

Abstract
Histone methylation, which is mediated by the histone lysine (K) methyltransferases (HKMTases), is a mechanism associated with many pathways in eukaryotes. Most HKMTases have a conserved SET (Su(var) 3‐9,E(z),Trithorax) domain, while the HKMTases with SET domains are called the SET domain group (SDG) proteins. In plants, only SDG proteins can work as HKMTases. In this review, we introduced the classification of SDG family proteins in plants and the structural characteristics of each subfamily, surmise the functions of SDG family members in plant growth and development processes, including pollen and female gametophyte development, flowering, plant morphology and the responses to stresses. This review will help researchers better understand the SDG proteins and histone methylation in plants and lay a basic foundation for further studies on SDG proteins.
Keywords: histone lysine (K) methyltransferase, histone methylation, HKMTases, SDG protein, SET domain group
1. INTRODUCTION
Histone methylation, a mechanism of modifying chromatin structures, is associated with stimulation from numerous pathways which are important for gene expression regulation both in animals and plants. This modification can alter the number of nucleosomes and affect its interactions with other proteins, particularly in regards to gene transcription processes. Methylation of histones is able to either increase or decrease gene transcription, whose processes mostly depend on the exact amino acid in the histone that is methylated, and how many methyl groups are attached. Also, histones can only be methylated on lysine (K) and arginine (R) residues, while the most commonly observed ones are on lysine residues of histone tails H3 and H4.1
The covalent modification of histone lysine methylation is mediated by histone lysine (K) methyltransferase (HKMTases). Most of HKMTases contain a conserved domain consisting of 130–150 amino acids with methyl transferring activity. This domain is named SET based on the three histone lysine methyltransferases, Suppressor of protein‐effect variegation 3–9 [Su(var) 3–9], Enhancer of zeste [E(z)], and Trithorax (trx), which were originally identified in Drosophila. The currently known HKMTases include SET domain group (SDG) proteins containing a conserved SET domain, as well as DOT1p and DOT1L which are exclusive of the SET domain. Although these two types of proteins have different methyltransfer domains, they both use S‐adenosylmethionine (SAM) as a methyl donor. Also, DOT1p and DOT1L which are present in animals and yeast, act on the methylation of H3K79.2, 3 In plants, however, only SDG proteins are so far believed to be serving as HKMTases.
2. CLASSIFICATION OF SDG PROTEINS
SDG proteins constitute a large protein family in plants. The first systematic study of the SDG family in plants was conducted by Baumbusch et al.4 They identified 37 genes containing SET domains in Arabidopsis by searching the whole Arabidopsis genome, and divided them into four sub‐families, namely E(Z), ASH1 (absent, small, or homeotic 1), TRX, and SU(VAR)3–9, with seven gene clusters named as E(Z) homologues, trx homologues, trx‐related, ash1 homologous, ash1‐related, Su(var)3–9 homologous, and Su(var)3‐9‐related, according to the similarities of the SET domains between these genes and SDGs in Drosophila. Later in Springer et al's study,5 22 SET domain proteins were analyzed in maize (Zea mays L.), comparing with 32 SET domain proteins in Arabidopsis by phylogenetic analysis and domain organization. The two proteins, ATXR5 and ATXR6, which only existed in yeast and plants, were separated from the TRX subfamily and considered to be a new separated IV subfamily. The original IV subfamily, the SU(VAR)3–9 group, was re‐annotated as class V based on its domain structure. In addition, a set of genes containing the split SET domains were found and the possibility of whether these genes could normally function as methyltransferase was discussed. After that, Ng et al6 incorporated the genes with the split SET domains into the SDG family based on the data from rice (Oryza sativa L.) and classified them into VI subfamily and VII subfamily according to the possibility of acting on nonhistone proteins. In 2012, a total of 5,536 genes containing SET domain were identified on the basis of the completed whole‐genome‐sequencing data from 165 species.7 According to the data in human and Arabidopsis, seven subfamilies were defined as E(z), Ash, Trx, PRDM, Suv, SMYD, and SETD. Among them, E(z), Ash, Trx, and Suv were the principal subfamilies. The six subfamilies, E(z), Ash, Trx, Suv, SMYD, and SETD corresponded to E(Z), ASH1, TRX, SU(VAR)3–9, VI, and VII subfamily, respectively.5 Due to the specificity of the PRDM subfamily in the classification of Zhang et al7 (existing only in human) and the independence of AtATXR5 and AtATXR6 out of all the branches in the clustering analysis, the classification of Ng et al5 could be applied in most of the studies including plant SDG genes. However, in 2013, Lu et al8 performed a genome‐wide identification and phylogenetic analysis of OsSET gene family in rice, and compared the OsSET with AtSET genes, ultimately dividing the OsSET gene family into five classes according to a different classification of Arabidopsis, which was based on the domain architectures and/or differences in enzymatic activity of SET‐domain containing proteins (see figure 2 in Reference 8). In this review, genes in Arabidopsis and rice were classified into seven classes based on the research of Ng et al6 and Liu et al8 (Table 1).
Table 1.
| Arabidopsis | Rice | ||||||
|---|---|---|---|---|---|---|---|
| Subfamily | Clan | Gene name | Accession number | Other names | Gene name | Accession number | Other names |
| I | 1 | MEA | At1G02580 | SDG5/EMB173 | |||
| E(z) | 2 | CLF | At2G23380 | SDG1 | OsSET24 | LOC_Os06g16390 | SDG711 |
| 3 | SWN | At4G02020 | SDG10/EZA1 | OsSET15 | LOC_Os03g19480 | SDG718 | |
| II | 1 | ASHH3 | At2G44150 | SDG7 | OsSET34 | LOC_Os09g13740 | SDG724 |
| Ash | ASHH4 | At3G59960 | SDG24 | ||||
| 2 | ASHR3 | At4G30860 | SDG4 | OsSET31 | LOC_Os08g34370 | SDG707 | |
| OsSET9 | LOC_Os02g39800 | SDG736 | |||||
| 3 | ASHH1 | At1G76710 | SDG26 | OsSET18 | LOC_Os04g34976 | SDG738 | |
| OsSET19 | LOC_Os04g45990 | SDG708 | |||||
| 4 | ASHH2 | At1G77300 | SDG8/EFS/LAZ2 | OsSET8 | LOC_Os02g34850 | SDG725 | |
| III | 1 | ATX1 | At2G31650 | SDG27 | OsSET33 | LOC_Os09g04890 | SDG723 |
| Trx | ATX2 | At1G05830 | SDG30 | ||||
| 2 | ATX3 | At3G61740 | SDG14 | OsSET37 | LOC_Os09g38440 | SDG732 | |
| ATX4 | At4G27910 | SDG16 | OsSET1 | LOC_Os01g11952 | SDG721 | ||
| ATX5 | At5G53430 | SDG29 | OsSET2 | LOC_Os01g46700 | SDG705 | ||
| 3 | ATXR3 | At4G15180 | SDG2 | OsSET27 | LOC_Os08g08210 | SDG701 | |
| 4 | ATXR7 | At5G42400 | SDG25 | OsSET43 | LOC_Os12g41900 | SDG717 | |
| IV | ATXR5 | At5G09790 | SDG15/PDE336 | OsSET6 | LOC_Os01g73460 | SDG720 | |
| ATXR5/6 | ATXR6 | At5G24330 | SDG34 | OsSET7 | LOC_Os02g03030 | SDG730 | |
| V | 1 | SUVH1 | At5G04940 | SDG32 | OsSET41 | LOC_Os11g38900 | SDG704 |
| OsSET16 | LOC_Os03g20430 | SDG713 | |||||
| SUVH3 | At1G73100 | SDG19 | OsSET22 | LOC_Os05g50980 | SDG728 | ||
| SUVH7 | At1G17770 | SDG17 | OsSET4 | LOC_Os01g65730 | SDG709 | ||
| SUVH8 | At2G24740 | SDG21 | OsSET40 | LOC_Os11g03700 | SDG733 | ||
| SUVH10 | At2G05900 | SDG11 | OsSET42 | LOC_Os12g13460 | SDG734 | ||
| Suv | 2 | SUVH4 | At5G13960 | SDG33/KYP | OsSET5 | LOC_Os01g70220 | SDG714 |
| 3 | SUVH2 | At2G33290 | SDG3 | OsSET25 | LOC_Os07g25450 | SDG726 | |
| SUVH9 | At4G13460 | SDG22 | OsSET32 | LOC_Os08g45130 | SDG715 | ||
| 4 | SUVR3 | At3G03750 | SDG20 | OsSET3 | LOC_Os01g59620 | SDG729 | |
| 5 | SUVH5 | At2G35160 | SDG9 | OsSET20 | LOC_Os04g53700 | SDG703 | |
| OsSET30 | LOC_Os08g30910 | SDG710 | |||||
| SUVH6 | At2G22740 | SDG23 | OsSET35 | LOC_Os09g19830 | SDG727 | ||
| 6 | SUVR1 | At1G04050 | SDG13 | ||||
| SUVR2 | At5G43990 | SDG18 | OsSET10 | LOC_Os02g40770 | SDG712 | ||
| SUVR4 | At3G04380 | SDG31 | |||||
| 7 | SUVR5 | At2G23740 | SDG6/CZS | OsSET11 | LOC_Os02g47900 | SDG706 | |
| VI | ASHR1 | At2G17900 | SDG37 | OsSET17 | LOC_Os03g49730 | SDG716 | |
| SMYD | ASHR2 | At2G19640 | SDG39 | OsSET28 | LOC_Os08g10470 | SDG740 | |
| ATXR1 | At1G26760 | SDG35 | OsSET14 | LOC_Os03g07260 | SDG739 | ||
| ATXR2 | At3G21820 | SDG36 | OsSET21 | LOC_Os05g41172 | SDG722 | ||
| ATXR4 | At5G06620 | SDG38 | OsSET38 | LOC_Os10g27060 | SDG741 | ||
| OsSET39 | LOC_Os10g36250 | ||||||
| VII | SETD1 | At1G01920 | OsSET12 | LOC_Os02g49326 | |||
| SETD | SETD2 | At1G14030 | LSMT‐L | OsSET13 | LOC_Os02g50100 | ||
| SETD3 | At1G24610 | OsSET23 | LOC_Os06g03676 | ||||
| SETD4 | At2G18850 | OsSET26 | LOC_Os07g28840 | SDG731 | |||
| SETD5 | At3G07670 | OsSET29 | LOC_Os08g14660 | ||||
| SETD6 | At3G55080 | OsSET36 | LOC_Os09g24530 | ||||
| SETD7 | At3G56570 | ||||||
| SETD8 | At4G20130 | PTAC14/TAC14 | |||||
| SETD9 | At5G14260 | ||||||
| SETD10 | At5G17240 | SDG40 | |||||
3. STRUCTURAL CHARACTERISTICS OF EACH SUBFAMILY OF SDG GENES
The SET domain is generally located at the C‐terminus of the protein, consisting of four highly conversed motifs: SET motif I (GxG), SET motif II (YxG), SET motif III (RFINHxCxPN), and SET motif IV (ELxFDY).9 These motifs can form a specific spatial structure including helixes and turns. The first half of motif III (RFINE) and the last Tyr (Y) of motif IV have the ability to bind with SAM, the Tyr of motif II is able to catalyze methyl‐transferring, while, the second half of motif III (CxPN) and motif IV (ELxFDY) can form the hydrophobic target lysine‐binding channel.
Usually, the SET domain sequence is the only sequence used to cluster the SDG family. However, each subfamily of SDG genes has distinct characteristics in its own domain structures and active sites. Subfamily E(z), Ash, Trx, and Suv can be further subdivided into different gene clusters on the basis of special domains besides of the SET domain (Figure 1).
Figure 1.
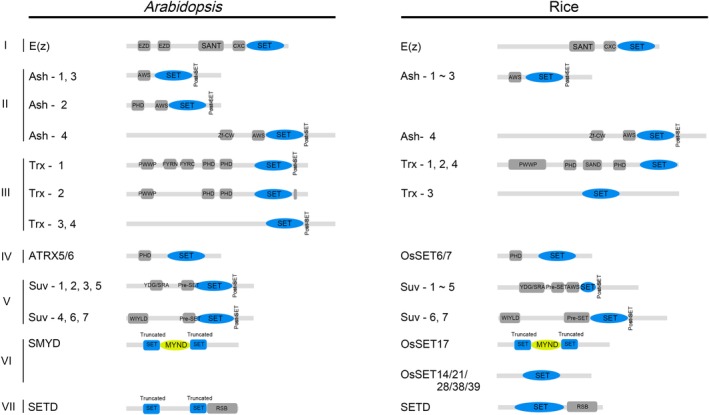
Domain architecture of SDG proteins from Arabidopsis and rice (Oryza sativa L.). EZD, E(z) domain; SANT, SWI3, ADA2, N‐CoR, and TFIIIB DNA‐binding domain; CXC, cysteine‐rich region; AWS, associated with SET domain; PHD, plant homeodomain; Zf‐CW, zinc‐finger with conserved Cys and Trp residues; PWWP, a domain names after a conserved Pro‐Trp‐Trp‐Pro motif; FYRN, F/Y‐rich N‐terminus; FYRC, F/Y‐rich C‐terminus; YDG, conserved Tyr‐Asp‐Gly motif; SRA, a conserved SET and a RING finger associated domain; RSB, Rubisco LSMT substrate‐binding domain
The E(z) subfamily in Arabidopsis includes three members, CURLY LEAF (CLF), MEDEA (MEA), and SWINGER (SWN), belonging to three gene clusters respectively. In addition to the SET domain, two other domains, the SWI3, ADA2, NCoR, TFIIIB DNA‐binding (SANT) domain and cysteine‐rich (CXC) domain, are also belonging to this subfamily. The SANT domain is able to bind the DNA sequences, while the CXC domain can specifically bind to the long noncoding RNAs (lncRNAs).10, 11
The E(z) subfamily belongs to Polycomb group (PcG) proteins, which function by forming a Polycomb Repressive Complex 2 (PRC2) protein complex to trimethylate H3K27 and inhibit gene expression. In Drosophila, the core PRC2 is formed by four important PcG proteins: Enhancer of Zeste [E(z)], Suppressor of Zeste12 [Su(z)12], Extra sex combs (Esc), and p55. Three homologous proteins of Su(z)12 were found in the Arabidopsis genome, including EMBRYONIC FLOWER2 (EMF2), FERTILIZATION INDEPENDENT SEED2 (FIS2), and Arabidopsis VERNALIZATION2 (VRN2). FERTILIZATION INDEPENDENT ENDOSPERM (FIE) is the single homolog of Esc, and MULTICOPY SUPPRESSOR OF IRA1 (MSI1) is the homolog of p55 found in Arabidopsis. These PcG proteins can form three kinds of PRC2 complexes through different combinations (see figure 8 in Reference 12 and thereby have distinct biological functions.13
In Drosophila, the PcG protein complex shares a large number of target sites with Trithorax Group (TrxG) protein complex. TrxG proteins fall into several functional categories including chromatin remodeling proteins, histone modifying methyltransferase, demethylase proteins, and DNA‐binding and accessory proteins14 because of the numerous steps in transcription activation. Among them, the histone modifying enzymes catalyze di‐ or trimethylation of H3K4me or H3K36 to affect the activity of PcG proteins.15 Therefore, they were considered to have antagonistic effects on gene expression regulation. Usually, the members of the Trx subfamily are thought to work through TrxG. In Arabidopsis, the Trx subfamily has a post‐SET domain consisting of three cysteines. These three cysteines, together with the cysteine at SET domain motif III, tetrahedrally coordinating a zinc ion near the active site, and could form a narrow channel to accommodate the target lysine side chain.9 The Trx subfamily can be subdivided into four gene clusters. Cluster III‐1 (ATX1, ATX2) contains Pro‐Trp‐Trp‐Pro (PWWP), F/Y rich N‐terminus (FYRN), F/Y rich C‐terminus (FYRC), and plant homeodomain (PHD) fingers. Cluster III‐2 (ATX3, ATX4, ATX5) includes PWWP domain and PHD fingers; and Cluster III‐3 (ATXR3) and III‐4 (ATXR7) do not contain domains other than SET and post‐SET domains. Among them, the PHD fingers and the PWWP domain can specifically bind to methylated lysine residues.16, 17 Also, FYRN and FYRC are often found in chromatin‐related proteins, and were shown to be involved in RNA silencing and flowering time regulation.18, 19
Members of the Ash subfamily may also function through TrxG.20 They act on H3K36 methylation and are also involved in H3K4 methylation.21 They are short in length and simple in structure, with the SET domain located in the middle of the sequence. In contrast, in other subfamilies the SET domain is close to the C‐terminus of the protein. Five Ash proteins are divided into four gene clusters. Apart from the post‐SET domain, an Associated With SET (AWS) domain is next to the N‐terminus of the SET domain in the Ash proteins. However, the specific function of AWS is unknown. In addition, the N‐terminus of the II‐2 (ASHR3) and II‐4 (ASHH2) proteins contains a PHD finger, and a cysteine and tryptophan conserved residue‐containing (CW) domain, which may evolve from a common ancestor with PHD fingers, respectively.22 ATXR5 and ATXR6 both contain a PHD finger and were considered as members of the Trx subfamily in the earliest classification. Nonetheless, they do not have a post‐SET domain at the C‐terminus of the SET domain, and according to the phylogenetic analysis based on SET domain, their SET domains are more similar to those of the Ash subfamily than the Trx subfamily.5 In addition, other than the Trx subfamily proteins, they could act on H3K27 methylation.23
The Suv subfamily has the largest number of genes and thus their structures are even more complicated. The subfamily members contain a pre‐SET and a post‐SET domain at their N‐terminus and C‐terminus, respectively. The pre‐SET domain includes nine invariant cysteine residues. These nine pre‐SET cysteines coordinate with three zinc ions to form an equilateral triangular cluster. This may be related to the transfer of zinc ions, but the specific function remains unclear.9 This subfamily is subdivided into seven clusters generally: V‐1, V‐2, V‐3, and V‐5 clusters, which were named as Suv homologs (SUVH) subfamilies; while V‐4, V‐6, and V‐7 clusters were called Suv‐related homolog (SUVR) subfamily. SUVH subfamily members contain a conserved SET and a RING finger associated (SRA) domain, which is able to bind the methylated DNA directly.24 In some members, another domain named AT‐hook can also bind with DNA. Unlike the SUVH subfamily, SUVR subfamily members do not contain the SRA domain but a WIYLD domain associated with ubiquitin‐binding activity.25 SUVH subfamily members primarily locate in heterochromatin with the ability to carry out mono‐and dimethylation of H3K9, dimethylation of H3K27, and methylation of H4K20. On the other hand, the SUVR subfamily members could act on heterochromatin, with a function of di‐ or trimethylation of H3K9.9
The SET domains in the SMYD and the SETD subfamilies are close to the N‐terminus of the gene, and they can be split into two segments by a fragment containing 50–120 amino acid residues. The inserted fragment of SMYD subfamily is a putative zinc‐finger motif that facilitates protein–protein interactions, like Myeloid, Nervy and DEAF‐1 (zf‐MYND).26 In the SMYD family, AtASHR1 has been shown to be related to demethylation of H3K4.27 Studies in animals have shown that the genes in the SMYD subfamily can methylate H3K4 and/or H3K36.28, 29 Mostly, members of the SETD subfamily are thought to play a role in the methylation of nonhistone proteins.6 However, in rice, only OsSET17, the homologue of AtASHR1, was predicted on the Pfam and SMART website to contain a MYND domain and a split SET domain (Figure 1). On the other hand, almost all the proteins in the SETD subfamily in rice (OsSET12, OsSET13, OsSET23, OsSET26, OsSET36) and most of the proteins in Arabidopsis (except AtSET1, AtSET4, AtSET7) all have a domain which has Rubisco LSMT substrate‐binding activity.
4. THE FUNCTION OF SDGs IN PLANT GROWTH AND ORGAN DEVELOPMENT
4.1. SDGs in pollen development
In flowering plants, both pistil and stamen play a vital role during plant reproductive processes. The pistil is composed of an ovary, a style and a stigma, while the stamen is consisted by anthers where the male gametophyte develops, and filaments which could provide anthers with structural support and nutrients.30
We screened the microarray data from the study of Honys and Twell31 which compared the transcript expression profiles throughout four successive stages of male gametophyte development: the uninucleate microspore stage, binucleate pollen stage, trinucleate pollen stage, and mature pollen grains stage in Arabidopsis and revealed that at least 22 of the 37 SDG genes were expressed during pollen development. Among them, 14 genes were shown to have different expression patterns among these four stages (fold change >2) (Table 2). In comparison with the expression patterns in seedlings,32 13 SDG genes accumulated extensively and 12 decreased in sperm cells. Five genes could only be detected in sperm cells, not in pollen and seedlings, while only one gene could be detected both in pollen and sperm cells with the signal intensity in sperm cells three times stronger than that in pollen (Table 2). This result indicates that many SDG genes are involved in pollen development. Some of them may be more specific to a certain developmental stage, and some of them may play important roles in the vegetative nucleus and in sperm cells. However, very few SDG genes have been characterized to directly function in pollen development.
Table 2.
The SDG genes differentially expressed during pollen development in Arabidopsis
| Data from Reference 31 | Data from Reference 32 | |||||||
|---|---|---|---|---|---|---|---|---|
| Gene name | Microspores | Bicellular pollen | Tricellular pollen | Mature pollen | FCa | HSb | SSc | SSd |
| CLF | 0 | 266 | 0 | 0 | −3.4 | |||
| SWN | 549.3 | 508 | 0 | 0 | −14.4 | |||
| ASHH3 | 0 | 0 | 0 | 0 | 1.5 | × | ||
| ASHR3 | 0 | 69.3 | 688.5 | 1,194 | 135.3 | × | ||
| ASHH1 | 195.8 | 180 | 141.7 | 140.6 | 9.4 | × | ||
| ASHH2 | 0 | 0 | 0 | 333.8 | 2.1 | × | ||
| ATX1 | 8.8 | |||||||
| ATX2 | −1.0 | |||||||
| ATX3 | 226.1 | 206 | 194.4 | 232.7 | 16.8 | × | ||
| ATX4 | 0 | 0 | 0 | 0 | 8 | × | × | |
| ATX5 | 170.1 | 137 | 369.4 | 270.2 | −0.8 | |||
| ATXR3 | 156.7 | 126 | 145.4 | 178.4 | −1.9 | |||
| ATXR7 | 213.6 | 198 | 76.66 | 0 | −3.7 | |||
| ATXR5 | 504.8 | 587 | 820.3 | 561.4 | ||||
| ATXR6 | 0 | 0 | 0 | 0 | 8.9 | × | × | |
| SUVH1 | 185.3 | 193 | 148.9 | 0 | −4.9 | |||
| SUVH3 | 154.9 | 161 | 204 | 196.3 | −1.2 | |||
| SUVH8 | 305.0 | 261 | 0 | 172.2 | 26 | × | × | |
| SUVH7 | 26 | × | × | |||||
| SUVH4 | 264.2 | 219 | 0 | 0 | 2.2 | × | ||
| SUVH6 | 274.3 | 292 | 449.2 | 449.4 | −1 | |||
| SUVH9 | 217.5 | 212 | 268.3 | 235.1 | ||||
| SUVR3 | 325.6 | 308 | 158.6 | 0 | −2.9 | |||
| SUVH5 | 598.8 | 487 | 163.2 | 201.7 | 22.2 | × | × | |
| SUVR1 | 128.6 | 111 | 0 | 0 | 4.9 | × | ||
| SUVR2 | 175.5 | 169 | 0 | 0 | −1.7 | |||
| SUVR5 | 194.0 | 177 | 159.5 | 164.4 | −3.1 | |||
| ASHR1 | 0 | 0 | 0 | 0 | 2 | × | × | |
| ASHR2 | 0 | 210 | 0 | 0 | ||||
Abbreviations: FC, fold change; HS, high expression; SS, specific expression.
Changed expression in sperm cells compared to seedlings.
Highly expressed in sperm cells (with an expression level at least 1.2‐folds more than in seedlings).
Specific to sperm cells (not expressed in seedlings and pollen).
Specific to sperm cells (with an expression level at least three times more than in pollen, but not expressed in seedlings).
ASHR3 is the first SDG gene that was found to be associated with male sterility in Arabidopsis. It is specifically expressed in flower buds and opened flowers.33 In the ashr3 mutant, the pollen morphology was normal, with normal vegetative nucleus and sperm cells. Although its pollen grain could germinate normally, its pollen tube was not able to elongate, thus leading to a large number of infertile ovules. Also, the covalently modified markers of demethylated H3K4 and trimethylated H3K36 in the vegetative nucleus of mature pollens were completely lost. This may inhibit the expression of genes which were involved in pollen tube elongation in the vegetative nucleus, and therefore lead to male sterility. At the same time, ASHR3 was found to be abundantly expressed in tapetum cells.34 Overexpression of ASHR3 resulted in defective stamens and aborted anthers as well as inhibited siliques growth. Yeast two‐hybrid screening revealed that the ASHR3 protein could interact with the putative basic helix–loop–helix (bHLH) transcription factor ABORTED MICROSPORES (AMS), which may function in anther and stamen development in Arabidopsis.34 Unfortunately, no further comparisons of the similarities and differences between transcriptome and phenotypes of ashr3 and ams single mutants and the ashr3/ams double mutants to confirm whether they would act on the same pathway were carried out. In addition, both studies have confirmed that ASHR3 was specifically localized in tapetum cells and pollen. Programmed cell death (PCD)–triggered degradation of tapetum was essential to pollen development, especially the exine formation. The reason why abnormal elongation of the pollen tube33 might be led by the loss of covalent modification of H3K4 trimethylation and H3K36 trimethylation in the vegetative nucleus and ultimately the depressing of genes related to pollen tube growth, or it might be caused by the abnormal development of the tapetum due to the loss of ASHR3 in tapetum cells. However, this question still needs further verification.
ASHH2, which belongs to the Ash subfamily, also acts on pollen development35 as approximately 90% of pollen grains were aborted in the ashh2 mutant. Abnormal phenotypes show up from the pollen mother cell stage, and after which tetrads formation was defective during meiosis, followed by the abnormally thickened and delayed degradation of tapetum cells. As a result, most of the pollen grains were abnormal with overall irregular shapes and exine patterns deviating from the even reticulation of the wild‐type plants. The expression of more than 600 genes was downregulated in ashh2 inflorescences, including DISRUPTION OF MEIOTIC CONTROL 1 (DMC1) associated with meiosis and AMS and MALE STERILE 2 (MS2) associated with tapetum development and anther dehiscence. The down‐regulation of these genes was found to have associations with a decrease in H3K36 trimethylation, but not in H3K4 trymethylation and H3K36 dimethylation. On the other hand, the downregulated genes in ashh2 were consistent with those in ms1 and/or sporocyteless (spl)/excess microsporocytes 1 (ems1), suggesting that ASHH2, MS1, and SPL may be involved in an overlapping pathway.
ATXR3 (SDG2), a member of Trx subfamily, is expressed in roots, stems, leaves and inflorescences, and functions in regulating the covalent trimethylation of H3K4 throughout the entire genome.36 The loss‐of‐function mutant of ATXR3 was dwarfed, with smaller rosettes throughout vegetative growth, and was completely sterile with collapsed pollen grains which were unable to rehydrate or germinate.36 In addition, the stamen filaments were too short to allow effective self‐pollination, and the anther contained only 2–3 locules. Though sporogenous cells developed and produced pollen grains in some locules, the pollen grains stuck to each other while the anther failed to dehiscence. Over 40% of pollen grains showed collapsed morphology and were larger in size. The male germ unit showed irregular positioning of the two sperm and single vegetative nucleus. Artificial pollination indicated that only 4% of pollen was fertile.37 Further studies have found that the mutant can only form 50% normal tetrad, suggesting that abnormal chromatin organization, nucleus degeneration, and DNA degradation had occurred during microspore formation. Transcriptome analyses of young flower buds revealed 452 genes downregulated by more than two‐fold in the mutant. Among them, 11 genes, including SPL and MS1, have been previously shown to be essential to male and/or female gametophyte development.37
It is worth noting that although the phenotypes of atxr3 and ashh2 were similar, less than 20% of differentially expressed genes were in common between the two mutants. These genes were primarily involved in metabolism and transport, indicating that ATXR3 and ASHH2 may regulate similar developmental events through distinct gene regulation networks.38
ATXR5 and ATXR6 have functional redundancy as they both can monomethylates at H3K27, and they were shown mainly involved in the replication of heterochromatins.39 Interestingly, their overexpression in transgenic plants showed different phenotypes. A large number of pollens aborted in the constitutive ATXR5 overexpressing plants and they were capable of normal meiosis and mitosis, forming a male gametophyte containing two trophic nucleus cells. The nucleus then began to expand and break, resulting in the formation of pollen grains without nucleus and DNA. Further analysis showed that male gametophytes of the transgenic plants underwent two pollinic mitoses, as they appear to contain two reproductive and a vegetative nucleus, which ultimately led to male sterility in plants.40 Meanwhile, the constitutive overexpression of ATXR6 was lethal. The overexpression of ATXR6 driven by its own promoter did not affect pollen development, but caused impairment of anther dehiscence which still could result in male sterility of the transgenic plants.40
In summary, only a few SDGs are known to play roles in pollen development. Further study is needed to reveal function of other SDGs as well as the relationship between different SDGs during pollen development process.
4.2. SDGs in female gametophyte development
In flowering plants, it is known that seed development begins with a double fertilization, during which process two sperm cells could be released from the pollen tube combining with egg cells and central cells. These cells then form embryos and endosperm. Among them, triploid endosperm provides nourishment for the developing embryo and seedlings. From the research of Wang et al,41 it is known that the E(z) subfamily gene MEA is specifically expressed in the late stage of central cell development and early stage of endosperm development, and the MEA allele from the male parent is silent, which showed obvious parental imprinting.42 A large number of central cells in the mea mutant proliferated excessively under unfertilized conditions, and this resulted in the seeds of mea having only endosperm without embryos. This process usually produced autonomous seeds and was preceded by the lack of repression of target genes such as PHERES1 (PHE1) due to the lack of H3K27me3.43 Also, although the single mutant of SWN may not affect the development of female gametophytes, the phenotype from losing both SWN and MEA was more severe than for the mea mutants.41 In addition, the DNA methyltransferase METHYLTRANSFERASE1 (MET1) has also been shown to directly participate in the repression of endosperm autonomic processes by binding to MEA.43
Likewise, the Ash subfamily gene ASHH2 and Trx subfamily gene ATXR3 could also contribute to the development of female gametophytes. In ashh2, nuclear degeneration and genomic DNA degradation may occur in both microspore and megaspore cells after meiosis, with only 20% of ovules fully developed and less than 25% of the remaining ovules eventually developed into seeds; the defects in atxr3 were even more intense, showing complete female sterility.37 Though the early‐stage premeiotic ovules contained normal single megaspore mother cells, more than half of the ovules cannot form the obvious embryo sac and the megaspores were degenerated during early gametophyte development. The remaining ovules contained a vacuolated embryo sac, whose nuclei and cells could degenerate before maturation. In conclusion, the loss‐of‐function of ASHH2 and ATXR3 may result in similar phenotypes during male gametophyte development on different pathways. They are also involved in similar developmental events of female gametophyte, but it is still uncertain whether they could overlap in the regulating mechanism during female gametophyte development. In addition, it is worth noting that the TrxG protein complex and PcG protein complex appear to act on different events during the female gametophyte development, in spite of the antagonism between them.
5. SDGS IN FLOWERING
Bolting and flowering are important symbols of annual or biennial herbaceous plants entering reproductive growth. They could be affected by a series of endogenous factors and external environmental factors. In Arabidopsis, this process is regulated by photoperiod, hormones, vernalization, and the autonomic pathway, as well as a series of secondary factors such as temperature, salinity, sugar content, light quality, and plant age. Studies have shown that this series of regulation processes can ultimately be attributed to the regulation of two core genes, FLOWERING LOCUS C (FLC) and FLOWERING LOCUS T (FT), and three key genes, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), LEAFY (LFY), and APETALA 1 (AP1), in Arabidopsis (see figure 1 in Reference 44). The expression activation of FLC was associated with the enrichment of H3K4 and H3K36 methylation at its promoter, and its expression inhibition was positively correlated with the degree of H3K9 and H3K27 methylation.45 In other words, a variety of histone covalent modifications jointly determine the expression level of FLC through a series of synergistic and antagonistic effects. For example, lesions in ASHH2 usually cause early flowering because of the decrease in H3K4 and H3K36 methylation levels at the FLC locus. Also, the decreasing expression of ATXR7 can promote flowering because of the suppression of FLC expression.46
5.1. The role of SDGs in activating FLC
The Trx subfamily methylates H3K4 through the TrxG protein complex to activate FLC transcription (Figure 2). The mutants of Trx subfamily genes ATX1‐5, ATXR3, and ATXR7 showed early flowering time in both long‐day and short‐day conditions.36, 47, 48, 49, 50 Among them, there was no significant difference between the flowering time of atx1/atx2 double mutants and atx1 or atx2 single mutants. Thus it could be inferred that they may act by the same mechanism;48 atx1/atx7 double mutants tended to flower earlier than atx1 or atx7 single mutants, which indicated that the function of the two genes may be redundant.50 The atxr3/atxr7 double mutants flowered earlier than atxr7, but they had basically the same flowering time as atxr3. This showed that ATXR3 and ATXR7 could act by the same mechanism, and ATXR3 may play a role in other mechanisms which are also related to flowering time.51 In atxr3, the level of H3K4me1 in the FLC locus increased, and the level of H3K4me2/3 decreased. While in atxr7, the level of covalent modification of H3K4me1/2/3 all declined. Yun et al believed that the reason may be the different methyltransferase activities of ATXR3 and ATXR7.51 Previous studies had shown that the amino acid at position 1,205 in human SDG protein G9a allowed the enzyme to perform mono‐, di‐, or trimethylation.52 This can be called the Phe/Tyr switch. The SDG gene, at which position was a tyrosine, was considered to have mono‐/dimethyltransferase activity while a phenylalanine at the same position resulted in di‐/trimethylation.52 As there is a phenylalanine at this position in ATXR3, this protein was inferred to have the di/trimethyl‐transferase activity. Thus, there was no doubt that the ATXR3 loss‐of‐function mutant would cause a decrease in the level of covalent modification of H3K4me2/3. Due to the loss of methyltransferase activity, the covalent modification of H3K4me1 could not be carried out, the mutant thus showed an increase H3K4me1 level. On the other hand, a tyrosine is at this position in ATXR7. Therefore, ATXR7 could be a mono‐/dimethyltransferase. Research showed that in atxr7, the level of H3K4me1/2 modification is reduced, resulting in the lack of substrate for SDG gene with trimethylation function, thus the level of H3K4me3 modification was passively reduced. In addition, ATX1 was a methyltransferase specific to H3K4me3, while in atx1, the level of H3K4me3 at the FLC locus reduced and the level of H3K4me2 increased.48 Although there was no further experimental verification, the situation in atx1 can still be explained by the theory of Yun et al.51
Figure 2.
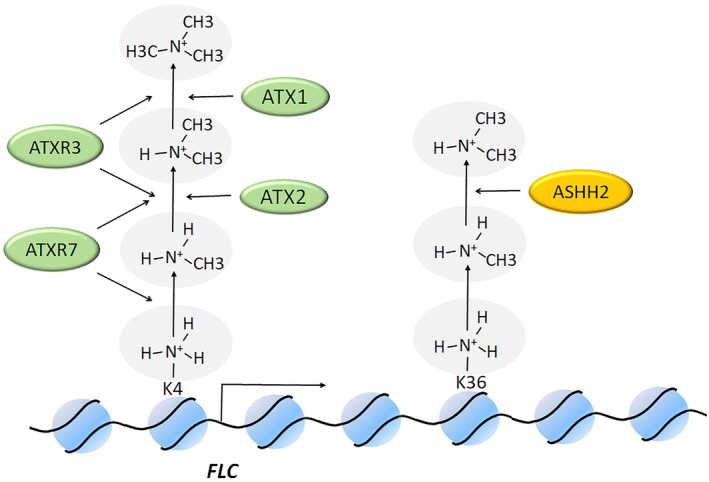
SDG genes activate the transcription of FLC. Trx genes promote the transcription by H3K4me, in which ATXR7 working on H3K4me1/2, ATXR3 acting on H3K4me2/3, ATX2 targeting H3K4me2 and ATX1 associated with H3K4me3. ASHH2 di‐methylates H3K36, impacting transcriptional elongation. FLC, FLOWERING LOCUS C
It is worth noting that the overexpression of ASH2R, one of the three structural proteins, can increase the level of H3K4me3 at the FLC locus and promote FLC transcription,53 indicating that the histone methyltransferase with H3K4me3 function near the FLC locus was sufficient, and the methyltransferase was not the major limitation of the H3K4me3 modification.
The H3K36 methylation is mainly accomplished by the Ash subfamily. ASHH2 was the representative gene of the Ash subfamily that acted on the flowering process (Figure 3). ASHH2 was also known as EARLY FLOWERING IN SHORT DAYS (EFS). ashh2 showed early flowering in both long‐ and short‐day conditions.54 In addition, when a homologous gene BnaASHH2 was knocked out by CRISPR‐Cas9 in Brassica napus, early flowering also appeared in the knockout mutant,55 which showed that the function of ASHH2 was conserved among different species. It was interesting that vernalization did not affect the flowering time of ashh2, indicating that the loss of function of ASHH2 disrupted the ability of plants to responding vernalization. However, vernalization does not affect the expression level of ASHH2 in wild type plants, it can then be inferred that ASHH2 does not act as an initiator of the vernalization response pathway. In addition, the loss of function of ASHH2 caused a remarkable reduction in H3K36me2 and a significant increase of H3K4me2 of the entire genome. Nonetheless, at the FLC locus, only a reduction in H3K36me2 and no significant increase of H3K4me2 was detected. This phenomenon indicated that ASHH2 was highly transcribed in the cell and may be involved in many processes other than flowering.
Figure 3.
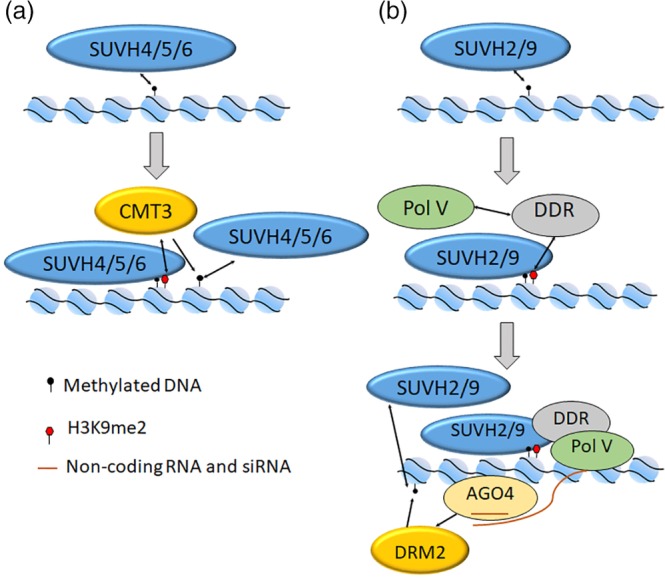
Model for the role of SUVH in heterochromatin formation and maintain. Two self‐reinforcing loops between maintenance of histone and DNA methylation are displayed. (a) SUVH4, SUVH5, and SUVH6 bind to methylated DNA and the H3K9me2 modification recruits CMT3 for DNA methylation. The methylated DNA further recruits SUVH4, SUVH5 and SUVH6. (b) SUVH2 and SUVH9 bind to methylated DNA at RdDM loci and the H3K9me2 modification recruits DDR complex at the loci. DDR leads Pol V to RdDM loci, producing noncoding RNAs to interact with 24 nt‐siRNAs bound by AGO4, which recruits DRM2 for DNA methylation. The methylated DNA is further bound by SUVH2 and SUVH9. AGO4, ARGONAUTE4; DRM2, DOMAINS REARRANGED METHYLTRANSFERASE 2; DDR complex, DMS3, DRD1, and RDM1; RdDM, RNA‐directed DNA methylation; SUVH, Suv homologs
Another Ash subfamily member, ASHH1, was also proven to have H3K36 methyltransferase activity. However, the flowering time was delayed in ashh1, which was different from other function‐known Trx subfamily genes and ASHH2, for which the loss‐of‐function mutants showed early flowering.56 In ashh1, the expression level of FLC increased, but no change was found in histone covalent modification levels.56 This implied that ASHH1 may not directly act on FLC. Also, the delayed flowering phenotype of ashh1 can be restored by vernalization, which also indicated that ASHH1 was not in the same pathway as ASHH2. Remarkably, the transcriptome of atxr7 was quite different from those of atx1, atx2, and ashh2, which shared the same early flowering phenotype with atxr7. On the contrary, it had a wide range of overlap with the transcriptome of ashh1, which displayed an opposite phenotype. This implicated a functional redundancy between ASHH1 and ATXR7 in regulating genes other than FLC.57
5.2. The role of SDGs in inhibiting FLC
After vernalization, the methylation levels of H3K4 and H3K36 at FLC decreased, with the levels of H3K9me2 and H3K27me2/3 increasing, so that the expression of FLC was repressed by allowing the transition to the reproductive phase.58 Usually, the decrease in methylation level of H3K4 is achieved by a protein complex composed of H3K4 demethylase FLOWERING LOCUS D, a histone deacetylase and a histone binding protein.59 COOLAIR, which is an antisense lncRNA that starts transcription from the 3′UTR of FLC, is also involved in this process.60, 61 SUVR5, and a SWIRM‐domain‐containing polyamine oxidase protein, AtSWP1, could interact with each other in plant cells and repress the expression of FLC by modifying H3K9me2 and H3K27me2 within the FLC locus. The loss‐of‐function of either SUVR5 or SWP1 resulted in demethylation of H3K9 and H3K27 within the FLC locus to promote the transcription FLC, and delaying flowering consequently.62
H3K27me3 is one of the most well‐studied modifications associated with FLC repression. H3K27me3 was determined by a VRN‐type PRC2 protein complex composed by CLF/SWN, and the E(z) subfamily member: VRN2, FIE, and MSI1. During vernalization, the lncRNA named COLDAIR, was transcribed from the first intron of FLC. Through specifically binding to the CXC domain in CLF, COLDAIR guided the PHD‐PRC2 protein complex which was formed by PRC2 and two PHD finger proteins, VERNALIZATION INSENSITIVE3 (VIN3) and VERNALIZATION5 (VRN5),63, 64, 65 to a specific region near the first intron of FLC. During this procedure, the protein complex which was guided by COOLAIR catalyzed the H3K27me3 modification and repressed the expression of FLC.11
When the temperature rises, the PRC2 protein complex functions at FLC via a pathway associated with DNA replication,66 which allowed the coverage of H3K27me3 modification across the FLC locus from the promoter to the 3′UTR. Subsequently, H3K27me3 modification recruited LIKE HETEROCHROMATIN PROTEIN 1 (LHP1) and bound with it, which allowed H3K27me3 to be stably retained at the FLC locus. This ensured that FLC was in a stable expression‐inhibited state.67 It is also worth noting that the FLC locus maintained a low level of H3K27me3 before vernalization, and was also regulated by the VRN–PRC2 complex.63
On the other hand, most crop plants flower after a long period of vegetative growth and this is caused by the inhibition from several regulators under LD conditions in summer. Thus, in most SD crops, the flower timing mechanisms are different from those in Arabidopsis. Take rice for example, HEADING DATE 3a (Hd3a) and Rice FT 1 (RFT1), the two FT homologues, are major florigen proteins with important roles in promoting flowering. The former acts under SD whereas the other functions under LD.68 Besides, there are also some upstream pathways of Hd3a and RFT1 in rice, including the Heading date1 (Hd1) pathway controlled by photoperiod and Early heading date 1 (Ehd1) pathway which encodes a B‐type response regulator that promotes flowering by controlling the expression of FT‐like genes under SD and LD.69 Moreover, some MADX‐BOX genes are also involved in controlling flowering by activating or suppressing Ehd1.70, 71
Usually, the promotion of flowering time requires the methylation of H3K4or H3K36. SDG701, which is related to the AtSDG2/AtATRX3, and could regulate the H3K4me3 at HD3a and RFT1.72 In 2015, RFT1 was found to be epigenetically controlled by OsSDG724 and OsSDG725. These two proteins, together with SDG708, belong to the SET domain family II. They could function in H3K36me2/3 modifications.68 On the other hand, a long day‐preferential flowering activator, the OsMADS50 chromatin, can also be methylated by these SDG proteins. OsSDG711 and OsSDG718, which function as methyltransferases, could downregulate the repressor of Hd1:OsLF. Also, the demethylase OsJMJ701 protein could delay plant flowering by suppressing RFT1 expression.45 The abnormal expression of SDG711 in rice could change the methylation levels of H3K4me3 and H3K27me3 in shoot apical meristem (SAM) during floral transition, which in turn affects the bolting and flowering time.73
Therefore, in addition to acting on FLC, histone covalent modification, especially H3K27me3 and H3K4me3 also played an important role in regulating the expression of other flowering‐related genes such as FT, LFY, and LHP1.74
6. SDGs IN PLANT MORPHOGENESIS
6.1. The role of SDGs in the vegetative stage
The root system comprises the underground world of a plant. Usually, its main functions are nutrient absorbing and water supplying under both normal and stressed environments. SDG family and histone methylation are essential for RAM maintenance and lateral roots formation during the development of roots.47 In sdg2, the H3K4me3 level is reduced with a loss of auxin gradient maximum in stem cell niche (SCN) cells of roots.75 The loss‐of‐function of mutant atx1‐1 has the phenotype of retarded growth of primary roots because of RAM activity reduction, as well as a disorganized cell patterns due to the disturbed cell division and proliferation. This has indicated that ATX1 plays a part in lateral root emergence and development.76
CLF, belonging to the E(z) subfamily, has functions in root meristem development and vascular cell proliferation and specification.77 The deletion of CLF can result in slightly elongated primary roots and enlarged root meristems. However, this protein usually controls the morphological structure of leaves and flowers by regulating the expression of homeotic genes such as AGAMOUS (AG), AP2, and SHOOTMERISTEMLESS (STM). The clf plant is dwarf with narrow and curled upwards leaves, and always shows early flowering. Loss of function of CLF can affect floral morphology as well, but this does not affect the development of hypocotyl or cotyledons.12, 78 The structure of SWN is similar to that of CLFs, and is also partially redundant in functions with CLF.12
In rice, SDG711 and SDG718 belong to the homologous genes of the E(z) subfamily. SDG711 is the homologue of CLF in Arabidopsis, while SDG718 is closer to SWN in Arabidopsis. These two genes can express in every part of the organs in rice, according to Liu et al,79 and the expression of these two genes can be induced by both long‐day light condition and short‐day light condition. Under these circumstances, the expression of OsLF can then be repressed so that the flowering time is affected, which in turn affects the vegetative growth.
In addition to the above processes, SDGs also participate in other plant growth and developmental processes such as regulating the circadian clock, determining the number of shoots, adjusting the growth and development of roots and germination of seeds.75, 80, 81, 82, 83
6.2. The role of SDGs in floral organ identity
In addition to regulating the flowering time of plants, ATX1 is also required for maintaining the expression of homeotic genes such as AP1, AP2, AG, and PISTILLATA (PI) by antagonizing the repressive activity of CLF, and participating in the establishment of plant morphology.84 Overexpression of ATX1 can result in no obvious phenotypes, however, the loss‐of‐function of ATX1 can cause various developmental defects. For example, atx1 mutants showed petals in various sizes, shapes, and different numbers, with the gynoecium lacking stigmatic papillae, as well as the carpels partially or completely unfused.84
Interestingly, the loss of both ATX1 and CLF functions can rescue each certain single‐mutant phenotype. Thus, it could be implied that these two genes have the same target site in regulating the expression of homeotic genes.85 Further research showed that ATX1 and CLF can directly bind to each other, and can generate H3K4me3 and H3K27me3 modifications at locus like AG and other related genes. The expression or silence of these genes could be then determined by the fine adjustments of the relative abundance of these two methylations.49 Besides the AG locus, this type of bivalent chromatin structure marker can also be detected at other loci, such as FLC, SUPERMAN (SUP), and AP1.49, 63 In mouse embryonic stem cells, the two markers, H3K4me3 and H3K27me3, have a broad co‐occurrence, and they both appear in the region where transcription factors are associated with cell differentiation. In response to this phenomenon, it could be then speculated that these transcription factors may keep a low level of expression in embryonic stem cells through bivalent chromatin structure markers. After the cells determine the direction of differentiation, a targeted removal of certain covalent modifications make the transcription factors to be transcriptionally activated or silenced, and then the cells can start to differentiate rapidly.86 However, in plants, studies about bivalent chromatin markers in regulating cell differentiation is still lacking. It is well known that plant cells have a more comprehensive totipotency than animal cells, which means a series of cells with differentiation and division ability, such as apical meristem, lateral meristem and intercalary meristem, are retained in mature plants. Hence, further studies are still needed to be carried out to determine whether bivalent domains have a wider range of effects in plants.
ATXR3 and ASHH2, which are the members in TrxG protein complex, also play a role in regulating the expression of homeotic genes. ashh2 has highly branched internodes and clusters flowers without pedicels. Its flowers mainly display sepals and carpels or carpeloid sepals and stamen‐like petals.35, 36 Except these two genes, there was no other homeotic gene with this kind of phenotype found in mutants of other members of the TrxG protein complex. It is worth noting that ASHH2 also could regulate the expression of PI, AP1, and AP2.35 However, the relationship between ASHH2 and CLF or ATX1 is still unclear. At least, yeast two hybrid screening has proven that the SET‐domain‐containing sequences from ATX1 and ASHH2 proteins can both associate and interact with ASHH1 protein.87
7. SDGs IN RESPONSE TO STRESS
Plants are exposed to a variety of biotic and abiotic stresses during their growth. On these occasions, covalent modifications of histone could form a transient response to various stresses. When the stress has disappeared, the changes in histones can return to normal levels. To survive repeated stresses, plants provide responses that may be different from their responses during the first encounter with the stress. Thus, “stress memory,” which means a different response to a similar stress,88 has appeared. Recent studies have suggested that histone modification plays an important role in plant stress memory, including biotic stresses and abiotic stresses such as drought, high salinity, heat, cold, and dark.89, 90, 91
Under drought stress, drought response factors and genes in the ABA biosynthesis pathway are activated by H3K4me3 modification.92 ATX1 can bind to the NINE‐CIS‐EPOXYCAROTENOID DIOXYGENASE 3 (NCED3) locus, a key enzyme in the ABA biosynthesis pathway, and increase the expression of NCED3. Thus, this initiates the ABA‐dependent drought response signaling pathway. ATX1 can also activate the expression of RESPONSIVE TO DESICCATION 29A (RD29A) and RD29B, and then participate in other drought response signaling pathways.93, 94 By training with multiple drought treatments, the enriched H3K4me3 levels in the RD29B and RESPONSIVE TO ABA 18 (RAB18) gene regions were maintained after rehydration. This means that ATX1 could participate in drought stress‐memory.95 However, in atx1, the stress memory of H3K4me3 did not completely disappear, indicating that ATX1 was not the core gene which was responsible for drought stress memory.95 With increasing time of treatment, H3K4me3 modifications increased gradually in the coding regions of the RD29A, RD29B, and RD20 genes, indicating that enrichment of H3K4me3 positively correlated with degree of drought.92, 93 On the other hand, CLF‐mediated H3K27me3 was involved in the drought stress response, but was not involved in stress memory.96 The study of Sani et al97 showed that Na+‐pretreated plants showed more drought tolerance than nontreated control plants after a Na+ stress‐free period. Also, there was a decrease in the H3K27me3 level around the high affinity K + transporter 1 (HKT1), which encoded a high‐affinity K (+) transporter. It is unclear which SET gene regulated the level of covalent modification of H3K27 at the HKT1 locus, but this change led to an increase in the expression of HKT1 and a better drought tolerance in plants.
Under low temperature, COLD‐REGULATED15A (COR15A) and GALACTINOL SYNTHASE3 (GOLS3) were activated by the decrease of H3K27me3 modification level on the gene region, and thereby helped plants to defend against coldness. Nonetheless, H3K27me3 modification at these two loci remain at a low level when the temperature rises.98 This indicated that covalent modification of H3K27me3 may play a role in the formation of low temperature stress memory. In addition, ASHH2 played a role in the stress memory of the touch‐inducing gene TOUCH3 (TCH3) caused by repeated mechanical stimulation.99 Under biotic stress, the loss‐of‐function of ATX1 affected the expression of many antibacterial and disease‐resistant genes including antifungal factors, lectins, chaperones, heat shock proteins and several WRKY family transcription factors.100 WRKY70 can be activated by Pseudomonas syringae pv. Tomato DC3000, and then regulated a set of antibacterial‐related genes such as PANOGENESIS‐RELATED GENE 1 (PR1), THIONIN 2.1 (THI2.1), VEGETATIVE STORAGE PROTEIN 2 (VSP2), PLANT DEFENSIN1.2 (PDF1.2), and HEVEIN‐LIKE PROTEIN (HEL) in the pathways of jasmonic acid (JA) and salicylic acid (SA).101 ASHH2 was directly implicated in the activation of defense marker genes that exerted functions in the JA/ET defense pathway to resist Botrytis cinereal in plants, such as the upstream disease resistance gene MITOGEN‐ACTIVATED PROTEIN KINASE KINASE 4 (MAPKK4), MAPKK5 and a series of downstream genes like ETHYLENE RESPONSE FACTOR 1 (ERF1), ERF2, MAPKK3, MYC2, VSP2, PDF1.2a. Further studies by syringing inoculation ashh2 and wild type plants with P. syringae pv. Tomato DC3000 showed that ASHH2 directly determines the expression level of Resistance (R) genes, such as RESISTANCE TO PSEUDOMONAS SYRINGAE 3 (RPS3/RPM1), RPS5, LAZARUS 5 (LAZ5), and could ensure the most basic disease resistance of plants.102
Although it was documented that the genes involved in drought stress, chilling or pathogens can be regulated by histone methylation, this may become controversial when it comes to high temperature conditions. H3K4me1 in Chlamydomonas reinhardtii and H3K9me2 of OsFIE1 were known to be sensitive to high temperature, while H3K9me2, H3K27me1/2/3 and H3K4me1 in Arabidopsis were not.100 Moreover, many SDGs were involved in high temperature responses. It can then be inferred that upon exposure to high temperature, up‐or‐downregulation of these genes might affect the status of methylation and further regulate the activity of target genes in response to high temperature.103
Light is essential to plant growth and development. Persistent darkness may cause dramatic gene expression changes, which in turn leads to abnormity in hypocotyl growth, petiole elongation, leaf‐area reduction, flowering time change and leaf senescence. According to the study of Yan et al,91 the level of H3K4me3 increased when Arabidopsis were exposed to long‐period darkness. Meanwhile, the number of H3K4me3‐increased genes was greater than that of H3K4me4‐diminished gene. The former genes were mainly associated with aging, senescence, response to absence of light and some processes closely related to hormones such as ABA, JA and ethylene. Thus, it can be indicated that the SDG protein may play a part during the responses to darkness.
By applying chromatin immunoprecipitation and deep sequencing (ChIP‐Seq) or whole‐genome tiling arrays technologies, the global histone modifications patterns in Arabidopsis were established. This include the detection of the levels of H3K9me3 and H3K27me3 under dark and light conditions; the levels of H3K4me1, H3K4me2, and H3K4me3 during watered and dehydration stress conditions; as well as the level of H3K27me3 in undifferentiated cells of the SAM and in differentiated leaf cells.92, 104, 105 In these studies, it was found that numerous genes targeted by histone lysine methylation were expressed in a tissue‐specific or condition‐specific manner. However, when we analyzed the expression of SDGs in the transcriptome data of these studies, only a few SDGs were found to be tissue‐specific or sensitive to the changes of growth condition. Although 40 SDG‐encoding genes were included in the transcriptome of dark/light‐grown plants, only three genes: SETD2, SETD5, and SETD7 displayed at least a twofold change in expression levels. The expression of 46 SDG encoding genes can be detected in the watered and dehydration stress groups, and only the expression of SETD2, SETD3, and ASHR1 was different between these two conditions. Forty‐seven SDG encoding genes showed expression in SAM and differentiated leaf, while only five genes, MEA, SUVR1, SUVR2, SUVH4, and SETD9, were enriched in a specific tissue. It was also worth noting that most of these SDG genes were not involved in the direct regulation of gene expression.
Together with the analysis of Trx and Ash subfamily gene expression during flowering, it can be inferred that there was a high possibility that the transcripts of SDGs are sufficient or excessive in the cell. Even if the environment changes, the expression of the SDG gene may not alter. SDGs can bind to the target gene locus, which may lead to the changes in the expression of downstream genes.47
8. SDGs IN FORMATION OF HETEROCHROMATIN
Except for ATXR5, ATXR6 and SUVR5, the other SDGs mentioned above all belong to the E(z), Ash, and Trx subfamilies. They mainly act on the euchromatin region and are related to the expression and repression of protein‐coding genes. The loss‐of‐function of these SDGs usually cause distinct developmental defects, which makes it easy to study their function. The Suv subfamily is the largest subfamily. In Arabidopsis, the Suv subfamily contains 15 members, which is equivalent to the sum of the gene numbers of the E(z), Ash, and Trx subfamilies. However, the Suv subfamily genes mainly act on the silencing of transposons and repeats in heterochromatin regions. Their single or double mutants often have no obvious phenotype.106 It is speculated that mutants of the Suv subfamily genes may show a distinct phenotype under abnormal conditions like extreme temperature, ultraviolet light and radiation, as the transposon of wild type plants would reactivate transcription under harsh conditions.21
The formation of heterochromatin needs a cooperation of histone lysine methylation and DNA methylation. In plants, DNA methylation occurs at cytosine of CG, CHG, and CHH (H = A, T or G). DNA methylation is established by DNA methyltransferase (DNMT), in which the methylation of CG is catalyzed by MET1, while the methylation of CHG is carried out by CHROMOMETHYLASE 3 (CMT3), and the methylation of CHH is mediated by CMT3 and DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2). In addition, DRM2 was involved in the de novo DNA methylation process guided by RNA‐directed DNA methylation (RdDM).
As early as 2002, researchers had discovered that DNA methylation in heterochromatin regions was dependent on the covalent modification of H3K9me2. After more than 10 years of research, it is now clear that SUVH4 is the main enzyme that acts on the covalent modification of H3K9me2 in the heterochromatin region.107 In addition, SUVH2, SUVH5, SUVH6, and SUVH9 also could act on the monomethylation and dimethylation of H3K9.21 These genes form two pathways according to whether they can participate in RdDM process. Among them, SUVH4, SUVH5, and SUVH6 bind to methylated CHH and CHG sites through the SRA domain and establish H3K9me2 modification.24 On the other hand, DNA methylase CMT3 methylated the CHG site via the bromo adjacent homology (BAH) and a chromo domain.108 The methylated CHG can in turn recruit histone methyltransferase such as SUVH4 (Figure 3a).
Both SUVH2 and SUVH9 are involved in the RdDM process related to DRM2. The SRA domain of SUVH2 has a higher affinity with methylated CG, and the SRA domain of SUVH9 mainly works in recognizing the methylated CHH.106 Also, SUVH2 and SUVH9 are able to bind the slightly methylated DNA near the RdDM site via the SRA domain. The only difference is that the H3K9me2 covalent modification they produced does not directly recruit DRM2. Instead, SUVH2 and SUVH9 were required for the occupancy of Pol V at the RdDM loci by DMS3, DRD1, and RDM1 (DDR) protein complexes and the production facilitation of noncoding RNAs. The noncoding RNAs can interact with 24‐nt siRNAs bound by ARGONAUTE4 (AGO4) and recruit the DRM2 to mediate DNA methylation. The methylated DNA can also attract the combination of SUVH2 and SUVH9.109 Although these two pathways differ in mechanism, a self‐reinforcing feedback loop is used to form a closed loop. By maintaining the methylation level of histones and DNA in the corresponding region, the stability of heterochromatin structures is enhanced (Figure 3b). However, when combining with an unmethylated DNA through a zinc‐finger domain, SUVH9 is capable of establishing DNA methylation and thus triggers gene silencing.110
The SUVR subfamily represented by SUVR4 has no SRA domain but a WIYLD domain. SUVR4 mainly acts on H3K9me1.34 The WIYLD domain can enhance the activity of SUVR4 by binding to ubiquitinated H2B, and enable the ability to dimethylate and trimethylate H3K9.110 SUVR4 is localized in both euchromatin and heterochromatin, but current studies only focused on its function on the methylation of transposons and pseudogenes in heterochromatin. Unlike the SUVH subfamily gene, the H3K9me3 covalent modification produced by the SUVR subfamily gene had no effect on DNA methylation, indicating that both SUVH and SUVR subfamilies act on heterochromatin, but function through different mechanisms.111
In addition, H3K27me1 covalent modifications mediated by ATXR5 and ATXR6 were also present in heterochromatin, but they could not directly act on DNA methylation and H3K9 lysine methylation covalent modification.21
9. CONCLUSION
Histone methylation and other histone modifications can play an important role in plant development, which was deeply involved in regulating gene expression. Nowadays, the gene expression regulation network is more complex than we have ever known. Besides the well‐known transcription factors, DNA methylation, miRNA, lncRNA, circle RNA, and histone modification are proven to be tightly linked with transcriptional and post‐transcriptional regulation. These regulation elements themselves are also regulated by other elements, which are interweaved to form a complex network.
In the past decade, studies focused mainly on the function of a single SDG gene in a certain histone methylation. Recently, more and more studies pay much attention to the function of SDG gene on nonhistone methylations and the crosslink of histone methylations and other modifications such as DNA/RNA methylations. This could help researchers establish a better and more integrated understanding on gene expression and its regulation networks.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
This work was funded by the National Natural Science Foundation of China (Nos. 31872109 and 31572126), and the Grand Science and Technology Special Project of Zhejiang Province (No. 2016C02051‐6).
Zhou H, Liu Y, Liang Y, et al. The function of histone lysine methylation related SET domain group proteins in plants. Protein Science. 2020;29:1120–1137. 10.1002/pro.3849
Funding information National Natural Science Foundation of China, Grant/Award Numbers: 31872109, 31572126; the Grand Science and Technology Special Project of Zhejiang Province, Grant/Award Number: 2016C02051‐6
Contributor Information
Heng Dong, Email: dongheng@hznu.edu.cn.
Li Huang, Email: lihuang@zju.edu.cn.
REFERENCES
- 1. Wang Y, Jia ST. Degrees make all the difference: The multifunctionality of histone H4 lysine 20 methylation. Epigenetics. 2009;4:273–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ng HH, Feng Q, Wang HB, et al. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Gene Dev. 2002;16:1518–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Leeuwen F, Gafken PR, Gottschling DE. Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell. 2002;109:745–756. [DOI] [PubMed] [Google Scholar]
- 4. Baumbusch LO, Thorstensen T, Krauss V, et al. The Arabidopsis thaliana genome contains at least 29 active genes encoding SET domain proteins that can be assigned to four evolutionarily conserved classes. Nucleic Acids Res. 2001;29:4319–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Springer NM, Napoli CA, Selinger DA, et al. Comparative analysis of SET domain proteins in maize and Arabidopsis reveals multiple duplications preceding the divergence of monocots and dicots. Plant Physiol. 2003;132:907–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ng DWK, Wang T, Chandrasekharan MB, Aramayo R, Kertbundit S, Hall TC. Plant SET domain‐containing proteins: Structure, function and regulation. Biochim Biophys Acta Gene Struct Expr. 2007;1769:316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang LS, Ma H. Complex evolutionary history and diverse domain organization of SET proteins suggest divergent regulatory interactions. New Phytol. 2012;195:248–263. [DOI] [PubMed] [Google Scholar]
- 8. Lu ZH, Huang XL, Ouyang YD, Yao JL. Genome‐wide identification, phylogenetic and co‐expression analysis of OsSET gene family in rice. PLoS One. 2013;8:e65426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng XD, Collins RE, Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomed. 2005;34:267–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aasland R, Stewart AF, Gibson T. The SANT domain: A putative DNA‐binding domain in the SWI‐SNF and ADA complexes, the transcriptional corepressor N‐CoR and TFIIIB. Trends Biochem Sci. 1996;21:87–88. [PubMed] [Google Scholar]
- 11. Heo JB, Sung S. Vernalization‐mediated epigenetic silencing by a long intronic noncoding RNA. Science. 2011;331:76–79. [DOI] [PubMed] [Google Scholar]
- 12. Chanvivattana Y, Bishopp A, Schubert D, et al. Interaction of polycomb‐group proteins controlling flowering in Arabidopsis . Development. 2004;131:5263–5276. [DOI] [PubMed] [Google Scholar]
- 13. Holec S, Berger F. Polycomb group complexes mediate developmental transitions in plants. Plant Physiol. 2012;158:35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xiao J, Lee US, Wagner D. Tug of war: Adding and removing histone lysine methylation in Arabidopsis . Curr Opin Plant Biol. 2016;34:41–53. [DOI] [PubMed] [Google Scholar]
- 15. Fletcher JC. State of the Art: trxG factor regulation of post‐embryonic plant development. Front Plant Sci. 2017;8:1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sanchez R, Zhou MM. The PHD finger: A versatile epigenome reader. Trends Biochem Sci. 2011;36:364–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Reddy B, Thompson J, et al. Regulation of set9‐mediated H4K20 methylation by a PWWP domain protein. Mol Cell. 2009;33:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garcia‐Alai MM, Allen MD, Joerger AC, Bycroft M. The structure of the FYR domain of transforming growth factor beta regulator 1. Protein Sci. 2010;19:1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang SB, Zhou B, Kang YY, et al. C‐terminal domains of histone demethylase JMJ14 interact with a pair of NAC transcription factors to mediate specific chromatin association. Cell Discov. 2015;1:15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tripoulas N, LaJeunesse D, Gildea J, Shearn A. The Drosophila ash1 gene product, which is localized at specific sites on polytene chromosomes, contains a SET domain and a PHD finger. Genetics. 1996;143:913–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorstensen T, Grini PE, Aalen RB. SET domain proteins in plant development. Biochim Biophys Acta Gene Regul Mech. 2011;1809:407–420. [DOI] [PubMed] [Google Scholar]
- 22. Hoppmann V, Thorstensen T, Kristiansen PE, et al. The CW domain, a new histone recognition module in chromatin proteins. EMBO J. 2011;30:1939–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacob Y, Feng SH, LeBlanc CA, et al. ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat Struct Mol Biol. 2009;16:763–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson LM, Bostick M, Zhang XY, et al. The SRA methyl‐cytosine‐binding domain links DNA and histone methylation. Curr Biol. 2007;17:379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thorstensen T, Grini PE, Mercy IS, et al. The Arabidopsis SET‐domain protein ASHR3 is involved in stamen development and interacts with the bHLH transcription factor ABORTED MICROSPORES (AMS). Plant Mol Biol. 2008;66:47–59. [DOI] [PubMed] [Google Scholar]
- 26. Leinhart K, Brown M. SET/MYND lysine methyltransferases regulate gene transcription and protein activity. Genes. 2011;2:210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. De‐La‐Pena C, Rangel‐Cano A, Alvarez‐Venegas R. Regulation of disease‐responsive genes mediated by epigenetic factors: Interaction of Arabidopsis‐Pseudomonas. Mol Plant Pathol. 2012;13:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown MA, Sims RJ, Gottlieb PD, Tucker PW. Identification and characterization of Smyd2: A split SET/MYND domain‐containing histone H3 lysine 36‐specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer. 2006;5:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu ST, Wu J, Sun BF, Zhong C, Ding JP. Structural and biochemical studies of human lysine methyltransferase Smyd3 reveal the important functional roles of its post‐SET and TPR domains and the regulation of its activity by DNA binding. Nucleic Acids Res. 2011;39:4438–4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang Y, Li Y, He SP, et al. A cotton (Gossypium hirsutum) WRKY transcription factor (GhWRKY22) participates in regulating anther/pollen development. Plant Physiol Biochem. 2019;141:231–239. [DOI] [PubMed] [Google Scholar]
- 31. Honys D, Twell D. Transcriptome analysis of haploid male gametophyte development in Arabidopsis . Genome Biol. 2004;5:R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Borges F, Gomes G, Gardner R, et al. Comparative transcriptomics of Arabidopsis sperm cells. Plant Physiol. 2008;148:1168–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cartagena JA, Matsunaga S, Seki M, et al. The Arabidopsis SDG4 contributes to the regulation of pollen tube growth by methylation of histone H3 lysines 4 and 36 in mature pollen. Dev Biol. 2008;315:355–368. [DOI] [PubMed] [Google Scholar]
- 34. Thorstensen T, Fischer A, Sandvik SV, et al. The Arabidopsis SUVR4 protein is a nucleolar histone methyltransferase with preference for monomethylated H3K9. Nucleic Acids Res. 2006;34:5461–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Grini PE, Thorstensen T, Alm V, et al. The ASH1 HOMOLOG 2 (ASHH2) histone H3 methyltransferase is required for ovule and anther development in Arabidopsis. PLoS One. 2009;4:e781710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo L, Yu YC, Law JA, Zhang XY. SET DOMAIN GROUP2 is the major histone H3 lysie 4 trimethyltransferase in Arabidopsis . Proc Natl Acad Sci U S A. 2010;107:22360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berr A, McCallum EJ, Menard R, et al. Arabidopsis SET DOMAIN GROUP2 is required for H3K4 trimethylation and is crucial for both sporophyte and gametophyte development. Plant Cell. 2010;22:3232–3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yao XZ, Shen WH. Crucial function of histone lysine methylation in plant reproduction. Chin Sci Bull. 2011;56:3493–3499. [Google Scholar]
- 39. Jacob Y, Stroud H, LeBlanc C, et al. Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature. 2010;466:987–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raynaud C, Sozzani R, Glab N, et al. Two cell‐cycle regulated SET‐domain proteins interact with proliferating cell nuclear antigen (PCNA) in Arabidopsis . Plant J. 2006;47:395–407. [DOI] [PubMed] [Google Scholar]
- 41. Wang DF, Tyson MD, Jackson SS, Yadegari R. Partially redundant functions of two SET‐domain polycomb‐group proteins in controlling initiation of seed development in Arabidopsis . Proc Natl Acad Sci U S A. 2006;103:13244–13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vielle‐Calzada JP, Thomas J, Spillane C, Coluccio A, Hoeppner MA, Grossniklaus U. Maintenance of genomic imprinting at the Arabidopsis medea locus requires zygotic DDM1 activity. Gene Dev. 1999;13:2971–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt A, Wohrmann HJP, Raissig MT, et al. The Polycomb group protein MEDEA and the DNA methyltransferase MET1 interact to repress autonomous endosperm development in Arabidopsis . Plant J. 2013;73:776–787. [DOI] [PubMed] [Google Scholar]
- 44. He YH. Chromatin regulation of flowering. Trends Plant Sci. 2012;17:556–562. [DOI] [PubMed] [Google Scholar]
- 45. Jeong HJ, Yang J, Yi J, An G. Controlling flowering time by histone methylation and acetylation in Arabidopsis and rice. J Plant Biol. 2015;58:203–210. [Google Scholar]
- 46. Sun CH, Fang J, Zhao TL, et al. The histone methyltransferase SDG724 mediates H3K36me2/3 deposition at MADS50 and RFT1 and promotes flowering in rice. Plant Cell. 2012;24:3235–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen DH, Huang Y, Jiang CH, Si JP. Chromatin‐based regulation of plant root development. Front Plant Sci. 2018;9:1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pien S, Fleury D, Mylne JS, et al. ARABIDOPSIS TRITHORAX1 dynamically regulates FLOWERING LOCUS C activation via histone 3 lysine 4 trimethylation. Plant Cell. 2008;20:580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Saleh A, Alvarez‐Venegas R, Yilmaz M, et al. The highly similar Arabidopsis homologs of trithorax ATX1 and ATX2 encode proteins with divergent biochemical functions. Plant Cell. 2008;20:568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamada Y, Yun JY, Woo SC, Amasino RM. ARABIDOPSIS TRITHORAX‐RELATED7 is required for methylation of lysine 4 of histone H3 and for transcriptional activation of FLOWERING LOCUS C . Plant Cell. 2009;21:3257–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yun JY, Tamada Y, Kang YE, Amasino RM. ARABIDOPSIS TRITHORAX‐RELATED3/SET DOMAIN GROUP2 is required for the winter‐annual habit of Arabidopsis thaliana . Plant Cell Physiol. 2012;53:834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Collins RE, Tachibana M, Tamaru H, et al. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem. 2005;280:5563–5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jiang DH, Kong NC, Gu XF, Li ZC, He YH. Arabidopsis COMPASS‐like complexes mediate histone H3 lysine‐4 trimethylation to control floral transition and plant development. PLoS Genet. 2011;7:e1001330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhao Z, Yu Y, Meyer D, Wu CJ, Shen WH. Prevention of early flowering by expression of FLOWERING LOCUS C requires methylation of histone H3K36. Nat Cell Biol. 2005;7:1256–1260. [DOI] [PubMed] [Google Scholar]
- 55. Jiang L, Li DH, Jin L, Ruan Y, Shen WH, Liu CL. Histone lysine methyltransferases BnaSDG8.A and BnaSDG8.C are involved in the floral transition in Brassica napus . Plant J. 2018;95:672–685. [DOI] [PubMed] [Google Scholar]
- 56. Xu L, Zhao Z, Dong AW, et al. Di‐ and tri‐ but not monomethylation on histone H3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana . Mol Cell Biol. 2008;28:1348–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Berr A, Xu L, Gao J, et al. SET DOMAIN GROUP25 encodes a histone methyltransferase and is involved in FLOWERING LOCUS C activation and repression of flowering. Plant Physiol. 2009;151:1476–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bastow R, Mylne JS, Lister C, Lippman Z, Martienssen RA, Dean C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature. 2004;427:164–167. [DOI] [PubMed] [Google Scholar]
- 59. Yu CW, Liu XC, Luo M, et al. HISTONE DEACETYLASE6 interacts with FLOWERING LOCUS D and regulates flowering in Arabidopsis . Plant Physiol. 2011;156:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Marquardt S, Raitskin O, Wu Z, Liu FQ, Sun QW, Dean C. Functional consequences of splicing of the antisense transcript COOLAIR on FLC transcription. Mol Cell. 2014;54:156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu Z, Ietswaart R, Liu FQ, Yang HC, Howard M, Dean C. Quantitative regulation of FLC via coordinated transcriptional initiation and elongation. Proc Natl Acad Sci U S A. 2016;113:218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Krichevsky A, Gutgarts H, Kozlovsky SV, et al. C2H2 zinc finger‐SET histone methyltransferase is a plant‐specific chromatin modifier. Dev Biol. 2007;303:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. De Lucia F, Crevillen P, Jones AME, Greb T, Dean C. A PHD‐polycomb repressive complex 2 triggers the epigenetic silencing of FLC during vernalization. Proc Natl Acad Sci U S A. 2008;105:16831–16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Greb T, Mylne JS, Crevillen P, et al. The PHD finger protein VRN5 functions in the epigenetic silencing of Arabidopsis FLC . Curr Biol. 2007;17:73–78. [DOI] [PubMed] [Google Scholar]
- 65. Sung SB, Amasino RM. Vernalization in Arabidopsis thaliana is mediated by the PHD finger protein VIN3. Nature. 2004;427:159–164. [DOI] [PubMed] [Google Scholar]
- 66. Finnegan EJ, Dennis ES. Vernalization‐induced trimethylation of histone H3 lysine 27 at FLC is not maintained in mitotically quiescent cells. Curr Biol. 2007;17:1978–1983. [DOI] [PubMed] [Google Scholar]
- 67. Mylne JS, Barrett L, Tessadori F, et al. LHP1, the Arabidopsis homologue of HETEROCHROMATIN PROTEIN1, is required for epigenetic silencing of FLC . Proc Natl Acad Sci U S A. 2006;103:5012–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jiang PF, Wang SL, Zheng H, et al. SIP1 participates in regulation of flowering time in rice by recruiting OsTrx1 to Ehd1. New Phytol. 2018;219:422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Doi K, Izawa T, Fuse T, et al. Ehd1, a B‐type response regulator in rice, confers short‐day promotion of flowering and controls FT‐like gene expression independently of Hd1 . Genes Dev. 2004;18:926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim SL, Lee S, Kim HJ, Nam HG, An G. OsMADS51 is a short‐day flowering promoter that functions upstream of Ehd1, OsMADS14, and Hd3a . Plant Physiol. 2007;145:1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matsubara K, Yamanouchi U, Nonoue Y, et al. Ehd3, encoding a plant homeodomain finger‐containing protein, is a critical promoter of rice flowering. Plant J. 2011;66:603–612. [DOI] [PubMed] [Google Scholar]
- 72. Liu KP, Yu Y, Dong AW, Shen WH. SET DOMAIN GROUP701 encodes a H3K4‐methytransferase and regulates multiple key processes of rice plant development. New Phytol. 2014;215:609–623. [DOI] [PubMed] [Google Scholar]
- 73. Zhou SL. Histone modification and DNA methylation function in epigenetic control of rice development. Doctoral dissertation, Huazhong Agriculture University, Wuhan, China 2017. [Google Scholar]
- 74. Srikanth A, Schmid M. Regulation of flowering time: All roads lead to Rome. Cell Mol Life Sci. 2011;68:2013–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yao XZ, Feng HY, Yu Y, Dong AW, Shen WH. SDG2‐mediated H3K4 methylation is required for proper Arabidopsis root growth and development. PLoS One. 2013;8:e56537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Napsucialy‐Mendivil S, Alvarez‐Venegas R, Shishkova S, Dubrovsky JG. Arabidopsis homolog of trithorax1 (ATX1) is required for cell production, patterning, and morphogenesis in root development. J Exp Bot. 2014;65:6373–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. de Lucas M, Pu L, Turco G, et al. Transcriptional regulation of Arabidopsis polycomb repressive complex 2 coordinates cell‐type proliferation and differentiation. Plant Cell. 2016;28:2616–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Goodrich J, Puangsomlee P, Martin M, Long D, Meyerowitz EM, Coupland G. A polycomb‐group gene regulates homeotic gene expression in Arabidopsis . Nature. 1997;386:44–51. [DOI] [PubMed] [Google Scholar]
- 79. Liu XY, Zhou C, Zhao Y, Zhou SL, Wang WT, Zhou DX. The rice enhancer of zeste[E(z)] genes SDG711 and SDG718 are respectively involved in long day and short day signaling to mediate the accurate photoperiod control of flowering time. Front Plant Sci. 2014;5:591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dong GF, Ma DP, Li JX. The histone methyltransferase SDG8 regulates shoot branching in Arabidopsis . Biochem Bioph Res Commun. 2008;373:659–664. [DOI] [PubMed] [Google Scholar]
- 81. Kumpf R, Thorstensen T, Rahman MA, et al. The ASH1‐RELATED3 SET‐domain protein controls cell division competence of the meristem and the quiescent center of the Arabidopsis primary root. Plant Physiol. 2014;166:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Malapeira J, Khaitova LC, Mas P. Ordered changes in histone modifications at the core of the Arabidopsis circadian clock. Proc Natl Acad Sci U S A. 2012;109:21540–21545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Muller K, Bouyer D, Schnittger A, Kermode AR. Evolutionarily conserved histone methylation dynamics during seed life‐cycle transitions. PLoS One. 2012;7:e51532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Alvarez‐Venegas R, Pien S, Sadder M, Witmer X, Grossniklaus U, Avramova Z. ATX‐1, an Arabidopsis homolog of trithorax, activates flower homeotic genes. Curr Biol. 2003;13:627–637. [DOI] [PubMed] [Google Scholar]
- 85. Saleh A, Al‐Abdallat A, Ndamukong I, Alvarez‐Venegas R, Avramova Z. The Arabidopsis homologs of trithorax (ATX1) and enhancer of zeste (CLF) establish 'bivalent chromatin marks' at the silent AGAMOUS locus. Nucleic Acids Res. 2007;35:6290–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bernstein BE, Mikkelsen TS, Xie XH, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. [DOI] [PubMed] [Google Scholar]
- 87. Valencia‐Morales MD, Camas‐Reyes JA, Cabrera‐Ponce JL, Alvarez‐Venegas R. The Arabidopsis thaliana SET‐domain‐containing protein ASHH1/SDG26 interacts with itself and with distinct histone lysine methyltransferases. J Plant Res. 2012;125:679–692. [DOI] [PubMed] [Google Scholar]
- 88. Avramova Z. Transcriptional 'memory' of a stress: Transient chromatin and memory (epigenetic) marks at stress‐response genes. Plant J. 2015;83:149–159. [DOI] [PubMed] [Google Scholar]
- 89. Kim JM, Sasaki T, Ueda M, Sako K, Seki M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front Plant Sci. 2015;6:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yadav CB, Muthamilarasan M, Dangi A, Shweta S, Prasad M. Comprehensive analysis of SET domain gene family in foxtail millet identifies the putative role of SiSET14 in abiotic stress tolerance. Sci Rep. 2016;6:32621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yan HY, Liu Y, Zhang K, Song J, Xu WY, Su Z. Chromatin state‐based analysis of epigenetic H3K4me3 marks of Arabidopsis in response to dark stress. Front Genet. 2019;10:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van Dijk K, Ding Y, Malkaram S, et al. Dynamic changes in genome‐wide histone H3 lysine 4 methylation patterns in response to dehydration stress in Arabidopsis thaliana . BMC Plant Biol. 2010;10:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Kim JM, To TK, Ishida J, Matsui A, Kimura H, Seki M. Transition of chromatin status during the process of recovery from drought stress in Arabidopsis thaliana . Plant Cell Physiol. 2012;53:847–856. [DOI] [PubMed] [Google Scholar]
- 94. Kim JM, To TK, Ishida J, et al. Alterations of lysine modifications on the histone H3 N‐tail under drought stress conditions in Arabidopsis thaliana . Plant Cell Physiol. 2008;49:1580–1588. [DOI] [PubMed] [Google Scholar]
- 95. Ding Y, Fromm M, Avramova Z. Multiple exposures to drought 'train’ transcriptional responses in Arabidopsis . Nat Commun. 2012;3:740. [DOI] [PubMed] [Google Scholar]
- 96. Liu N, Fromm M, Avramova Z. H3K27me3 and H3K4me3 chromatin environment at super‐induced dehydration stress memory genes of Arabidopsis thaliana . Mol Plant. 2014;7:502–513. [DOI] [PubMed] [Google Scholar]
- 97. Sani E, Herzyk P, Perrella G, Colot V, Amtmann A. Hyperosmotic priming of Arabidopsis seedlings establishes a long‐term somatic memory accompanied by specific changes of the epigenome. Genome Biol. 2013;14:R59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kwon CS, Lee D, Choi G, Chung WI. Histone occupancy‐dependent and ‐independent removal of H3K27 trimethylation at cold‐responsive genes in Arabidopsis. Plant J. 2009;60:112–121. [DOI] [PubMed] [Google Scholar]
- 99. Cazzonelli CI, Nisar N, Roberts AC, Murray KD, Borevitz JO, Pogson BJ. A chromatin modifying enzyme, SDG8, is involved in morphological, gene expression, and epigenetic responses to mechanical stimulation. Front Plant Sci. 2014;5:553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Alvarez‐Venegas R, Sadder M, Hlavacka A, et al. The Arabidopsis homolog of trithorax, ATX1, binds phosphatidylinositol 5‐phosphate, and the two regulate a common set of target genes. Proc Natl Acad Sci U S A. 2006;103:6049–6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Alvarez‐Venegas R, Al Abdallat A, Guo M, Alfano JR, Avramova Z. Epigenetic control of a transcription factor at the cross section of two antagonistic pathways. Epigenetics. 2007;2:106–113. [DOI] [PubMed] [Google Scholar]
- 102. Palma K, Thorgrimsen S, Malinovsky FG, et al. Autoimmunity in Arabidopsis acd11 is mediated by epigenetic regulation of an immune receptor. PLoS Pathog. 2010;6:e1001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Huang Y, Mo YJ, Chen PY, et al. Identification of SET domain‐containing proteins in Gossypium raimondii and their response to high temperature stress. Sci Rep. 2016;6:32729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Charron JBF, He H, Elling AA, Deng XW. Dynamic landscapes of four histone modifications during deetiolation in Arabidopsis . Plant Cell. 2009;21:3732–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lafos M, Kroll P, Hohenstatt ML, Thorpe FL, Clarenz O, Schubert D. Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 2011;7:e1002040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Johnson LM, Law JA, Khattar A, Henderson IR, Jacobsen SE. SRA‐domain proteins required for DRM2‐mediated de novo DNA methylation. PLoS Genet. 2008;4:e100280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Jackson JP, Johnson L, Jasencakova Z, et al. Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana . Chromosoma. 2004;112:308–315. [DOI] [PubMed] [Google Scholar]
- 108. Du JM, Zhong XH, Bernatavichute YV, et al. Dual binding of chromomethylase domains to H3K9me2‐containing nucleosomes directs DNA methylation in plants. Cell. 2012;151:167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Liu ZW, Shao CR, Zhang CJ, et al. The SET domain proteins SUVH2 and SUVH9 are required for pol V occupancy at RNA‐directed DNA methylation loci. PLoS Genet. 2014;10:e1003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang HM, Lang ZB, Zhu JK. Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol. 2018;19:489–506. [DOI] [PubMed] [Google Scholar]
- 111. Veiseth SV, Rahman MA, Yap KL, et al. The SUVR4 histone lysine methyltransferase binds ubiquitin and converts H3K9me1 to H3K9me3 on transposon chromatin in Arabidopsis . PLoS Genet. 2011;7:e1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]