

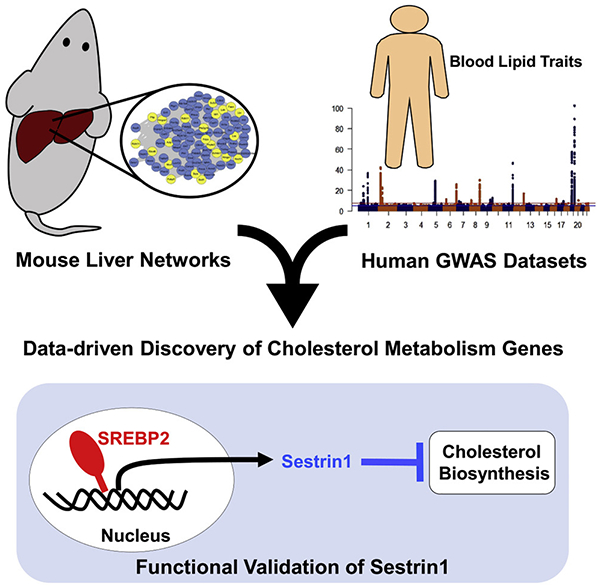
SUMMARY
Identifying the causal gene(s) that connect genetic variation to a phenotype is a challenging problem in genome-wide association studies (GWASs). Here, wse develop a systematic approach that integrates mouse liver co-expression networks with human lipid GWAS data to identify regulators of cholesterol and lipid metabolism. Through our approach, we identified 48 genes showing replication in mice and associated with plasma lipid traits in humans and six genes on the X chromosome. Among these 54 genes, 25 have no previously identified role in lipid metabolism. Based on functional studies and integration with additional human lipid GWAS datasets, we pinpoint Sestrin1 as a causal gene associated with plasma cholesterol levels in humans. Our validation studies demonstrate that Sestrin1 influences plasma cholesterol in multiple mouse models and regulates cholesterol biosynthesis. Our results highlight the power of combining mouse and human datasets for prioritization of human lipid GWAS loci and discovery of lipid genes.
Graphical Abstract
INTRODUCTION
Low-density lipoprotein (LDL)-cholesterol and related plasma lipids are heritable risk factors that contribute to the development of coronary artery disease (Hegele, 2009). Because of their strong heritability, human genome-wide association studies (GWASs) with plasma levels of total cholesterol (TC), LDL-cholesterol, high-density lipoprotein (HDL)-cholesterol, and triglyceride (TG) levels, have discovered more than 150 genome-wide significant loci (Teslovich et al., 2010; Willer et al., 2013). However, despite the wealth of genetic data there are only a handful of examples whereby a locus has translated to the validation of an unknown gene influencing plasma lipid levels (Bauer et al., 2015; Hsieh et al., 2016; Musunuru et al., 2010). The lack of success translating GWAS data to gene discovery is at least partly due to the high rate of false positives in prioritizing and identifying causal candidate genes.
There are many established approaches to facilitate GWAS data translation into causal gene discovery. The most common method of causal gene prioritization is integration of expression Quantitative Trait Locus (eQTL) data to identify genes that show strong local regulation (commonly referred to as cis eQTL) within an associated locus (Willer et al., 2013). The eQTL-based approach is supported by data suggesting that most loci associated with complex traits, including plasma lipid levels, are believed to act through modulation of gene expression (Nicolae et al., 2010). Integration of GWAS data with genomic feature data, such as transcription factor binding sites, DNase hypersensitive sites, histone modification, and chromatin organization data has also allowed for improved dissection of GWAS loci and causal gene identification (Claussnitzer et al., 2015; Smemo et al., 2014; Wang et al., 2016). In addition to genome-wide significant loci, recent studies have demonstrated that associations below the commonly used genome-wide significant P-value (5 × 10−8) cutoff can be biologically valuable and replicated in independent cohorts (Nelson et al., 2017; Wang et al., 2016). Commonly referred to as sub-threshold loci or suggestive loci, these loci represent another source for gene discovery.
A key research goal in the post-GWAS era is to identify the causal genes and biological pathways through which associated loci operate to contribute to alterations in traits. This research is essential to identify new therapeutic targets that can lead to clinically relevant changes. In fact, recent evidence indicates that selecting therapeutic targets that have strong human genetic evidence can potentially double the success rate of drug development (Nelson et al., 2015). Given the difficulties in identifying causal genes in human lipid GWASs, we developed a flexible and scalable approach that could aid in the discovery and prioritization of causal lipid genes as well as identify relevant sub-threshold loci within existing data. To this end, we developed an integrative data-driven method leveraging mouse liver co-expression networks and human lipid GWAS data to identify, prioritize, and validate novel cholesterol and lipid genes (Figure 1A). We validate our approach with the systematic prioritization of Sestrin1 (Sesn1). Sesn1 is replicated across mouse liver co-expression networks and is associated in all human lipid GWAS datasets we analyzed. Our results demonstrate Sesn1 can modulate cholesterol biosynthesis in cells and mice through cholesterol feedback inhibition distinct from previously known mechanisms. Overall, our resource provides a useful method for identification of genes relevant to human variation in plasma lipid levels and highlights the value of cross-species integration for lipid gene discovery.
Figure 1. Identification of conserved liver cholesterol module across diverse mouse populations.
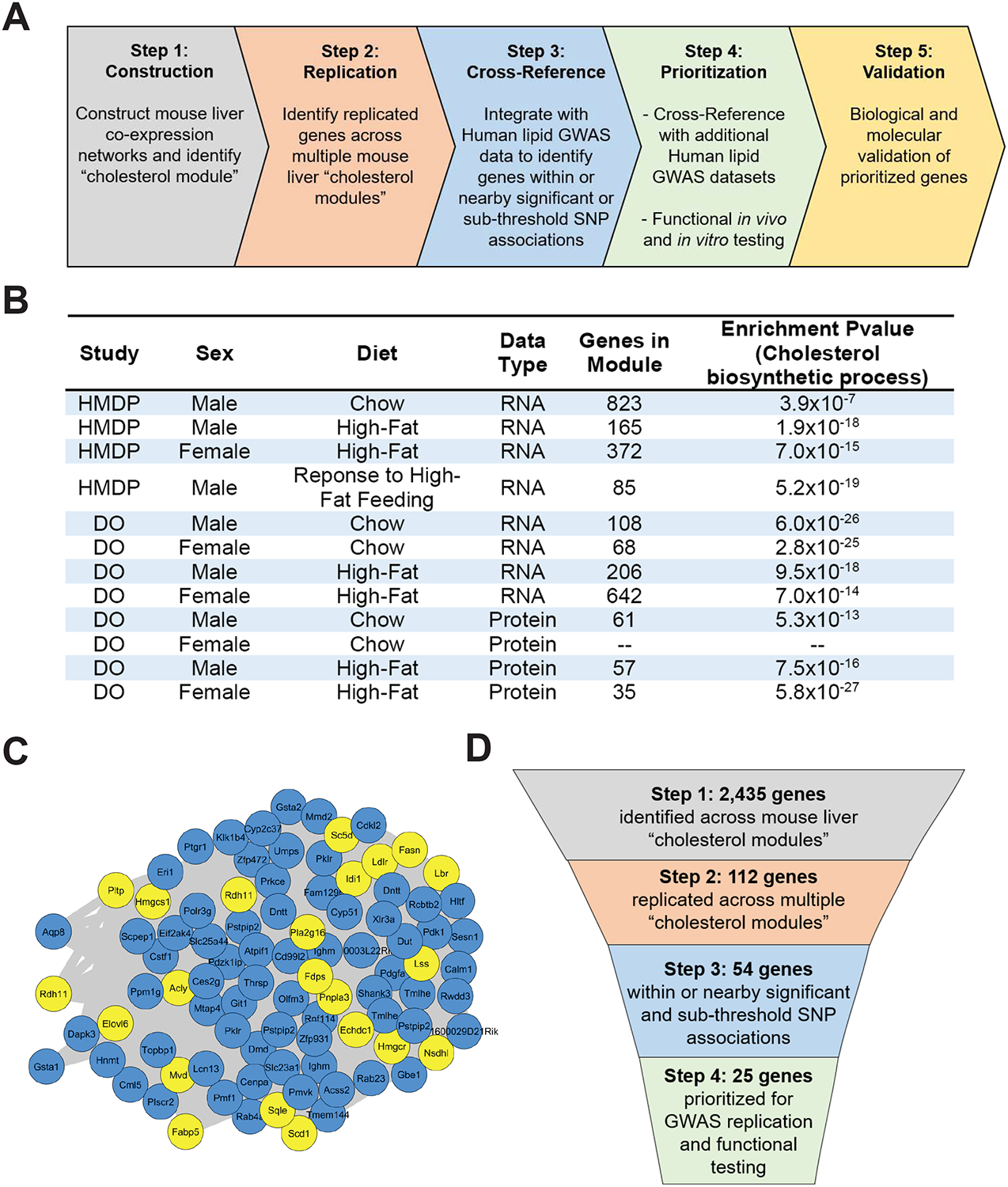
(A) Schematic diagram of stepwise approach to construct liver co-expression networks, identify replicated genes, cross-reference with human lipid GWAS datasets, prioritize genes, and validate candidate genes.
(B) Table showing liver datasets used for WGCNA analysis with indicated mouse population, sex, diet, data type, total number of genes present within the identified cholesterol module, and GO enrichment P-value for cholesterol biosynthetic process.
(C) Representative network view of cholesterol module from the HMDP male mice in response to high-fat feeding. Yellow nodes represent genes with known role in cholesterol and lipid metabolism, and blue nodes represent genes with no known role in cholesterol and lipid metabolism.
(D) Schematic diagram of our integrative stepwise approach with indicated number of genes prioritized at each step.
RESULTS
Genome-wide mouse liver co-expression network analysis
We obtained publicly available mouse liver genome-wide transcriptomic and proteomic data from two mouse genetic reference populations, the Hybrid Mouse Diversity Panel (HMDP) and the Diversity Outbred (DO) population (Chick et al., 2016; Parks et al., 2013; Parks et al., 2015). These data are collected from more than 900 unique mice and are composed of 12 distinct datasets, based on sex, dietary intervention, and molecular trait (transcript or protein) (Table S1). With each individual dataset, we constructed global co-expression networks, using Weighted Gene Correlation Network Analysis (WGCNA), which clusters correlated genes across individuals into modules of genes (Langfelder and Horvath, 2008). In 11 out of 12 datasets, we were able to identify a module of genes significantly enriched for the Gene Ontology (GO) term, cholesterol biosynthetic process (Figures 1B and C), which we term the “cholesterol module”. The cholesterol module ranges in size from 35 to 823 genes depending on the dataset and collectively represents 2,435 unique genes across all 11 identified cholesterol modules (Table S2). With the 2,435 unique genes, we identified genes that were replicated across cholesterol modules in more than three datasets at the transcript level, more than two datasets at the protein level, or present in both transcript and protein datasets. From this analysis, we were able to identify 112 replicated genes (Table S3). With these 112 replicated genes, we were able to recover all the genes involved in the cholesterol biosynthetic pathway (e.g. Hmgcr and Hmgcs1), cholesterol uptake (Ldlr and Pcsk9), cholesterol transcriptional regulation (Srebf2 and Insig1), and lipid metabolism (e.g. Acly and Acss2) (Figure S1A–B). We also intersected the 112 replicated genes with publicly available Sterol regulatory element-binding proteins 1/2 (SREBP1 and SREBP2) genome-wide chromatin immunoprecipitation sequencing (CHIP-seq) data and found 43 genes with significant CHIP-seq peaks (Table S4) (Seo et al., 2009; Seo et al., 2011). Collectively, through mouse liver co-expression network analysis we identified a conserved cholesterol module and our replication analysis identified 112 genes that capture known cholesterol and lipid metabolism genes, providing proof of concept.
Cross-reference with human lipid GWAS data
Next, we cross-referenced the 112 replicated genes from the mouse co-expression networks with the publicly available Global Lipid Genetics Consortium (GLGC) human GWAS data for plasma TC, LDL-cholesterol, HDL-cholesterol, and TG levels (Willer et al., 2013). Projecting the 112 replicated genes from the mouse onto the human genome, we identified single nucleotide polymorphisms (SNPs) within 200 kilobases (kb) of the gene start and stop position that showed association. Using a 5% false discovery rate (FDR) as a sub-threshold significant cutoff (TC, P = 2.4 × 10−4; LDL-cholesterol, P = 1.7 × 10−4; HDL-cholesterol, P = 1.8 × 10−4; TG, P = 1.6 × 10−4) and a genome-wide significant cutoff (P = 5 × 10−8), we were able to identify 48 genes that met our criteria (Nelson et al., 2017; Wang et al., 2016). Among the 48 genes, 18 genes had SNPs at a genome-wide significant level and 30 genes had SNPs at a sub-threshold significant level (Table S5). In total, we identified 48 genes showing replication in mouse liver networks and association with plasma lipid traits in humans (Table 1). We also retained six genes that were on the X chromosome, which is often omitted from most GWASs (Wise et al., 2013). Among these 48 replicated and cross-referenced genes and the six X chromosome genes, 25 have no previously identified role in cholesterol and lipid metabolism. Out of these 25 genes, 21 genes show genome-wide significant or sub-threshold association with lipid traits and four genes are on the X chromosome. In the end, 25 total genes were prioritized for further human GWAS replication analysis and functional testing (Figure 1D and S1C–E).
Table 1.
Cross-reference with GLGC to prioritize 54 genes.
| Human Association | Gene Symbol | Mouse Replication NO. (transcript : protein: both) | Human GWAS Lipid Traits (*sub-threshold) | Lipid and Cholesterol Metabolism Relevant Pathways |
|---|---|---|---|---|
| Genome-wide | Fads1 | 3 ; - ; - | TC, LDL, HDL, TG | Fatty acid and triacylglycerol metabolism |
| Fads2 | 3 ; - ; - | TC, LDL, HDL, TG | Fatty acid metabolism | |
| Acsl5 | 3 ; - ; - | TC, LDL, HDL, TG | Fatty acid metabolism | |
| Gpam | 3 ; - ; - | TC, LDL, HDL, TG | Metabolism of lipids and lipoproteins | |
| Hmgcr | 6 ; 3 ; 2 | TC, LDL | Cholesterol Biosynthesis | |
| Ldlr | 6 ; - ; - | TC, LDL, HDL | Plasma lipoprotein clearance | |
| Mmab | 8 ; 2 ; 2 | TC, HDL | Transcript adjacent to Mvk | |
| Mvk | 7 ; 3 ; 2 | TC, HDL | Cholesterol biosynthesis | |
| Acacb | 3 ; - ; - | TC, HDL | Fatty Acid Biosynthesis | |
| Pcsk9 | 4 ; - ; - | TC, LDL | Plasma lipoprotein clearance | |
| Dhcr24 | 3 ; - ; 2 | TC, LDL | Cholesterol biosynthesis | |
| Echdc1 | 5 ; - ; - | HDL, TG | Fatty Acid Biosynthesis | |
| Aacs | 3; - ; - | TC*, HDL, TG | Fatty acid and triacylglycerol metabolism | |
| Fdft1 | 8 ; 3 ; 2 | TG | Cholesterol biosynthesis | |
| Sub-Threshold | Samm50 | 3 ; - ; - | TC* | Transcript adjacent to Pnpla3 |
| Mvd | 8 ; 3 ; 2 | LDL* | Cholesterol Biosynthesis | |
| Sqle | 8 ; 3 ; 2 | TG* | Cholesterol Biosynthesis | |
| Cyp2r1 | 3 ; - ; - | HDL* | Fatty Acid Biosynthesis | |
| Tmem97 | 5 ; - ; 2 | TC*, LDL* | Cholesterol Regulation | |
| Abcb4 | - ; - ; 2 | LDL* | Fatty acid and triacylglycerol metabolism | |
| Fdps | 8 ; 3 ; 2 | TC*, LDL* | Cholesterol Biosynthesis | |
| Acly | 3 ; - ; 0 | TC*, LDL*, HDL* | Fatty Acid Biosynthesis | |
| Acss2 | 7 ; - ; 2 | TC*, HDL*, TG* | Fatty Acid Biosynthesis | |
| Thrsp | 3 ; - ; - | TC*, LDL* | Fatty acid and triacylglycerol metabolism | |
| Acat2 | 7 ; - ; - | TC*, LDL* | Cholesterol biosynthesis | |
| Tm7sf2 | 5 ; 2 ; 2 | TC*, LDL*, HDL* | Cholesterol biosynthesis | |
| Insig1 | 4 ; - ; - | TC* | Regulation of cholesterol biosynthesis | |
| X -chromosome | Nsdhl | 8 ; 3 ; 2 | Cholesterol Biosynthesis | |
| Ebp | 3 ; - ; - | Cholesterol Biosynthesis | ||
| Genome-wide | Lrpap1 | - ; - ; 2 | TC, LDL, TG | |
| Prss8 | 4 ; - ; - | TC*, LDL*, TG | ||
| Hdgf | 3 ; - ; - | HDL | ||
| Ptprj | 3 ; - ; - | TC, HDL | ||
| Sub-Threshold | Bcap29 | - ; 2 ; - | LDL*, HDL* | |
| Utp18 | - ; - ; 2 | TC*, LDL* | ||
| Sesn1 | 4 ; - ; - | TC*, LDL*, HDL* | ||
| Nol9 | - ; - ; 2 | LDL* | ||
| Aldoc | 3 ; 2 ; 2 | TC*, LDL* | ||
| Cnp | - ; - ; 2 | TC*, LDL*, HDL* | ||
| Nudt2 | 3 ; - ; - | TG* | ||
| Scpep1 | 4 ; - ; - | TG* | ||
| Gramd3 | 4 ; - ; - | TC* | ||
| Pold2 | 5 ; - ; - | TG* | ||
| Naglu | - ; - ; 2 | TC*, LDL*, HDL* | ||
| Rras | 3 ; - ; - | TC*, TG* | ||
| Tlcd1 | 4 ; - ; - | TC*, LDL* | ||
| Pgd | 3 ; - ; - | TC* | ||
| Napa | 3 ; - ; - | TC*, LDL*, HDL* | ||
| Anxa6 | 3 ; - ; - | TC* | ||
| Sigmar1 | 3 ; - ; - | TC* | ||
| X -chromosome | Cetn2 | 4 ; - ; - | ||
| Mospd2 | - ; - ; 2 | |||
| Haus7 | 5 ; - ; 2 | |||
| Slc16a2 | - ; - ; 2 |
Replicated genes were cross-referenced with human Global Lipids Genetics Consortium (GLGC) dataset to prioritize 48 genome-wide or sub-threshold significant genes and six X-chromosome genes.
TC: total cholesterol; LDL: low density lipoprotein; HDL: high density lipoprotein; TG: triglyceride.
Human GWAS replication for prioritized genes
With the 25 prioritized genes, we looked for replication of association in two additional human lipid GWAS datasets, Million Veteran Program (MVP) and the UK-Biobank (UKBB) (Table S6) (Bycroft et al., 2018; Klarin et al., 2018). In the UKBB GWAS for self-reported high-cholesterol, we identified seven genes with SNPs below genome-wide (P = 5 × 10−8) and sub-threshold significance (using 5% FDR as cutoff). In the MVP GWAS data for plasma TC, LDL-cholesterol, HDL-cholesterol, and TG, we identified eight genes with SNPs below genome-wide (P = 5 × 10−8) and sub-threshold significance (P < 1 × 10−6). Seven genes (SESN1, BCAP29, LRPAP1, TLCD1, PRSS8, PTPRJ, and RRAS) out of the 25 genes were associated with at least one human plasma lipid trait in all GWAS datasets we cross-referenced. As most GWAS loci are a result of regulatory variation, we also integrated the 25 genes with three human liver expression quantitative trait locus (eQTL) datasets (Consortium, 2013; Innocenti et al., 2011; Schadt et al., 2008). Out of the 25 prioritized genes, 13 genes show significant cis eQTL in human livers, however, none showed significant co-localization with the GWAS signal (Table S7).
Functional testing of prioritized genes
We performed six functional tests on the 25 prioritized genes to determine their involvement in cholesterol metabolism. In response to cholesterol feeding in mice, 13 and 12 genes showed transcriptional repression in livers after one week and eight weeks of feeding, respectively (Figure 2A, and S2A–C). In contrast, when mice were treated with 0.02% Lovastatin for one week, 12 gene transcripts were upregulated (Figure 2B and S2D). In AML12 hepatocyte cells, depletion of cholesterol with methyl-β-cyclodextrin (MBCD) for three or six hours resulted in transcript upregulation of seven and four genes, respectively (Figure S2E, F). We also determined if gene silencing by siRNA would affect cellular cholesterol levels. Using Hmgcs1 gene expression as an indicator of cellular cholesterol status, we found Hmgcs1 expression significantly altered after 10 genes were silenced (Figure 2C). Based on functional studies, we identified three genes (Aldoc, Sesn1, Sigmar1) that showed significant response across all six functional tests performed (Figure 2D), indicating their functional involvement in cholesterol metabolism in mice and cells.
Figure 2. Functional testing and human GWAS replication of 25 prioritized genes.
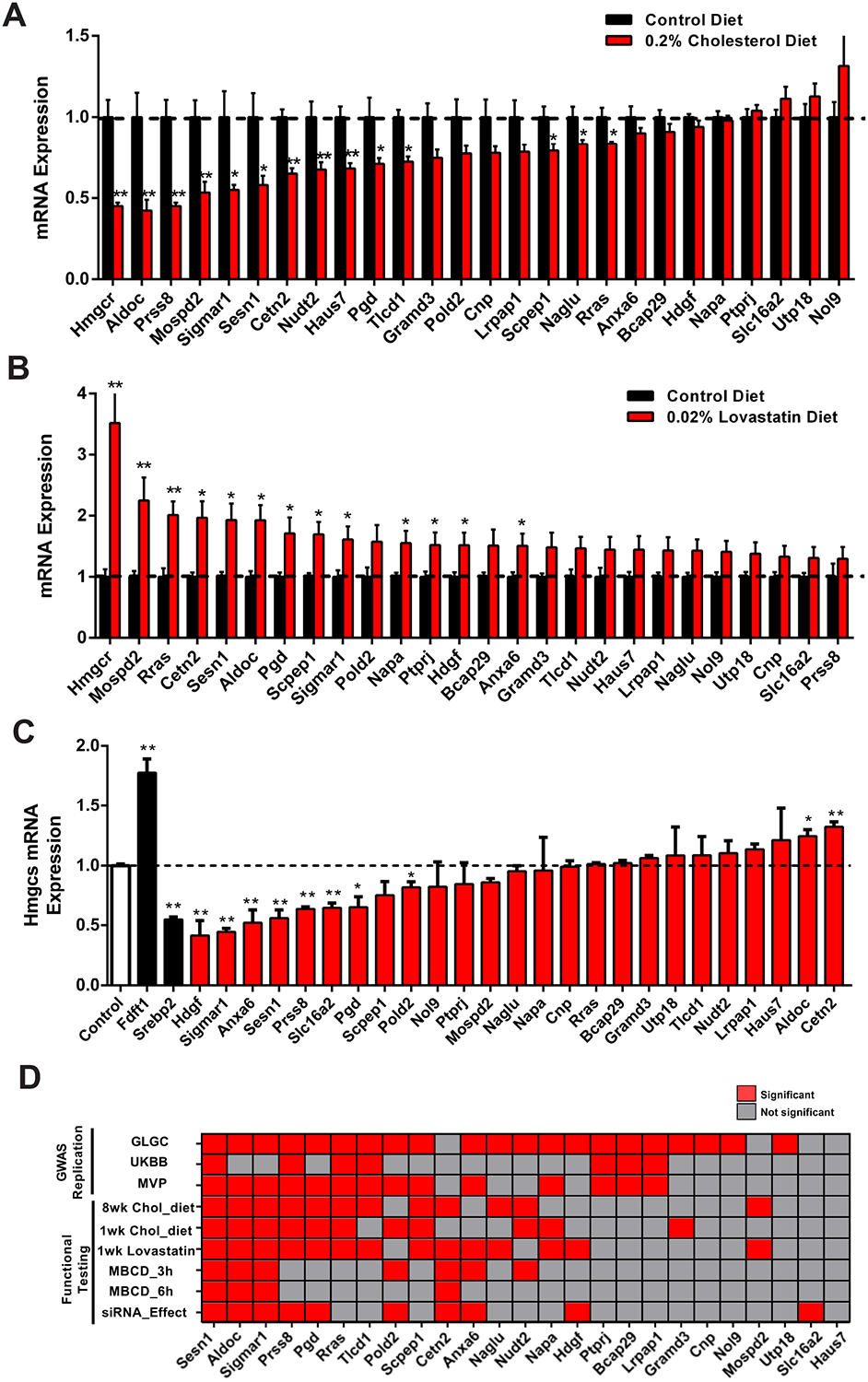
(A) Liver mRNA expression of 25 prioritized genes in male mice fed a 0% (black) or 0.2% (red) cholesterol diet for eight weeks. n = 9.
(B) Liver mRNA expression levels of 25 prioritized genes after one week of 0.02% lovastatin (red) or control (black) treatment in male mice. n = 5.
(C) Hmgcs1 mRNA expression levels after silencing each of the 25 prioritized genes (red) in AML12 cells. Fdft1 and Srebp2 (black) silence was used as positive and negative control. n = 3.
(D) GWAS replication and Functional Testing. Graphical representation of 25 prioritized genes showing association in three human lipid GWAS (GLGC, MVP and UKBB-500K), red indicates significant or sub-threshold association, gray indicates no association. For functional testing, graphical representation of prioritized genes showing significance, red indicates statistical significance (P < 0.05) and gray indicates not significance.
Data represented is mean ± SE. **P < 0.01, *P < 0.05.
Identification and prioritization of Sesn1
Combining human lipid GWAS replication and functional testing results, the only common gene is SESN1 (Sestrin1) (Figure 2D). SESN1 belongs to the Sestrin protein family (SESN1, SESN2, and SESN3), and has never been implicated or reported to be involved in cholesterol metabolism. In human lipid GWAS data, SNPs within or nearby SESN1 are associated with at least one plasma lipid trait across all GWAS datasets we analyzed (Figure 3A, B and Table S6). Extending the association of SESN1 with human plasma lipid traits, a recently published GWAS dataset based on electronic healthcare records identified SNPs nearby SESN1 associated with plasma HDL-cholesterol (rs6243660, P = 5.70 × 10−6) and TG (rs1327472, P = 2.70 × 10−5) (Hoffmann et al., 2018) (Table S8). Using the UKBB data from 500,000 individuals (UKBB-500K) for plasma lipids traits there are multiple SNPs within and nearby SESN1 associated with plasma TC (rs3734649, P = 4.89 × 10−7), LDL-cholesterol (rs720698, P = 4.34 × 10−7), HDL-cholesterol (rs2768541, P = 2.06 × 10−10) and TG (rs2768541, P = 1.68 × 10−10) (Table S8) (McInnes et al., 2019). In the UKBB-500K data, we also identified SNPs within SESN1 (rs720698, P = 6.49 × 10−8) associated with plasma Apolipoprotein B levels, the primary apolipoprotein found on LDL (Table S8). Taken together, the association of SESN1 with plasma lipid traits is highly replicated across multiple independent human GWAS datasets.
Figure 3. The association of SESN1 with human lipid traits in human GWAS datasets.
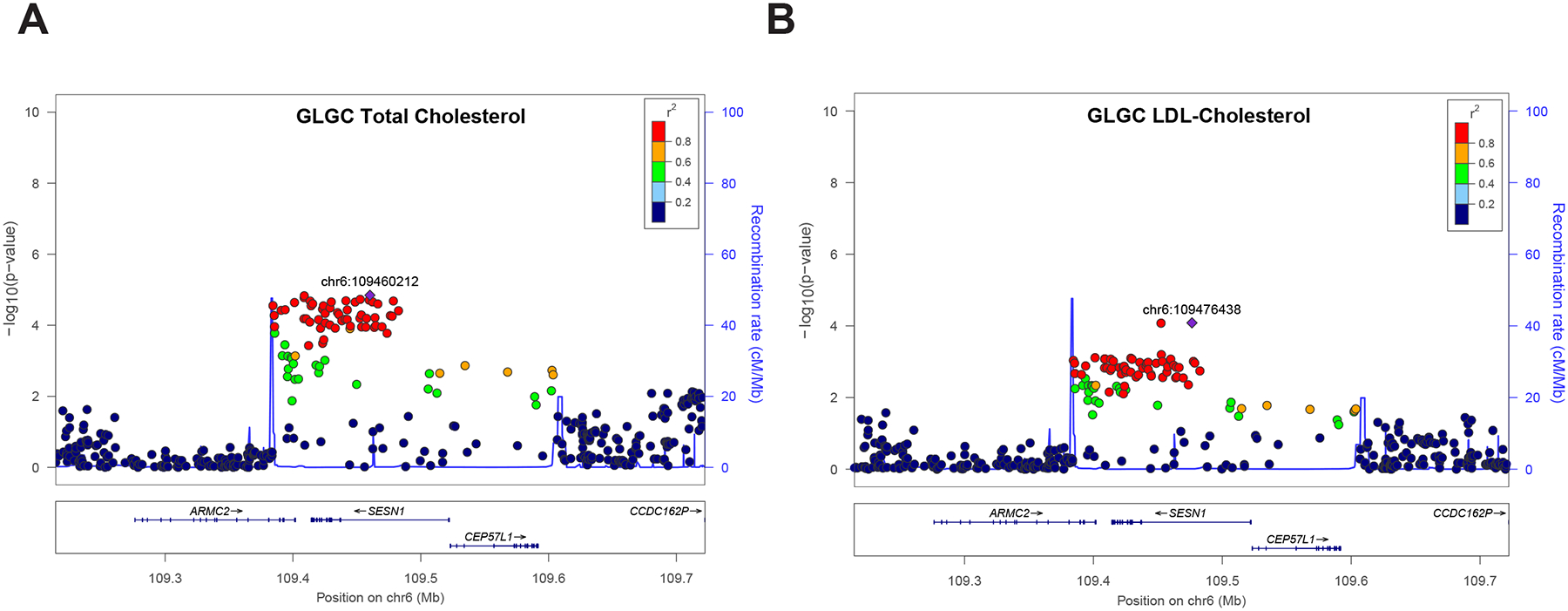
(A) Regional association plot for SESN1 with plasma total cholesterol (TC) in Global Lipids Genetics Consortium (GLGC) 2013 dataset.
(B) Regional association plot for SESN1 with plasma total cholesterol (LDL) in Global Lipids Genetics Consortium (GLGC) 2013 dataset. The left y-axis shows the significance of the association, and the right y-axis shows the recombination rate across the region (blue line). The purple diamond with p-value in red indicates the most associated SNP and adjacent SNPs are colored according to level for linkage disequilibrium (LD) with the most associated SNP.
We next investigated the association of SESN1 with plasma lipid traits using available exome datasets from the GLGC and UKBB. In the GLGC exome analysis of 300,000 individuals, there was one common SNP (rs2273668, P = 9.16 × 10−7, MAF = 0.11) associated with plasma TC (Figure S3A) (Liu et al., 2017; Lu et al., 2017). This common SNP is also associated with plasma TC in both the GLGC GWAS (P = 2.19 × 10−5) and UKBB-500K GWAS (P = 7.99 × 10−7) datasets (Bycroft et al., 2018; McInnes et al., 2019; Willer et al., 2013). In the UKBB exome analysis of 50,000 individuals, we did not detect any SNPs associated with plasma TC (Figure S3B). It worth noting, in the UKBB exome dataset of 50,000 individuals, we did not detect an association with the common SNP (rs2273668), which may be due to the relatively small sample size compared to the other human lipid GWAS datasets. In summary, using the GLGC and UKBB exome datasets, we did not identify any low frequency or rare exome SNPs within SESN1 associated with plasma TC. We also analyzed three publicly available liver eQTL datasets, but did not identify a significant cis eQTL for SESN1 expression (Consortium, 2013; Innocenti et al., 2011; Schadt et al., 2008). One reason for this could be the relatively low expression of SESN1 in human livers (Figure S3C, D).
Sesn1 is transcriptional regulated by cholesterol
Based on our systematic stepwise approach, we pinpoint SESN1 as a candidate causal gene and then initiated biochemical studies focused on the role of SESN1 in cholesterol metabolism. In mice, liver expression levels of Sesn1, but not the other Sestrin family members (Sesn2 and Sesn3), are repressed after seven days of cholesterol feeding (Figure 4A). Moreover, in AML12 hepatocytes, Sesn1 expression is induced upon cholesterol depletion with MBCD and repressed upon addition of cholesterol complexes, while Sesn2 and Sesn3 fail to respond (Figure 4B, C). In other cell types, such as CHO, NIH-3T3, 293T, and HepG2 cells, Sesn1 is also transcriptionally regulated by cholesterol manipulation (Figure S4A–E). Transcriptional regulation of Sesn1 by cholesterol is characteristic of a SREBP2 target gene (Horton et al., 1998; Seo et al., 2011). Consistent with SREBP2 regulation, overexpression of a truncated constitutively active SREBP2 upregulated Sesn1 expression, while silencing or inhibition of SREBP2 blocked the response of Sesn1 to cellular cholesterol depletion (Figure 4D, E and S4F–H) (Toth et al., 2004). Confirming our finding that Sesn1 transcription is regulated by SREBP2 in cells, Sesn1 has been found to contain a SREBP2 CHIP-seq peak (Table S4), supporting that Sesn1 is a SREBP2 target gene.
Figure 4. Sesn1 is required to maintain normal plasma cholesterol levels in mice.
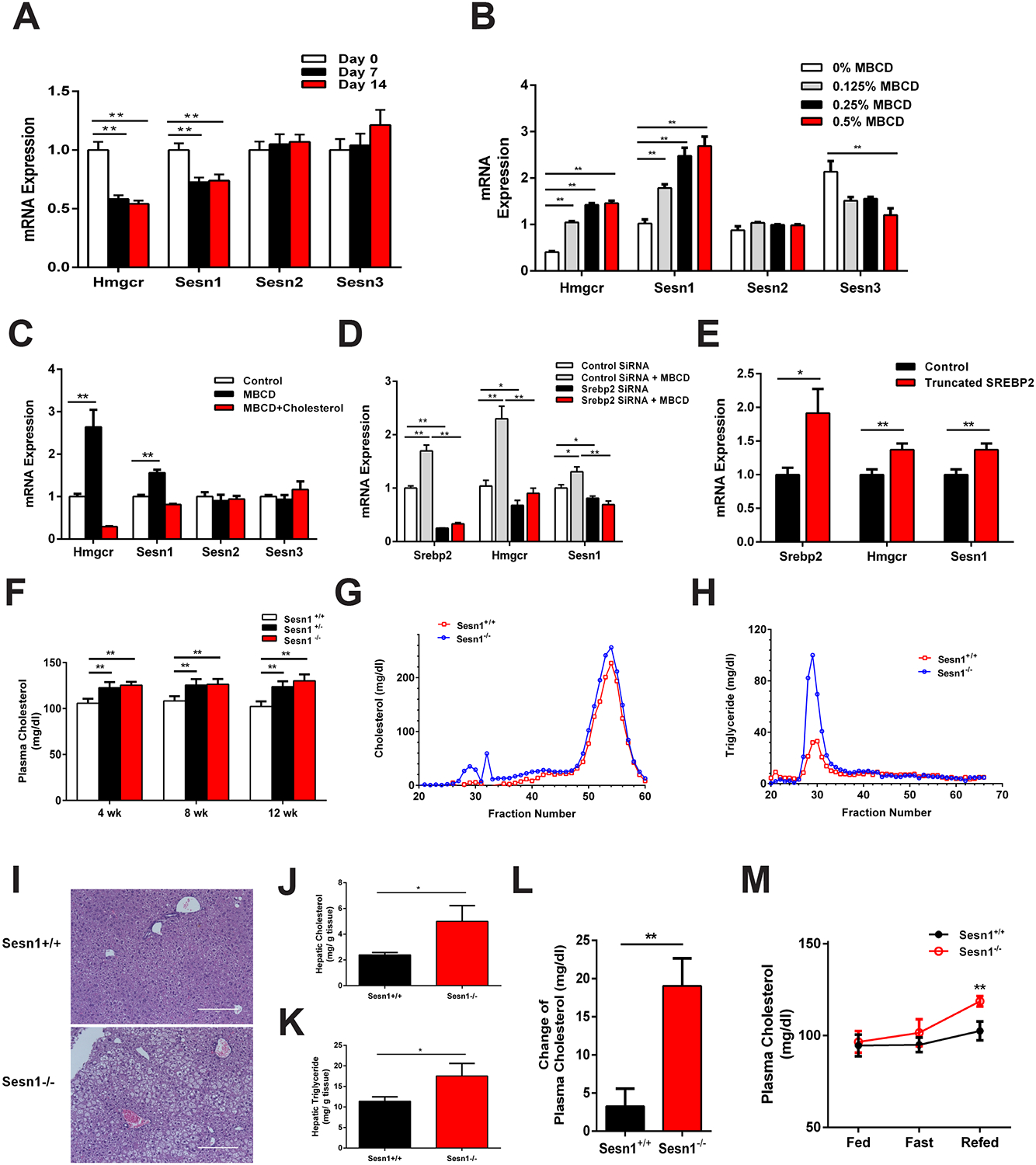
(A) Liver mRNA expression level in mice fed cholesterol diet for 0 (white), 7 (black), and 14 days (red). n = 9.
(B) mRNA expression level in AML12 cells treated with 0% (white), 0.125% (gray), 0.25% (black) or 0.5% MBCD (red). n = 3.
(C) Gene expression of Sesn1 in response to cellular cholesterol. AML12 cells treated with MBCD (black) or MBCD with cholesterol complexes (red). n = 3.
(D) mRNA expression level in SREBP2 siRNA transfected AML12 cells, with or without MBCD treatment. n = 3.
(E) mRNA expression levels in control (black) or truncated Srebp2 plasmid (red) transfected AML12 cells. n = 3.
(F) Total plasma cholesterol levels in Sesn1+/+ (white), Sesn1+/− (black) or Sesn1−/− (red) mice after cholesterol diet feeding for 4, 8, and 12 weeks. n > 14.
(G-H) Fast protein liquid chromatography (FPLC) analysis of plasma cholesterol (G) and triglyceride (H) in Sesn1+/+ (red) and Sesn1−/− (blue) mice. n = 6
(I, J, K) H&E staining (I), hepatic cholesterol (J), and triglyceride content (K) in 12 weeks cholesterol diet fed Sesn1+/+ (top panel) and Sesn1−/− (bottom panel) mouse livers.
(L) Change of plasma cholesterol levels in Sesn1+/+ (black) and Sesn1−/− (red) mice after three days of cholesterol diet feeding. n > 13.
(M) Plasma total cholesterol levels in female Sesn1+/+ (black) and Sesn1−/− (red) mice under random-fed, fasted or refed. n = 5.
Data represented is mean ± SE, **P < 0.01, *P < 0.05.
Sesn1 is required to maintain plasma cholesterol levels
To determine the physiological role of Sesn1, we used a Sesn1 knockout mouse and bred Sesn1 homozygous (Sesn1−/−), heterozygous (Sesn1+/−), and wildtype (Sesn1+/+) mice (Figure S4I). When fed a cholesterol diet, both Sesn1−/− and Sesn1+/− mice have higher levels of plasma TC compared to Sesn1+/+ mice at 4, 8, and 12 weeks (Figure 4F). Fast performance liquid chromatography (FPLC) analysis of plasma showed higher very low-density lipoproteins (VLDL) containing cholesterol and triglyceride particles in Sesn1−/− mice (Figure 4G, H). Sesn1−/− mice also had higher hepatic cholesterol and TG content and histological analysis of the livers showed more lipid droplets, which is suggestive of fatty liver development (Figure 4I–K and S4J). When fed a diet without cholesterol, we observed no difference in plasma TC and TG levels between Sesn1+/+ and Sesn1−/− mice (Figure S4K and S4L).
To investigate adaptation to nutrient status, we measured the ability of Sesn1 to maintain plasma cholesterol levels after consuming a cholesterol diet or during fasting/refeeding cycle. After three days of cholesterol feeding, Sesn1−/− mice have an increase in plasma TC and TG, while Sesn1+/+ mice are able to maintain normal plasma TC and TG levels (Figure 4L and S4M). Moreover, Sesn1−/− mice fail to maintain plasma TC and TG levels during refeeding after a 24 hour fast (Figure 4M and S4N and S4O). Our data show that Sesn1 is required to control adaptation during cholesterol feeding and after refeeding.
Sesn1 functions independent of cholesterol uptake or SREBP2 signaling
Next, we investigated the molecular mechanism through which Sesn1 regulates plasma TC levels. We measured liver LDL receptor (LDLR) and proprotein convertase subtilisin/kexin type 9 (PCSK9) protein expression in cholesterol fed mice and found no difference (Figure 5A). We also crossed Sesn1−/− mice to the Ldlr knockout (Ldlr−/−) background and generated Ldlr−/−Sesn1+/− and Ldlr−/−Sesn1+/+ mice. When fed a cholesterol diet, Ldlr−/−Sesn1+/− mice showed elevated plasma TC levels compared to Ldlr−/−Sesn1+/+ mice (Figure 5B), demonstrating that Sesn1 regulates plasma cholesterol levels independent of the LDLR. We next investigated SREBP2 signaling, which is a major transcriptional regulator of cholesterol metabolism (Sakai et al., 1996). Transcript abundance of SREBP2 target genes (Hmgcr and Hmgcs) and Srebp1c target genes (Fasn and Scd1) were unchanged or lower in Sesn1−/− mouse livers after fasting/refeeding, or after three days and 12 weeks of cholesterol feeding (Figure 5C and S5A–C), indicating intact SREBP2 and SREBP1c signaling in Sesn1 deficient livers. In vitro silencing of Sesn1 in AML12 hepatocytes results in higher cellular cholesterol levels in lipoprotein deficient serum (LPDS) medium, which excludes cholesterol uptake by LDLR (Figure 5D). Importantly, silencing Sesn2 or Sesn3 genes did not alter cellular cholesterol levels (Figure S5D and S5E), demonstrating that Sesn1 is the only Sestrin protein member that affects cholesterol. Consistent with our findings in Sesn1−/− mice, we observed unchanged LDLR protein level and repression of SREBP2/SREBP1c target gene expression after silencing Sesn1 in AML12 cells (Figure 5E and F), which is likely a result of higher cellular cholesterol levels. Moreover, the expression levels of Abca1 and Abcg5/8 were unchanged in Sesn1 deficient livers and when Sesn1 is silenced in AML12 cells, indicating intact liver X receptors (LXR) signaling (Figure S5F and S5G). Taken together, Sesn1 affects plasma and cellular cholesterol independent of LDLR, SREBP2, and LXR.
Figure 5. Sesn1 regulates cellular and plasma cholesterol content independent of LDLR and SREBP2 signaling.

(A) Liver PCSK9 and LDLR protein levels in mice after 0.2% cholesterol diet feeding for three days. n = 4.
(B) Plasma total cholesterol levels in Ldlr−/− Sesn1+/+ (black) and Ldlr−/− Sesn1+/− (red) male mice after 4 and 12 weeks of cholesterol diet feeding. n > 9.
(C) Liver mRNA expression levels of cholesterol and lipid genes in male Sesn1+/+ (black) and Sesn1−/− (red) mice fed a cholesterol diet for three days. n = 4.
(D) Cellular cholesterol levels in AML12 cells after Sesn1 silencing (red) or control (black) incubated in FBS or lipoprotein depleted serum (LPDS) medium. n = 4.
(E) LDLR protein levels in Sesn1 siRNA or control siRNA treated AML12 cells with FBS, n = 4.
(F) mRNA expression levels of cholesterol and lipid metabolism genes in AML12 cells after Sesn1 silencing with FBS or LPDS medium. n = 3.
Data represented is mean ± SE, **P < 0.01, *P < 0.05.
Liver-specific function of Sesn1
To determine which tissue Sesn1 exerts its effects on cholesterol metabolism, we generated a tissue specific knockout mouse model. As the liver is a central regulator of plasma cholesterol levels, we tested the role of Sesn1 in the liver by crossing floxed Sesn1 mice (Sesn1fl/fl) with Albumin-Cre (Alb-Cre+/−) transgenic mice (Figures S6A, B). When fed a high-cholesterol diet for three days, Sesn1 liver-specific heterozygous deficient mice (Alb-cre+/Sesn1fl/wt) significantly increase plasma cholesterol levels compared to control mice (Alb-cre-/Sesn1fl/wt) (Figure 6A). Furthermore, deficiency of Sesn1 in liver resulted in higher plasma cholesterol levels after refeeding (Figure 6B). Taken together, liver specific deletion of Sesn1 impairs plasma cholesterol adaptation to cholesterol feeding and after refeeding.
Figure 6. Sesn1 has a liver-specific role and regulates cholesterol biosynthesis.
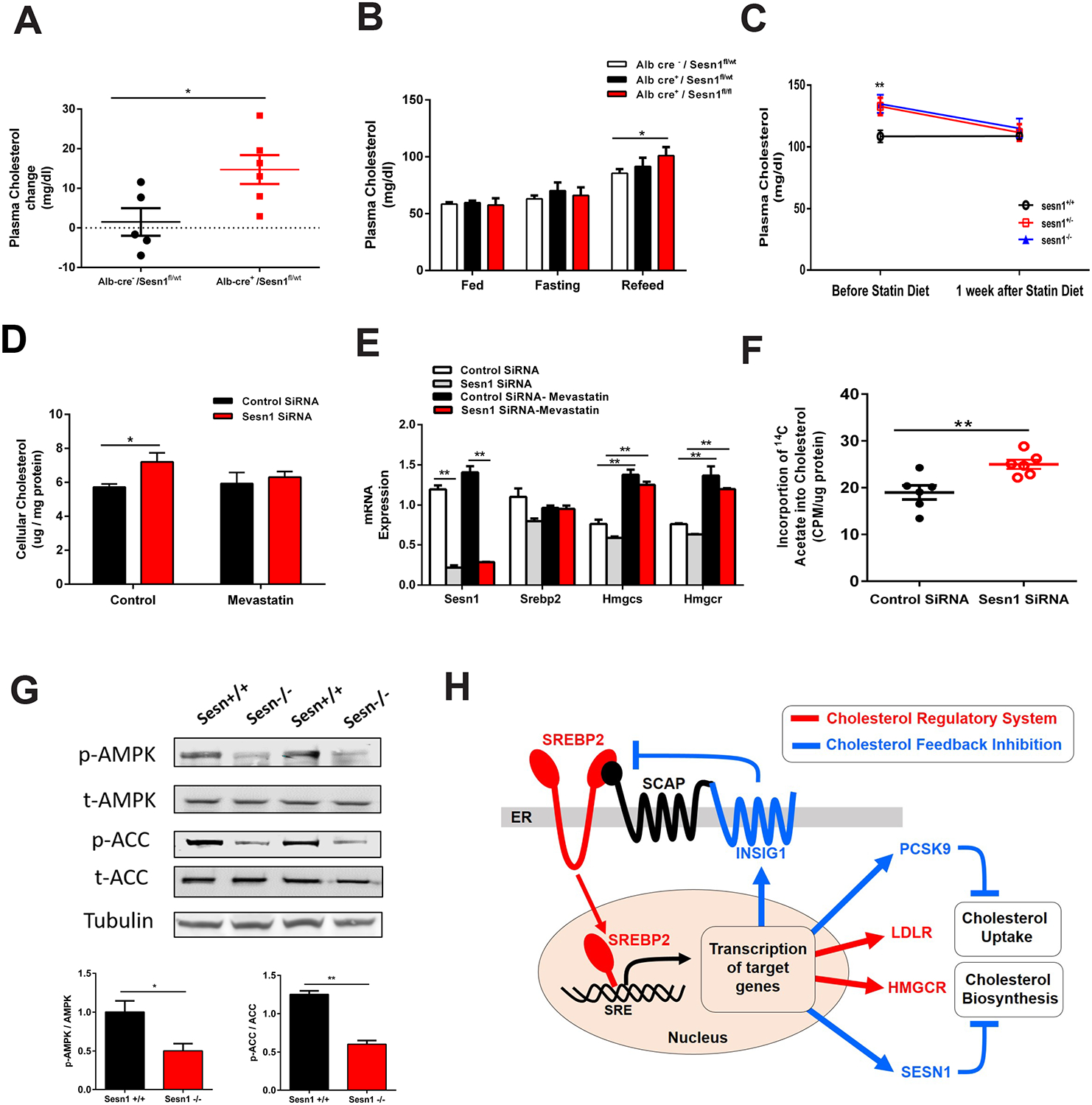
(A) Plasma total cholesterol levels change in three days cholesterol diet fed Alb-cre−/Sesnfl/wt (black) and Alb-cre+/Sesnfl/wt male mice (red). n > 6.
(B) Plasma total cholesterol levels in random fed, fasted, and high-carbohydrate diet refed Alb-cre−/Sesnfl/wt (white), Alb-cre+/Sesnfl/wt (black) and Alb-cre+/Sesnfl/fl (red) female mice (red). n > 3. (C) Plasma total cholesterol levels in Sesn1+/+ (black), Sesn1+/− (red), and Sesn1−/−(blue) mice before and after lovastatin treatment. Mice were fed a cholesterol diet three days. n > 4.
(D) Quantification of total cellular cholesterol levels in AML12 cells after Sesn1 silencing (red) or control (black), with mevastatin. n = 4.
(E) mRNA expression levels in control siRNA (white or black bars) or Sesn1 siRNA (gray or red bars) treated AML12 cells with mevastatin. n = 3.
(F) Measurement of incorporation of radiolabeled 14C-acetate into cholesterol in AML12 cells after Sesn1 silencing (red) or control (black).
(G) Liver protein expression levels and quantification of AMP kinase (AMPK), phosphorylated AMPK, acetyl-CoA carboxylase (ACC) and phosphorylated ACC in Sesn1+/+ and Sesn1−/− mice fed cholesterol diet for 12 weeks.
(H) Cholesterol biosynthesis is controlled by a feedback inhibition mechanism primarily through the action of Insig1 (Insulin induced gene 1). Under cholesterol depletion, SREBP2 is activated for transcriptional activated of cholesterol biosynthesis by HMGCR and/or cholesterol uptake by LDLR, termed as cholesterol regulation (red line). Insig1 is up-regulated, leading to retention of the Scap-SREBP complex and causing SREBP2 inactivation. This is called cholesterol feedback inhibition (blue line). In our study, Sesn1, was found to be a target of SREBP2, and can also inhibit cholesterol biosynthesis. Our finding establishes Sesn1 as a regulator of cholesterol feedback inhibition
Data represented is mean ± SE, **P < 0.01, *P < 0.05.
Sesn1 regulates cholesterol biosynthesis
We next determined the molecular mechanism through which Sesn1 regulates cholesterol metabolism. To determine if Sesn1 influences hepatic cholesterol biosynthesis, we treated mice with Lovastatin to inhibit Hmgcr activity and block cholesterol biosynthesis. In this experiment, we observed a significant reduction in plasma cholesterol levels in cholesterol fed Sesn1+/− and Sesn1−/− mice but not in control mice (Sesn1+/+) (Figure 6C). In AML12 cells, treatment with Mevastatin prevented the effect of Sesn1 on increasing cellular cholesterol levels, without altering SREBP2 or its target genes (Figure 6D and 6E). Moreover, quantification of cholesterol biosynthesis by measuring the incorporation of radiolabeled acetate into cholesterol showed an increase in cholesterol biosynthesis rate when Sesn1 is silenced (Figure 6F). Overall, the data demonstrate that the effect of Sesn1 on plasma cholesterol is through regulation of cholesterol biosynthesis in a liver-specific manner.
As Sesn1 is reported to activate AMP kinase (AMPK) and inhibit the mammalian target of rapamycin complex 1 (mTORC1), we investigated both pathways as potential mechanisms through which Sesn1 could operate (Budanov and Karin, 2008; Kim et al., 2015; Parmigiani et al., 2014). After cholesterol feeding, Sesn1−/− mouse livers had significantly reduced phosphorylated AMPK and phosphorylated ACC, which is an AMPK target (Figure 6G), whereas mTORC1 signaling was unchanged (Figure S6C and S6D). Altogether, our studies indicate Sesn1 functions to repress cholesterol biosynthesis likely through activating AMP kinase, which can phosphorylate Hmgcr and inhibit its activity (Clarke and Hardie, 1990; Loh et al., 2019).
DISCUSSION
Here we present an approach that integrates mouse liver co-expression network analysis with human plasma lipid GWAS data to identify causal genes that drive associations with plasma levels of TC, LDL-cholesterol, HDL-cholesterol, and TG. From the mouse genetic reference population liver datasets, our global co-expression network analysis was able to identify a module of genes significantly enriched for cholesterol biosynthesis in both transcriptomic and proteomic datasets. This cholesterol module was identified in all gene expression and transcriptomic datasets we analyzed and was identified in three out of the four global liver proteomic datasets. One possible explanation for our ability to identify this module across mouse populations is that we are simply identifying gene targets of the SREBP1 and SREBP2 transcription factors. We investigated this possibility and out of the 112 replicated genes that we prioritized from the mouse liver co-expression networks, only 43 genes have SREBP1 or SREBP2 CHIP-seq peaks in existing datasets (Seo et al., 2009; Seo et al., 2011). This result indicates that our cholesterol module is not exclusively composed of SREBP1 and SREBP2 target genes. We were unable to identify a cholesterol module in human liver datasets, which may reflect the high degree of variability and tissue quality collected in human studies versus well-controlled mouse studies.
Through replication across mouse datasets, we identified 112 genes and projected these genes onto the human genome and looked for SNPs that were associated below a 5% FDR cutoff within 200 kb of the gene start and end position. A 200 kb window was used to avoid missing associations that were not directly within the gene region. A 5% FDR cutoff was used to include associations that were below the traditionally employed genome-wide significant threshold (P = 5 × 10−8). With this approach, we were able to identify genes within genome-wide and sub-threshold loci. We identified 48 genes associated with plasma lipid traits below a 5% FDR cut-off and six genes located on the X chromosome. More than half (29 out of 54) of the genes we identified are well-described genes in lipid and cholesterol metabolism, providing evidence that our approach can identify genes known in lipid and cholesterol metabolism pathways. Interestingly, 13 of the known genes are within sub-threshold associations, indicating that sub-threshold loci contain relevant genes to plasma lipid and cholesterol metabolism.
Of the 54 genes we prioritized through human lipid GWAS integration, 25 have no previously recognized role in lipid or cholesterol metabolism. With these 25 candidate lipid genes, we looked for replication of association in two additional human plasma lipid GWAS datasets. We also performed functional tests to screen for genes regulated by cholesterol and affecting cellular cholesterol levels in cells. One drawback of our functional tests is that we are biasing results towards the identification of SREBP2 target genes, as we screened in both mice and cells for transcriptional activity in response to dietary or cellular cholesterol manipulation. However, one of the 25 genes, Slc16a2 (located on the X chromosome), was not responsive to cholesterol in our studies but was identified as a cholesterol regulator gene in our silencing studies in AML12 cells. This gene was recently targeted by the International Mouse Phenotyping Consortium (IMPC). Confirming our finding that Slc16a2 affects cellular cholesterol in cells, Slc16a2 knockout mice have significantly reduced plasma LDL-cholesterol levels (Meehan et al., 2017). Our successful identification of Slc16a2 demonstrates that our integrative approach is not restricted to the identification of SREBP2 target genes.
Another advantage of our approach is the ability to prioritize candidate genes within existing genome-wide significant loci. In this regard, we identified and prioritized PRSS8 as gene within a genome-wide significant loci for TG and sub-threshold loci for TC and LDL-cholesterol. This locus was previously prioritized using eQTL data and the CTF1 (Cardiotrophin 1) gene was prioritized to be the likely causal gene (Willer et al., 2013). However, the locus is large and contains more than a dozen genes within linkage disequilibrium of the lead SNP association. Our data demonstrate that PRSS8 is transcriptionally regulated by cholesterol in mice and can influence cellular cholesterol levels in cells. Taken together, our data indicates that PRSS8 would be a likely causal gene for this locus.
Through lipid GWAS integration and functional tests, we pinpoint Sesn1 as a causal gene associated with human variation in plasma total cholesterol levels. Our identification of Sesn1 was systematic and completely data-driven. Other members of the Sestrin family, in particular Sesn2 have been studied extensively. Sesn2 binds leucine and influences mTORC1 activity through upstream interactions with GATOR2 (Chantranupong et al., 2014; Saxton et al., 2016; Wolfson et al., 2016). In mice, deletion of Sesn2 causes insulin resistance and glucose intolerance which can be exacerbated by deletion of Sesn3 (Lee et al., 2012). However, the role of Sesn1 in cholesterol metabolism has never been reported or studied to our knowledge. In our studies we could find no effect of Sesn2 or Sesn3 on cholesterol metabolism, indicating that Sesn1 is distinct from the other Sestrin family members in regulating cholesterol. Our mouse data showing that liver-specific loss of Sesn1 in mice results in the inability to adapt to cholesterol feeding and refeeding indicates an essential role for Sesn1 in regulation of cholesterol metabolism in vivo.
In our analysis of the SESN1 association in humans, we show that SNPs within or nearby SESN1 are reproducibly associated with plasma lipid traits in multiple independent human GWAS datasets. In both the GLGC and UKBB GWAS plasma lipid datasets there are dozens of common SNPs from Chromosome 6 position 109.28 Mb to 109.38 Mb that are in strong linkage disequilibrium (LD) (r2 > 0.9). The strong LD of SNPs at this region is observed across ethnic populations, which makes fine mapping strategies difficult to narrow down the association at the SESN1 locus. To address the influence of exome variants, we also investigated the contribution of low frequency and rare exome variants in SESN1 and found none associated with plasma cholesterol in the GLGC or UKBB exome datasets. However, there is one common protein-coding variant (rs2273668) for SESN1 associated with plasma cholesterol levels in both the GLGC and UKBB-500K datasets. This variant, which is a missense variant (Leu103Ile), is in strong LD (r2 > 0.9) with dozens of non-coding SNPs, making discerning the contribution of the non-coding SNPs versus the protein-coding SNP difficult. In the future, it will be interesting to test and identify the causal SNP that contributes to SESN1 function or expression regulation that ultimately contributes to plasma cholesterol levels in humans
Our data demonstrate that partial loss of Sesn1 in heterozygous knockout mice is sufficient to disturb the cholesterol biosynthetic pathway and alter plasma cholesterol levels. These results indicate that expression level differences in SESN1 between individuals might drive the association in humans. However, our analysis of human liver eQTL datasets did not identify a significant cis eQTL for SESN1. One possible explanation might be due to the relatively low expression level of SESN1 in liver compared with other tissues. Another possible explanation is that existing liver eQTL datasets do not provide sufficient power to detect a cis eQTL for SESN1.
Taken together, the data would indicate that intact Sesn1 is necessary to maintain cellular and plasma cholesterol levels and that higher expression of Sesn1 leads to lower plasma cholesterol levels during consumption of cholesterol containing foods. We performed detailed biological investigation of Sesn1 and show that Sesn1 is regulated by cholesterol in a SREBP2-dependant manner and that Sesn1 can regulate cholesterol biosynthesis. Cholesterol biosynthesis is highly controlled by feedback regulation through the action of Insig1 (Insulin induced gene 1), which functions to retain SREBP2 in the endoplasmic reticulum (Engelking et al., 2005). Our results suggest that during cholesterol feeding, Sesn1 can also control feedback regulation of cholesterol biosynthesis (Figure 6H). In support of this model, silencing Sesn1 in cells results in higher cholesterol biosynthesis rate without influencing the transcriptional activity of SREBP2 or its target genes. Furthermore, loss of Sesn1 in mice results in impaired ability to adapt to cholesterol feeding and refeeding conditions. Our work establishes Sesn1 as a regulator of cholesterol feedback inhibition independent of Insig1.
Taken together, our systematic stepwise approach combining mouse and human data highlights the power of cross-species analysis and provides a useful approach for the identification of causal lipid GWAS genes. Through our work we were able to prioritize genes within existing lipid loci, identify genes within sub-threshold loci, and validate genes that are associated with plasma lipid and cholesterol levels in humans. Moreover, our biochemical validation studies with Sesn1 identify a new gene that functions in feedback regulation of cholesterol biosynthesis.
Limitations of Study
In our study, we cannot definitively conclude that SESN1 is causally connected to human lipid phenotypes. Our mouse and biological data strongly indicate that expression level differences in SESN1 influences plasma cholesterol levels. However, there is no identifiable cis eQTL for SESN1 in currently available human liver datasets. In the future, larger human liver eQTL datasets with greater detection power may clarify this issue. There is also a lack of evidence to support protein-coding variants affect the function of SESN1 to influence lipid phenotypes in humans. Therefore, testing specific protein-coding variants to determine their effect on SESN1 function will help clarify the causal role of SESN1 in humans.
STAR METHODS
Lead contact and materials availability
Further information and requests for resources and reagents should be directed and will be fulfilled by the Lead Contact, Brian W. Parks (brian.w.parks@wisc.edu).
Experimental model and subject details
Cell Culture Model
AML12 cells were maintained in F12/DMEM 50/50 medium (Corning, Corning, NY and 10% FBS (Corning, Corning, NY) with supplementation with Insulin-Transferrin-Selenium (Corning, Corning, NY). HepG2 cells were maintained in Eagle’s Minimum Essential Medium (EMEM) with 10% FBS. CHO-K1 cells were maintained in F12 medium (Corning, Corning, NY) and 10% FBS. For Betulin (Sigma) experiments, AML12 cells were pre-treated with 6 μm Betulin for 3 hours before experiment and then changed to medium with 0.5% Methyl-beta-cyclodextrin (MBCD) (Alfa Aesar, Havervill, MA) for additional 6 hours. For siRNA knockdown studies, AML12 cells were plated at 50% confluence one day before transfection. Cells were transfected with 50–80 nM scramble or targeted siRNAs together with lipofectamine 2000 (Life Technologies, Eugene, OR) for 6 hours in reduced serum MEM. AML12 cells were incubated with lipoprotein depleted serum (LPDS) (Alfa Aesar, Havervill, MA) medium for additional 24 hours before harvest. For truncated Srebp2, Srebp2 plasmid was transfected into AML12 cells for 48 hours. For MBCD dose response, AML12 cells were treated with 0%, 0.125%, 0.25% or 0.5% MBCD in 0.5% LPDS medium for 6 hours. For statin treatment cell culture experiments, 6 hours after SiRNA transfection, AML12 cells were treated with 2 μM Mevastatin for additional 16 hours.
Mouse Model
Sesn1 global knockout mice (Sesn1tm1b(EUCOMM)wtsi) on the C57BL/6nJ background were generated by the European Conditional Mouse Mutagenesis Program (EUCOMM) and obtained from the Jackson Laboratories (Bar Harbor, Maine). Sesn1 knockout (Sesn1−/−) were maintained as heterozygotes (Sesn1+/−) and interbred to generate experimental control (Sesn1+/+), Sesn1+/−, and Sesn1−/− mice. For double knockout Sesn1 and LDL receptor (LDLR) knockout mice (B6.129S7-Ldlrtm1Her/J), Sesn1−/− mice were bred to generate compound LDLR−/−Sesn1+/+ and LDLR−/−Sesn1+/− experimental mice. For liver-specific Sesn1 knockout mice, Sesn1fl/l (Sesn1tm1c(EUCOMM)wtsi) mice were crossed with Albumin-Cre (B6.Cg-(Alb-cre)/J) mice to generate liver-specific Sesn1 heterogynous (Alb-cre+ /Sesn1fl/wt) and homozygous (Alb-cre+ /Sesn1fl/fl) knockout mice. For cholesterol feeding studies, 8–10 week old experimental mice were fed with 0.2% cholesterol diet (Envigo TD.150322) or 0% cholesterol diet (Envigo TD.110180) for 12 weeks. Bleeding was performed at 3 days, 4, 8 and 12 weeks after 4 hour fast. For fasting-refeeding study, mice were deprived of food for 24 hours or refed with 70% carbohydrate diet (Envigo TD.98090) for 16 hours after a 24 hour fast. For statin treatment in mice, 8–12 week old mice were fed 0.2% cholesterol diet for 3 days, and then fed with 0.2% cholesterol mixed with 0.02% lovastatin for 7 days. Unless specified otherwise all mice used in the study are male. Mice were housed on a 12:12 h light:dark cycle at 25°C with ad libitum access to water and standard laboratory chow diet (Envigo 2019) and were healthy. All experimental procedures were performed with approval from the Institutional Care and Use Committee (IACUC) at the University of Wisconsin-Madison.
Method details
Mouse Genome-Wide Gene Expression, Transcriptomic, and Proteomics Datasets
We obtained publicly available mouse liver data from the Hybrid Mouse Diversity Panel (HMDP) and the Diversity Outbred (DO) mouse genetic reference populations from the following resources: 288 (101 inbred strains) male HMDP mice maintained on a normal chow diet (Gene Expression Omnibus (GEO)-GSE16780; Gene Expression Microarrays, Affymetrix HT Mouse Genome 430A Array)(Bennett et al., 2010), 227 (113 inbred strains) male and 206 (102 inbred strains) female HMDP mice fed a high-fat diet for 8 weeks (GEO-GSE64769), Gene Expression Microarrays, Affymetrix HT Mouse Genome 430A Array)(Parks et al., 2015). Microarray data was RMA normalized in R and data from the same strains were averaged. For the response to high-fat diet data the average gene expression in the liver of high-fat fed strains versus the same strain fed a chow diet was calculated. DO mice liver RNA sequencing data from 48 male and 50 female mice fed a normal chow diet (GEO-GSE72759, Illumina HiSeq 2000), 46 male and 48 female mice fed a high-fat diet for 23 weeks (GEO-GSE72759, Illumina HiSeq 2000) (Chick et al., 2016). DO mice liver proteomic data from 48 male, 50 female mice fed a normal chow diet and 46 male, 48 female mice fed a high-fat diet for 23 weeks is deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD002801 (http://www.proteomexchange.org/). Processed gene-level RNA-seq and proteomic data for DO mice was obtained at (http://churchill-lab.jax.org/website/proteomics/).
Weighted Gene Co-expression Network Analysis (WGCNA)
Network construction was performed using the WGCNA package in R (Langfelder and Horvath, 2008; Zhang and Horvath, 2005). For each of the 12 datasets, all quantified transcripts or proteins were used to construct the network. The pairwise adjacency matrix between genes was used to determine topological overlap between gene pairs. The topological overlap matrix was raised to a power of three to separate the transcripts with many connections and transcripts with few connections. Next, the topological overlap matrix was used to identify modules of significantly co-expressed genes, and the clustering dendogram of the topological overlap matrix was cut to define modules. Defined modules of genes were then subjected to gene set enrichment analysis on Gene Ontology (GO) categories using the runGO function within the GO.db R package, which uses Fisher’s exact test to test for significant enrichment on GO categories (Carlson M (2018). GO.db: A set of annotation maps describing the entire Gene Ontology. R package version 3.7.0.). Network visualization was performed in Cytoscape.
Cross-reference with Human Lipid GWAS Datasets
We obtained summary statistic data (www.lipidgenetics.org) for genotyped and imputed SNPs in the joint meta-analysis described in Willer et. al. (2013) for plasma TC, LDL-cholesterol, HDL-cholesterol, and TG (Willer et al., 2013). For each plasma lipid trait, we calculated a 5% false discovery rate (FDR) using all SNPs using Q-value estimation for false discovery rate control with the qvalue R package (John D. Storey with contributions from Andrew J. Bass, Alan Dabney and David Robinson (2018). qvalue: Q-value estimation for false discovery rate control. R package version 2.14.0.). The 5% FDR value was used as a sub-threshold level for significance and the threshold for which we conducted our cross-reference analysis. For each gene that met our replication criteria in the mouse, we projected onto the human genome (build hg19) and identified SNPs below a 5% FDR threshold within a 200kb window of the gene start and end position. If we identified a SNP below 5% FDR within the gene window, we prioritized the gene. We used data from the GLGC Exome GWAS to confirm our finding in GLGC dataset. The GLGC Exome GWAS data described in Liu et. al. (2017) for plasma TC, LDL-cholesterol, HDL-cholesterol, and TG was analyzed using LocusZoom and a 5% FDR cutoff was used as indication of replication of association within 200kb window of the prioritized gene(Liu et al., 2017; Pruim et al., 2010).
For replication of association in independent lipid GWAS datasets, we used two more plasma lipid GWAS data: Million Veterans Program (MVP) and the UK biobank (Bycroft et al., 2018; Klarin et al., 2018; Liu et al., 2017). The MVP lipid GWAS data described in Klarin et. al. (2018) for plasma TC, LDL-cholesterol, HDL-cholesterol, and TG was analyzed using published results and a 5×10−8 p-value cutoff was used as indication of replication of association within 200 kb window of the prioritized gene (Klarin et al., 2018). The UK biobank self-reported high cholesterol data phenotype was analyzed using publicly available GWAS results at http://www.nealelab.is/uk-biobank/. Genotypes were imputed from HRC plus UK10K & 1000 Genomes reference panels as released by UK Biobank in March 2018. The non-cancer illness code, self-reported high_cholesterol (variable type: binary, n_controls: 317184, n_cases: 43957) was selected. A 5% FDR cutoff was used as indication of replication of association within 200 kb window of the prioritized gene.
For electronic healthcare records based GWAS dataset analysis (Hoffmann et al., 2018), SNPs within 200kb of SESN1 were analyzed for association with plasma TC, LDL-cholesterol, HDL-cholesterol and TG. Subthreshold (P = 5 × 10−5) were used for significance. For UKBB lipid trait analysis, SNPs within 200kb of SESN1 region were analyzed for their association with TC, LDL-cholesterol, HDL-cholesterol, TG as well as Apolipoprotein B using Global Biobank Engine (McInnes et al., 2019): https://biobankengine.stanford.edu/. The p-value cutoffs used for UKBB lipid traits analysis are 5.0 × 10−5.
For UK Biobank exome sequencing analysis, we obtained whole exome sequencing data from the publicly available subset of UKBB participants (Bycroft et al., 2018) (https://www.biorxiv.org/content/10.1101/572347v1). We filtered samples by white British ancestry that had blood lipid data, matched reported sex, and had a greater than 90% genotype call rate as calculated by Plink 1.9 (Chang et al., 2015). Variants with less than 90% genotype call rate or Hardy-Weinberg P less than 1 × 10−15. We then phased with Eagle v2.4.1 to impute missing genotype calls (Loh et al., 2016). We performed single-variant tests for all blood lipid traits in SAIGE and group-based burden and SKAT tests in SAIGE-gene (Zhou et al., 2018) (https://www.biorxiv.org/content/10.1101/583278v1). In both cases, we regressed out covariates age, sex, batch, and principal components. For the group-based tests, gene coding regions were obtained from Gencode v32 (Frankish et al., 2019). Variants in the group-based tests were filtered by minor allele frequency (variants with MAF < 1% or MAF < 5%) and annotations from VEP 97.3 (moderate and high impact or high impact only) (Frankish et al., 2019).
Human eQTL analysis
We performed liver eQTL analysis of prioritized genes in three human liver datasets: Genotype-Tissue Expression (GTEx), European Descent and RNA-Chip Liver eQTL. The gene with following statistical significance (Bonferroni-corrected P < 0.05 for GTEx dataset, p < 1.6 × 10−12 for European Descent dataset, and chi-squared P < 2 × 10−16 for RNA-Chip Liver eQTL) were selected. The colocalization analysis of human GWAS lipid signaling and eQTL signaling were performed using the Bayesian statistical method “Sherlock” at http://sherlock.ucsf.edu. The GLGC lipid datasets, GTEx and European Descent eQTL datasets were selected for signaling colocalization analysis. The Log Bayes Factor (LBF) and P were reported.
Quantification of Cholesterol Synthesis
AML12 cells were cultured for 48 hours in six-well plates (24 hours incubation with 0.5% lipoprotein depleted serum (LPDS) with/without overnight incubation with 2 uM mevastatin), at which time the media were replaced. Then cells were incubated with 1 μCi of [14C] acetate (ARC, 0158A) for 24 hours, after which time the cells were washed twice with phosphate-buffered saline, harvested by trypsinization and scraping, resuspended in PBS. Lipids were extracted into 5 ml of chloroform/methanol (2:1), the solvent was removed by evaporative centrifugation, and the lipids were resuspended in 50 μl of chloroform/methanol and spotted onto silica thin layer plates (Whatman, Florham Park, NJ). Chromatography was carried out in hexane/diethyl ether/acetic acid (80:20:1). Cholesterol was identified by co-chromatography of an authentic standard visualized by iodine vapor staining and quantified by electronic autoradiography. Samples were scraped from the TLC plates and re-suspended in 6 ml of scintillation cocktail and counted in liquid scintillation counter (PerkinElmer).
Plasma Total Cholesterol, Triglyceride Measurement and Fast Protein Liquid Chromatography (FPLC) Analysis
Male Sesn1+/+ and Sesn1−/− mice were fed with 0.2% cholesterol diet for 12 weeks and plasma was collected after 4 hours of fasting before bleeding. Plasma total cholesterol and triglyceride were measured using enzymatic reagent according to manufacturer’s instructions. In all studies, 3 μl of plasma were used for measurement. Concentration of plasma cholesterol and triglyceride was calculated based on standard curve. For FPLC analysis, mouse plasma was collected in EDTA containing tubes, and then centrifuged at 4 °C for 10 min. Plasma was collected and its density adjusted with KBr to d = 1.225 g/ml. The total lipoprotein fraction was isolated following density ultracentrifugation. Following centrifugation the d < 1.225 g/ml fraction was applied to Superose 6 10/300GL columns (GE Healthcare) and separated into lipoprotein classes using size-exclusion fast protein liquid chromatography (FPLC). Each fraction was assayed for total plasma cholesterol (Wako Diagnostics Cholesterol E) and triglyceride (Infinity™Thermo Sci) as described in Pollard, RD et. al. (2015)(Pollard et al., 2015).
Cell and Tissue Cholesterol Quantification
Extraction of lipids was carried out from 3 × 106 cells. Briefly, cells were collected and re-suspended by 1 mL PBS. 100 μL of suspended cells were kept for measurement of protein concentration. The remaining cells were centrifuged and 1 mL of 2:1 chloroform/methanol was added to cell pellet. The cell pellet with organic reagent were vortexed at room temperature for 20 min. After addition of saline (0.9% NaCl), the lower layer was collected for analysis, air-dried, and re-suspended in 100 μL ethanol. The lipid in livers were extracted based on Folch method (Folch et al., 1957). Briefly, mouse liver tissue (~ 40 mg) was homogenized with 2:1 chloroform/methanol and shaken for 20 minutes at room temperature. The solvent was washed with 0.2 volume of 0.9% NaCl solution. After vortexing, the mixture was centrifuged to separate the two phases and the lower organic phase was collected, dried, and re-suspended in 100 μL ethanol. Extracted cholesterol and triglyceride was measured using enzymatic reagent (Pointe Scientific, MI) according to manufacturer’s instructions.
Quantitative PCR
Total RNA was extracted in QIAzol reagent (Qiagen, CA) according to manufacturers’ recommendations. 1000 ng of total RNA was reversed transcribed to cDNA by High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, IL). qPCR assay was performed using KAPA-SYBR-FAST qPCR master mix kit (Sigma-Aldrich, IL) in Roche 480 real time PCR machine (Roche, CT). RNA quantity was normalized to reference genes (RPL4 or GAPDH) and quantification was performed based on standard curve method.
Immunoblotting
Mouse liver (~ 20 mg) was homogenized in RIPA protein extraction buffer (Sigma, IL) with freshly added protease and phosphatase inhibitors (Thermo Fisher, IL) using sonication method. The lysate was then centrifuged at 12,000g for 20 minutes and supernatant was collected. Protein concentration was determined by Pierce Protein Reagent Assay BCA Kit (Thermo Fisher, IL). For immunoblotting, extracted proteins were resolved on SDS-PAGE and analyzed by western blotting using the LI-COR Odyssey Imager (LI-COR Lincoln, NE).
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 6. All data are presented as mean ± SEM. Statistical significance (P < 0.05) was determined by one-way ANOVA then followed by unpaired Student’s t-test. Mice were randomized to study groups and sample size (n) can be found in the legends for the graphs. No power calculations were conducted to predetermine appropriate mouse sample sizes. The number of animal replicates needed was chosen based on similar experiments in the laboratory investigating metabolic phenotypes. We did not exclude any outliers from our work.
Supplementary Material
Table S1: Mouse liver gene expression, transcriptomic, and proteomic datasets used in study, Related to Figure 1.
Table S2: Genes present in cholesterol modules across 11 datasets and 2,435 unique genes across all datasets, related to Figure 1.
Table S3: Replicated genes across 11 cholesterol modules, related to Figure 1.
Table S4: Srebp1/2 CHIP-seq analysis of replicated genes, related to Figure 1.
Table S5: Cross-Reference replicated genes with GLGC human lipid GWAS data to identify 54 genes, related to Figure 1.
Table S6: Cross-reference replicated genes with GLGC-Exome, MVP and UKBB Lipid datasets, related to Figure 2.
Table S7: Liver eQTL analysis of 25 prioritized genes, related to Figure 2.
Table S8: SESN1 is associated with lipid traits in UKBB and electronic health records datasets, related to Figure 3.
ACKNOWLEDGEMENTS
This work was supported in part by NIH HL123021 (BWP), HL147097 (BWP), HL138907 (MGS), AG050135 (DWL), AG051974 (DWL), AG056771 (DWL), and AG062328 (DWL). ZL is supported by NIH Molecular and Applied Nutrition Training Program (T32-DK007665). Parts of this research has been conducted using the UK Biobank Resource under application number 24460. This research was conducted while DWL was an American Federation for Aging Research (AFAR) grant recipient. DWL is supported in part by the U.S. Department of Veterans Affairs (I01-BX004031), and aspects of this work was supported using facilities and resources from the William S. Middleton Memorial Veterans Hospital. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work does not represent the views of the Department of Veterans Affairs or the United States Government. We would like to thank Cristen Willer and Mete Civelek for helpful suggestions. We would like to thank the Rivas lab for making the Global Biobank Engine resource available.
Footnotes
COMPETING INTERESTS
D.W.L has received funding from, and is a scientific advisory board member of, Aeonian Pharmaceuticals, which seeks to develop novel, selective mTOR inhibitors for the treatment of various diseases.
REFERENCES
- Bauer RC, Sasaki M, Cohen DM, Cui J, Smith MA, Yenilmez BO, Steger DJ, and Rader DJ (2015). Tribbles-1 regulates hepatic lipogenesis through posttranscriptional regulation of C/EBPalpha. J Clin Invest 125, 3809–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, et al. (2010). A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res 20, 281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budanov AV, and Karin M (2008). p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. (2018). The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, and Lee JJ (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, and Sabatini DM (2014). The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chick JM, Munger SC, Simecek P, Huttlin EL, Choi K, Gatti DM, Raghupathy N, Svenson KL, Churchill GA, and Gygi SP (2016). Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PR, and Hardie DG (1990). Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 9, 2439–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. (2015). FTO Obesity Variant Circuitry and Adipocyte Browning in Humans. N Engl J Med 373, 895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT (2013). The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelking LJ, Liang G, Hammer RE, Takaishi K, Kuriyama H, Evers BM, Li WP, Horton JD, Goldstein JL, and Brown MS (2005). Schoenheimer effect explained--feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J Clin Invest 115, 2489–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, and Sloane Stanley GH (1957). A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem 226, 497–509. [PubMed] [Google Scholar]
- Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res 47, D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele RA (2009). Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet 10, 109–121. [DOI] [PubMed] [Google Scholar]
- Hoffmann TJ, Theusch E, Haldar T, Ranatunga DK, Jorgenson E, Medina MW, Kvale MN, Kwok PY, Schaefer C, Krauss RM, et al. (2018). A large electronic-health-record-based genome-wide study of serum lipids. Nat Genet 50, 401–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, and Shimano H (1998). Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest 101, 2331–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Koseki M, Molusky MM, Yakushiji E, Ichi I, Westerterp M, Iqbal J, Chan RB, Abramowicz S, Tascau L, et al. (2016). TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature 535, 303–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti F, Cooper GM, Stanaway IB, Gamazon ER, Smith JD, Mirkov S, Ramirez J, Liu W, Lin YS, Moloney C, et al. (2011). Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet 7, e1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Ro SH, Kim M, Park HW, Semple IA, Park H, Cho US, Wang W, Guan KL, Karin M, et al. (2015). Sestrin2 inhibits mTORC1 through modulation of GATOR complexes. Sci Rep 5, 9502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarin D, Damrauer SM, Cho K, Sun YV, Teslovich TM, Honerlaw J, Gagnon DR, DuVall SL, Li J, Peloso GM, et al. (2018). Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat Genet 50, 1514–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW, Bandyopadhyay G, Li N, Aghajan M, Jang I, et al. (2012). Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab 16, 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu DJ, Peloso GM, Yu H, Butterworth AS, Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, et al. (2017). Exome-wide association study of plasma lipids in >300,000 individuals. Nat Genet 49, 1758–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh K, Tam S, Murray-Segal L, Huynh K, Meikle PJ, Scott JW, van Denderen B, Chen Z, Steel R, LeBlond ND, et al. (2019). Inhibition of Adenosine Monophosphate–Activated Protein Kinase–3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Signaling Leads to Hypercholesterolemia and Promotes Hepatic Steatosis and Insulin Resistance. Hepatol Commun 3, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh PR, Danecek P, Palamara PF, Fuchsberger C, Y AR, H KF, Schoenherr S, Forer L, McCarthy S, Abecasis GR, et al. (2016). Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 48, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Peloso GM, Liu DJ, Wu Y, Zhang H, Zhou W, Li J, Tang CS, Dorajoo R, Li H, et al. (2017). Exome chip meta-analysis identifies novel loci and East Asian-specific coding variants that contribute to lipid levels and coronary artery disease. Nat Genet 49, 1722–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes G, Tanigawa Y, DeBoever C, Lavertu A, Olivieri JE, Aguirre M, and Rivas MA (2019). Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. Bioinformatics 35, 2495–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan TF, Conte N, West DB, Jacobsen JO, Mason J, Warren J, Chen CK, Tudose I, Relac M, Matthews P, et al. (2017). Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat Genet 49, 1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, et al. (2010). From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, Zeng L, Ntalla I, Lai FY, Hopewell JC, et al. (2017). Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet 49, 1385–1391. [DOI] [PubMed] [Google Scholar]
- Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, Floratos A, Sham PC, Li MJ, Wang J, et al. (2015). The support of human genetic evidence for approved drug indications. Nat Genet 47, 856–860. [DOI] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, and Cox NJ (2010). Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet 6, e1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. (2013). Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab 17, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks BW, Sallam T, Mehrabian M, Psychogios N, Hui ST, Norheim F, Castellani LW, Rau CD, Pan C, Phun J, et al. (2015). Genetic architecture of insulin resistance in the mouse. Cell Metab 21, 334–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, and Budanov AV (2014). Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep 9, 1281–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard RD, Blesso CN, Zabalawi M, Fulp B, Gerelus M, Zhu X, Lyons EW, Nuradin N, Francone OL, Li XA, et al. (2015). Procollagen C-endopeptidase Enhancer Protein 2 (PCPE2) Reduces Atherosclerosis in Mice by Enhancing Scavenger Receptor Class B1 (SR-BI)-mediated High-density Lipoprotein (HDL)-Cholesteryl Ester Uptake. J Biol Chem 290, 15496–15511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, Boehnke M, Abecasis GR, and Willer CJ (2010). LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 26, 2336–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Duncan EA, Rawson RB, Hua X, Brown MS, and Goldstein JL (1996). Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell 85, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, Schwartz TU, and Sabatini DM (2016). Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science 351, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. (2008). Mapping the genetic architecture of gene expression in human liver. PLoS Biol 6, e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YK, Chong HK, Infante AM, Im SS, Xie X, and Osborne TF (2009). Genome-wide analysis of SREBP-1 binding in mouse liver chromatin reveals a preference for promoter proximal binding to a new motif. Proc Natl Acad Sci U S A 106, 13765–13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, and Osborne TF (2011). Genome-wide localization of SREBP-2 in hepatic chromatin predicts a role in autophagy. Cell Metab 13, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. (2014). Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. (2010). Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth JI, Datta S, Athanikar JN, Freedman LP, and Osborne TF (2004). Selective coactivator interactions in gene activation by SREBP-1a and −1c. Mol Cell Biol 24, 8288–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Tucker NR, Rizki G, Mills R, Krijger PH, de Wit E, Subramanian V, Bartell E, Nguyen XX, Ye J, et al. (2016). Discovery and validation of sub-threshold genome-wide association study loci using epigenomic signatures. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. (2013). Discovery and refinement of loci associated with lipid levels. Nat Genet 45, 1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise AL, Gyi L, and Manolio TA (2013). eXclusion: toward integrating the X chromosome in genome-wide association analyses. Am J Hum Genet 92, 643–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, and Sabatini DM (2016). Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, and Horvath S (2005). A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4, Article17. [DOI] [PubMed] [Google Scholar]
- Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, LeFaive J, VandeHaar P, Gagliano SA, Gifford A, et al. (2018). Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Mouse liver gene expression, transcriptomic, and proteomic datasets used in study, Related to Figure 1.
Table S2: Genes present in cholesterol modules across 11 datasets and 2,435 unique genes across all datasets, related to Figure 1.
Table S3: Replicated genes across 11 cholesterol modules, related to Figure 1.
Table S4: Srebp1/2 CHIP-seq analysis of replicated genes, related to Figure 1.
Table S5: Cross-Reference replicated genes with GLGC human lipid GWAS data to identify 54 genes, related to Figure 1.
Table S6: Cross-reference replicated genes with GLGC-Exome, MVP and UKBB Lipid datasets, related to Figure 2.
Table S7: Liver eQTL analysis of 25 prioritized genes, related to Figure 2.
Table S8: SESN1 is associated with lipid traits in UKBB and electronic health records datasets, related to Figure 3.