

Abstract
Myelodysplastic syndrome (MDS) is a group of heterogeneous disorders caused by ineffective hematopoiesis and characterized by bone marrow dysplasia and cytopenia. Current treatment options for MDS are limited to supportive care, hypomethylating agents, and stem cell transplant. Most patients eventually succumb to the disease or progress to leukemia. Previously, we have shown that CD123 can be used to delineate MDS stem cells in high-risk MDS patients and that the CD123 positive population is biologically distinct from normal hematopoietic stem cells. Furthermore, selective targeting of MDS stem cells can dramatically reduce tumor burden in preclinical models. Based on these findings, we propose CD123 as a candidate target for CAR T cell therapy in high-risk MDS patients. To test this concept, we employed a chimeric antigen receptor (CAR) lentiviral vector containing a CD123-specific single-chain variable fragment in combination with the CD28 costimulatory domain, CD3ζ signaling domain and truncated EGFR (EGFRt). Utilizing this system, we show that CD123 CAR can be expressed on both healthy donor and MDS patient derived T lymphocytes with high efficiency leading to the successful elimination of MDS stem cells both in vitro and in patient-derived xenografts. These results provide concept for the use of CD123 targeted CAR T cells as a therapeutic option for patients with MDS.
INTRODUCTION
Myelodysplastic syndrome (MDS) is a hematological disorder resulting from stem cell driven clonal growth of pathological hematopoietic progenitors and ineffective hematopoiesis1. The annual incidence of MDS is in excess of 20 per 100,000 people2. The pathological progression of MDS is described with each distinct stage of disease evolution and characterized by increasingly aberrant biological features. The international prognostic scoring system (IPSS-R) is utilized to define the life expectancy and leukemic progression3,4. It incorporates degree of pancytopenia, cytogenetic abnormalities, and number of blasts. Early stage, also known as very low and low-risk MDS is characterized by low IPSS-R scores. Intermediate, high, and very high-risk MDS patients display high IPSS-R scores with profound pancytopenia, unfavorable cytogenetic abnormalities, and increased blast count. Approximately 25% of high and very high-risk patients will progress to AML within a year5. Low risk MDS patients are typically managed with supportive care. Upon acquisition of high-risk features patients are typically treated with hypomethylating agents (azacitidine or decitabine), but response rates are only about 30% and resistance develops within two years. Thus, development of new and improved therapies is critical.
Like most myeloid malignancies, MDS is thought to arise from mutations in early hematopoietic stem or progenitor cells. Thus, in order to investigate more effective MDS therapies, we have focused on properties inherent to malignant hematopoietic stem cells (HSCs). Notably, while malignant stem cells in more advanced diseases such as AML have received significant attention, MDS stem cells remain much less characterized6–8. This is likely due to the lack of good experimental models that adequately mimic human disease, as well as the relatively low abundance of tumor cells that can be isolated from patients. Nonetheless, defining therapies that effectively target MDS stem cells is of undeniable importance, and has been the focus of our efforts. We recently reported a detailed study of high-risk MDS patients in which specific molecular and cellular characteristics were described6. Of particular importance to the present study, we demonstrated that the cell surface antigen CD123 is up-regulated on MDS stem cells. This is a notable observation because, while CD123 is expressed at low level on normal HSCs, its expression was also shown to be up-regulated in AML, implying that the antigen is activated during pathogenesis9. Indeed, previous studies have shown a gradual increase of CD123 in low, intermediate, and high-risk MDS10. Our studies demonstrate that expression of CD123 coincides with major changes in the molecular biology of MDS stem cells, which include a strong increase in protein synthesis machinery and changes in energy metabolism6. These findings indicate a clear progression towards the emergence of AML. Thus, expression of CD123 not only provides a means to identify and target MDS stem cells, it also represents a strategy for eradicating the most clinically problematic stage of the disease.
Based on the findings outlined above, we sought to develop therapies that could directly target MDS cells expressing CD123. Namely, we chose to pursue the cell therapy-based approach of Chimeric Antigen Receptor (CAR) T cells. CARs are synthetic constructs that, when introduced into T cells, enable recognition and killing of target cells in a highly specific and MHC independent manner. This is accomplished through an antigen recognition motif that is typically derived from a target-specific antibody which, in turn, is fused to an intracellular CD3 signaling domain (CD3ζ). Incorporation of a T cell costimulatory (e.g. CD28, 4–1BB) domain dramatically increases the potency of CAR T cells11–14.
For the present study, we generated a CD123 CAR consisting of a single-chain variable fragment derived from a monoclonal antibody specific for CD1239. This fragment is fused in-frame to the human immunoglobulin G4 Fc region followed by the CD28 costimulatory domain, CD3ζ signaling domain, and truncated EGFR (EGFRt)9,15. CD123 CAR T cells have demonstrated efficacy in eradicating AML in preclinical studies16 and are currently being evaluated in a clinical trial to treat patients with relapsed/refractory AML and BPDCN (NCT:02159495)17. Given the expression of CD123 in MDS stem cells, we investigated targeting CD123 as a potential strategy for treating patients with high-risk MDS.
METHODS
Generation of CAR lentiviral vector
CD123 CAR and control CD19 CAR constructs were described previously9. CD123 and CD19 lentiviral vectors were produced in 293T cells. In short, 293T cells were plated one day prior to transfection in DMEM with 10% FBS to achieve 80% confluence on the day of transfection. On day of transfection, appropriate amount of lentiviral DNA and CAR vector in 2XHBS CaCl2 were added to the 293T culture. Three days after transfection, viral supernatant was harvested, centrifuged at 2000RPM for 10 minutes at 4°C, filtered through 0.45μm vacuum filter, and mixed with 25% volume of PEG-it™ Virus Precipitation Solution 5X (System Biosciences, CA) (4°C), and stored at 4°C. Next day, the solution was, centrifuged at 2540RPM at 4°C for 30 minutes, aspirated, and centrifuged again at 2540RPM at 4°C for 20 minutes. All trace fluid was carefully removed, and the pellet was resuspended in 1/300 of original volume with cold sterile 1x PBS and stored at −80C until ready for use.
Healthy donor and patient derived T cell transduction
The University of Colorado Institutional Review Board approved the tissue bank protocol (IRB Protocol 06–0720), and all subjects gave informed consent in accordance with the Declaration of Helsinki. Healthy donor derived T cells were obtained from fresh peripheral blood mononuclear cells (PBMCs) using Ficoll-Paque PREMIUM (GE Healthcare, Chicago, IL). Patient derived T cells were obtained from cryopreserved peripheral blood from our MDS patient tissue bank. T cells were selected from the tissue samples using human CD3 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Purified CD3+ cells were cultured in X-VIVO™15 media (Lonza Group, Basel, Switzerland) with 10% heat-inactivated fetal bovine serum (Corning Inc., Corning, NY) and 50 unit/ml IL-2 (PeproTech, NJ) and 0.5ng/ml IL-15 (PeproTech). Cultured T cells were stimulated by CD3/CD28 Dynabeads (Gibco™ Biosciences) then transduced with CD123 CAR or left untransduced (mock). After 6~8 days, CD3/CD28 Dynabeads were removed from T cells, and CD123 CAR expression in transduced T cells was determined by flow with anti-CD3-FITC (BioLegend, CA), anti-human EGFRt (BioLegend), biotin-Protein L (GenScript, China), Straptevidin-PE (BD Biosciences, CA), and Live/Dead-APC/Cy7 viability dye (Invitrogen™ Thermo Fisher Scientific, MA).
CAR T cell cytotoxicity and cytokine assays
To assess CAR T cell mediated cytotoxicity, 5×104 EGFR+ CD123 CAR T cells were cocultured with equal number of MDS-L cells or bone marrow cells derived from MDS patients in X-VIVO™ 15 media with 10% FBS for 48 hours (hrs) at 37°C with 5% CO2. For MDS-L cell line killing assays in vitro, CD123, CD3 and EGFRt expressions were evaluated by flow cytometry to determine the number of MDS-L cells and CAR T cells in coculture using following antibodies: anti-CD123-APC (BD Biosciences), anti-CD3-FITC (BioLegend), and anti-EGFRt-BV421 (Biolegend). For primary MDS cell killing assays, number of MDS stem cell and CAR T cells were assessed by flow cytometry with following antibodies: anti-CD34-PE/Cy7 (BD Biosciences), anti-CD38-PE-CF594 (BD Horizon), anti-CD123-APC (BD Biosciences), anti-CD3-FITC (BioLegend) and anti-EGFRt-BV421 (BioLegend). Viability of each cell population was determined by Live/Dead-APC/Cy7 (Invitrogen™ Thermo Fisher Scientific, MA). For proinflammatory cytokine release assay, coculture supernatants were collected at 48hrs analyzed by V-PLEX Plus Proinflammatory Panel1 (human) Kit (Meso Scale Diagnostics LLC, MA). To assess CAR T cell cytokine response to target cells, intracellular TNF-α expressions and degranulation were measured in CAR T cells that were cocultured with target cells. In brief, equal number of CD123 CAR T cells and target tumor cells (E:T ratio of 1:1), either MDS-L cells or MDS patient bone marrow samples, were cocultured in X-VIVO™ 15 media with 10% FBS, Golgistop™ Protein Transport Inhibitor (BD Biosciences), and anti-human CD107A-BV510 (BD Horizon) for 5hrs at 37°C with 5% CO2. The cells were then harvested and labelled with anti-CD123-BV711 (BD Biosciences), anti-CD3-FITC (BioLegend), anti-CD4-PE/Cy7 (BioLegend) and anti-CD8-APC (BioLegend) as well as viability staining by Live/Dead Far Red-APC/Cy7 (Life technologies). After fixing the cells with Cytofix/Cytoperm™ (BD Biosciences), intracellular staining for cytokines were performed using anti-TNF-α-V450 (BD Biosciences).
Colony formation assay
CD123 CAR T cells, CD19 CAR T cells, and untransduced mock T cells were generated from a healthy donor PBMC and cocultured with autologous CD34+ cells in X-VIVO™ 15 media with 10% FBS for 48hrs at E:T ratio 1:1. Cells were washed once, resuspended in cell resuspension solution (R&D Systems), added to human methycellulous complete media (R&D Systems), and cultured with 2000 CD34+ cells per ml per plate. 8 days later, colonies of BFU-E, CFU-GEMM and CFU-GM were counted manually. Images with all plates were captured with BIO-RAD ChemiDoc™ MP Imaging System with 0.8 second exposure.
Generation of MDS xenograft model
Animal experiments were performed under protocols approved by the University of Colorado Institutional Animal Care and Use Committee (Protocol# B-103416(09)1E). To generate patient derived xenograft (PDX) mice, each NSG-S mouse was conditioned with 1.5μg busulfan in 100ul sterilized FACS buffer (10% heat-inactivated fetal bovine serum (Corning™) in PBS) via intraperitoneal injection 24hrs prior to xenograft transplant. Xenograft cells were resuspended in sterile PBS with OKT3 (BioXCell, NH, 1ug/10×6 cells) prior to intravenous tail vein injection. High-risk MDS patient bone marrow cells (1.5×106 cells from MDS#1 or 0.5×106 cells from MDS#6, per mouse) were used as sources of xenograft. Engraftment was confirmed in randomly chosen mouse that was adoptively transferred with high-risk MDS patient bone marrow cells starting 8 weeks after MDS cell injection. Mouse femurs were flushed. Collected cells were assessed by flow cytometry using anti-human CD45-Alexa Fluor 700 (BD Biosciences), anti-CD34-PE/Cy7, anti-CD38-PE-CF594, and anti-CD123-APC. Xenograft engraftment was determined as greater than 50% hCD45+ in total flushed PDX mouse bone marrow cells and the presence of hCD45+/CD34+/CD38+/CD123+.
Flow cytometry
All flow cytometry analyses were performed by BD FACSCelesta™ flow cytometer (BD Biosciences) and analyzed by FlowJo™ software (FlowJo LLC, OR).
CD123 epitope density quantification
Quantum™ MESF beads were purchased from Bangs Laboratories Inc, IN. Staining and quantification of CD123 expressions was performed following manufactures instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism v5.04 (GraphPad Software Inc., CA). Unpaired Student t tests were used to identify significant differences between treatment groups.
RESULTS
Generation of CD123 CAR T cells
For these studies we utilized a previously characterized CD123 CAR construct that has been shown to be effective in eliminating AML cells both in vitro and in vivo9. CD123 CAR contains a scFv that recognizes CD123, followed by CD28 costimulatory domain, CD3ζ signaling domain, and a truncated EGFR (EGFRt) domain that serves as an epitope tag to identify successfully transduced T cells. As a control, we used an scFv that recognizes CD199,15, cloned into the same vector backbone. In order to generate CAR T cells, we utilized the method outlined in Supplemental Figure 1A. Briefly, CD3 positive cells were isolated from healthy donor peripheral blood mononuclear cells followed by T cell stimulation with IL-2, IL-15, and CD3/CD28 dynabeads. T cells were then transduced in the presence of protamine sulfate and the lentiviral vector overnight before 7 to 10 days culture in IL-2 and IL-15. We evaluated transduction efficiency via flow cytometry through measurement of EGFRt and protein-L. Transduction efficiencies of up to 74% were obtained (MOI 1.0) (Supplemental Figure 1B). Both CD4+ and CD8+ subpopulations were effectively transduced with CD123 and CD19 CAR and confirmed in two different healthy donors (Supplemental Figure 1A–B).
CD123 CAR T cells eradicate CD123+ MDS-L cells in vitro
To examine the anti-tumor function of CD123 CAR T cells, we first examined the tumoricidal function of healthy donor derived CD123 CAR T cells against the MDS-L cell line18. MDS-L was derived from MDS patient with high proportion of cells exhibiting CD34+/CD123+ immunophenotype. CD123+ MDS-L cells were effectively eliminated after coculture with CD123 CAR T cells for 48hrs at an effector to target (E:T) ratio of 1:1. In contrast, no killing was observed when MDS-L cells were cocultured with CD19 CAR T cell, mock T cells, or without any T cells (Figure 1A–B). These results indicate efficient elimination of CD123+ MDS-L cells by CD123 CAR T cells. To further examine the anti-tumor function of CD123 CAR T cells, intracellular production of the inflammatory cytokine TNF-α, was examined in CD123 CAR T cells, CD19 CAR T cells and mock T cells after coculture with MDS-L at E:T ratio of 1:1 or without tumor cell for 5hrs. A significant and robust increase in TNF-α+ cells was detected in response to the CD123 CAR T cell product after coculture with MDS-L cells. This was not observed in CD19 CAR T cells or mock T cells (Figure 1C, Supplemental Figure 1C). In addition, an increased number of CD123 CAR T cells showed surface expression of CD107A+, a surrogate marker for degranulation, suggesting activation of CD123 CAR T cells in the presence of CD123 expressing cells (Figure 1C, Supplemental Figure 1C).
Figure 1. Healthy donor derived CD123 CAR T cells target and eliminate CD123+ MDS-L cell line.
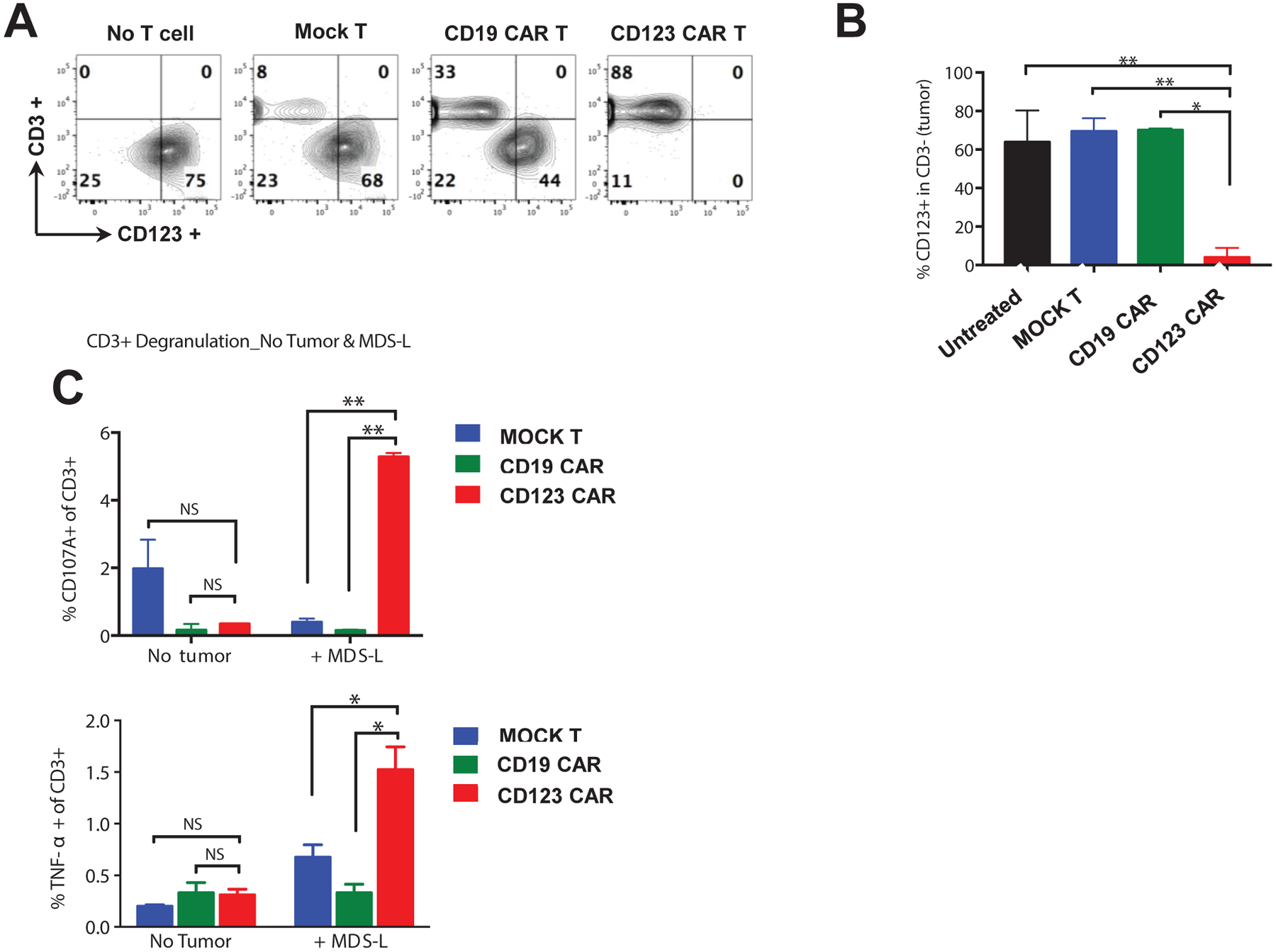
(A) MDS-L cells were cocultured for 48hrs at E:T ratio of 1:1 with mock T cells, CD19 CAR T cells, or CD123 CAR T cells, stained for CD3 and CD123 and analyzed by flow cytometry (mean ± SD of duplicates, n = 2 healthy donors with 3–5 technical duplicates). (B) Statistical analyses of flow cytometry showing elimination of CD123+ MDS-L cells by CD123 CAR T cells (red), but not mock T cells (blue), or CD19 CAR T cells (green). (C) Mock T cells (blue), CD19 CAR T cells (green), CD123 CAR T cells (red) were culture with and without MDS-L cells for 5hrs at E:T ration of 1:1. Presence of CD123 antigen on MDS-L cells leads to increase of CD107A+ and TNF-α+ in CD123 CAR T cells, but not in mock T cells and CD19 CAR T cells (mean ± SD of duplicates, unpaired parametric t-test. NS: p > 0.05, * p ≤ 0.05, ** p ≤ 0.01).
Targeting of CD123 MDS stem cells in patient specimens
Given the efficacy of CD123 CAR T cells to target CD123+ cell lines, we next investigated the activity of CD123 CAR T cells against primary MDS specimens (Table 1). We have recently reported that CD123 is up-regulated during MDS pathogenesis and indicates a stage of disease evolution in which malignant stem cells acquire multiple molecular properties associated with AML19. Primary MDS stem cells are characterized as CD34+/CD38−/CD123+. Initial studies were performed using specimens from four different patients (pt#1–4) with high-risk MDS (Figure 2A). CD123 CAR T cells generated from healthy donors were cocultured with each specimen (pt#1–4) for 48hrs. We observed 50–100% elimination of primary MDS stem cells by calculating the proportion of MDS stem cells (CD34+/CD38−/CD123+) after normalizing to mock treatment (Figure 2B–C). These data suggest that CD123 CAR T cells exhibit efficient and specific anti-tumor activity against CD123 expressing cell lines and primary MDS cells.
Table 1. Characterization of primary high-risk MDS samples.
Age, mutational, cytogenetic profile and % of blast in marrow of 8 high-risk MDS patients used in this study.
| Patient# | Age/ Sex | Mutation | Karyotype | Blasts% |
|---|---|---|---|---|
| 1 | 77 M | IDH2, ASXL1, U2AF | Gain of 1q21 sequences | 10 |
| 2 | 82 M | NRAS | Gain of 8 | 5 |
| 3 | 92 M | DNMT3A, BCOR, TET2 | Normal karyotype | 15 |
| 4 | 55 F | SF3B1 | Normal karyotype | 8 |
| 5 | 62 F | ASXL1, KDM6A, STAG2, TET2 | Normal karyotype | 6 |
| 6 | 65 M | ASXL1, PTPN11, TET2 | Monosomy 7 | 5 |
| 7 | 79 M | RUNX1, TET2 | Normal karyotype | 10 |
| 8 | 71 M | NA | Deletion of 7q31,deletion of 3q21, deletion of 20q12,gain of 11q23 | 5 |
Figure 2. Healthy donor derived CD123 CAR T cells target and eliminate bone marrow derived primary CD123+ MDS stem cells in vitro.
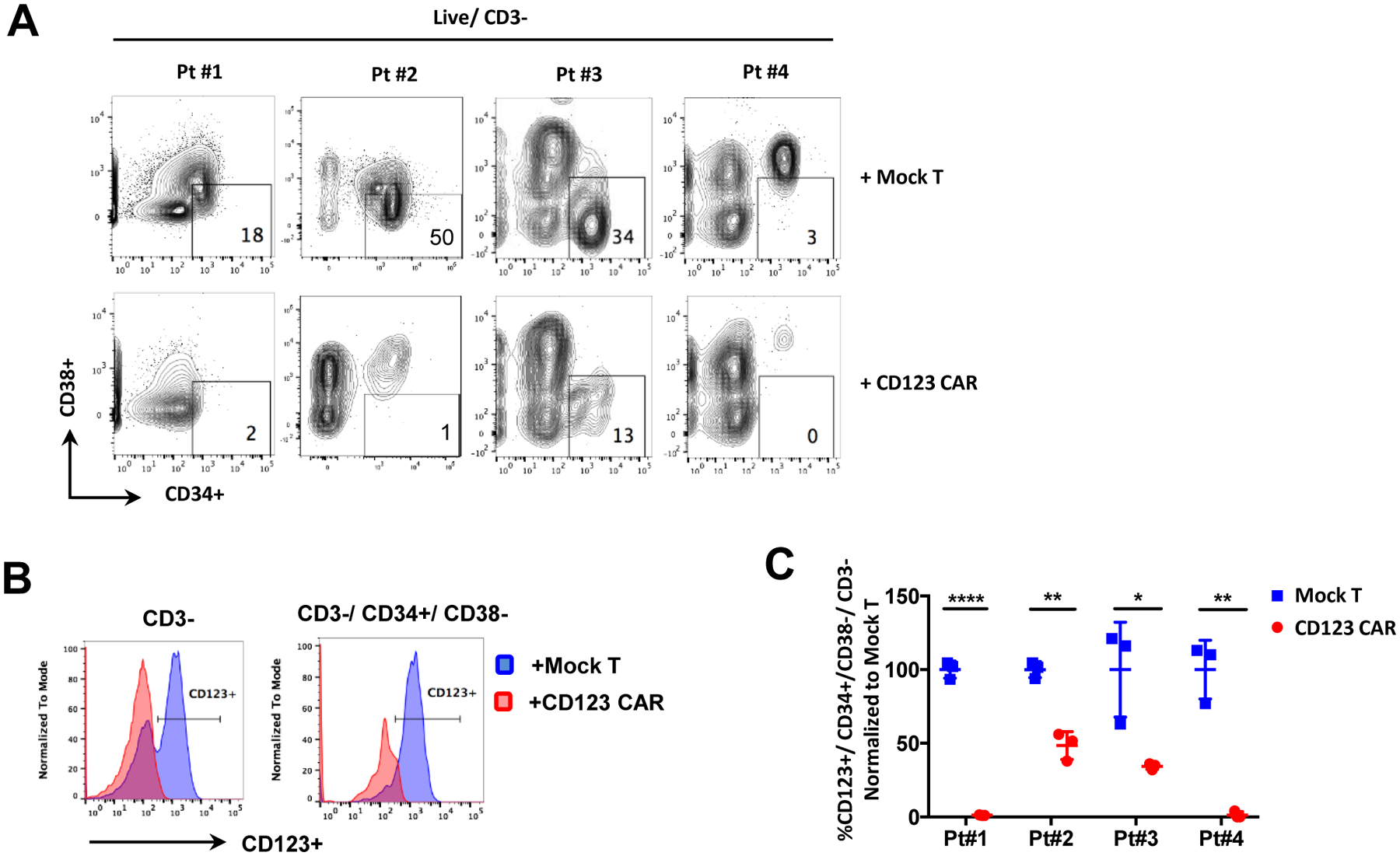
(A) Primary MDS cells from 4 high-risk MDS patients were cocultured with healthy donor derived mock T cells or CD123 CAR T cells for 48hrs at E:T ratio of 1:1. Primitive cells (CD34+ CD38-) in MDS patient bone marrow samples were then analyzed by flow cytometry. (B) Representative flow analyses showing elimination of MDS stem cells (CD34+/CD38−/CD123+) and MDS bone marrow cells (CD3−/CD123+) by CD123 CAR T cells (red) and not by mock T cells (blue), indicated by disappearance of CD123+ subpopulation. (C) Statistical analyses of flow cytometry showing elimination of MDS stem cells in bone marrow samples from 4 different patients by healthy donor derived CD123 CAR T cells (red) compared to mock T cells (blue). Shown by percentage of CD123+CD34+CD38, normalized to mock T cells (n = 3, mean ± SD of duplicates, unpaired parametric t-test. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Autologous CD123 CAR T cells successfully eliminate high-risk MDS disease in vitro
While previous experiments suggest the specificity of CD123 CAR T cells20, the possibility of allogeneic, non-specific targeting of CD123 expressing cells cannot be excluded from the experiments shown in Figures 1–2. In addition, use of allogeneic T cells significantly complicates any potential clinical application. Therefore, we sought to determine if autologous T cells derived from high-risk MDS patient T cells can also be used to generate effective CD123 CAR T cells to eliminate CD123+ MDS cells. To our knowledge, CAR T cells have never been created from MDS patients, thus we sought to test the suitability of autologous lymphocytes.
Peripheral blood T cells were derived from four high-risk MDS patients to generate CD123 CAR T cells as described above. Greater than 50% transduction efficiency was achieved with three patients (pt#2, #5, and #6 after 7 days of transduction, and 9% transduction efficiency was observed with T cells from the fourth patient (pt#1) (Figure 3A). Both CD4+ and CD8+ T cell subpopulations were transduced (Figure 3 A–B). These efficiencies are similar to those achieved in T cells from AML patients transduced with CD123 vectors9.
Figure 3. High-risk MDS patient T cells successfully transduced to express CD123 CAR and target matched bone marrow derived MDS stem cells in vitro.
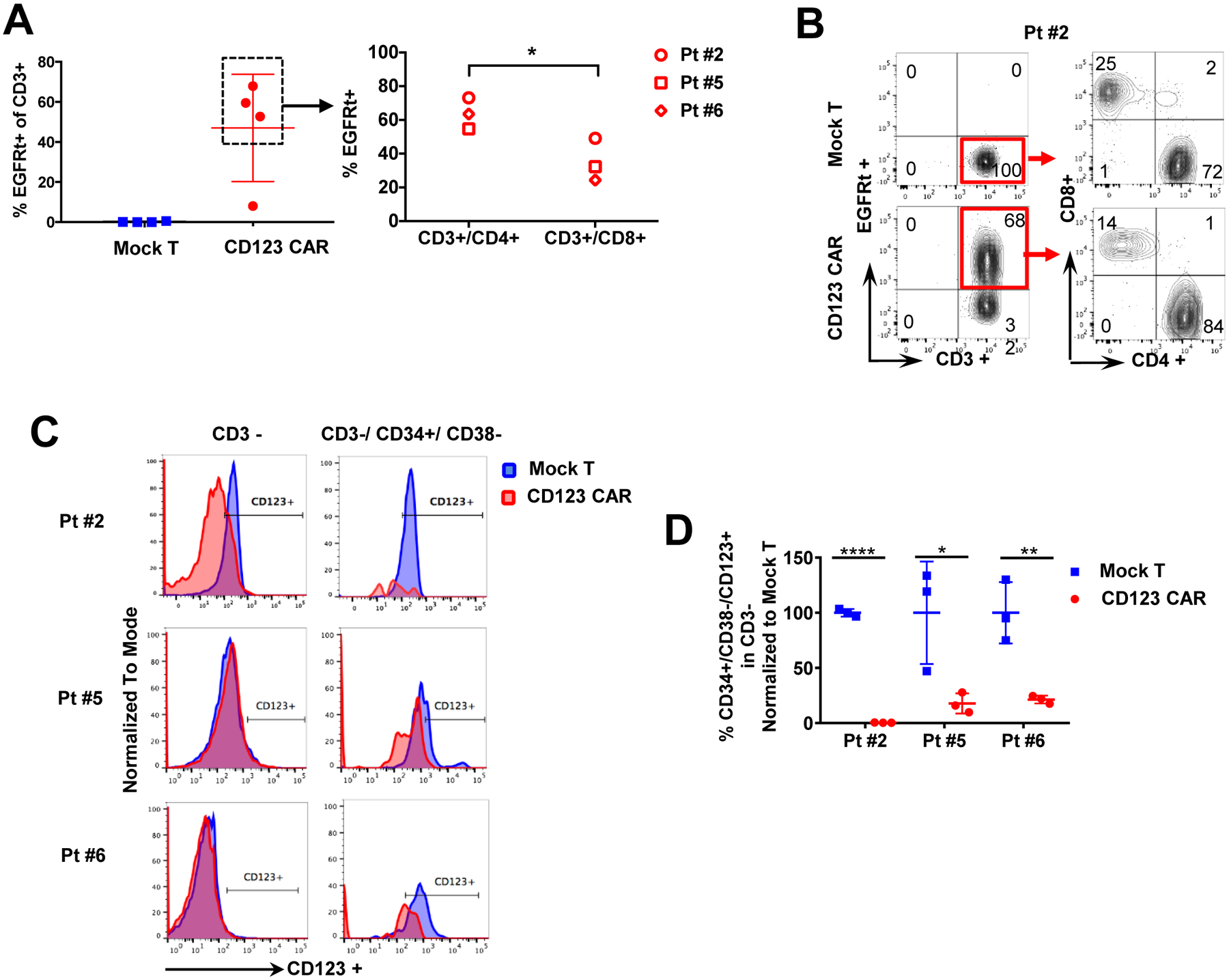
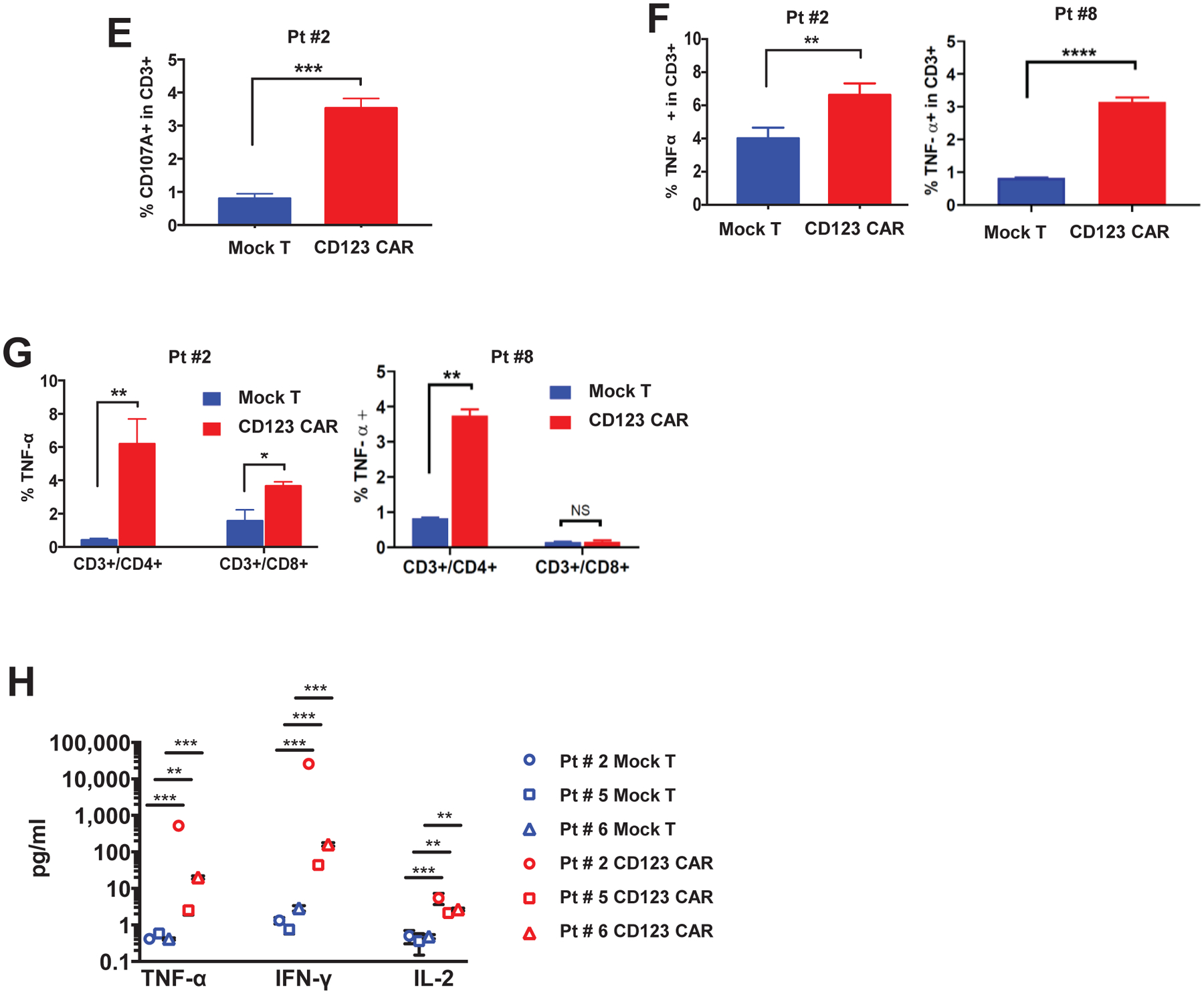
(A) Peripheral blood T cells from 4 high-risk MDS patients (pt#1, #2, #5, #6) were transduced with CD123 CAR. CD123 CAR T cells (red) or mock T cells (blue) were analyzed by flow cytometry. Successful >50% transduction (%EGFRt+ cells) was obtained in 3 patients (pt#2, #5, #6) derived T cells. One patient (pt#1) had 9% transduction efficiency. CD123 CAR T cells from pt#2, #5, #6 were further analyzed for CD4 and CD8. Both CD4 and CD8 populations expressed CD123 CAR. (B) Representative flow cytometry showing transduction efficiency of CD4+ and CD8+ subpopulations in CAR T cells (EGFRt+) and non-transduced mock T cells for pt#2. Red box denotes populations further gated for CD4 and CD8 expression. (C) Histograms of CD123+ cells in pt #2, #5, #6 MDS bone marrow (CD3-) and MDS stem cells (CD3−/CD34+/CD38−) after coculture with autologous CD123 CAR T cells (red) or mock T cells (blue) for 48hrs. (D) Primary MDS cells from pt #2, #5, #6 were cocultured with autologous mock T cells (blue) or CD123 CAR T cells (red) for 48hrs at E:T ratio of 1:1. MDS stem cells (CD34+/CD38−/CD123+) in bone marrow (CD3-) are significantly decreased after coculture with autologous CD123 CAR T cells but not mock T cells. Percentage normalized to mock T cell treatment (n=3, mean ± SD). (E) Degranulation as detected by cell surface CD107A is significantly higher in CD123 CAR T cells (red) compared to mock T cells (blue) after coculture with autologous bone marrow sample at E:T ratio 1:1 for 5hrs (n=3, mean ± SD) in pt#2. (F-G) TNF-α production as detected by intracellular flow cytometry is significantly higher in CD123 CAR T cells (red) compared to mock T cells (blue) after coculture with autologous bone marrow sample in the total CD3+ T cell population (F) and both CD4+ and CD8+ subpopulations (G) at E:T ratio 1:1 for 5hrs (n=3, mean ± SD) in pt#2, #8. (H) Secretion of TNF-α, IFN-γ, and IL-2 in culture supernatant of MDS bone marrow sample with autologous CD123 CAR T cells (red) is significantly higher compared to mock T cells (blue) when cocultured for 48hrs at E:T ratio of 1:1 for pt#2 and pt#6, and 72hrs and E:T ratio 2:1 for pt#5 (n=3, mean ± SD, unpaired parametric t-test, *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001).
Next, to test the functional activity of autologous CAR T cells, we cocultured CD123 CAR and mock T cells with autologous MDS bone marrow samples for 48hrs at an E:T ratio of either 1:1 or 2:1. We observed elimination of both bulk tumor (labeled CD3−/CD123+) as well as MDS stem cells (labeled CD34+/CD38−/CD123+)(Figure 3C, D). Notably, as shown in Figure 3C–D, overall cytotoxicity for MDS stem cells ranged between 80–100% with residual cells remaining at 0.37 ± 1.97% in pt#2, 17.83 ± 27.34% in pt#5, and 21.33 ± 16.2% in Pt#6 when normalized to mock T cell treatment. Additionally, mock T cells did not present significant killing efficacy against autologous target cells when compared to bone marrow only group in either of the two patient samples tested (Figure 3D). Epitope densities of CD123 in remaining CD34+/ CD38−/ CD123+ cells after coculture were analyzed for pt#6. A significant decrease in the CD123 epitope density was observed after the treatment of CD123 CAR T cells in comparison to mock T cells or no T cells. The CD123 CAR T cell treatment resistant cells demonstrated average CD123 for specimens expressing 10–15,000 CD123 molecules/cell, indicating a threshold of CD123 CAR T cell activity at about 10,000 CD123 molecules/cell (Supplemental Figure 2).
To further evaluate the functional activity of autologous generated CAR T cells, we next examined degranulation and secretion of cytokines. Degranulation, as detected by cell surface CD107A expression, was elevated in CD123 CAR T cells, but not in mock T cell after 5hrs coculture in representative patient sample after incubation with autologous MDS bone marrow samples (Figure 3E). Similarly, intracellular levels and secretion of TNF-α were also increased in both patient samples (Figures 3F–G). The increase in TNF-α production was found in both CD4+ and CD8+ subpopulations (Figure 3G). In addition, a panel of human proinflammatory cytokines was analyzed from the supernatant of the cytotoxicity assay, where autologous CD123 CAR T cells were cocultured with MDS bone marrow samples for 5hrs. Significant increases of TNF-α, IFN-γ, and IL-2 were observed in all three patient-derived CD123 CAR T cell products compared to mock T cells (Figure 3H). However, no significant difference in intracellular IFN-γ was observed by flow cytometry (Supplemental Figure 3). Taken together, these results show that autologous CD123 CAR T cells are effective at eliminating MDS stem cells in patient derived specimens in vitro.
Autologous CD123 CAR T cells successfully eliminate high-risk MDS disease in patient derived xenograft model in vivo
To evaluate the activity of CD123 CAR T cells in the context of a preclinical in vivo model, we next established a patient-derived xenograft (PDX) to assess the eradication of MDS stem cells. Recently, we demonstrated that high-risk MDS specimens can successfully engraft immune deficient NSG-S mice when the animals are conditioned via pretreatment with busulfan and T cells depleted from the specimen prior to engraftment19. Using this approach, we established cohorts of mice engrafted with primary MDS marrow derived from patients #1 and #6 (Figure 4A–B). Notably, both specimens were able to engraft into mouse marrow with high efficiency as assessed by the expression of human CD45 in comparison to absence of hCD45 in the pretransplant mice. MDS pt#1 specimen represented 69% of marrow cells at 8 weeks post-transplant with 7% comprised of CD34+ cells. Additionally, 12% of hCD45+ MDS cells and 15% of hCD45+/CD34+ progenitor cells were CD123+, respectively (Figure 4A). MDS pt#6 specimen achieved a level of 68% engraftment at 21 weeks post-transplant, with 25% of cells expressing CD34. CD123 expression was higher in this specimen, with 40% and 52% positive cells in the CD45+ and CD45+/CD34+ populations respectively (Figure 4B).
Figure 4. Eradication of MDS tumor cells in patient derived xenograft by autologous CD123 CAR T cells.
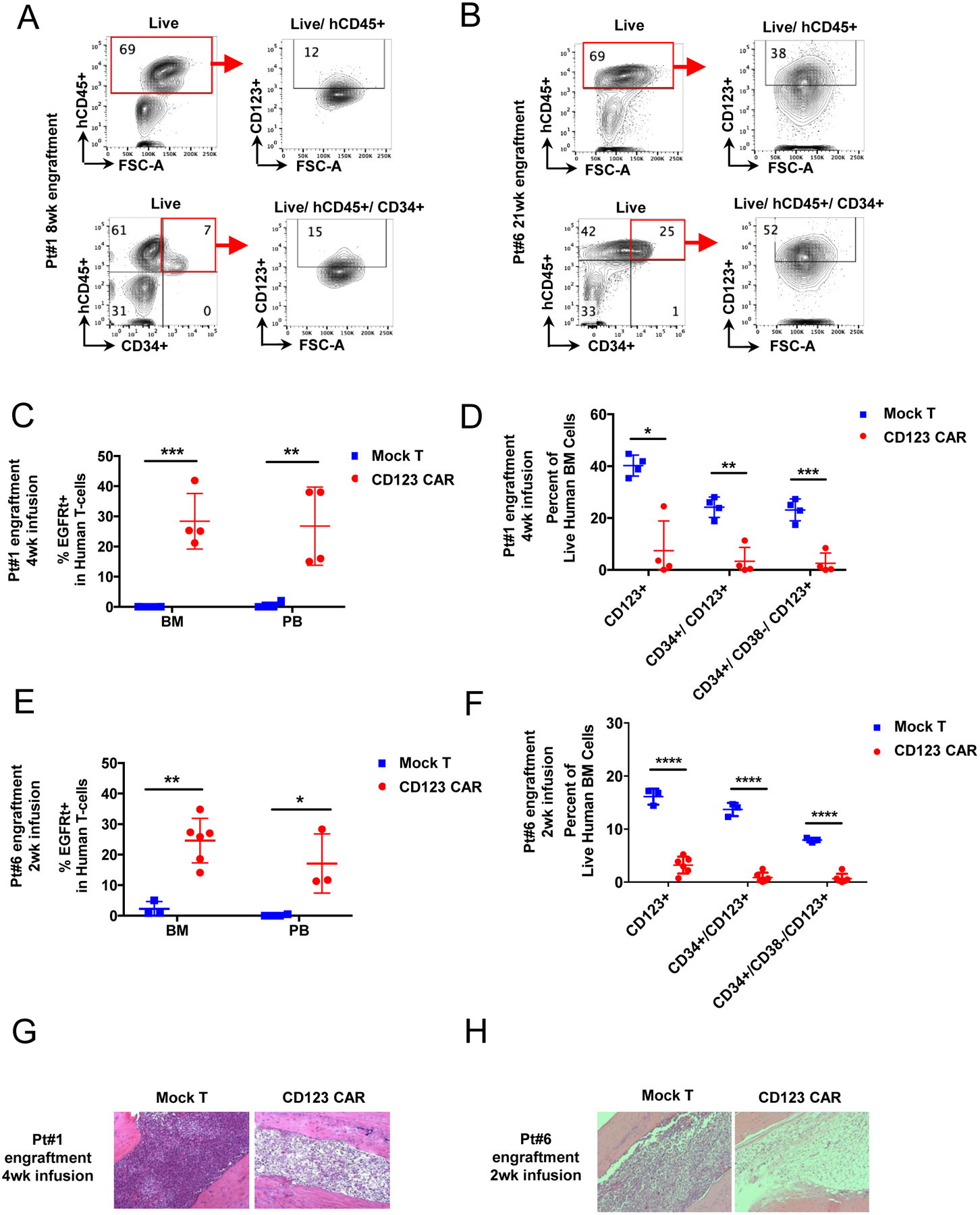
(A-B) Primary MDS cells from 2 high-risk MDS patients (pt#1, #6) were used to generate PDX mice. Representative flow cytometry analysis of each patient sample for CD123+ population in both hCD45+ and hCD45+/CD34+ populations after 8 or 22 weeks of engraftment is shown in pt#1(A) and pt#6 (B). Red boxes denote engrafted human cells (hCD45+). (C-D) PDX from pt#1 were treated with autologous CD123 CAR T cells (red) or mock T cells (blue). 4 weeks later mice were analyzed for persistence of CD123 CAR T cells in bone marrow and peripheral blood (C) and elimination of MDS stem cells (D). (E-F) PDX from pt#6 were treated with autologous CD123 CAR T cells (red) or mock T cells (blue). 2 weeks later mice were analyzed for persistence of CD123 CAR T cells in bone marrow and peripheral blood (E) and elimination of MDS stem cells (F). (mean ± SD, unpaired parametric t-test, n=4 for each group of mock or CD123 CAR T cells for pt#1 xenografts, n = 3 for group of mock and n = 5 for group of CD123 CAR T cells for pt#6 xenografts. (G-H) Representative morphology of MDS xenograft bone marrow showing MDS tumor eradication at 2 or 4 weeks post CD123 CAR T cell infusion in MDS PDX mice pt#1 (G) and pt#6 (H). (Mean ± SD, unpaired parametric t-test. * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001)
For animals transplanted with MDS pt#1 specimen, autologous CAR T cells were administered at 10 weeks post-engraftment. Mice received either autologous CD123 CAR T cells or mock T cells (n=10 of each group, 7×104 CD123 CAR T cells or mock T cells for each animal). At four weeks after T cell infusion, 26.75 ± 6.52% of human CD45+ cells in the mouse peripheral blood and 28.38 ± 4.61% of human CD45+ cells in bone marrow were CD123 CAR T cells as assessed by the expression of the EGFRt marker (Figure 4C). Analysis of bulk MDS cells (hCD45+/CD123+), MDS progenitors (hCD45+/CD34+/CD123+), and MDS stem cells (hCD45+/CD34+/CD38−/CD123)+ demonstrated significantly decreased MDS tumor burden after CD123 CAR T cell treatment (Figure 4D). The decreased MDS tumor burden was further corroborated by performing histological analyses of bone marrow from MDS PDX mice 4 weeks after CD123 CAR T cell infusion (Figure 4G). The images show reduction in tumor burden when mice were treated with CD123 CAR T cells when compared to mock T cells.
The cohort of animals engrafted with the second high-risk MDS patient specimen (#6) exhibited a similar immunophenotype as specimen #1, as shown in Figure 4A and B. At 2 weeks post-transplant, all animals were sacrificed to assess MDS tumor cell eradication, and CAR T cell persistence in xenograft mouse bone marrows (n=3 of mock group, n=6 of CAR T group). CAR T cells represented 24.6 ± 4.44% and 17 ± 4.69% in bone marrow and peripheral blood respectively (Figure 4E), demonstrating CD123 CAR T cell persistence 2 weeks post infusion. CD123 CAR T cell persistence was accompanied by the eradication of MDS cells as shown by significant decreases in the percentage of bulk tumor cells (hCD45+/CD123+), MDS progenitors (hCD45+/CD34+/CD123+), and MDS stem cells (hCD45+/CD34+/CD38−/CD123+) (Figure 4F). Histological examination confirmed the elimination of MDS cells in mouse treated with CD123 CAR T cells, but not mock T cells (Figure 4H).
Normal CD34+ cells retain colony forming capacity after treatment with CD123 CAR T cells
To examine the potential toxicity of CD123 CAR T cells treatment of normal hematopoietic stem cells, healthy donor derived CD123 CAR T cells, CD19 CAR T cells, and untransduced mock T cells were cocultured with autologous matching CD34+ cells for 48hrs at an E:T ratio of 1:1. Cells were then plated in human methycellulose complete media to examine colony formation with 2000 CD34+ cells per plate. 8 days later, colony units of BFU-E, CFU-GEMM, and CFU-GM were manually counted and statistical analyses were performed. Exposure to CD123 CAR T cells did not have any adverse effect on CD34+ cell stemness (Figure 5 A). This result suggests normal stem/progenitor cells are not significantly affected by treatment with autologous CD123 CAR T cells.
Figure 5. Normal CD34+ cells retain colony forming capacity after treatment with CD123 CAR T cells.
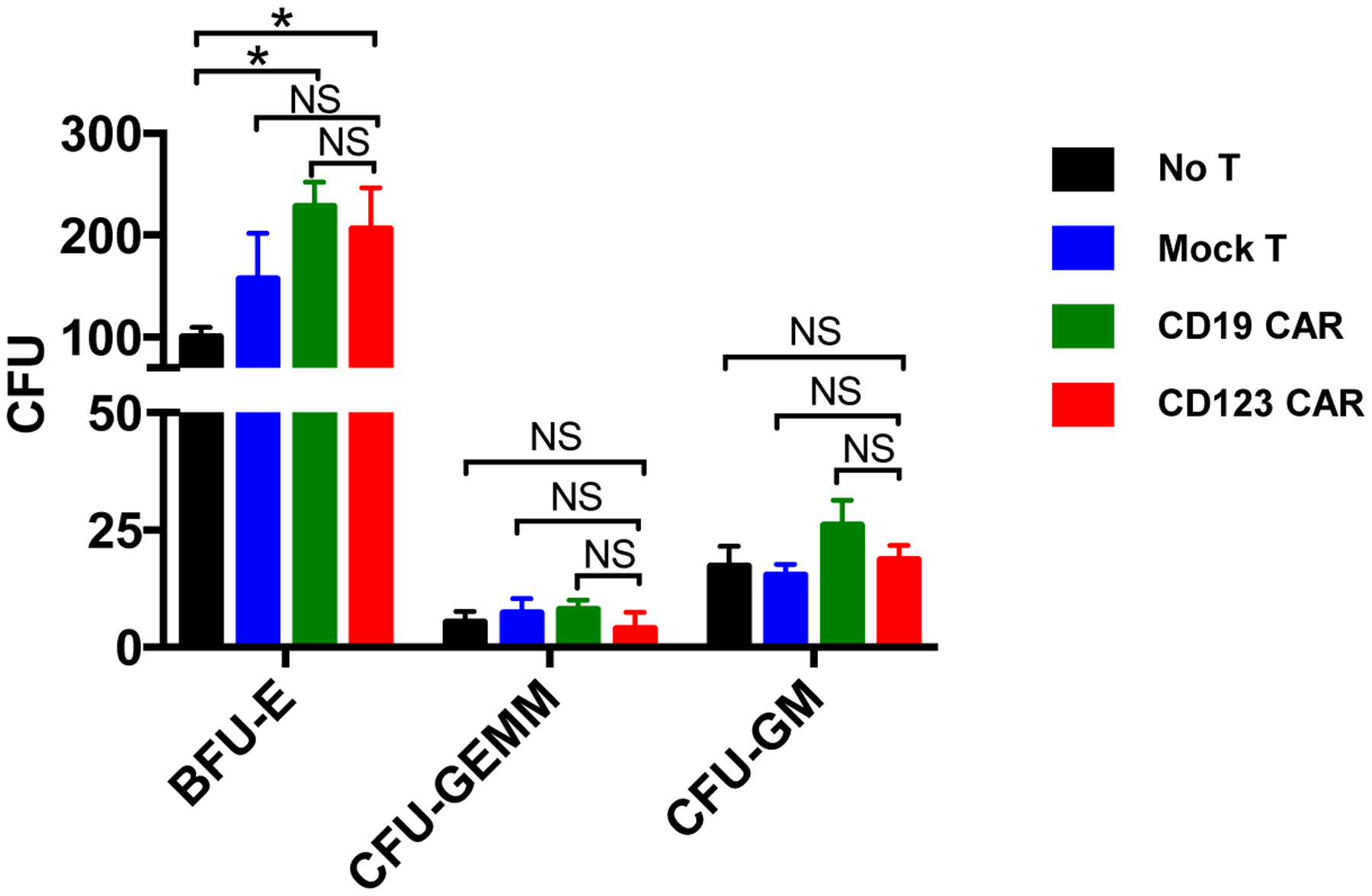
Healthy donor CD34+ cells were cultured with autologous mock T cells (blue), CD19 CAR T cells (green), or CD123 CAR T cells (red) for 48hrs at E:T ratio of 1:1. CD34+ cells were then analyzed for colony formation. No significant adverse effect was observed in BFU-E, CFU-GEMM, or CFU-GM colony formation after coculture with CD123 CAR T cells, CD19 CAR T cells, or mock T cell after 8 days. (N = 3, mean ± SD of triplicates, unpaired parametric t-test, NS: p > 0.05, * p ≤ 0.05.)
DISCUSSION
Developing strategies to treat high-risk MDS and prevent progression to acute leukemia remains a major challenge. To date, MDS therapies have largely been limited to the use of hypomethylating agents, which are not curative and do not prevent progression to AML. MDS is a disease of stem cell origin, and the relative paucity of good models to study MDS pathogenesis and malignant stem cell biology has also been a significant challenge. However, relatively recent studies have begun to define the properties of MDS stem cells derived from primary patient specimens7,8,21–23. Studies have demonstrated MDS stem cells can be identified immunophenotypically including the expression of CD997 and IL1RAP24 on the surface and that these epitopes can be therapeutically targeted. Furthermore, MDS stem cells have been shown to exhibit unique and targetable molecular properties including increased PAK1 and IL-8 signaling25,26. Our work has described specific molecular changes that accompany late stages of MDS stem cell pathogenesis and identified up-regulation of CD123 as a coincident event in the evolution of MDS stem cells19. CD123 has previously been the subject of numerous studies in the AML field, due to its selective expression on AML stem cells. Thus, multiple approaches based on targeting CD123 are being investigated16,27–29.
In the present study, we utilized a CAR T cell strategy to target CD123. CAR T cell therapy has previously demonstrated high remission rate with CD19-directed CAR T cells, namely with pediatric and adult acute lymphoblastic leukemia (ALL), with complete remission (CR) of near 90%30 and near 60% with NHL31. CAR T cell therapy may offer a better prognosis for relapsed patients with hematologic malignancies than chemotherapy. In addition to CD19, there are growing numbers of targets that have been validated with Ab-based therapy, and are being translated as targets for CAR-based therapy for patients with various malignancies.
The findings in this study demonstrate several important scientific principles. First, we show for the first time that autologous T cells derived from MDS patients can be successfully transduced and are highly active in multiple functional assays. Importantly, we obtained effective transduction of both CD4 and CD8 positive T cells from MDS patient specimens. Second, we developed and characterized a xenograft model in which high-risk MDS specimens engraft to high levels and thus provide an in vivo model for characterization of MDS stem cell behavior. Third, we demonstrate that autologous T cells are highly active in our xenograft model and selectively target the CD123 positive population. Equally important, CD123 CAR T cells did not show appreciable targeting of normal hematopoietic stem and progenitor cells. While at least one previous study has reported harm to normal primitive cells32, it appears that the specific design and affinity of a given CAR construct can be tuned for a level of reactivity that maintains sufficient selectivity to be functionally useful. Indeed, a threshold of antigen density for CAR T cells has been reported previously33, hence, it is perhaps not surprising that malignant cells can be differentiated from normal progenitors given that CD123 expression is known to be substantially higher on malignant populations19,27,34,35. In our studies, we observed some surviving CD123+ cells. Using epitope density analysis, we show that CD123 CAR T cells are able to eliminate autologous target cells with CD123 epitope density at about 10 × 103 or higher. This threshold is consistent with reports from other groups, and likely explains the survival of CD123dim non-primitive healthy progenitors36.
It is important to note that while eradicating CD123 positive MDS stem cells may substantially reduce disease burden and pathogenesis, one would expect that residual CD123 negative MDS stem cells might remain. Indeed, Li et al demonstrated progressive increase in CD123 expression as diseases evolves from low to intermediate to high-risk stages10, indicating that MDS stem cells in low risk disease will not be targeted by CD123 CAR T cells. However, given the fact that most MDS patients are generally advanced in age, reducing the risk of progression to AML and decreasing symptoms associated with high-risk MDS may still have highly significant clinical impact.
In summary, the preclinical studies herein provide strong rationale for testing CD123 CAR T cells for the treatment of high-risk MDS patients. Expression of CD123 provides a validated target antigen for cell-based therapy, and also serves as an excellent biomarker to assess the efficacy of CAR T therapy. Furthermore, as a somewhat less acute disease than AML, the setting of MDS provides a clinical context in which various strategies to optimize CAR T use may be evaluated.
Supplementary Material
Supplemental Figure 1. Production of CD123 and CD19 CAR T cells. (A) Schema of CD123 and CD19 CAR T cell production. (B) Representative flow analysis and statistical analyses of CD123 CAR and CD19 CAR transduced healthy donor T cells or mock T cells from healthy donor and average of transduction efficiencies in two different healthy donors (mean ± SD, unpaired parametric t-test). Gating strategies are indicated by the red boxes and arrows.
Supplemental Figure 2. CD123 epitope density required for CD123 CAR T cell efficacy. Primary MDS cells from pt #6 were cocultured with autologous mock T cells or CD123 CAR T cells for 48hrs at E:T ratio of 1:1. MDS stem cells (CD34+/CD38−/CD123+) were analyzed by flow cytometry. Residual CD34+/CD38−/CD123+ cells detected in bone marrow cells after coculture with autologous CD123 CAR T cells, mock T cells or no T cells were analyzed for epitope density of CD123. Epitope density of CD123 in CD34+/CD38−/CD123+ cells after coculture with autologous CD123 CAR T cells is significantly decreased compared to coculture with mock T cells or no T cell, indicating a threshold of CD123 CAR T cell affinity at about 10,000 CD123 molecules/cell (n = 3, mean ± SD, unpaired parametric t-test, * p ≤ 0.05, ** p ≤ 0.01.).
Supplemental Figure 3. Intracellular IFN-γ production in CD123 CAR T cells and mock T cells after coculture with autologous MDS bone marrow sample. pt#2 and pt#8 derived CD123 CAR or mock T cells were cocultured with autologous MDS samples at E:T ratio 1:1 for 5hrs, stained for INF-γ and analyzed by flow cytometry. Intracellular IFN-γ was increased in CD3+/CD4+ subpopulation, but not in the CD3+/CD8+ subpopulation or the total CD3+ cells in CD123 CAR T cells compared to mock T cells derived from pt#2. No significant differences found between T cell compartment when comparing CD123 CAR T cells and mock T cells derived from pt#8. (n = 3, mean ± SD, unpaired parametric t-test. p > 0.05, **** p ≤ 0.0001)
Supplemental Figure 4. Representative histograms of flow cytometry analyses of persisting CD123 CAR T cells in peripheral blood (PB) and bone marrow (BM) of xenografted mice.
ACKNOWLEDGEMENTS
This work was funded by an Edward P Evans foundation translational grant. B.M.S is supported by an Evans P. Evans Young Investigator Award. C.T.J. is supported by R01CA200707.
Footnotes
Conflict-of-interest disclosure: The authors declare no competing final interest.
REFERENCES
- 1.Heaney M, DW DG. Myelodysplasia. N Engl J Med. 1999;340(21):1649–1659. [DOI] [PubMed] [Google Scholar]
- 2.Rollison D, Howlader N, Smith M, et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood. 2008;112(1):452–459. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PL, Tuechler H, Schanz J, et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood. 2012;120(12):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voso MT, Fenu S, Latagliata R, et al. Revised International Prognostic Scoring System (IPSS) Predicts Survival and Leukemic Evolution of Myelodysplastic Syndromes Significantly Better Than IPSS and WHO Prognostic Scoring System: Validation by the Gruppo Romano Mielodisplasie Italian Regional Database. Journal of Clinical Oncology. 2013;31(21):2671–2677. [DOI] [PubMed] [Google Scholar]
- 5.Germing U, Hildebrandt B, Pfeilstöcker M, et al. Refinement of the international prognostic scoring system (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia. 2005;19(12):2223–2231. [DOI] [PubMed] [Google Scholar]
- 6.Stevens BM, Khan N, D’Alessandro A, et al. Characterization and targeting of malignant stem cells in patients with advanced myelodysplastic syndromes. Nature Communications. 2018;9(1):3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung SS, Eng WS, Hu W, et al. CD99 is a therapeutic target on disease stem cells in myeloid malignancies. Science translational medicine. 2017;9(374):eaaj2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barreyro L, Will B, Bartholdy B, et al. Overexpression of IL-1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012;120(6):1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mardiros A, Santos CD, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li LJ, Tao JL, Fu R, et al. Increased CD34+CD38−CD123+ cells in myelodysplastic syndrome displaying malignant features similar to those in AML. International Journal of Hematology. 2014;100(1):60–69. [DOI] [PubMed] [Google Scholar]
- 11.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and Development of Therapies using Chimeric Antigen Receptor-Expressing T cells. Immunol Rev. 2014;257(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savoldo B, Ramos CA, Liu E, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. The Journal of Clinical Investigation. 2011;121(5):1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stegen SJCvd, Hamieh M, Sadelain M. The pharmacology of secondgeneration chimeric antigen receptors. Nature Reviews Drug Discovery. 2015;14:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sadelain M, Brentjens R, Rivière I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discovery. 2013:388–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Chang WC, Wong CW, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood. 2013;122(18):3138–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lihua Budde JYS, Kim Young, Blanchard Suzette, Wagner Jamie, Stein Anthony S, Weng Lihong, Del Real Marissa, Hernandez Rochelle, Marcucci Emanuela, Shepphird Jennifer K, Wang Xiuli, Wood Brent, Marcucci Guido, Brown Christine E, Forman Stephen J. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. American Society of Hematology 2017. 2017. [Google Scholar]
- 18.Nakamura S, Ohnishi K, Yoshida H, et al. Retrovirus-mediated gene transfer of granulocyte colony-stimulating factor receptor (G-CSFR) cDNA into MDS cells and induction of their differentiation by G-CSF. Cytokines, Cellular and Molecular Therapy. 2000;6(2):61–70. [DOI] [PubMed] [Google Scholar]
- 19.Stevens B, Khan N, D’Alessandro A, et al. Characterization and Targeting of Malignant Stem Cells in Patients with Advanced Myelodysplastic Syndromes (MDS). Nature Communications. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mardiros A, Dos Santos C, McDonald T, et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and anti-tumor effects against human acute myeloid leukemia. Blood. 2013:blood-2012-2012-474056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taggart J, Ho T-C, Amin E, et al. MSI2 is required for maintaining activated myelodysplastic syndrome stem cells. Nature Communications. 2016;7:10739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mossner M, Jann J-C, Wittig J, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016;128(9):1246–1259. [DOI] [PubMed] [Google Scholar]
- 23.Woll PS, Kjallquist U, Chowdhury O, et al. Myelodysplastic syndromes are propagated by rare and distinct human cancer stem cells in vivo. Cancer cell. 2014;25(6):794–808. [DOI] [PubMed] [Google Scholar]
- 24.Barreyro L, Will B, Bartholdy B, et al. Overexpression of interleukin 1 receptor accessory protein in stem and progenitor cells and outcome correlation in AML and MDS. Blood. 2012:blood-2012-2001-404699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pandolfi A, Stanley RF, Yu Y, et al. PAK1 is a therapeutic target in acute myeloid leukemia and myelodysplastic syndrome. Blood. 2015:blood-2014-2012-618801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schinke C, Giricz O, Li W, et al. IL8-CXCR2 pathway inhibition as a therapeutic strategy against MDS and AML stem cells. Blood. 2015:blood-2015-2001-621631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin L, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor α chain, eliminates human acute myeloid leukemic stem cells. Cell stem cell. 2009;5(1):31–42. [DOI] [PubMed] [Google Scholar]
- 28.Li F, Sutherland MK, Yu C, et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody-Drug Conjugate for Acute Myeloid Leukemia. Molecular cancer therapeutics. 2018;17(2):554–564. [DOI] [PubMed] [Google Scholar]
- 29.Jordan CT, Upchurch D, Szilvassy SJ, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777–1784. [DOI] [PubMed] [Google Scholar]
- 30.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. New England Journal of Medicine. 2018;378(5):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schuster SJ, Svoboda J, Chong EA, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. New England Journal of Medicine. 2017;377(26):2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor–modified T cells. Blood. 2014;123(15):2343–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe K, Terakura S, Martens AC, et al. Target Antigen Density Governs the Efficacy of Anti–CD20-CD28-CD3 ζ Chimeric Antigen Receptor–Modified Effector CD8+ T Cells. The Journal of Immunology. 2015;194(3):911–920. [DOI] [PubMed] [Google Scholar]
- 34.Jordan C, Upchurch D, Szilvassy S, et al. The interleukin-3 receptor alpha chain is a unique marker for human acute myelogenous leukemia stem cells. Leukemia. 2000;14(10):1777. [DOI] [PubMed] [Google Scholar]
- 35.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645. [DOI] [PubMed] [Google Scholar]
- 36.Mackall CL. Enhancing the Efficacy of CAR T Cells. American Society of Hematology; 2017; Atlanta, GA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Production of CD123 and CD19 CAR T cells. (A) Schema of CD123 and CD19 CAR T cell production. (B) Representative flow analysis and statistical analyses of CD123 CAR and CD19 CAR transduced healthy donor T cells or mock T cells from healthy donor and average of transduction efficiencies in two different healthy donors (mean ± SD, unpaired parametric t-test). Gating strategies are indicated by the red boxes and arrows.
Supplemental Figure 2. CD123 epitope density required for CD123 CAR T cell efficacy. Primary MDS cells from pt #6 were cocultured with autologous mock T cells or CD123 CAR T cells for 48hrs at E:T ratio of 1:1. MDS stem cells (CD34+/CD38−/CD123+) were analyzed by flow cytometry. Residual CD34+/CD38−/CD123+ cells detected in bone marrow cells after coculture with autologous CD123 CAR T cells, mock T cells or no T cells were analyzed for epitope density of CD123. Epitope density of CD123 in CD34+/CD38−/CD123+ cells after coculture with autologous CD123 CAR T cells is significantly decreased compared to coculture with mock T cells or no T cell, indicating a threshold of CD123 CAR T cell affinity at about 10,000 CD123 molecules/cell (n = 3, mean ± SD, unpaired parametric t-test, * p ≤ 0.05, ** p ≤ 0.01.).
Supplemental Figure 3. Intracellular IFN-γ production in CD123 CAR T cells and mock T cells after coculture with autologous MDS bone marrow sample. pt#2 and pt#8 derived CD123 CAR or mock T cells were cocultured with autologous MDS samples at E:T ratio 1:1 for 5hrs, stained for INF-γ and analyzed by flow cytometry. Intracellular IFN-γ was increased in CD3+/CD4+ subpopulation, but not in the CD3+/CD8+ subpopulation or the total CD3+ cells in CD123 CAR T cells compared to mock T cells derived from pt#2. No significant differences found between T cell compartment when comparing CD123 CAR T cells and mock T cells derived from pt#8. (n = 3, mean ± SD, unpaired parametric t-test. p > 0.05, **** p ≤ 0.0001)
Supplemental Figure 4. Representative histograms of flow cytometry analyses of persisting CD123 CAR T cells in peripheral blood (PB) and bone marrow (BM) of xenografted mice.