I(A) ratios < 0.75 are considered to be “candidates” for μs-ms dynamics. Additionally, spectrum A can be used to calculate R2* ~ R2, the exchange-minimized transverse relaxation rate, which is sensitive to ps-ns timescale rotational diffusion and N-H bond vector librational dynamics.
The two-point experiments did not show any difference in signal intensity between the A and B spectra for asparagine ladder residues in any variant, indicating that the asparagine ladder is rigid on the μs-ms timescale. Thus, fast to intermediate exchange dynamics of 15N nuclei are unlikely to cause the decreased intensities observed for ladder NH2 resonances. Rather, the CPMG experiments reveal that asparagine 74 and 98 side chain NH(D) R2* values are among the largest relaxation rates of the asparagine side-chains in pp32 (Figure 5A). In fact, asparagines 74 and 98 side chains have R2* values equal to or greater than most backbone R2* values (Figure 5A, compare black outlined points to dashes). This is true for all variants at both 20 and 35 °C, suggesting that the side chains of asparagines 74 and 98 have rigidity (and hence, rotational and correlation times) comparable to hydrogen-bonded backbone NH groups. In contrast, R2* values for the N123 side chain NH(D) groups in the C123N variant are much lower (both at 20 and 35°C) than for the 74 and 98 side chains.
Figure 5. Transverse relaxation rates and 1H-15N NOEs for asparagine NHD and backbone NH groups in wild-type pp32 and stabilizing variants.

Circles indicate asparagine side-chain NHD values, dashes to the left of circles indicate backbone amide values. Experiments were performed at 20 °C and 35 °C (light and dark colors, respectively). Values from N74 and N98 side chains are outlined in black. Samples were 15N-labelled and fully exchanged into 50% D2O to improve signal-to-noise of ladder side chain resonances. (A) R2* measurements in wild-type pp32, T49L, and C123N. R2* measurements were determined from two-point CPMG experiments 33 with where T is the constant CPMG time period used for the experiment, Imax is peak intensity with νcpmg set at the maximum value during the T period, and I0 as the reference peak intensity. (B) 1H-15N NOE values for wild-type pp32, T49L, and C123N. Although the NOE value for the wild-type asparagine 98 Hz appears to exceed the theoretical maximum of ~0.85 at 20 °C 47, its value at 35 °C is within the expected range for a protein of this size. Conditions: 20 mM NaPO4, 50 mM NaCl, and 0.1 mM TCEP, pH 6.8 (after accounting for D2O).
To explore rigidity and dynamics on a faster timescale, we measured 1H-15N NOEs for wild-type pp32 and the T49L and C123N variants, again using 50% deuterated samples. All ladder asparagines NH(D) groups have 1H-15N NOE values of approximately one. In contrast, non-ladder asparagine NH(D)s span a broad range of 1H-15N NOE values with an average of 0.29. As with R2* Values, the 1H 15N NOE values of asparagine 74 and 98 protons are similar to values for rigid backbone amides (Figure 5B, compare black outlined points to dashes).
Temperature coefficients of asparagine 74 and asparagine 98 side chain NH2 groups.
The dynamics experiments above confirm the ladder is rigidly structured, consistent with formation of strong hydrogen bonds. Strong hydrogen bonding involving the asparagine ladder Hz protons is also suggested from the chemical shift values. To further probe the hydrogen bond strength of the asparagine ladder side chains, we measured the magnitude of the change in proton chemical shifts with increasing temperature (so-called “temperature coefficients”, ΔδNH/ΔT) for all asparagine side chain NH2z groups for wild-type pp32, T49L, L69A, C123N, and YD (Figure 6). ΔδNH/ΔT values can be used to identify intramolecular hydrogen bonding 48 and have been shown to correlate with hydrogen bond length 49–51 and local unfolding 32. Amides engaged in intramolecular hydrogen bonds have ΔδNH/ΔT values greater (i.e., less negative) than −4.5 ppb K−1 52; within this range, short strong hydrogen bonds tend to have more negative ΔδNH/ΔT values 51.
Figure 6. Temperature coefficients for asparagine side chains in wild-type pp32 and peripheral variants.

HE and HZ temperature coefficients for each asparagine side chain from wild-type pp32 and peripheral variants are shown as gray and red lines, respectively. The two ladder positions are outlined in black. Temperature coefficients were determined from linear fits to changes in proton chemical shift at four temperatures (283, 288, 293, 303 K). Conditions: ~600 μM protein, 20 mM NaPO4, 50 mM NaCl, 0.1 mM TCEP, pH 6.8.
The non-ladder asparagine ΔδNH/ΔT values are all negative, with mean values of −3.5 ± 1.4 ppb K−1 (Figure 6). For some but not all of these non-ladder asparagines, HZ and HE ΔδNH/ΔT values are distinct, and are largely independent of the variant background (for example, N59 and N114). The ladder asparagines have more positive ΔδNH/ΔT values than most non-ladder asparagines (Figure 6). This indicates greater resilience to temperature perturbation, as expected for stable intramolecular hydrogen bonds. Asparagine 74 HZ ΔδNH/ΔT values are generally more positive than those of HE; the sole exception is that of the T49L variant. In contrast, asparagine 98 HE ΔδNH/ΔT values are larger than those of HZ, with the former showing positive ΔδNH/ΔT values. This positive ΔδNH/ΔT value suggests that the hydrogen bond involving asparagine 98 HE is weak, consistent with its measured 1H chemical shift. In contrast, the asparagine 98 HZs (measured only for C123N and T49L variants) are among the most negative ladder ΔδNH/ΔT values, suggesting that the hydrogen bond involving asparagine 98 HZ is unusually strong, consistent with its measured 1H chemical shift.
Hydrogen exchange of asparagine ladder NH2 groups.
Chemical shift values, dynamics, and temperature coefficients provide convincing evidence for the existence of a rigid network of bonds connecting the side-chains of asparagines 74 and 98, but they do not provide estimates for the stability of the asparagine ladder structure. The rates of hydrogen exchange of amide groups with solvent are influenced by the stability of hydrogen-bonds involving amide NH groups, since exchange requires disruption of intramolecular hydrogen bonding 53. Thus, NH groups that are strongly hydrogen bonded exchange slowly. For particularly stable hydrogen bonds, exchange rates may decrease to the limit set by global stability. Although global protection from exchange is often observed for a subset of backbone NH groups that are buried and stably hydrogen bonded into secondary structures, large protection factors are not typically seen for labile side-chain protons 54.
To probe the local stability of the asparagine ladder, we monitored hydrogen-deuterium exchange rates (HDX) by rapidly changing the solvent from H2O to D2O, and collecting 1H-15N HSQC spectra over time (Figure 7). The NH2-filtered HSQC pulse sequence was used to reduce spectral overlap between asparagine 74 and 98 NH2 groups and backbone amides. As a result, exchange curves report on the decay of the NH2 isotopomer to either the NHEDZ or the NDEHZ isotopomer. Therefore, the fitted exchange rate constant is the sum of the individual HE and HZ exchange rate constants, and can be regarded as an upper limit for exchange. If one hydrogen exchanges faster than the other, the rate constant approximates the fast exchange process, whereas if the two protons exchange at the same rate, the fitted rate constant is twice the individual exchange rate constants.
Figure 7. Side-chain hydrogen exchange data and protection factors for N74 and N98 side-chain NH2 groups in wild-type pp32 and variants.

(A) N74 peak heights as a function of exchange time for wild-type pp32 and peripheral variants (wild-type, gray; T49L, purple; C123N, orange; L69A, green; and YD, blue). HE heights are shown as unfilled circles with dashed fitted curves. HZ heights are shown as filled circles with solid fitted curves. (B) N98 peak heights as a function of exchange time as in (A). (C) Logarithmic protection factors for N74 NH2 protons versus folding free energies calculated from urea denaturation experiments (Figure 2). Circles are from NH2-filtered HSQCs are colored as in (A); squares are from unfiltered HSQC spectra of wild-type pp32 21 (Hz, filled gray square; HE, empty gray squares). The dashed line represents global exchange (slope = 1, intercept = 0). (D) Logarithmic protection factors for N98 NH2 protons as in (B). Conditions: 150 mM NaPO4, 50 mM NaCl, 0.1 mM TCEP, pH 6.8 (after accounting for D2O) at 30 °C. Note that although these conditions are slightly different than those in Figure 2D, these differences have no effect on fitted thermodynamic parameters (not shown).
In the first spectrum obtained after exchange is initiated (25 to 90 minutes), all of the solvent-exposed asparagines have fully exchanged, leaving only cross peaks for N74 and N98 NH2 groups (Figure S7, note that rapid exchange during sample preparation is also observed for the potential ladder extending asparagine 123 in the C123N variant). The side-chain NH2 protons of asparagine 74 are highly protected, with lifetimes ranging from hours to days depending on the variant (Figure 7A). The side-chain NH2 protons of asparagine 98 are even more protected than those of asparagine 74, with lifetimes up to two months for the most stable variants (Figure 7B). The exchange rates of asparagine 74 and 98 are several orders of magnitude smaller than those measured for solvent exposed asparagines 54.
Though the individual HE and HZ exchange rates could not be determined from the NH2-filtered exchange data, we were able to determine HE and Hz exchange rates for both ladder asparagines in wild-type pp32 using unfiltered 1H-15N-HSQC spectra that were obtained in a previous study of backbone amide hydrogen exchange 21. Using these spectra, we determined the HE and HZ exchange rates for the side chains of asparagine 74 and 98 by monitoring cross peaks from the NHD species (Figure S8). The asparagine 74 HZ exchanges more slowly than the HE (Table 2), consistent with Hz and HE exchange rates in model compounds 36 and with an EX2 exchange process. The asparagine 98 HE and HZ exchange at nearly the same rate, which may be an indication that for this highly protected group, exchange has some EX1 character. The overall slower rates of exchange determined from the unfiltered versus filtered HSQC spectra is likely to result from the lower temperature of the former (20 °C versus 30 °C).
Table 2.
Hydrogen exchange rates for ladder asparagine side chains in pp32 variants.
| Variant | NH2a | Asparagine 74 HZb | HEb | NH2a | Asparagine 98 HZb | HEb |
|---|---|---|---|---|---|---|
| wild-type | 41.7 ± 1.7 × 10−6 | 3.0 ± 0.3 × 10−6 | 7.7 ± 1.3 × 10−6 | 2.7 ± 0.1 × 10−6 | 1.4 ± 0.2 × 10−7 | 1.2 ± 0.2 × 10−7 |
| T49L | 4.3 ± 0.1 × 10−6 | – | – | 2.3 ± 0.1 × 10−7 | – | – |
| L69Ac | 845 ± 92 × 10−6 | – | – | 301 ± 58 × 10−6 | – | – |
| C123N | 12.4 ± 0.2 × 10−6 | – | – | 2.3 ± 0.1 × 10−7 | – | – |
| YDc | 38 ± 9 × 10−6 | – | – | 17 ± 8 × 10−6 | – | – |
Units for hydrogen exchange rate constants are s−1. Uncertainties estimates are from the square-root of the diagonal elements of the covariance matrix obtained from nonlinear least-squares fitting.
Hydrogen exchange rates were measured from NH2 filtered HSQC spectra at 30 °C with 150 mM NaPO4, 50 mM NaCl, 0.1 mM TCEP, pH 6.8 (after correcting for D2O).
Hydrogen exchange rates were calculated using unfiltered HSQC spectra from 21 collected at 20 °C with 20 mM NaPO4, 50 mM NaCl, 0.1 mM EDTA, 0.2 mM TCEP, pH 6.7 (after correcting for D2O).
Only partial decay curves were obtained due to rapid exchange; thus, uncertainties in rate constants are comparatively large.
To determine whether exchange of the asparagine ladder NH2 protons requires complete unfolding for exchange, we computed local stabilities from protection factors (PFs) and compared these values to unfolding free energies measured from urea denaturation experiments. The ladder extending (C123N) and peripheral substitutions (T49L, L69A, YD) were used to modulate global stability, and potentially alter hydrogen exchange rates. For asparagine 74, local stabilities estimated from hydrogen exchange measurements are lower than the global stability limit for each variant, particularly for variants with increased global stability (C123N and T49L; Figure 7C). This suggests that exchange of the N74 side-chain NH2s involves a sub-global mechanism, although the partial correlation to global unfolding free energies indicates that the exchange-competent forms are influenced to some degree by overall stability. In contrast, local stabilities estimated from asparagine 98 hydrogen exchange rates are close to values expected from global exchange (Figure 7D) over a broad range of global stabilities. Only the most highly stable T49L variant falls off the unit slope line, exchanging with a rate that is roughly equivalent to that of the C123N variant despite being 1.6 kcal mol−1 more stable. These very slow hydrogen exchange rates are consistent with strong hydrogen bonding for both ladder asparagine side-chains, especially that of asparagine 98.
Discussion
The asparagine ladder is a highly conserved feature of many LRR proteins. Despite conjecture about the importance of the ladder to LRR proteins 39, few studies have directly probed the role of the asparagine ladder in LRR protein structure and stability. The unique properties shared by asparagine 74 and 98 (i.e. inverted HE and HZ chemical shift, large protection factors, large contributions to ΔG°H2O, etc.) derive from their chemical environment, which crystal structures show to be common among ladder asparagines across the LRR family 38,39,55. Thus, it is likely that the features observed in the short asparagine ladder of pp32 are representative of residues in longer ladders like those of YopM. The present study shows the asparagine ladder is a key component of LRR structure that forms a rigid network of hydrogen bonds providing repeat-to-repeat coupling.
Asparagine ladder hydrogen bonds.
Crystal structures have shown that asparagine ladder side chains form hydrogen bonds with local backbone atoms 39. The urea-induced unfolding transitions measured here provide compelling experimental evidence that these hydrogen bonds contribute favorably to stability (Figure 2). Substitution of either ladder residue increases the folding free energy of pp32 by about 4.5 kcal mol−1 compared to wild-type pp32, Table 1. Ladder asparagine side chains also behave like structured backbone amides in NMR relaxation experiments (Figure 5). Additionally, ladder side chains are highly protected from hydrogen exchange (Figure 6). To our knowledge, the only comparable level of protection of exchangeable side chains are asparagines 43 and 44 of BPTI 56. However, unlike the ladder asparagine 98 in pp32, the protected asparagines in BPTI exchange more rapidly than the global exchange limit, based on the stability of BPTI 57. Given these data, it is clear that ladder side chains form stable interactions integral to the LRR motif.
The high degree of protection of asparagine ladder NH2 groups from hydrogen exchange demonstrates that these groups are completely sequestered from solvent. Thus, variations in the temperature coefficients (ΔδNH/ΔT values) of ladder NH2 proton resonances result from changes in the native protein structure. The asparagine 98 HE and HZ ΔδNH/ΔT values are particularly notable given their opposite signs compared to surface exposed asparagines, with HE protons showing positive ΔδNH/ΔT values (Figure 6). The large difference between in ΔδNH/ΔT values for the HE and HZ protons of asparagine 98 suggests different levels of hydrogen bonding for the HE and HZ proton.
As with ΔδNH/ΔT values, the HE and HZ protons of asparagine 98 also differ significantly in their proton chemical shifts, which are not only inverted compared to unstructured asparagine NH2 groups but are significantly separated from each other (by about 1.35 ppm in the proton dimension, Figure 4A). The downfield shifts in the HZ asparagine 98 protons and the upfield shifts of the HE protons relative to surface asparagines suggests an asymmetric degree of hydrogen bonding by the ladder side-chains, in which the HZ hydrogen forms a strong hydrogen bond to the previous repeat, whereas the HE proton hydrogen bonds weakly. This is supported by the ΔδNH/ΔT values, which shows that the asparagine 98 HZ is more negative than that of HE consistent with a shorter HZ hydrogen bond 50.
Though the HE/HZ chemical shift differences for asparagine 74 are similar to those for asparagine 98, they are less pronounced, consistent with the overlapping distribution of ΔδNH/ΔT values for the HZ and HE resonances of asparagine 74. Interestingly, the T49L variant is exceptional in this regard, showing an HZ/HE proton chemical shift separation (1.380 ppm) that is similar to those for asparagine 98 (~1.48 ppm); likewise, the asparagine 74 HE ΔδNH/ΔT value in the T49L variant is larger than that of HZ. It is possible that the strongly stabilizing T49L substitution strengthens the asparagine 74 HZ hydrogen bond to the backbone CO group of residue 49 (the site of the substitution) so that it resembles that of asparagine 98.
The unique spectroscopic features of asparagine 98 (in particular, chemical shifts, ΔδNH/ΔT values), and the convergence of asparagine 74 to these features in a variant in which the donor residue to the asparagine 74 HZ is substituted to the consensus sequence (Figure S1) suggest an archetypal LRR ladder bonding pattern in which the inter-repeat HZ hydrogen bond is strong, and the intra-repeat HE hydrogen bond is weak. This pattern of bonding is consistent with the absence of a detectable isotope effect between asparagine 74 HE and asparagine 98 HZ despite their sharing a hydrogen bond acceptor (Figure 1A). Such isotope effects have been observed between hydrogen bond pairs to a single acceptor 58,59; the absence of such an effect here, along with the upfield chemical shift of HE, is consistent with a weak HE hydrogen bond.
Asparagine ladder structural features.
Above, we described the evidence for the asparagine ladder’s formation of stable hydrogen bonds. As a result, the highly structured side chains likely experience increased dipolar relaxation from other nearby structured protons, which would explain why the ladder HE/Z protons have low signal intensity. High-resolution structures of pp32 show that asparagine 74 and 98 HE/Z are indeed surrounded by a larger number of ordered protons than non-ladder asparagines (Figure S9A). In addition, backbone amide signal intensity is inversely correlated with the number of interproton contacts (Figure S9B). Since the ladder side chains seem to behave like the structured backbone amides, we view the asparagine ladder as a “second backbone”, providing a hydrogen bonding network that extends through the hydrophobic core. In large LRR proteins such as YopM, extended asparagine ladders may provide coupling over long distances, and may contribute to the high degree of cooperativity seen in those proteins20,60.
The importance of this second backbone to the LRR motif is exemplified by the far-UV CD spectra of wild-type pp32 and ladder substituting variants (Figure 2A). These variants show progressively larger disruptions in native secondary structure as one and then both asparagines are substituted. YopM, another LRR protein, shows similar changes in its far-UV CD spectrum after deletion of stabilizing repeats, which results from unfolding of multiple repeats that are adjacent to the site of deletion 61. These partial unfolding transitions highlight the importance of interfaces in stabilizing leucine-rich repeats. The pp32 data extends this observation, showing that only the conserved asparagine need be removed to elicit a similar disruption in secondary structure. This suggests that the asparagine ladder is a major contributor to LRR interfaces, which are integral to LRR secondary structure.
Despite the demonstrated importance of the asparagine ladder to LRR structure, ladder-extending substitutions were either destabilizing (S27N, V52N; Figure S3) or only marginally stabilizing (C123N, Figure 2D). For C123N, the substituted asparagine has small R2*, undergoes rapid hydrogen exchange (Figures 5A and 6), is highly dynamic on the ps-ns timescale (Figure 5B), and lacks strong NOEs to protons expected to be in close proximity (data not shown), features more similar to the solvent-exposed asparagines than to ladder asparagines 74 and 98. The inability to extend the ladder to adjacent repeats highlights the importance of sequence context for the ladder architecture. It appears that LRRs lacking an asparagine at the ladder position have additional sequence differences that stabilize the non-asparagine residue (e.g., valine 52 in pp32), and that these sequence differences are incompatible with an asparagine. However, multiple sequence alignments of tandem LRRs show little pairwise covariance between the ladder position and other positions, suggesting that any such covariance involves multiple positions.
The asparagine ladder and cooperativity.
As a result of the asparagine ladder’s role at repeat interfaces, it is likely to contribute to cooperativity during protein folding. The chemical denaturation of the N98A variant of pp32 supports this hypothesis, showing a significantly shallower unfolding transition than wild-type pp32 while retaining native-like structure. This behavior differs from that of pp32 variants that replace the conserved leucine residues that define the LRR motif. Previous studies have shown that substitutions to conserved leucines are similarly destabilizing but do not affect cooperativity 20,21. Coupling is also demonstrated in the non-additivity of ΔG°H2O values for the asparagine ladder substitutions. Substituting either ladder asparagine is strongly destabilizing (ΔΔG°H2O ≈ +3.5 kcal mol−1), but substitution of the second ladder asparagine is modestly stabilizing ΔΔG°H2O ≈ −0.4 kcal mol−1 relative to the single variant). This indicates an energetic coupling between the two positions 62,63 of around −5 kcal mol−1; in other words, ladder asparagines are stabilized by their neighboring asparagines by 5 kcal mol−1. Since individual LRRs are likely unfolded 64,65, the coupling provided by the asparagine ladder is important for LRR protein folding.
Although the double mutant cycles reveal coupling between ladder asparagines, they do not reveal how coupling is achieved. A possible mechanism is suggested from the pattern of CSPs in the stabilizing pp32 variant C123N. This substitution produces significant CSPs for asparagine 74 backbone NH and side chain HZ despite their distance from the substitution site (> 9 Å). Since asparagine 98 directly contacts the substituted residue 123, residue 74 CSPs may be transmitted through structural rearrangements of asparagine 98. This would imply the asparagine ladder residues can propagate structural changes between adjacent ladder positions. In addition to the hydrogen bonds between the asparagine side chain NH2 and backbone carbonyl oxygens at positions i-27 and i-3, a second possible conduit for propagation is hydrogen bonding between the backbone amide of the ladder residue in repeat i-1 and the ladder side chain Oδ1 in repeat i (Figure 1A). Perhaps the modest sensitivity of the HE chemical shift to substitution is related to the comparatively weak hydrogen bond involving the HE proton. Small structural changes would result in large chemical shift changes for the strongly hydrogen bonded HZ (consistent with large negative ΔδNH/ΔT values) but small chemical shift changes for the weakly bonded HE proton. This explanation is consistent with the observation that the HZ CSP resulting from C123N substitution is in the upfield direction (Figure S6).
Supplementary Material
Acknowledgements
We thank Katherine Tripp at The Center for Molecular Biophysics, and the Johns Hopkins University Biomolecular NMR Center for providing facilities and resources. This research is supported by National Institutes of Health Grant GM068462 (to D. B.). S. A. K. was supported by the NIH training grant T32-GM008403.
Footnotes
Supporting Information
Sequence conservation; additional raw and normalized titration curves; additional NMR spectra for C123N and T49L variants from HSQC, NH2-filtered HSQC, and E.COSY experiments; side-chain CSPs; hydrogen exchange NMR spectra and fits from 21; and comparison of crystallographic and NMR peak intensity data
References
- (1).Worth CL, and Blundell TL (2010) On the evolutionary conservation of hydrogen bonds made by buried polar amino acids: The hidden joists, braces and trusses of protein architecture. BMC Evol. Biol 10, 1–11. doi: 10.1186/1471-2148-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yang X, Kathuria SV, Vadrevu R, and Matthews CR (2009) βα-Hairpin clamps brace βαβ modules and can make substantive contributions to the stability of TIM barrel proteins. PLoS One 4, 1–9. doi: 10.1371/journal.pone.0007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Basanta B, Chan KK, Barth P, King T, Sosnick TR, Hinshaw JR, Liu G, Everett JK, Xiao R, Montelione GT, and Baker D. (2016) Introduction of a polar core into the de novo designed protein Top7. Protein Sci. 25, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Perutz MF (1978) Electrostatic effects in proteins. Science (80-. ). 201, 1187–1191. [DOI] [PubMed] [Google Scholar]
- (5).Cunningham BC, and Wells JA (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science (80-. ). 244, 1081–1085. [DOI] [PubMed] [Google Scholar]
- (6).Wheeler LC, Lim SA, Marqusee S, and Harms MJ (2016) The thermostability and specificity of ancient proteins. Curr. Opin. Struct. Biol 38, 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sailer ZR, and Harms MJ (2017) Molecular ensembles make evolution unpredictable. Proc. Natl. Acad. Sci 114, 11938–11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Martin LC, Gloor GB, Dunn SD, and Wahl LM (2005) Using information theory to search for co-evolving residues in proteins. Bioinformatics 21, 4116–24. [DOI] [PubMed] [Google Scholar]
- (9).Anishchenko I, Ovchinnikov S, Kamisetty H, and Baker D. (2017) Origins of coevolution between residues distant in protein 3D structures. Proc. Natl. Acad. Sci 114, 9122–9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Nakano S, Motoyama T, Miyashita Y, Ishizuka Y, Matsuo N, Tokiwa H, Shinoda S, Asano Y, and Ito S. (2018) Benchmark analysis of native and artificial NAD+-dependent enzymes generated by a sequence-based design method with or without phylogenetic data. Biochemistry 57, 3722–3732. [DOI] [PubMed] [Google Scholar]
- (11).Goyal VD, and Magliery TJ (2018) Phylogenetic spread of sequence data affects fitness of SOD1 consensus enzymes: Insights from sequence statistics and structural analyses. Proteins Struct. Funct. Bioinforma 86, 609–620. [DOI] [PubMed] [Google Scholar]
- (12).Morcos F, Pagnani A, Lunt B, Bertolino A, Marks DS, Sander C, Zecchina R, Onuchic JN, Hwa T, and Weigt M. (2011) Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proc. Natl. Acad. Sci 108, E1293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Harms MJ, and Thornton JW (2014) Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature 512, 203–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Salinas VH, and Ranganathan R. (2018) Coevolution-based inference of amino acid interactions underlying protein function. Elife 7, 1–20. doi: 10.7554/eLife.34300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Faiman GA, and Horovitz A. (1996) On the choice of reference mutant states in the application of the double-mutant cycle method. Protein Eng. Des. Sel 9, 315–316. [DOI] [PubMed] [Google Scholar]
- (16).Baase WA, Liu L, Tronrud DE, and Matthews BW (2010) Lessons from the lysozyme of phage T4. Protein Sci. 19, 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Canale AS, Cote-Hammarlof PA, Flynn JM, and Bolon DN (2018) Evolutionary mechanisms studied through protein fitness landscapes. Curr. Opin. Struct. Biol 48, 141–148. [DOI] [PubMed] [Google Scholar]
- (18).Eisenberg DS, and Sawaya MR (2017) Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem 86, 69–95. [DOI] [PubMed] [Google Scholar]
- (19).Kajava AV (1998) Structural diversity of leucine-rich repeat proteins. J. Mol. Biol 277, 519–527. [DOI] [PubMed] [Google Scholar]
- (20).Courtemanche N, and Barrick D. (2008) The Leucine-Rich Repeat Domain of Internalin B Folds along a Polarized N-Terminal Pathway. Structure 16, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dao TP, Majumdar A, and Barrick D. (2015) Highly polarized C-terminal transition state of the leucine-rich repeat domain of PP32 is governed by local stability. Proc. Natl. Acad. Sci 112, E2298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dao TP, Majumdar A, and Barrick D. (2014) Capping motifs stabilize the leucine-Rich repeat protein PP32 and rigidify adjacent repeats. Protein Sci. 23, 801–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jeng MF, and Englander SW (1991) Stable submolecular folding units in a non-compact form of cytochrome c. J. Mol. Biol 221, 1045–1061. [DOI] [PubMed] [Google Scholar]
- (24).Pace CN (1986) Determination and Analysis of Urea and Guanidine Hydrochloride Denaturation Curves. Methods Enzymol. 131, 266–280. [DOI] [PubMed] [Google Scholar]
- (25).Street TO, Courtemanche N, and Barrick D. (2008) Protein Folding and Stability Using Denaturants. Methods Cell Biol. 84, 295–325. [DOI] [PubMed] [Google Scholar]
- (26).Masse JE, and Keller R. (2005) AutoLink: Automated sequential resonance assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. J. Magn. Reson 174, 133–151. [DOI] [PubMed] [Google Scholar]
- (27).Chimenti MS, Castañeda CA, Majumdar A, and García-Moreno EB, (2010) Structural Origins of High Apparent Dielectric Constants Experienced by Ionizable Groups in the Hydrophobic Core of a Protein. J. Mol. Biol 405, 361–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).McIntosh LP, Brun E, and Kay LE (1997) Stereospecific assignment of the NH2 resonances from the primary amides of asparagine and glutamine side chains in isotopically labeled proteins. J. Biomol. NMR 9, 306–312. [DOI] [PubMed] [Google Scholar]
- (29).Löhr F, and Rüterjans H. (1997) H2NCO-E. COSY, a Simple Method for the Stereospecific Assignment of Side-Chain Amide Protons in Proteins. J. Magn. Reson 124, 255–258. [DOI] [PubMed] [Google Scholar]
- (30).Cai M, Huang Y, and Clore GM (2001) Accurate orientation of the functional groups of asparagine and glutamine side chains using one-and two-bond dipolar couplings. J. Am. Chem. Soc 123, 8642–3. [DOI] [PubMed] [Google Scholar]
- (31).Goddard TD, and Kneller DG SPARKY 3. University of California San Francisco. [Google Scholar]
- (32).Tomlinson JH, and Williamson MP (2012) Amide temperature coefficients in the protein G B1 domain. J. Biomol. NMR 52, 57–64. [DOI] [PubMed] [Google Scholar]
- (33).Mulder FAA, Skrynnikov NR, Hon B, Dahlquist FW, and Kay LE (2001) Measurement of slow (μs-ms) time scale dynamics in protein side chains by 15N relaxation dispersion NMR spectroscopy: Application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc 123, 967–975. [DOI] [PubMed] [Google Scholar]
- (34).Kay LE, Torchia DA, and Bax A (1989) Backbone Dynamics of Proteins As Studied by 15N Inverse Detected Heteronuclear NMR Spectroscopy: Application to Staphylococcal Nuclease. Biochemistry 28, 8972–9. [DOI] [PubMed] [Google Scholar]
- (35).Molday RS, Englander SW, and Kallen RG (1972) Primary Structure Effects on Peptide Hydrogen Exchange. Biochemistry 11, 150–158. [DOI] [PubMed] [Google Scholar]
- (36).Krishna NR, Sarathy KP, Huang DH, Stephens RL, Glickson JD, Smith CW, and Walter R. (1982) Primary Amide Hydrogen Exchange in Model Amino Acids: Asparagine, Glutamine, and Glycine Amides. J. Am. Chem. Soc 104, 5051–5053. [Google Scholar]
- (37).Bai Y, Milne JS, Mayne L, and Englander SW (1993) Primary structure effects on peptide group hydrogen exchange. Proteins Struct. Funct. Bioinforma 17, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Evdokimov AG, Anderson DE, Routzahn KM, and Waugh DS (2001) Unusual molecular architecture of the Yersinia pestis cytotoxin YopM: A leucine-rich repeat protein with the shortest repeating unit. J. Mol. Biol 312, 807–821. [DOI] [PubMed] [Google Scholar]
- (39).Kobe B, and Deisenhofer J. (1995) Proteins with leucine-rich repeats. Curr. Opin. Struct. Biol 5, 409–416. [DOI] [PubMed] [Google Scholar]
- (40).Huyton T, and Wolberger C. (2007) The crystal structure of the tumor suppressor protein pp32 (Anp32a): Structural insights into Anp32 family of proteins. Protein Sci. 16, 1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zamora-Caballero S, Šiaučiunaite-Gaubard L, and Bravo J. (2015) High-resolution crystal structure of the leucine-rich repeat domain of the human tumour suppressor PP32A (ANP32A). Acta Crystallogr. Sect. F Struct. Biol. Commun 71, 684–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Fossat MJ, Dao TP, Jenkins K, Dellarole M, Yang Y, McCallum SA, Garcia AE, Barrick D, Roumestand C, and Royer CA (2016) High-Resolution Mapping of a Repeat Protein Folding Free Energy Landscape. Biophys. J 111, 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Matilla A, and Radrizzani M. (2005) The Anp32 family of proteins containing leucine-rich repeats. Cerebellum 4, 7–18. [DOI] [PubMed] [Google Scholar]
- (44).Jenkins KA, Fossat MJ, Zhang S, Rai DK, Klein S, Gillilan R, White Z, Gerlich G, McCallum SA, Winter R, Gruner SM, Barrick D, and Royer CA (2018) The consequences of cavity creation on the folding landscape of a repeat protein depend upon context. Proc. Natl. Acad. Sci 115, E8153–E8161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Harsch T, Schneider P, Kieninger B, Donaubauer H, and Kalbitzer HR (2017) Stereospecific assignment of the asparagine and glutamine sidechain amide protons in proteins from chemical shift analysis. J. Biomol. NMR 67, 157–164. [DOI] [PubMed] [Google Scholar]
- (46).Cordier F, and Grzesiek S. (1999) Direct observation of hydrogen bonds in proteins by interresidue (3h)J(NC’) scalar couplings. J. Am. Chem. Soc 121, 1601–1602. [DOI] [PubMed] [Google Scholar]
- (47).Gong Q, and Ishima R. (2007) 15N-{1H} NOE experiment at high magnetic field strengths. J. Biomol. NMR 37, 147–157. [DOI] [PubMed] [Google Scholar]
- (48).Baxter NJ, and Williamson MP (1997) Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 9, 359–369. [DOI] [PubMed] [Google Scholar]
- (49).Cordier F, and Grzesiek S. (2002) Temperature-dependence of protein hydrogen bond properties as studied by high-resolution NMR. J. Mol. Biol 317, 739–752. [DOI] [PubMed] [Google Scholar]
- (50).Cierpicki T, Zhukov I, Byrd RA, and Otlewski J. (2002) Hydrogen bonds in human ubiquitin reflected in temperature coefficients of amide protons. J. Magn. Reson 157, 178–180. [DOI] [PubMed] [Google Scholar]
- (51).Hong J, Jing Q, and Yao L. (2013) The protein amide1HNchemical shift temperature coefficient reflects thermal expansion of the N-H···O=C hydrogen bond. J. Biomol. NMR 55, 71–78. [DOI] [PubMed] [Google Scholar]
- (52).Cierpicki T, and Otlewski J. (2001) Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 21, 249–261. [DOI] [PubMed] [Google Scholar]
- (53).Skinner JJ, Lim WK, Bédard S, Black BE, and Englander SW (2012) Protein dynamics viewed by hydrogen exchange. Protein Sci. 21, 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Rajagopal P, Jones BE, and Klevit RE (1998) Solvent exchange rates of side-chain amide protons in proteins. J. Biomol. NMR 11, 205–212. [DOI] [PubMed] [Google Scholar]
- (55).Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, Enkhbayar P, Matsushima N, Lee H, Yoo OJ, and Lee JO (2007) Crystal Structure of the TLR4-MD-2 Complex with Bound Endotoxin Antagonist Eritoran. Cell 130, 906–917. [DOI] [PubMed] [Google Scholar]
- (56).Tüchsen E, and Woodward C. (1987) Hydrogen Exchange of Primary Amide Protons in Basic Pancreatic Trypsin Inhibitor: Evidence for NH2 Group Rotation in Buried Asparagine Side Chains. Biochemistry 26, 8073–8078. [DOI] [PubMed] [Google Scholar]
- (57).Makhatadze GI, Kim K-S, Woodward C, and Privalov PL (1993) Thermodynamics of bpti folding. Protein Sci. 2, 2028–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Liu A, Lu Z, Wang J, Yao L, Li Y, and Yan H. (2008) NMR detection of bifurcated hydrogen bonds in large proteins. J. Am. Chem. Soc 130, 2428–2429. [DOI] [PubMed] [Google Scholar]
- (59).Preimesberger MR, Majumdar A, Aksel T, Sforza K, Lectka T, Barrick D, and Lecomte JTJ (2015) Direct NMR detection of bifurcated hydrogen bonding in the α-helix N-caps of ankyrin repeat proteins. J. Am. Chem. Soc 137, 1008–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kloss E, and Barrick D. (2008) Thermodynamics, Kinetics, and Salt dependence of Folding of YopM, a Large Leucine-rich Repeat Protein. J. Mol. Biol 383, 1195–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Kloss E, and Barrick D. (2009) C-terminal deletion of leucine-rich repeats from YopM reveals a heterogeneous distribution of stability in a cooperatively folded protein. Protein Sci. 18, 1948–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).LiCata VJ, and Ackers GK (1995) Long-Range, Small Magnitude Nonadditivity of Mutational Effects in Proteins. Biochemistry 34, 3133–3139. [DOI] [PubMed] [Google Scholar]
- (63).Di Cera E. (1995) Thermodynamic theory of site-specific binding processes in biological macromolecules. Cambridge University Press, Cambridge, England. [Google Scholar]
- (64).Krantz DD, Zidovetzki R, Kagan BL, and Zipursky SL (1991) Amphipathic β structure of a leucine-rich repeat peptide. J. Biol. Chem 266, 16801–16807. [PubMed] [Google Scholar]
- (65).Gay NJ, Packman LC, Weldon MA, and Barna JCJ (1991) A leucine-rich repeat peptide derived from the Drosophila Toll receptor forms extended filaments with a β-sheet structure. FEBS Lett. 291, 87–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.