

Abstract
Swine acute diarrhea syndrome coronavirus (SADS‐CoV) is a novel coronavirus that is involved in severe diarrhea disease in piglets, causing considerable agricultural and economic loss in China. The emergence of this new coronavirus increases the importance of understanding SADS‐CoV as well as antivirals. Coronaviral proteases, including main proteases and papain‐like proteases (PLP), are attractive antiviral targets because of their essential roles in polyprotein processing and thus viral maturation. Here, we describe the biochemical and structural identification of recombinant SADS papain‐like protease 2 (PLP2) domain of nsp3. The SADS‐CoV PLP2 was shown to cleave nsp1 proteins and also peptides mimicking the nsp2|nsp3 cleavage site and also had deubiquitinating and deISGynating activity by in vitro assays. The crystal structure adopts an architecture resembling that of PLPs from other coronaviruses. We characterize both conserved and unique structural features likely directing the interaction of PLP2 with the substrates, including the tentative mapping of active site and other essential residues. These results provide a foundation for understanding the molecular basis of coronaviral PLPs' catalytic mechanism and for the screening and design of therapeutics to combat infection by SADS coronavirus.
Keywords: active site, crystal structure, DUB, PLP2, SADS‐CoV, ubiquitin
Short abstract
PDB Code(s): http://firstglance.jmol.org/fg.htm?mol=6L5T;
1. INTRODUCTION
Swine acute diarrhea syndrome coronavirus (SADS‐CoV), a novel member of the genus Alphacoronavirus, first detected and identified as the aetiological agent of a devastating swine disease outbreak in southern China in 2017, which caused death to 24,693 piglets and huge economic and agricultural losses. 1 Clinical signs of infected piglets include acute vomiting and severe and acute diarrhea, leading to the death. The genome sequence of SADS‐CoV shares high homology with a bat coronavirus detected in a cave near the pig farm, implying SADS‐CoV most likely originated from bats.2, 3 It is in urgent need to develop effective antivirals for SADS‐CoV, since the high lethality of this new emerging coronavirus.
Two classes of cysteine proteases are encoded by coronaviruses, either a chymotrypsin fold (resides in nsp5) or a papain‐like fold (contained in nsp3).4, 5 These proteases contribute to the processing of the two precursor polyproteins (pp1a and pp1ab) of the viral replicase complex and are thought to regulate the host cell functions to promote viral infection. The viral papain‐like protease (PLP) domain is contained in nsp3 of coronaviruses and participates in the proteolytic processing of the N‐terminal region of the polyproteins. It could cleave nsp1|nsp2, nsp2|nsp3, and nsp3|nsp4 sites, while all sites downstream nsp4 are processed by nsp5.6, 7 Besides cleaving the polyproteins, PLPs also possess a related enzymatic activity to promote virus replication: deubiquitinating (DUB) and de‐ISGylating activities, although their physiological functions have not been completely clarified.8, 9 Both ubiquitin and ISG15 are involved in preventing viral infection, and several viruses have utilized Ubl‐conjugating pathways to counter the pro‐inflammatory properties of Ubl‐conjugated host proteins.10, 11 For coronaviruses, one such mechanism relies on evading the host immune response via the action of viral isopeptidase.12, 13
Currently, there are no licensed vaccines or antiviral drugs against SADS‐CoV. Therefore, the rapid identification of targets for the development of antivirals is a high priority. SADS‐CoV genome was annotated to contain main protease (Mpro) and PLPs. However, the structural characteristics and the detailed catalytic mechanism of SADS‐CoV papain‐like protease 2 (PLP2) are still unclear, as are antiviral strategies.
Here, we describe the enzymatic activities of the SADS‐CoV PLP2, and determine its crystal structure to 1.72 Å. In in vitro assays, we characterized the purified protease enzymatically, that is, PLP2 efficiently cleaves nsp1, ubiquitin‐AMC, ISG15‐AMC, and two peptide‐AMC substrates. Structural analysis, site‐directed mutagenesis and kinetic studies were conducted to identify key active site residues involved in the substrate binding and catalysis. The model of PLP2‐Ub complex and kinetic studies facilitates the identification of residues and regions important for ubiquitin binding. A detailed description of the SADS‐CoV PLP2 presented in this study is critical for the understanding of the mechanism of this enzyme in the biogenesis of the coronavirus replicase complex and evading host immune responses, and also critical for the development of antiviral drugs against SADS‐CoV.
2. RESULTS
2.1. Cleavage of nsp1 fusion protein by SADS‐CoV PLP2
Currently, there is no available biochemical data about SADS‐CoV PLP2. To determine whether it could recognize and process polyproteins encoded by the SADS‐CoV genome, we incubated purified recombinant PLP2 with purified nsp1‐eGFP in a time‐course assay, and then the reaction products were analyzed by SDS‐PAGE. When the trans‐cleavage assay was done at its optimum temperature (23°C), the PLP2 proteins showed the cleavage of nsp1‐eGFP to some extent and the free nsp1 and eGFP were released (Figure 1). It showed that the peptide connecting eGFP to nsp1 had been hydrolyzed by the protease activity of PLP2. This assay demonstrated that SADS‐CoV PLP2 could recognize and process the cleavage site KRGG of nsp1|nsp2.
FIGURE 1.
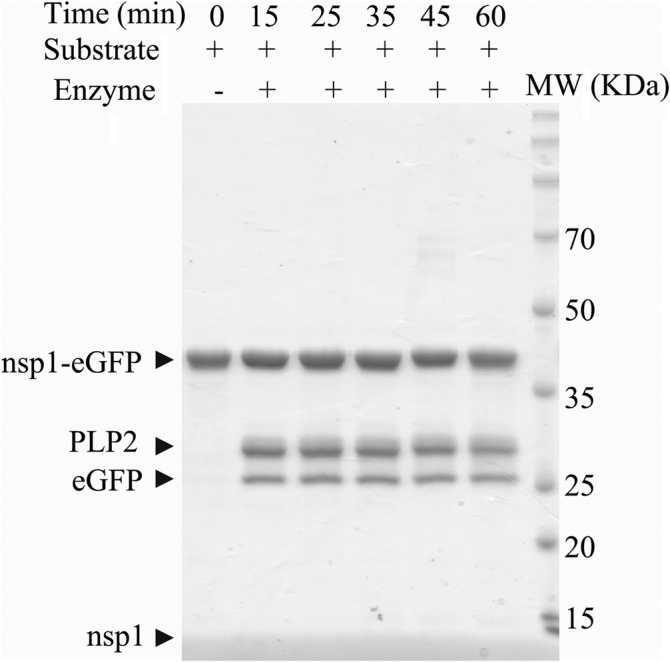
SADS‐CoV PLP2 cleaves nsp1‐eGFP. Time‐dependent cleavage of nsp1‐eGFP by SADS‐CoV PLP2. A 200 nM of nsp1‐eGFP was incubated with 20 nM of PLP2 at 23°C for the incubated time and then the mixture was analyzed by SDS‐PAGE. The protease activity was indicated by the release of free nsp1 and eGFP
2.2. Crystal structure of PLP2
A sequence encoding 293 amino acids (1588–1880) PLP2 domain of nsp3 of SADS‐CoV was purified and crystallized and the structure was solved by the Bromide single wavelength anomalous dispersion (SAD) method by collecting a dataset of potassium bromide soaked SADS‐CoV PLP2 crystals (P212121 spacegroup) at bromide peak wavelength. One PLP2 molecule occupies the asymmetric unit. For simplicity, we renumber PLP2 to 1–293. Clear electron density permitted unambiguous modeling of all residues except the ubiquitin‐like domain and the loop corresponds to the so‐called BL2 loop (250–255 aa). The final model was refined to 1.72 Å resolution (R work = 17.93% and R free = 21.16%) with a good stereochemistry. Final statistics for the data collection and refinement are shown in Table 1. The first 52 residues composing N‐terminal ubiquitin‐like domain was not modeled into the structure because of none electronic density, suggesting it was highly flexible. So the structure mainly composed of the core catalytic part. The overall structure resembles a right hand composed of three domains, the thumb, palm, and fingers (Figure 2a). The secondary structure arrangement of the catalytic domain has 7 α‐helices (6 in the thumb domain, 1 in the fingers domain), 13 total β‐strands (2 in the thumb domain, 4 β‐strands in the fingers domain, and 7 β‐strands in the palm domain), and 1 310‐helix (η) in the thumb domain and another 310‐helix (η) in the fingers domain.
TABLE 1.
Data collection and refinement statistics of SADS‐CoV PLP2
| Wavelength (Å) | 0.9206 (KBr) | 0.9709 |
|---|---|---|
| Space group | P212121 | P212121 |
| Cell parameters | ||
| a, b, c (Å) | 34.04, 69.20, 104.84 | 34.23, 68.51, 104.35 |
| α, β, γ (°) | 90.00 90.00 90.00 | 90.00 90.00 90.00 |
| Resolution (Å) a | 34.95–1.67 (1.71–1.67) | 41.51–1.72 (1.75–1.72) |
| Reflections | 478,565 (35,732) | 345,942 (17,073) |
| Unique reflections | 29,590 (2,160) | 26,961 (1,362) |
| Rmerge | 0.093 (1.405) | 0.116 (1.636) |
| Rmeas | 0.100 (1.495) | 0.121 (1.705) |
| Rpim | 0.033 (0.504) | 0.034 (0.476) |
| CC1/2 | 0.999 (0.735) | 0.999 (0.623) |
| Completeness (%) | 99.8 (99.6) | 100 (99.5) |
| Multiplicity | 16.2 (16.5) | 12.8 (12.5) |
| I/σ | 18.4 (2.2) | 16.7 (1.7) |
| Rwork/Rfree | 0.1793/0.2116 | |
| RMS (bonds) (Å) | 0.013 | |
| RMS (angels) (°) | 1.130 | |
| Ramachandran favored (%) | 97.39 | |
| Ramachandran allowed (%) | 2.61 |
Values in parentheses are for the highest resolution shell.
FIGURE 2.
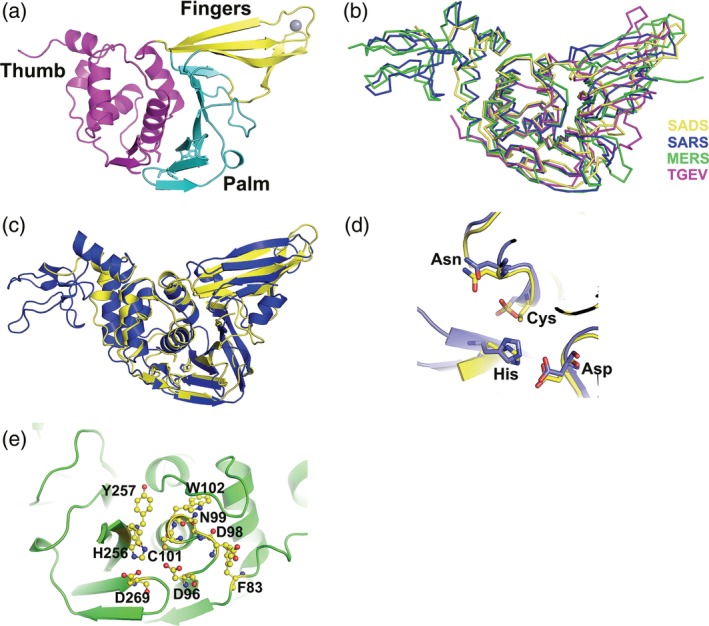
Overall structure of SADS‐CoV PLP2, the comparisons with SARS‐CoV and MERS‐CoV PLPros and TGEV PLP1 and the proposed active site of SADS‐CoV PLP2. (a) The overall structure of SADS‐CoV PLP2, locations of the thumb (magenta), palm (cyan), and fingers (yellow) domains are indicated by different colors. The zinc atom (red) is shown in gray sphere representation. (b) Comparison of the structure of SADS‐CoV PLP2 with those of SARS‐CoV PLPro (PDB ID: https://bioinformatics.org/firstglance/fgij//fg.htm?mol=4OVZ), MERS‐CoV PLpro (PDB ID: https://bioinformatics.org/firstglance/fgij//fg.htm?mol=4P16) and TGEV PLP1 (PDB ID: https://bioinformatics.org/firstglance/fgij//fg.htm?mol=3MP2). A ribbon diagram shows SADS‐CoV PLP2 in yellow, SARS‐CoV PLpro in blue, MERS‐PLpro in green, and TGEV PLP1 in magenta. (c) Superimposition of the structures of SADS‐CoV PLP2 (yellow schematic) over SARS‐CoV PLPro (blue schematic) shown in cartoon. Catalytic triad residues are shown in ball‐and‐stick representations. The orientation of structures shown in (b) and (c) are similar to that of (a). (d) Close‐up view of the active site segments for SADS‐CoV PLP2 (carbon atoms in yellow) and SARS‐CoV PLpro (carbon atoms in blue). The Cys‐His‐Asp triad and the asparagine as the oxyanion hole residue are conserved in the three‐dimensional structures. (e) Proposed SADS‐CoV PLP2 active site. Catalytic residues, C101, H256 and D269, and other important active‐site residues such as F83, D96, D98, N99, W102, and Y257 are shown in ball‐and‐stick
The thumb domain is predominantly α‐helical and contains six α‐helices and two short β‐strands that are arranged in a β‐hairpin. The thumb domain makes contacts with the palm domain using this β‐hairpin. Amino acids 167–293 fold into palm and fingers domains. This region consists of mostly β‐strands. Although the fingers domain is composed of three long and one short β‐strands, the palm is made up of seven β‐strands. One Zn2+ ion is coordinated by three cysteine residues (Cys180, Cys183, and Cys209) and one histidine residue (His211) from the loops connecting four β‐strands in the fingers domain. The strand and helix numbering used here are different from those of SARS‐CoV PLpro, 14 because the present structure model does not have the N‐terminal Ubl domain and also because of structural differences (Figure 2b,c). The three long β‐strands of the fingers domain protrude out of the protein, which facilitates the formation of a hydrophobic cavity at the junction of thumb and fingers domains, whereas the palm slopes into the cavity (Figure 2a).
BL2 loop was proposed previously to regulate the activity, and two glycine residues flanking this loop are conserved in PLPs (and several other ubiquitin‐specific proteases [USPs]), and might be responsible for the flexibility of this region.14, 15, 16 It was previously shown that in the SARS PLpro structure, this loop significantly changes conformation upon binding of the inhibitor‐GRL0617. 17 And BL2 loop of SADS‐CoV PLP2 was not modeled because of none electron density, suggesting its high flexibility.
SADS‐CoV PLP2 includes the spatial proximity Cys101, H256, and Asp269 (Figure 2c,d), which are implicated in the catalysis, referred to as the catalytic triad. Cys101 is positioned at the N‐terminus of the α3 of the thumb domain, while both His256 and Asp269 residues are located in the palm domain. The distance between Cys101 and His256 is 3.21 Å, and that between His256 and Asp269 is 3.82 Å.
2.3. Comparison of the overall fold
We searched the Protein Data Bank (PDB) for structural homologs of SADS‐CoV PLP2 using the DALI server (http://echidna.biocenter.helsinki.fi/dali_server). 18 The search identified the structure of SARS‐CoV PLpro as the top match (Z score = 21.5) with a root mean square deviation (RMSD) of 2.7 Å for 225 overlapping Ca atoms and MERS‐CoV PLpro as the second match (Z score = 18.6), with the RMSD of 3.1 Å for 216 Ca atoms. In addition, it is also similar to cellular DUBs (such as human ubiquitin‐specific proteases [USPs] USP4, 5, 7, 8, and 21).
The overall superposition of the structure is excellent in the thumb and palm regions, whereas the β‐strands of the fingers do not superimpose as well (Figure 2b), when SADS‐CoV PLP2 is compared to PLpro from SARS, 19 MERS CoVs, 20 and TGEV PLP1. 21 The overall architectures of these enzymes are similar, and the proposed catalytic triad is very well aligned (Figure 2c,d). But there are several regions with significant differences observed from the overlay, such as the fingers domains have different conformations and extents of closure/opening relative to the palm domains. Since TGEV PLP1 and MERS‐CoV PLpro are very similar to SARS‐CoV PLpro,21, 22 and the biochemical and structural studies of SARS PLpro are much detailed, we will focus on the comparison of SADS‐CoV PLP2 and SARS‐CoV PLpro to get insights into the molecular mechanism of SADS‐CoV PLP2.
Superimposition analysis revealed some regions with significant differences between SADS‐CoV PLP2 and SARS‐CoV PLpro. The palm domain aligns much more well than the thumb and fingers domain (Figure 2c). The first significant difference is observed in the thumb domain. The arrangement of the six α‐helixes is different, in which six α‐helixes of the SARS‐CoV PLpro thumb domain are folded slightly more compact than SADS‐CoV PLP2 thumb domain, even though the two β‐strands forming β‐hairpins in the thumb domain align well. The second significant difference is observed in the fingers domain. The fingers domain of SADS‐CoV PLP2 is much closer to the palm domain in the space, while SARS‐CoV PLpro fingers domain tilts slightly away from the palm domain. This results in a relatively different orientation of this region of the structure. Thirdly, there is a big difference between the spatial arrangement between the palm and fingers domain, which is specifically highlighted by the fact that the whole zinc finger domain was shifted approximately 3.68 Å, when the Zinc location was measured. It is notable that this difference between the SADS‐CoV PLP2 and SARS‐CoV PLpro structure maps to a region that is distant from the active site, including the substrate‐binding region.
Comparison of the structure of SADS‐CoV PLP2 with that of MERS‐CoV PLpro revealed significant differences in the fingers domain between the two PLPs and also the extent of opening/closure of the fingers domain and palm domain, like the significant difference to SARS‐CoV PLpro. Comparison of structures from SADS‐CoV PLP2 and TGEV PLP1 shows that they are more similar, when 179 residues Cα are aligned, and the RMSD is 1.9 Å. This is reasonable because both of SADS‐CoV and TGEV could infect pigs and the P4‐P1 KMGG sequence of TGEV PLP1, which is similar to KXGG consensus sequence of SADS‐CoV PLP2. But there are some differences due to that TGEV PLP1 is much shorter than SADS‐CoV PLP2 (Figure 2b).
PLPs from SADS, SARS, and MERS CoVs and TGEV show some structural differences that would give rise to different substrate specificities and catalytic efficiencies. Besides the structural differences among the four PLPs, they share similarity at the assembly of the three domains and the regions surrounding the active site, which could explain why all of them could function as a PLP.
2.4. Model of ubiquitin bound to PLP2
The structural similarity of SADS‐CoV PLP2 to SARS‐CoV or MERS‐CoV PLPro and TGEV PLP1 suggests that SADS‐CoV PLP2 may also have signature substrate‐binding site to support DUB activity in order to target ubiquitinated host factors to promote virus replication. In the presence of the structure of an enzyme‐substrate complex, the molecular basis of its catalysis and the binding mode of peptidyl substrates or ubiquitin to SADS‐CoV PLP2 could be better understood, while attempts to co‐crystallization of wild type or mutant forms of PLP2 with different forms of ubiquitin were unsuccessful. But since SADS‐CoV PLP2 could cleave at the nsp1|nsp site and nsp2|nsp3 site and whose P1–P4 positions are occupied by the KRGG peptide and KAGG peptide, respectively (KXGG consensus site), resembling the LXGG sequence recognized by SARS‐CoV PLpro thus enables using SARS‐CoV PLpro bound with Ub (4 mm3) to model the ubiquitin molecule into the corresponding region of SADS‐CoV PLP2 (Figure 3). Because there are no loops blocking the active site of apo‐form PLP2, the C‐terminal tail of ubiquitin is easily accommodated in the active site. The β‐barrel of ubiquitin is mainly engaged by the fingers domain of PLP2, whereas the extreme C‐terminal tail containing the diglycine motif extends into a groove located at the junction of thumb and palm domains (Figure 3b,c).
FIGURE 3.
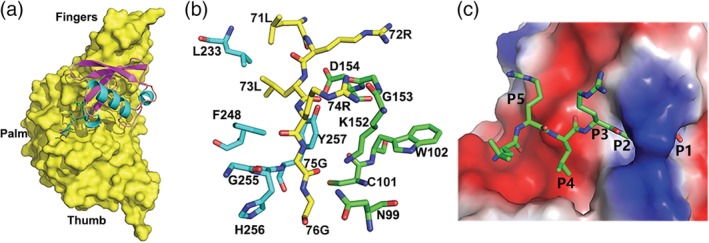
Hypothetical model of the interaction of ubiquitin with the SADS‐CoV PLP2 active site based on the structure of SARS‐CoV PLpro bound with Ub. (a) The model of SADS‐CoV PLP2 bound with ubiquitin. A surface representation of the SADS‐CoV PLP2 is shown complexed with modeled ubiquitin. The C‐terminal of ubiquitin is shown by a ball‐and‐stick representation. (b) Modeled interactions between the C‐terminal tail of ubiquitin and the SADS‐CoV PLP2. Ubiquitin residues are colored in yellow carbon, while PLP2 residues are shown in cyan or green carbons. (c) A surface representation of the SADS‐CoV PLP2 with a tunnel of the active site bound by C‐terminal five residues of ubiquitin. The P1–P5 positions of ubiquitin are labeled
It has been demonstrated that an LXGG motif at the P4–P1 positions of the substrate is essential for the recognition and cleavage by SARS‐CoV PLpro, and it appears to have no preferences for the P′ positions or for residues N‐terminal to P4.23, 24 A positively charged arginine at the P3 position of the ubiquitin substrates correlates well with one positively charged lysine at one of three P3 positions in SADS‐CoV PLP2 polyprotein substrates. MERS‐CoV or SARS‐CoV PLpro appears to have loosely defined pockets in their active sites, even though the crystal structures of both MERS‐CoV and SARS‐CoV PLpros bound with one mono‐Ub show a network of hydrogen bonds stabilizing the C‐terminus of ubiquitin in the active site.25, 26
More importantly, the di‐glycine is in proximity to key catalytic residues and BL2 that are conserved across PLPs (Figure 4). Although the overall pattern of binding the ubiquitin is similar in those from SADS‐CoV PLP2 and SARS‐CoV or MERS‐CoV PLPros, the nature of amino acids of PLPs from these CoVs to bind ubiquitin is somewhat different. These differences are illustrated in the following example involving one salt bridge or hydrogen‐bonding interactions. The side‐chain of Arg42 of ubiquitin forms a salt bridge with the side‐chain of Glu168 of SARS‐CoV PLpro, whereas Arg42 might interact with Ser160 in SADS‐CoV PLP2.
FIGURE 4.
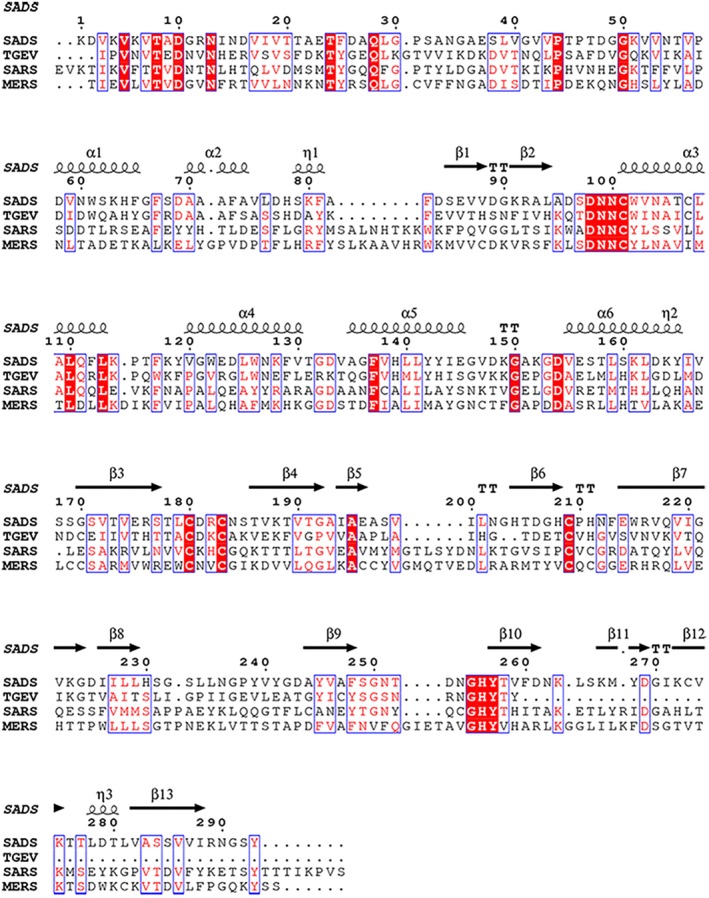
Sequence alignments of SAD‐CoV PLP2, TGEV PLP1, SARS‐CoV, and MERS‐CoV PLPros. Secondary structural elements of SADS‐CoV PLP2 are shown at the top. Absolutely conserved residues are highlighted with red background while conserved residues are shown in red
The present predicted model reveals that two bulky aromatic residues (W102 and Y257) are partially responsible for the strict requirement for glycines at the P1 and P2 positions of PLPs' substrates (Figure 3b). Aromatic residues in proximity to the catalytic cysteine are very common in PLPs because they can aid to increase the nucleophilicity of the catalytic cysteine residue. 27 Residue N99 and K152 also appear to contribute P1 specificity by hovering above the active site in a position that could occlude residues larger than a glycine (Figure 3b). Stabilization of the P3 backbone is contributed by an additional aromatic residue, F248, which corresponds to histidine in both USP14 and HAUSP, and Y265 of SARS‐CoV PLpro, Y269 of MERS‐CoV PLpro, and Y175 of TGEV PLP1. This residue sitting below the P3 subsite may stabilize the backbone of the substrate by interacting with the P3 residue, as well as orient the bulky P3 side chain out of the cleft (Figure 3b,c). Due to spatial requirements, the P5 arginine is oriented upward and appears to be stabilized in part by a negative patch on the thumb domain created by two residues, D154 and S157, which are also conserved in USP14 and HAUSP, and SARS‐CoV or MERS‐CoV PLpro. S157 could form a hydrogen bond with the P5 arginine side chain, whereas D154 hydrogen bonds to the P4 backbone amide as in HAUSP and USP14 and SARS‐CoV or MERS‐CoV PLpro and TGEV PLP1.
The allowance for a lysine or a leucine residue at the P4 position of PLP2 substrates can be explained by the presence of a small hydrophobic crevice formed by F248, S249, and S284, which lie underneath the bound ubiquitin (Figure 3c). Many of the interactions that can potentially anchor ubiquitin in the catalytic groove of PLPs originate from hydrogen bonding to the backbone atoms of ubiquitin. The same interactions are also observed in the active sites of HAUSP and USP14.15, 16
Recently, potential small compound inhibitors which have been screened out for SARS‐CoV based on their ability to inhibit SARS‐CoV PLpro do not inhibit MERS‐CoV PLpro, 28 suggesting that finding a general anti‐coronaviral drug based on PLPs would be a great challenge.
2.5. Proposed active‐site conformation
Little is known about the catalytic mechanism of SADS‐CoV PLP2, so we gleaned some insights by comparing it with PLPs from SARS‐CoV and MERS‐CoV or TGEV. Based on the conservation between the overall fold and the active site residues (Figures 2 and 4), the active site in SADS‐CoV PLP2 is proposed (Figure 2e). The three residues of SADS‐CoV PLP2 (C101‐H256‐D269) form the canonical catalytic triad, consistent with the catalytic triad found in many PLPs, including SARS‐CoV or MERS‐CoV PLpro and TGEV PLP1 (Figure 2).14, 26, 28 The catalytic cysteine (C101) is located at the base of the palm domain (Figure 2d). The aspartate acid residue (D269) is located in a classic triad formation, within hydrogen‐bonding distance to His256. Asp269 might have only a stabilizing role for the Cys101 and His256 during catalysis.
An important feature of cysteine and serine protease enzyme mechanism involves the formation an oxyanion hole to stabilize the negative charged tetrahedral intermediate following nucleophilic attack by the cysteine side chain during the hydrolysis. Generally, the oxyanion hole is close to the catalytic center. The amino groups of side‐chains of asparagine or glutamine residues in some PLPs could provide the stabilization forces required for the oxyanion hole.14, 29 The oxyanion hole contains a N99 in the corresponding position is found in SADS‐CoV PLP2 structure. Besides N99, W102, which is conserved among coronavirus PLPs, is located over the catalytic cysteine, and may also contribute to the stabilization of oxyanion hole, because the side chain of W102 could form a hydrogen bond with an intermediate in the active site (Figure 2e). And also several important structural features, including a series of loops surrounding the area, dictate the access to the narrow active site. One such loop is situated at the mouth of the active site and is composed of amino acid residues 92–99. And W102, which we propose to also stabilize the oxyanion hole, protrudes over this loop into the active site (Figure 2e). And an important interaction between D98 on the loop and F83 behind the loop appears to direct this loop into the active site.
It was shown previously that the oxyanion hole of SARS‐CoV PLpro is stabilized by W107 and the replacement of this residue by alanine abolished the protease activity (5). In SADS‐CoV PLP2, the spatially equivalent position is occupied by D96 (2.96 Å away from Cys101, 3.82 Å away from His256 and 4.80 Å away from D269). But D96 cannot act to stabilize the oxyanion in a manner similar to W107 of SARS‐CoV PLpro and Q19 in papain because it would be negatively charged and so on could not be the hydrogen donator. Instead, since it is so close to C101 (2.96 Å from the sulfur atom of the side chain of C101), it might promote Cys101 to perform the catalysis. And D96 is poorly conserved among CoVs (Figure 4), implying that the interaction between D96 and the catalytic cysteine (C101) might be unique to SADS‐CoV PLP2. Meanwhile, in MERS‐CoV PLpro, the Trp is replaced by a Leu, one residue who could not be a hydrogen bond donor, and wild‐type MERS‐CoV PLpro has a lower kcat/Km than the SARS PLpro, whereas Leu to Trp actually increased the kcat/Km 23‐fold.6, 30
BL1 and BL2 block the access to the active site and palm regions when USP14 is in a free state, and these loops are hypothesized to modulate the DUB activity of USP14. 16 The present PLP2 structural model does not contain BL2 because of none electronic density. The BL2 of PLP2 (residues 250–255) is similar in length to the BL2 of both HAUSP and USP14, all of which contain two glycines on either side.
The cleft formed between the thumb and palm domains could direct the C‐terminal of the substrate into the active site. The oxyanion hole of the PLP2 active site appears to be composed of N99, C101, and W102, which appear suitably arranged to stabilize the negative charge intermediate that forms during the catalysis. So the catalytic triad, oxyanion hole‐stabilizing residues and other residues are likely to form the active site and essential for the cleavage of substrates by SADS‐CoV PLP2 (Figure 2e).
2.6. Enzymatic activities of mutants of PLP2
Based on the sequence and structure alignments of SADS‐CoV PLP2 with the well‐identified SARS‐CoV or MERS‐CoV PLpro (Figures 2 and 4), some mutants are designed and produced and their enzymatic activities toward various commercially available substrates were determined to probe their importance. For the cleavage assay using the fusion protein as the substrate, some of the mutants showed reduced activity on nsp1‐eGFP (Figure S1). The cleavage products indicated to some extent that alanine substitution at the sites had some effects on the processing of nsp1‐eGFP. To quantify the kinetics of the mutants, we turned to fluorogenic model peptide‐AMC, Ub‐AMC, or ISG15‐AMC, where the AMC fluophore is conjugated to the end of the peptide or Ub or ISG15, enabling kinetic quantification of SADS‐CoV PLP2 by monitoring fluorescence during hydrolysis of the –AMC amide bond. All of the kinetic parameters are summarized in Table 2.
TABLE 2.
Kinetic parameters of SADS‐CoV PLP2 wild type and mutants with different fluorescent substrates
| Substrate | Kinetic parameter | PLP2 (WT) | ΔN49 | C101A | H256A | D269A | F83A | D96A | D98A | W102A | N99A | Y257A |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Z‐KAGG‐AMC | kcat (min−1) | 0.92 | 2.18 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.7047 | 0.0000 | 0.4388 | 0.5158 |
| Km (μM) | 8.79 | 17.57 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 35.600 | 0.0000 | 28.000 | 31.990 | |
| kcat/Km (μM−1 min−1) | 0.104 ± 0.017 | 0.124 ± 0.0116 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0000 | 0.0198 ± 0.003 | 0.0000 | 0.0157 ± 0.002 | 0.0161 ± 0.003 | |
| Z‐LRGG‐AMC | kcat (min−1) | 0.6871 | 1.0180 | 0.0000 | 0.0000 | 0.7341 | 0.5095 | 0.0000 | 0.0925 | 0.1858 | 0.0000 | 0.2332 |
| Km (μM) | 37.340 | 42.0500 | 0.0000 | 0.0000 | 44.170 | 17.7700 | 0.0000 | 19.140 | 28.1700 | 0.0000 | 16.650 | |
| kcat/Km (μM−1 min−1) | 0.0184 ± 0.003 | 0.0242 ± 0.004 | 0.0000 | 0.0000 | 0.0166 ± 0.002 | 0.0287 ± 0.004 | 0.0000 | 0.0048 ± 0.001 | 0.0066 ± 0.001 | 0.0000 | 0.0140 ± 0.003 | |
| Ub‐AMC | kcat (min−1) | 1.2850 | 1.5050 | 0.0000 | 0.5957 | 0.5396 | 2.1230 | 0.0000 | 0.0000 | NA | NA | NA |
| Km (μM) | 0.3416 | 0.3206 | 0.0000 | 0.4732 | 0.3692 | 0.4532 | 0.0000 | 0.0000 | NA | NA | NA | |
| kcat/Km (μM−1 min−1) | 3.76 ± 0.45 | 4.69 ± 0.8 | 0.0000 | 1.25 ± 0.48 | 1.46 ± 0.38 | 4.68 ± 0.50 | 0.0000 | 0.0000 | NA | NA | NA | |
| ISG15‐AMC | kcat (min−1) | 3.9330 | 5.3680 | 0.0000 | 2.5180 | NA | NA | NA | NA | NA | NA | NA |
| Km (μM) | 0.6199 | 0.6467 | 0.0000 | 0.6308 | NA | NA | NA | NA | NA | NA | NA | |
| kcat/Km (μM−1 min−1) | 6.3 ± 0.2 | 8.3 ± 0.1 | 0.0000 | 3.9 ± 0.11 | NA | NA | NA | NA | NA | NA | NA |
Abbreviation: NA, not available.
After mutated to Ala, the catalytic triad (C‐H‐D) was completely inactive with KAGG‐AMC, a peptide substrate as the predicted cleavage sequence of the nsp2‐nsp3 of SADS‐CoV, which is consistent with their roles as catalytic sites. And N49Δ could be gained by enzymatic activity, indicating that this region might have a regulatory role. It has been reported that N52 Δ of SARS‐CoV PLpro and N62 Δ of MERS‐CoV PLpro also had increased enzymatic activity.12, 30, 31 D96 that are close to C101, abolished its activity, might be due to its critical role in promoting C101 during the catalysis. F83A also totally lost its activity. For the D98A mutant (D109 in SARS), the Km is increased about fourfold and kcat is reduced a little, and the kcat/Km is about 1/5 of the wild‐type protein, suggesting that it might play an important role in the substrate binding. W102A (Y113 in SARS‐CoV PLpro) abolished the activity, which might be due to the substrate binding or the stabilization of the oxyanion hole. N99 (N110 for SARS‐CoV PLpro) reduced its kcat/Km to about 1/6 of the wild‐type protein. This result accordingly suggests that N99 would participate in forming the oxyanion hole which is critical for the enzymatic activity, which was proposed and postulated and identified to some extent for SARS‐CoV or MERS‐CoV PLpros.14, 30 Thus, consistent with the structural model, N99 may play a role in positioning the intermediate for the nucleophilic attack by Cys101, and the interaction between N99 and the substrate may further the substrate binding. Y257 (Y274 in SARS) is close to H256 and faces the substrate P4 subsite, which might be a part of the substrate binding regulation, also reduced the PLP2's activity when mutated to alanine.
KXGG is the consensus cleavage site for SADS‐CoV PLP2, which bears strong similarity to the C‐terminal tail of ubiquitin and ISG15 (consensus sequence LRGG). So we used LRGG‐AMC for measuring the DUB and DeISG15 activity in in vitro assay. The Δ 49N also has slightly increased activity. C101A and H256A abolished their activity. Whereas, D269A has slightly reduced kcat/Km, in which kcat is a little higher but Km is increased, which is consistent with its proposed role in aiding C101 and H256 during the catalysis. N99A abolished its enzymatic activity, suggesting its critical role in catalysis. F83 has slightly increased activity, where Km is about half of that of wild‐type protein with a slightly reduced kcat. D96A abolished the activity, highlighting its special role to promote the catalytic center. D98A has greatly reduced activity, in which Km is 19.14 μM but kcat is 0.0928 min−1 and kcat/km is about 26.09% of that of the wild type PLpro. For W102A, the activity is about 35.87% of that of the wild‐type protein. For Y257A, Km is 16.65 μM and kcat is 0.2332 min−1, and kcat/Km is almost the same as the wild‐type protein due to the increased affinity for the substrate even though the catalytic activity is decreased.
Since the structure of SADS‐CoV PLP2 is similar to previously identified PLpro from SARS‐CoV or MERS‐CoV and TGEV PLP1, which have DUB activity, we used Ub‐AMC as the substrate to determine the effects of the mutants to investigate its DUB activity in detail. When Ub‐AMC is used as the substrate, kcat is 1.285 min−1, Km is 0.3416 μM, and kcat/Km is 3.76 μM−1 min−1, which shows that SADS‐CoV PLP2 cleaves the Ub‐AMC 35‐fold more efficiently than KAGG‐AMC and 203‐fold more efficiently than LRGG‐AMC, in which kcat for Ub‐AMC is a little higher than that for KAGG‐AMC, and 1.8‐fold of LRGG‐AMC, while Km for Ub‐AMC is just 3.9% of that for KAGG‐AMC and 0.91% of Km for LRGG‐AMC. This indicates that the regions outside binding to P1–P4 would increase the binding affinity of the enzyme to the substrate. The N49 Δ also had increased enzymatic activity, in which the Km does not change much whereas the kcat increased slightly (Table 2). C101A abolished the enzymatic activity, which confirmed its essential role in the catalysis. H256A and D269A retain only 33.25% and 38.83% of the enzymatic activity, respectively, which confirms their catalytic roles. F83A has slightly increased activity (1.25 fold), showing that the bulky side chain might block the optimal orientation of the substrate to some extent. D96A and D98A abolished the DUB activity, which confirmed the critical interaction with C101 for the catalysis and the regulatory role of the loop of amino acid residue 92–99, respectively (Figure 3c).
To investigate whether SADS‐CoV PLP2 could recognize and cleave ISG15‐AMC to show DeISG activity, we used available human ISG15‐AMC because the pig ISG15 is about 68% identical to hISG15 and also the extreme C‐terminal tail sequence of both of them is LRLRGG. SADS‐CoV PLP2 could cleave hISG15‐AMC with kcat of 4.281 min−1 and Km of 0.4602 μM. The kcat/Km is 6.3 μM− 1 min−1, showing that the catalytic efficiency is about 1.68 fold of Ub‐AMC as the substrate and also much higher than that of Z‐KAGG‐AMC or Z‐LRGG‐AMC. It demonstrates that SADS‐CoV PLP2 has better cleavage efficiency for ISG15 than ubiquitin. N49Δ also has slightly increased activity (Table 2). C101A totally lost the activity, suggesting its essential role in catalysis. H256A has 61.90% activity, suggesting its aiding Cys101 during catalysis. These results show that SADS‐CoV PLP2 may also have DeISGylating activity and the triad C‐H‐D might also be the catalytic center.
In summary, the results of kinetic studies of these mutants are consistent with the critical roles of the active site and other residues of SADS‐CoV PLP2 predicted in ubiquitin binding model and the proposed active site. Furthermore, the significant differences in the catalytic efficiency of SADS‐CoV PLP2 to hydrolyze tetrapeptide‐AMC (KAGG‐AMC or LRGG‐AMC) and Ub‐AMC or ISG15‐AMC suggest that there are additional and important interactions between PLP2 and Ub or ISG15 outside of the enzyme's catalytic center, which needs further research.
3. DISCUSSION
In this study, we demonstrate that SADS‐CoV PLP2 could cleave nsp1‐eGFP and also function as DUB and DeISGylating activity, as MERS‐CoV PLpro, SARS‐CoV PLpro and TGEV PLP1, even though the sequence identity of SADS‐CoV PLP2 to them is low and there are some structural differences among them. The crystal structure shows that the overall structure is similar to the structures of PLpro from SARS‐ or MERS‐CoVs and TGEV PLP1. The active site and important residues for catalysis are also highly conserved in the primary sequence and three‐dimensional structure, showing that they might share similar molecular mechanism for catalysis. Based on the structural similarity to SARS‐CoV PLpro and the biochemical data, SADS‐CoV PLP2 functions through a proposed cysteine protease catalytic cycle where Cys101 acts as nucleophile, His256 functions as a general acid–base, and Asp269 is paired with the histidine helping to align it and promote the deprotonation of Cys101. The catalytic cysteine (Cys101) is situated at the foot (N‐terminus) of α‐helix3 in the thumb domain, as the helix dipole could contribute to the microenvironment that stabilizes the thiolate of Cys during catalysis and this structural feature may also contribute to the nucleophilicity of Cys101.14, 29 The side chain sulfur atom of Cys101 is positioned 3.21 Å from the side‐chain nitrogen atom of the catalytic‐triad histidine (His256), which is located at the foot of the palm domain, and adjacent to the flexible loop BL2. One of the oxygen atoms of the side chain of catalytic aspartic acid (Asp269) is located 2.75 Å from the side‐chain nitrogen of the catalytic histidine at the foot of the palm domain (Figure 2e).
Analysis of the SADS‐CoV PLP2 structure reveals that the oxyanion hole contains an N99 in the corresponding positions (Figure 2d). N99, which is highly conserved among coronavirus PLPs, is situated above the catalytic cysteine. N99A abolished the enzymatic activity, implicating its crucial contribution to oxyanion hole stabilization and thus catalytic function, as the side‐chain amine group of N99 might form H‐bond with the negatively charged intermediate's oxyanion. D96A abolished the enzymatic activity for all of the four substrates used in this study, while the side chain of D96 is located very closely to the C101. And the unique interaction between D96 with the catalytic cysteine might account for the differences of the substrate specificity or the catalytic efficiency. But SADS‐CoV PLP2 oxyanion hole may not be complete compared to SARS‐CoV PLpro, as Y113 in SARS‐CoV PLpro is replaced to W102 in SADS‐CoV PLP2 and W107 is replaced to D96.
A series of loops surrounding the area dictate access to the narrow active site. One such loop comprised of amino acid residues 92–99 is situated at the mouth of the active site. W102 that is close to C101 protrudes over this loop into the active site, consistent with its proposed role to stabilize the oxyanion hole or the substrate (Figures 2d and 3c). An interaction between D98 of the loop with F83 behind the loop is likely to anchor this loop into the active site. F83A mutant is inactive in enzymatic assays (Table 2), indicating that the interaction of F83 with D98 is very likely important for preventing it from moving to block the active site access (Figure 3b). And the equivalent residues of SARS‐CoV PLPro are W94 and D109, which formed the hydrogen bond essential for the catalytic activity.23, 29
Based on the consensus sequence of PLP2 for the three cleavage sites in the SADS‐CoV polyprotein and biochemical studies addressing the substrate preference, it is proposed that a KXGG motif at the P4‐P1 positions of the substrate is essential for the recognition and cleavage and also could explain why SADS‐CoV PLP2 could cleave LRGG‐AMC which is both the consensus sequences of SARS‐CoV nsp1‐4 cleavage sequence and the C‐terminal sequences of ubiquitin and ISG15, even though with a lower kcat/Km, compared with KAGG‐AMC.
The kcat/Km of PLpro from SARS‐ or MERS‐CoVs and TGEV PLP1 for LRGG‐AMC are 4.41 ± 0.50, 0.05 ± 0.01, and 0.67 ± 0.11 S−1 mM−1, respectively;7, 21 while that of SADS‐CoV PLP2 is 3.07 × 10−7 ± 5 × 10−8 S−1 mM−1, which is much lower than that of SARS‐ or MERS‐CoV PLpro and TGEV PLP1. Differences in the cleaving ability of Ub‐AMC by SARS‐ or MERS‐CoV PLpros, TGEV PLP1 and SADS PLp2 may be due to the sequence and structural differences, which affect binding affinities for ubiquitin. The potential of the PLPs to cleave the peptide bond after LXGG has been shown to be dependent on residues flanking the cleavage sites.5, 29 The nature of P3 and P4 amino acids of the peptide substrates KAGG‐AMC and LRGG‐AMC cleaved by SADS‐CoV PLP2 is significantly different; especially, the amino acid occupying the P4 position that is bound in the active site during catalysis is very different in both substrates, lysine or leucine. Despite this, SADS‐CoV PLP2 can cleavage both substrates, conceivably with different efficiencies. Thus, the active site pocket offers promiscuity but at the cost of catalytic efficiency, in which LRGG‐AMC was cleaved less efficiently than KAGG‐AMC.
Based on the structural and biochemical analysis, it is reasonable that KAGG is the preferred substrate even though it could also accommodate LRGG. For the P4 site, the surrounding region of SADS‐CoV PLP2 has a slightly negative charged patch, which would interact with lysine as a positive charged residue, while leucine is a kind of hydrophobic residue. And this would explain why SADS‐CoV PLP2 has a higher catalytic efficiency of KAGG‐AMC over LRGG‐AMC. Furthermore, the model of PLP2 in complex with ubiquitin supported by the mutagenesis studies provides insights into the molecular interactions between the enzyme and substrate. The extensive interactions predicted between PLP2 and Ub beyond the extreme C‐terminal sequence (LRLRGG) may account for the significantly higher activity of PLP2 with Ub‐AMC than with the Z‐LRGG or Z‐KAGG tetrapeptide. So the other residues flanking the active site may have potential changes upon the substrate binding that influence the intrinsic catalytic activity of PLPs, which would be validated through the determination of the structure of the enzyme‐substrate complex.
The N49Δ against all the substrates tested had slightly increased cleavage activity, indicating that the Ubl may not be essential for the activity. It has been reported that the Ubl domains of MERS‐CoV or SARS‐CoV PLpro are dispensable for their ubiquitin chain recognition and processing activities.12, 30 It needs further work to investigate whether the Ubl of PLPs may instead be a unique hub for protein– protein interactions between the replicase components.
The differences between the sequence and the structure, especially the spatial arrangement close to the active site could explain the enzymatic differences in substrate specificity and catalytic efficiencies observed for the PLPs from SADS‐CoV, SARS‐CoV and MERS CoV, and TGEV, which might also reflect the difference of the host cell factors, which needs further research. This may also offer the good basis for designing PLPs with directed shifts in substrate specificities.
Most viruses hijack the cellular ubiquitinating system, but some encode their own ubiquitin ligases and DUBs. 10 However, the identification of the natural substrates of viral enzymes remains an ongoing challenge. The SADS‐CoV PLP2 structure and enzymatic activities toward various substrates reported here will facilitate this quest. Here the enzymatic activities of PLP2 are explicitly identified in vitro investigation using purified recombinant proteins, so further research is needed to explore its function in cells and animals. The relatively low amino acid sequence identities among them suggest that there are unique mechanistic aspects for each enzyme from different coronaviruses. The differences in substrate recognition and cleavage efficiency between these closely related PLPs suggest that none of the enzymes can be used as a generalized model to explain the kinetic behaviors of all CoV PLPs.
In summary, the biochemical and structural characterization of SADS‐CoV PLP2 in this study would provide a powerful tool for understanding the molecular mechanism for the processing of viral polyprotein and also ubiquitin‐ or ISG15‐conjugated proteins, and also lays the foundation for the design and optimization of antivirals that block viral replication and pathogenesis of SADS‐CoV.
4. MATERIALS AND METHODS
4.1. Protein expression and purification
The cDNA of the SADS‐CoV PLP2 (1588–1880) was synthesized by the GenScript, and cloned into pET28a‐SUMO to create pET28a‐SUMO‐SADS‐PLP2, encoding for a N‐terminal (His)6‐SUMO tag. The pET28a‐SUMO‐SADS‐PLP2 plasmids were transformed into Escherichia coli BL21(DE3) and single colonies were inoculated into LB with Kanamycin (35 μg/ml) incubated at 37°C. When the optical density (600 nm) reached 0.6–0.8, cultures were cooled on ice before induction with 0.2 mM IPTG at 16°C for 20 hr. Cell pellets were resuspended in the buffer (20 mM phosphate buffer [pH 8.0], 0.3 M NaCl, 10 mM imidazole, 2 mM β‐mercaptoethanol, 0.5 U DNaseI, 0.5 mM PMSF), and lysed by ultra‐high pressure cell disruptor at 58 MPa for 5 min at 6°C. Then, the lysate was clarified by centrifugation (25,000g for 50 min at 4°C) and then the supernatants were loaded onto a Ni‐NTA column (Ni Sepharouse™ 6 Fast Flow, GE Healthcare) equilibrated with the lysis buffer. And the column was washed with the wash buffer (20 mM phosphate buffer [pH 8.0], 0.3 M NaCl, 30 mM imidazole). Finally, the column was eluted with the elution buffer (20 mM PB [pH 8.0], 0.3 M NaCl, 300 mM imidazole). The eluted proteins were concentrated and replaced with the digesting buffer which included 20 mM phosphate buffer (pH 8.0), 150 mM NaCl, 150 mM imidazole, and then ULP1 protease was added to the SUMO‐PLpro solution with the molar ratio of 1:200 to cleave off the sumo‐tag at 4°C and the SUMO‐tag was removed by loading the sample onto the Ni‐NTA column. Then, the protein was applied onto by the Hiload™ 16/600 superdex™ 75 (GE Healthcare) with the gel‐filtration buffer (50 mM Tris–HCl [pH 8.0], 50 mM NaCl, 2 mM β‐mercaptoethanol). The pure PLP2 sample was flash‐frozen with liquid nitrogen and then stored at −100°C.
4.2. Site‐directed mutagenesis
The primers were designed using Snapgene program. Mutagenesis reactions were performed using Phanta max super‐Fidelity DNA polymerase (Vazyme). After the reaction was completed, it was incubated with 1 μl of DpnI (Monad) at 37°C for 1 hr. The digested mutagenesis product was then transformed into competent bacterial cells. All mutations were verified by sequencing. The expression constructs were transformed into BL21(DE3).
4.3. Expression and purification of the fusion protein nsp1‐eGFP and cleavage assay
For the preparation of recombinant (His)6‐tagged nsp1‐eGFP fusion protein, the nsp1‐eGFP fusion gene was generated by overlapping PCR and cloned into pET30a with NdeI and XhoI restriction sites. (His)6‐nsp1‐eGFP protein was expressed in E. coli BL21(DE3) and purified with Ni‐NTA column and then with the Hiload™ 16/600 Superdex™ 75 (GE Healthcare). The fusion protein processing was performed at 50 mM HEPES (pH 7.5), 150 mM NaCl. In the 100 μl reaction solution, including 3 μM substrate and 1 μM enzymes, the mix solution was reacted for various times at 23°C then analyzed by SDS‐PAGE.
4.4. Ubiquitin‐, ISG15‐AMC, and peptide‐AMC kinetics
The enzyme catalyzed reaction rate of the SADS‐CoV PLp2 was determined using four different fluorescent substrate with the 7‐amino‐4‐methylcoumarin (AMC) to determine apparent kcat/Km for SADS‐CoV PLP2 and its mutants, including Ub‐AMC or ISG15‐AMC (ENZO), Z‐KAGG‐AMC, or Z‐LRGG‐AMC (GL Biochem Ltd, Shanghai, People's Republic of China), whose hydrolysis cleaves at Gly‐AMC bound and to release free AMC, which can be monitored with the increase of fluorescence signal of the AMC in excitation wavelength at 360 nm and emission wavelength at 460 nm, while the amount of the AMC released can be determined using a stand curve with the analytical‐grade AMC. The reaction was incubated at 24 ± 1°C, and the fluorescence signal was monitored by the SYNERGY™ H1 Hybrid Multi‐Mode Microplate Reader. For the initial reaction rate, the fluorescence increase per minute (AFU/min), was replaced by the amount of AMC released (μM/min). The enzyme activity assay was performed at 50 mM HEPES (pH 7.5), 150 mM NaCl in black, flat‐bottom 96‐well plates (Corning). For Z‐LRGG‐AMC, the enzyme reaction concentration was 25 nM, and the substrate work concentrations were from 1.25 to 50 μM. For Z‐KAGG‐AMC assay, the work concentration of the PLpro (WT) was 25 nM and all the mutants at 50 nM, the final concentrations of the substrate were from 1.25 to 50 μM. For Ub‐AMC or ISG15‐AMC, the enzyme concentration was 1 nM, and the work concentrations of the substrate were 0.1 to 1 μM. The steady‐state enzyme‐kinetic parameters were determined by fitting the initial velocity (μM/min) to the Michaelis–Menten equation based on a free AMC standard curve. The program GraphPad Prism6 was used for the data analysis. Experiments were performed in triplicate and error bars indicate SEM.
4.5. Crystallization and data collection
Crystals of SADS PLp2 were grown using a reservoir solution of 0.1 M MES (pH 6.5), 0.1 M CaCl2, 16% PEG20000 at 16°C for 2 weeks by the sitting‐drop vapor‐diffusion technique. The crystal was fished out and transferred to cryo‐protection solution (reservoir solution supplemented with 20% ethylene glycol). X‐ray diffraction data were collected at 100 K on the beamline 17U or 19U1 at the National Synchrotron Radiation Research Center, China (λ = 0.9206 or 0.9709 Å). Data were processed using XDS 32 and AIMLESS as part of the CCP4 suite. 33
4.6. Structure determination and refinement
The phase was solved by using SAD with Bromide using 0.9206 Å wavelength as the peak data. Then, the initial model was produced by Autosol in PHENIX package. 34 And then the model was used with molecular replacement for the native structure. Model building was carried out by Coot. 35 The refinement of the structure was done with PHENIX suite. Finally, stereochemical quality and final validation of the model were performed using MolProbity. 36 The final statistics of data collection and structural refinement are shown in Table 1. Figures were prepared in PyMOL (http://www.pymol.org/). 37 Atomic coordinates and experimental structure factors for SADS‐PLpro were deposited in the PDB with accession code 6L5T. Protein sequences were retrieved from the NCBI GenPept database. Multiple sequence alignments were conducted with ClustalW (http://npasa-pbil.ibpc.fr) 38 with default settings and alignment outputs were generated with ESPript v.2.2 (http://espript.ibcp.fr). 39
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
chengpeng fan: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; resources; supervision; validation; visualization; writing‐original draft; writing‐review and editing. Lu Wang: Data curation; methodology; validation. Weihua Hu: Data curation; methodology.
Supporting information
Figure S1. Enzymatic activity of SADS‐CoV PLP2 mutants.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant #: 31570762 to F. C. P.). The contents of this paper are only the responsibility of the authors and do not necessarily represent the official view of the National Natural Science Foundation of China. The diffraction data were collected at the beamline BL17U and BL19U1 of Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, People's Republic of China.
Wang L, Hu W, Fan C. Structural and biochemical characterization of SADS‐CoV papain‐like protease 2. Protein Science. 2020;29:1228–1241. 10.1002/pro.3857
Funding information National Natural Science Foundation of China, Grant/Award Number: 31570762
REFERENCES
- 1. Pan Y, Tian X, Qin P, et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet Microbiol. 2017;211:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P, Fan H, Lan T, et al. Fatal swine acute diarrhoea syndrome caused by an hku2‐related coronavirus of bat origin. Nature. 2018;556:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gong L, Li J, Zhou Q, et al. A new bat‐HKU2‐like coronavirus in swine, China, 2017. Emerg Infect Dis. 2017;23:1607–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anand K, Palm GJ, Mesters JR, Siddell SG, Ziebuhr J, Hilgenfeld R. Structure of coronavirus main proteinase reveals combination of a chymotrypsin fold with an extra alpha‐helical domain. EMBO J. 2002;21:3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Harcourt BH, Jukneliene D, Kanjanahaluethai A, et al. Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain‐like protease activity. J Virol. 2004;78:13600–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin MH, Chuang SJ, Chen CC, et al. Structural and functional characterization of MERS coronavirus papain‐like protease. J Biomed Sci. 2014;21:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kong L, Shaw N, Yan L, Lou Z, Rao Z. Structural view and substrate specificity of papain‐like protease from avian infectious bronchitis virus. J Biol Chem. 2015;290:7160–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lindner HA, Fotouhi‐Ardakani N, Lytvyn V, Lachance P, Sulea T, Menard R. The papain‐like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol. 2005;79:15199–15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clementz MA, Chen Z, Banach BS, et al. Deubiquitinating and interferon antagonism activities of coronavirus papain‐like proteases. J Virol. 2010;84:4619–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Isaacson MK, Ploegh HL. Ubiquitination, ubiquitin‐like modifiers, and deubiquitination in viral infection. Cell Host Microbe. 2009;5:559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Devaraj SG, Wang N, Chen Z, et al. Regulation of irf‐3‐dependent innate immunity by the papain‐like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282:32208–32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain‐like protease ubiquitin‐like domain and catalytic domain regulate antagonism of irf3 and nf‐kappab signaling. J Virol. 2009;83:6689–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lindner HA, Lytvyn V, Qi H, Lachance P, Ziomek E, Menard R. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain‐like protease. Arch Biochem Biophys. 2007;466:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ratia K, Saikatendu KS, Santarsiero BD, et al. Severe acute respiratory syndrome coronavirus papain‐like protease: Structure of a viral deubiquitinating enzyme. Proc Natl Acad Sci U S A. 2006;103:5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu M, Li P, Li M, et al. Crystal structure of a UBP‐family deubiquitinating enzyme in isolation and in complex with ubiquitin aldehyde. Cell. 2002;111:1041–1054. [DOI] [PubMed] [Google Scholar]
- 16. Hu M, Li P, Song L, et al. Structure and mechanisms of the proteasome‐associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ratia K, Pegan S, Takayama J, et al. A noncovalent class of papain‐like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc Natl Acad Sci U S A. 2008;105:16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Holm L. Benchmarking fold detection by dalilite v.5. Bioinformatics. 2019;35:5326–5327. [DOI] [PubMed] [Google Scholar]
- 19. Baez‐Santos YM, Barraza SJ, Wilson MW, et al. X‐ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain‐like proteases. J Med Chem. 2014;57:2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lei J, Mesters JR, Drosten C, Anemuller S, Ma Q, Hilgenfeld R. Crystal structure of the papain‐like protease of MERS coronavirus reveals unusual, potentially druggable active‐site features. Antiviral Res. 2014;109:72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wojdyla JA, Manolaridis I, van Kasteren PB, et al. Papain‐like protease 1 from transmissible gastroenteritis virus: Crystal structure and enzymatic activity toward viral and cellular substrates. J Virol. 2010;84:10063–10073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baez‐Santos YM, Mielech AM, Deng X, Baker S, Mesecar AD. Catalytic function and substrate specificity of the papain‐like protease domain of nsp3 from the middle east respiratory syndrome coronavirus. J Virol. 2014;88:12511–12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. The papain‐like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J Virol. 2005;79:15189–15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ratia K, Kilianski A, Baez‐Santos YM, Baker SC, Mesecar A. Structural basis for the ubiquitin‐linkage specificity and deisgylating activity of SARS‐CoV papain‐like protease. PLoS Pathog. 2014;10:e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chou CY, Lai HY, Chen HY, Cheng SC, Cheng KW, Chou YW. Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain‐like protease. Acta Crystallogr. 2014;D70:572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bailey‐Elkin BA, Knaap RC, Johnson GG, et al. Crystal structure of the middle east respiratory syndrome coronavirus (MERS‐CoV) papain‐like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J Biol Chem. 2014;289:34667–34682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnston SC, Larsen CN, Cook WJ, Wilkinson KD, Hill CP. Crystal structure of a deubiquitinating enzyme (human UCH‐l3) at 1.8 a resolution. EMBO J. 1997;16:3787–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee H, Lei H, Santarsiero BD, et al. Inhibitor recognition specificity of MERS‐CoV papain‐like protease may differ from that of SARS‐CoV. ACS Chem Biol. 2015;10:1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baez‐Santos YM, St John SE, Mesecar AD. The SARS‐coronavirus papain‐like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clasman JR, Baez‐Santos YM, Mettelman RC, O'Brien A, Baker SC, Mesecar AD. X‐ray structure and enzymatic activity profile of a core papain‐like protease of MERS coronavirus with utility for structure‐based drug design. Sci Rep. 2017;7:40292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaudhuri R, Tang S, Zhao G, Lu H, Case DA, Johnson ME. Comparison of SARS and NL63 papain‐like protease binding sites and binding site dynamics: Inhibitor design implications. J Mol Biol. 2011;414:272–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kabsch W. XDS. Acta Crystallogr. 2010;D66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the ccp4 suite and current developments. Acta Crystallogr. 2011;D67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams PD, Afonine PV, Bunkoczi G, et al. Phenix: A comprehensive python‐based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Emsley P, Cowtan K. Coot: Model‐building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 36. Davis IW, Leaver‐Fay A, Chen VB, et al. Molprobity: All‐atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. DeLano WJ. The pymol molecular graphics system. New York: Schrödinger, LCC, 2002. [Google Scholar]
- 38. Larkin MA, Blackshields G, Brown NP, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 39. Robert X, Gouet P. Deciphering key features in protein structures with the new endscript server. Nucleic Acids Res. 2014;42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Enzymatic activity of SADS‐CoV PLP2 mutants.