

Abstract
Mitogen‐activated protein (MAP) kinase signaling is central to multiple cellular responses and processes. MAP kinase p38α is the best characterized member of the p38 MAP kinase family. Upstream factors and downstream targets of p38α have been identified in the past by conventional methods such as coimmunoprecipitation. However, a complete picture of its interaction partners and substrates in cells is lacking. Here, we employ a proximity‐dependent labeling approach using biotinylation tagging to map the interactome of p38α in cultured 293T cells. Fusing the advanced biotin ligase BioID2 to the N‐terminus of p38α, we used mass spectrometry to identify 37 biotin‐labeled proteins that putatively interact with p38α. Gene ontology analysis confirms known upstream and downstream factors in the p38 MAP kinase cascade (e.g., MKK3, MAPKAPK2, TAB2, and c‐jun). We furthermore identify a cluster of zinc finger (ZnF) domain‐containing proteins that is significantly enriched among proximity‐labeled interactors and is involved in gene transcription and DNA damage response. Fluorescence imaging and coimmunoprecipitation with overexpressed p38α in cells supports an interaction of p38α with ZnF protein XPA, a key factor in the DNA damage response, that is promoted by UV irradiation. These results define an extensive network of interactions of p38α in cells and new direct molecular targets of MAP kinase p38α in gene regulation and the DNA damage response.
Keywords: BioID, biotinylation, interactome, MAP kinase, p38, p38α, protein–protein interaction, proximity‐dependent labeling
1. INTRODUCTION
Mitogen‐activated protein (MAP) kinases are proline‐directed serine/threonine kinases involved in signaling networks within multiple cell types and regulation of diverse cellular processes, including cell migration and proliferation, gene transcription and translation of messenger RNA.1, 2 There are four p38 MAP kinases encoded in the mammalian genome, p38α, p38β, p38γ, and p38δ, which share high sequence similarity, yet differ in their expression pattern.3, 4, 5 The best characterized member, p38α (MAPK14), is ubiquitously expressed and was initially described downstream of inflammatory stimuli in macrophages. 6 Various stimuli activate p38α, including UV irradiation, oxidative, and osmotic stress.7, 8, 9, 10 Activation of p38α follows either a conventional three‐tiered mechanism resulting in dual phosphorylation within the activation loop of p38α11, 12 or direct activation.13, 14 Molecular scaffolding proteins facilitate the interactions of upstream activation partners including MAP kinase kinase kinases (MAP3Ks) (e.g., TAK1, ASK1, MLK3), MAP2Ks MKK3 and MKK6 and p38α itself. 4 The nonconventional direct activation of p38α is mediated by protein–protein interaction with TGF‐β‐activated kinase 1‐binding protein 2 (TAB2) followed by autophosphorylation.13, 15 Thus, distinct interactions of p38α with different protein partners are critical for activation mechanisms of p38α.
Activation of p38α cross‐regulates the c‐Jun N‐terminal kinase (JNK) MAP kinase pathway. Activity of p38α inhibits the activation and physiologic function of JNK in cultured cells and in multiple cell types in vivo.16, 17 UV‐stimulated p38α activation induces transcription of MKP1/DUSP1 phosphatase, to inhibit JNK activity and prevent apoptosis.18, 19 More indirect pathways of p38‐regulated JNK antagonism are through GRAP2 and HPK1 proteins, 3 suggesting additional direct or indirect interactions of molecular components within the MAPK cascades. 20
Under physiological conditions, p38α localizes to both the cytoplasm and the nucleus in most cell types studied.21, 22, 23 There are several cytoplasmic substrates of p38α, interactions of which are often transient in nature. There are, however, more stable complexes of p38α with cytosolic targets such as MAPKAPK2 (MK2).24, 25 Nuclear substrates of p38α include transcription factors such as AP1 family members ATF2, 26 c‐Fos, 27 MEF2A/C, 28 or STAT1. 29 Other nuclear targets include transcriptional co‐regulator PGC‐1α 30 and heterogeneous ribonucleoprotein particle A1 (hnRNP‐A1), a cofactor in mRNA splicing. 31 Whether p38α engages in complexes with these or other cytoplasmic or nuclear targets or whether these interactions are transient is not known. Previous studies relied on affinity purification of p38α to define complexes with interaction partners.32, 33 These traditional methods of studying p38α interactions were limited to stable complexes, or relatively high affinity, rather than functionally important, though likely more dynamic, transient interactions with substrates. Thus, a large proportion of the p38α interactome may not be accurately defined or even unknown.
The regulation and function of p38α in cellular responses such as gene transcription, cell cycle regulation, and stress response are linked to interactions with other proteins. 3 Thus, a more detailed and extensive coverage of the p38α interactome would result in a better understanding of processes involving this MAP kinase. We aimed to identify new interactions of p38α using proximity‐dependent labeling (PDL) with biotin. Biotinylation tagging uses bait proteins fused to the modified promiscuous bacterial biotin ligase BioID2 to label proteins in close proximity on lysine side chains with a biotin moiety. 34 The resulting BioID2‐p38α interactome in cultured cells confirmed interactions with upstream components of the classical and alternative activation pathway as well as of downstream substrates of p38α. In addition, components of the JNK cascade were identified. Furthermore, a cluster of Zn‐finger (ZnF) proteins, which was significantly enriched, suggests new targets of p38α in gene transcription and the UV‐induced DNA damage response. Thus, we present a new molecular tool to study both stable and transient interactions of protein kinases that may lead to novel functional insights into signal transduction.
2. RESULTS
2.1. Biotinylation proximity labeling of the p38α interactome in cultured cells
Interactions of p38α with other proteins have thus far been identified by traditional methods, such as coimmunoprecipitation or pull‐down approaches that are limited by antibody affinity or lysis and solubility, respectively.32, 33 To gain insight into p38α interactions in situ in living cells, we employed proximity‐dependent biotinylation tagging. 35 We fused wild‐type p38α to the advanced promiscuous biotin ligase BioID2 in a construct for expression in 293T cells, a standard human cell line used for protein biochemistry and cell biology (Figure 1a). A construct for expression of BioID2 alone served as a control construct (Figure 1a). Upon transfection into 293T cells, expression of either BioID2‐p38α or BioID2 was readily detectable by immunoblot (Figure 1b). Treating cells with growth medium supplemented with additional biotin (200 μM) resulted in a strong pattern of biotinylated proteins of a wide range of molecular weights as detected by probing Western blots with streptavidin (SA; Figure 1b). Biotinylated protein signal intensities correlated with expression levels of BioID2 and BioID2‐p38α fusion protein. No biotinylated proteins were detectable in lysates from cells expressing enhanced green fluorescence protein (eGFP) (Figure 1b). These results showed that BioID2 expression with and without in‐frame fusion to p38α results in abundant biotinylation of proteins in cultured HEK 293T cells.
Figure 1.
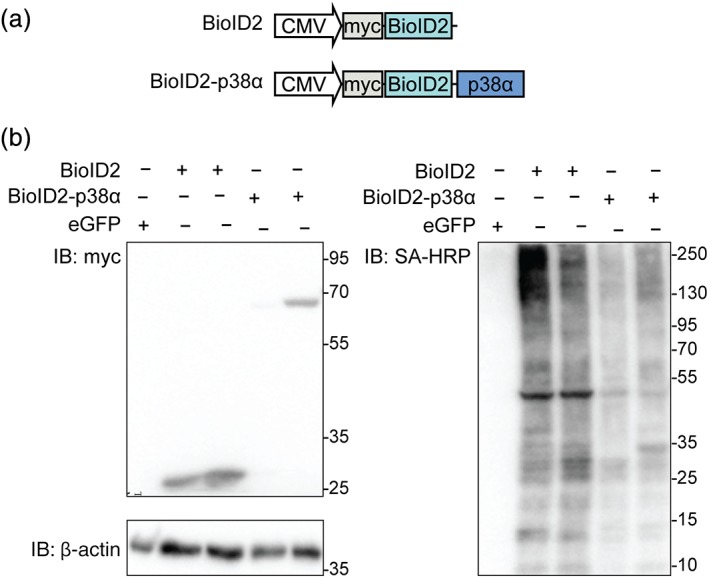
BioID2‐p38α fusion protein for proximity labeling in cultured cells. (a) Schematic of expression constructs for myc‐BioID2 control and myc‐BioID2‐p38α. (b) Immunoblots of 293T cells expressing either BioID2 (n = 2), BioID2‐p38α (n = 2), or enhanced green fluorescence protein (eGFP) (n = 1) and incubated with biotin (0.2 mM) for 24 hr. Protein expression was detected by immunoblot probed for myc tag or β‐actin as loading control. Biotinylated polypeptides were detected by probing with streptavidin–horseradish peroxidase (HRP) conjugate
We next aimed to address the identity of biotinylated proteins to establish a list of interaction partners of p38α in HEK 293T cells. Lysates from proliferating HEK 293T cells at subconfluency and transfected with constructs expressing either eGFP, myc‐BioID2, or myc‐BioID2‐p38α were subjected to methanol extraction of proteins, tryptic digest, and neutravidin‐mediated enrichment of biotinylated peptides (Figure 2a). Enriched peptides were buffer exchanged for detection by tandem MS–MS (Figure 2a). Mass spectrometry (MS) in combination with Mascot LCMSMS searching (Swissprot Database human proteome) identified 4,723 individual biotinylated peptides that mapped to 1,289 known polypeptides/proteins with high confidence (peptide false discovery rate [FDR] = 1.3%, protein FDR = 0.5%) (Tables 1, S1, and S2). No biotinylated peptides were detected in samples from eGFP‐expressing cells (Table 1). Comparing the degree of overlap in the proteins identified between myc‐BioID2 and myc‐BioID2‐p38α samples (479 of 1,290 total biotinylated proteins overlap; ∼37% of total proteins; ∼84% of proteins in myc‐BioID2‐p38α) suggested a substantial background of biotinylation in this experimental setting (Figure 2b, Table S3). An initial comparison of interactome profiles showed a significant between‐group difference in the abundance of each biotinylated protein as measured by median spectral counts (SC; Figure 2c). Rigorous background subtraction resulted in lists of proteins exclusive to either myc‐BioID2 or myc‐BioID2‐p38α (Tables 1, [Link], [Link], and S6). Analysis for gene ontology of the common background proteins labeled in both experimental groups in HEK293T cells identified mainly nuclear proteins and factors involved in RNA metabolism (Table S4). This suggests that BioID2 biotinylates several nuclear proteins independently of fusion to a bait protein in proliferating HEK293T cells under the experimental conditions used. A list of biotinylated proteins overrepresented in myc‐BioID2‐p38α‐expressing samples were used for further analysis (Table S5). Gene ontology profiling of biotinylated proteins detected above threshold when comparing myc‐BioID2‐p38α‐expressing samples and the background proteins common to both myc‐BioID2‐ and myc‐BioID2‐p38α‐expressing cells showed significantly different biological process enrichment, corroborating the robustness of our background subtraction method.
Figure 2.
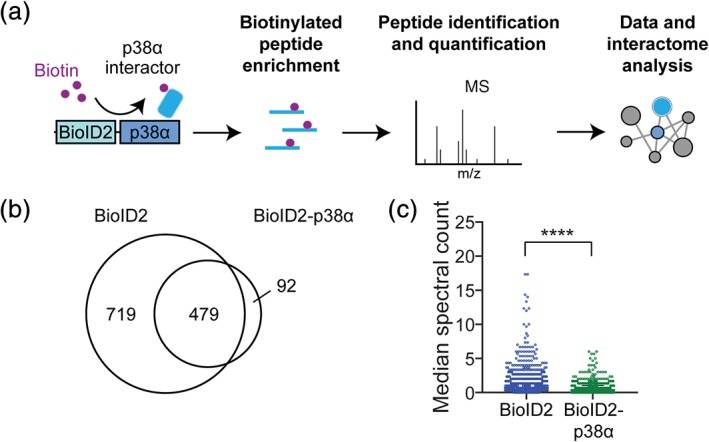
Quantification and evaluation of proteins biotinylated by BioID2 and BioID2‐p38α. (a) Experimental schematic of intracellular proximity‐dependent ligation with fusion protein BioID2‐p38α. Samples enriched for biotinylated peptides were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Nonredundant proteins were subjected to STRING network analysis for known protein–protein interactions and to DAVID gene ontology (GO) analysis. (b) Venn diagram of unique biotinylated proteins identified in the control BioID2 and/or BioID2‐p38α samples. (c) Quantification and comparison of median spectral count (SC) per biotinylated protein for samples from BioID2‐p38α‐ and BioID2‐expressing cells. Data are represented as median ± confidence interval of three independent experiments (n = 3) ****p < .0001 (Wilcoxon paired test)
Table 1.
Unique protein identities from neutravidin‐enriched samples and biotinylated nonredundant proteins detected by MS
| Sample | Biological replicates | Total biotinylated peptides detected | Unique biotinylated proteins | Average biotinylated peptides per protein | Interactome proteins (nonredundant) | Proportion nonredundant |
|---|---|---|---|---|---|---|
| eGFP | 1 | 1,157 total proteins identified by MS (no biotinylated peptide hits) | ||||
| BioID2 | 3 | 3,730 (±91) | 1,086 (±265) | 3.43 | NA | NA |
| BioID2‐p38α | 3 | 993 (±71) | 436 (±110) | 2.28 | 37 (±5) | 8.5% |
Abbreviations: eGFP, enhanced green fluorescence protein; MS, mass spectrometry.
Expression from plasmid may result in protein levels of BioID2‐p38α different from those of endogenous p38α, which may impact on the proximity labeling. We compared expression levels by immunoblot for p38α in BioID2‐p38α‐expressing cells, BioID2‐expressing, and GFP transfection control cells (Figure 3a). Expression of exogenous BioID2‐p38α was slightly, yet not significantly lower as compared to endogenous p38α in BioID2‐p38α‐expressing cells (Figure 3a,b). Levels of endogenous p38α were comparable across all conditions (Figure 3a,c). We addressed localization of p38α fused to BioID2 by immunofluorescence to compare it to endogenous p38α (Figure 3d). In BioID2 and GFP control cells, p38α showed punctate cytoplasmic signal of comparable intensities (Figure 3d). As expected, endogenous and exogenous p38α expression added up to higher signal intensity in BioID2‐p38α‐expressing cells, yet the localization appeared comparable to controls (Figure 3d). Hence, expression of BioID2‐p38α has no significant impact on levels and localization of p38α. Exogenous BioID2‐p38α is not markedly overexpressed in our experiments, adding less than existing endogenous expression.
Figure 3.
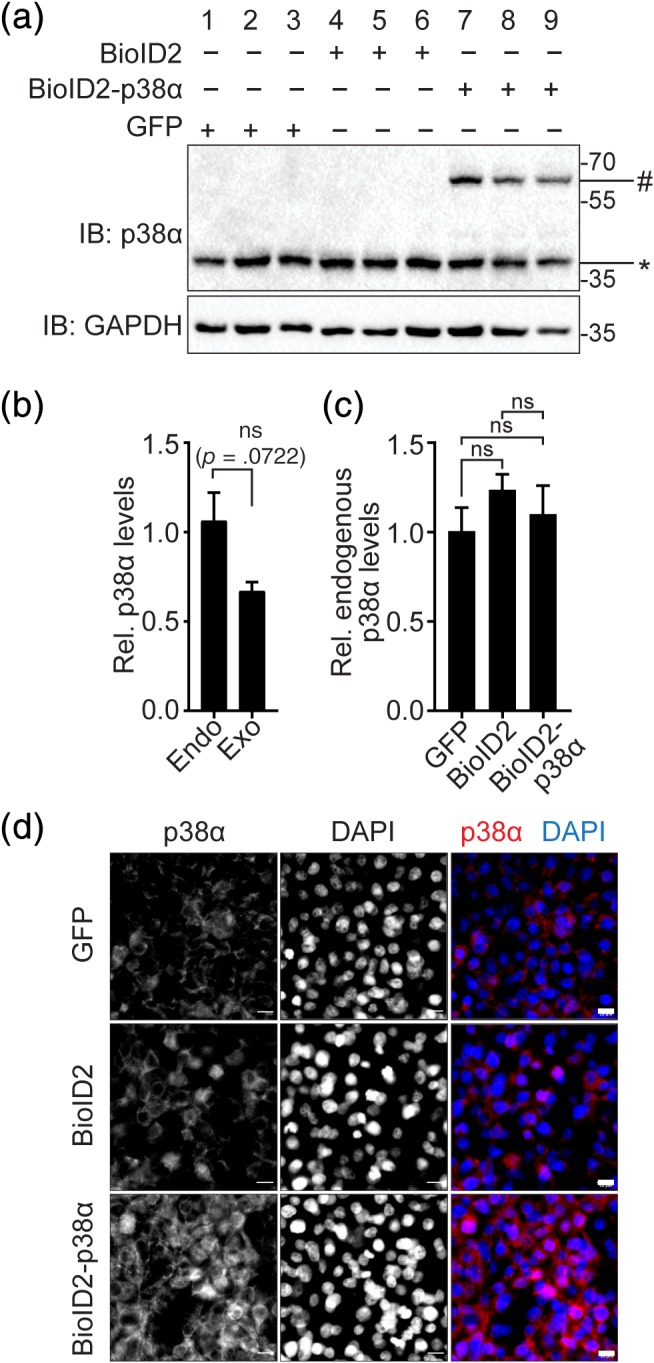
Comparison of expression of BioID2‐p38α and endogenous p38α in cultured cells. (a) Immunoblot of lysates from 293T cells expressing BioID2‐p38α or BioID2. Immunoblots were probed for p38α and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) as loading control. #, BioID2‐p38α (∼70 kD); *, endogenous p38α (38kD) (n = 3). (b) Quantification and comparison of endogenous (Endo) and exogenous (Exo; BioID‐p38α) p38α expression from immunoblots in (a) for Lanes 7–9 in cells expressing BioID‐p38α. Expression is expressed relative to GAPDH (n = 3). ns, not significant (Student's t test). (c) Quantification and comparison of endogenous p38α expression from immunoblots in (a) for Lanes 1–9. Expression is expressed relative to GAPDH (n = 3) ns, not significant (analysis of variance [ANOVA] with Bonferroni posttest). (d) Immunofluorescence with anti‐p38α in 293T cells transfected with mycBioID2‐p38α or BioID2. DAPI, nuclei. Scale bar, XYZ μm. Representative images of three independent experiments are shown
2.2. p38α interactome confirms the MAP kinase cascade and alternative activation by TAB2 in cultured cells
Biotinylated proteins overrepresented in myc‐BioID2‐p38α samples were mapped onto a network of interactions using the STRING database of known associations (Figure 4a, Tables S6 and S8). We added the putative interactions identified through our proximity‐labeling approach (Figure 4a). STRING search revealed a network surrounding p38α of 37 proteins (average local clustering coefficient = 0.456; protein–protein interaction enrichment p = 2.6 × 10−6), including two strongly interconnected clusters of proteins additional to a group of novel interaction partners without known interconnections (Figure 4a). Distribution of proteins by association to subcellular regions included 31 nuclear (FDR = 2.0 × 10−10) and 20 cytoplasmic (FDR = 5.0 × 10−3), with 14 proteins of both nuclear and cytoplasmic localization. Gene ontology analysis identified strong enrichment of MAP kinase (p = 1.19 × 10−6) and toll‐like receptor signaling pathways (p = 7.48 × 10−5). MAP kinase signaling‐associated proteins formed a large part of one of the two interconnected clusters and comprised of direct upstream factors of canonical p38 and JNK MAP kinase signaling MAP3K7, MAP4K4, MAP2K4, and additional MAP kinase MAPK3 (ERK1) (Figure 4b). Notably, this cluster contained TAB2, a direct binding partner and alternative activation factor of p38α 13 (Table 2). These results identify interactions of known regulators of MAP kinase activity connected with p38α in cultured cells.
Figure 4.
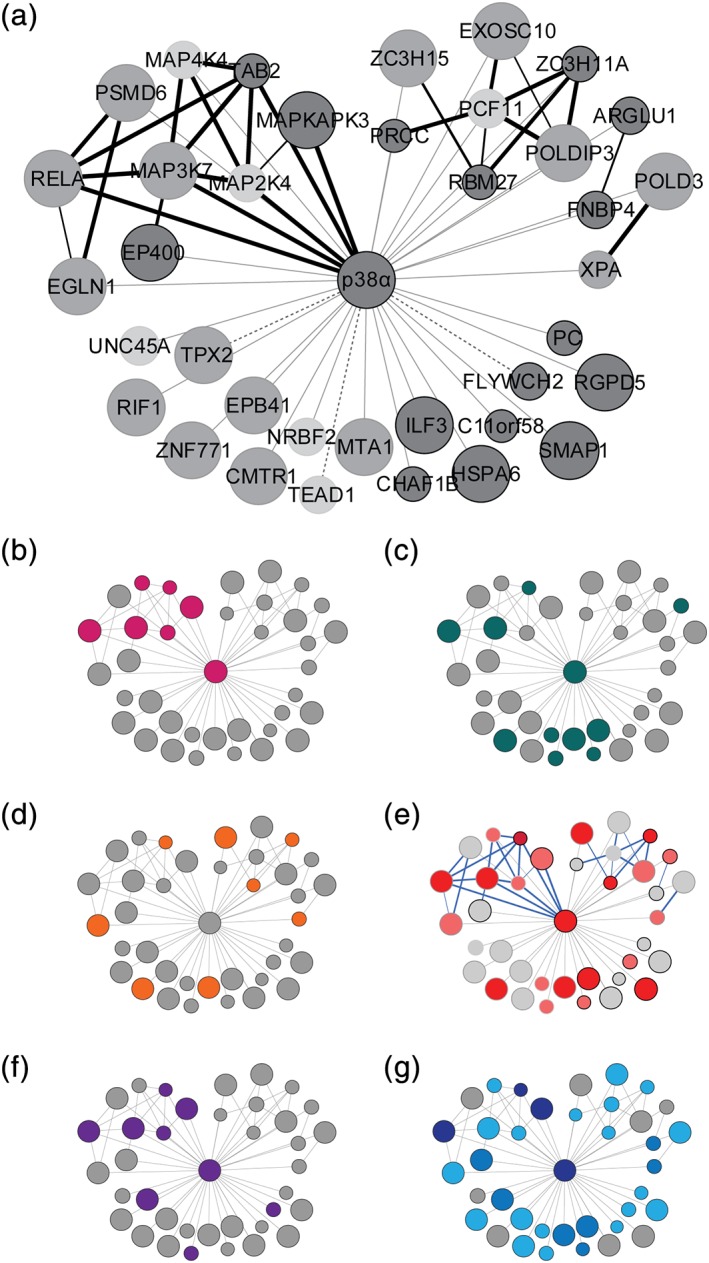
Biotinylation‐derived p38α interaction network and gene ontology in cultured cells. (a) Protein network interactome of BioID2‐p38α generated using STRING (v10.5). Nodes represent nonredundant biotinylated proteins from BioID2‐p38α samples. Associations annotated by STRING are blue nodes connected by red edges. Edge thickness, confidence of annotated association. Dashed edges, known interactions not registered in the STRING database. Node size, relative protein abundance. Node shading, criterion met for network inclusion: significant compared with BioID2 background (dark gray), exclusive in BioID2‐p38α with ≥2 SCs (gray), fold change over BioID2 background ≥3 (light gray). (b–d) Node annotation of highest scoring functional annotation clusters. (b) MAPK signaling: 22% total network coverage, p = 1.2 × 10−6. (c) Transcription regulation: 30% total network coverage, p = 1.7 × 10−3. (d) Zinc‐finger domain‐containing proteins: 22% total network coverage, p = 4.9 × 10−3 (Fisher's exact test). (e) Node overlap of highest scoring functional annotation clusters in all three (dark red), two clusters (red), or one cluster (pink). Blue edges, high confidence associations. (f) Network annotation of known p38α interaction partners: 21.6% total network coverage. (g) Network annotation of known (dark blue) and putative p38α phosphorylation targets. Putative targets were predicted by KEA2 (blue) and GPS3.0 (light blue) prediction. Network coverage: 10.8% known, 78% KEA2 + GPS predicted, 21.6% KEA2‐only predicted, 75.7% GPS‐only predicted
Table 2.
Functional annotation cluster analysis of unique protein identities from neutravidin‐treated samples and biotinylated nonredundant proteins detected by MS
| Biological process | Gene count | p‐Value | Genes | Fold change | FDR |
|---|---|---|---|---|---|
| Annotation cluster 1. Enrichment score: 2.33 | |||||
| MAPK signaling pathway | 8 | 1.2 × 10−6 | MAPK14, MAPK3, MAP2K4, MAP3K7, HSPA6, MAP4K4, RELA, TAB2 | 12.0 | 1.3 × 10−3 |
| Epstein–Barr virus infection | 6 | 8.4 × 10−6 | RELA, PSMD6, MAP2K4, MAP3K7, TAB2, MAPK14 | 18.8 | 9.1 × 10−3 |
| Toll‐like receptor pathway | 5 | 7.5 × 10−5 | RELA, MAP2K4, MAP3K7, TAB2, MAPK14 | 17.8 | 7.7 × 10−2 |
| Annotation cluster 2. Enrichment score: 1.17 | |||||
| Transcription regulation | 11 | 1.7 × 10−3 | MAPK14, ARGLU1, NRBF2, MTA1, TEAD1, ILF3, MAP3K7, CHAF1B, RELA, ZNF771, TAB2 | 2.8 | 1.7 |
| Transcription | 10 | 2.0 × 10−2 | CHAF1B, RELA, NRBF2, ARGLU1, ANF771, ILF3, MAP3K7, MAPK14, TEAD1, MTA1 | 2.3 | 2.0 |
| Annotation cluster 3. Enrichment score: 0.95 | |||||
| ZnF‐associated | 8 | 3.7 × 10−2 | XPA, ZNF771, TAB2, EGLN1, MTA1, RBM27, ZC3H11A, ZC3H15 | 2.4 | 2.4 |
Note: Top 3 DAVID DB annotation clusters (n = 37 proteins from three biological replicates). Fold change refers to enrichment in comparison to the whole human proteome.
Abbreviations: FDR, false discovery rate; MS, mass spectrometry; ZnF, zinc finger.
Analysis of the BioID2‐p38α interactome for the biological process ontology identified a set of 10 proteins associated with transcriptional regulation (Figure 4c; p = 1.7 × 10−3). The proteins encompassed transcription factors and transcriptional coregulators, including ARGLU1, metastasis‐associated gene 1 (MTA1), ILF3, TEA domain family member 1 (TEAD1), and overlapped partially with MAP kinase signaling factors (MAP3K7 and MAPK14 [p38α], p = 1.2 × 10−6; Table 2) and with JNK signaling components (MAP2K4, TAB2, and MAP3K7, p = 3.9 × 10−3; Table S7).
Analysis of the p38α interactome by protein domain ontology identified an enriched set of proteins containing zinc finger (ZnF) domains (Figure 4d; DAVID INTERPRO domain enrichment cluster, p = 4.9 × 10−3). ZnF domain‐containing proteins constituting this group were XPA, TAB2, ZNF771, MTA1, ZC3H11A, RBM27, EGLN1, and ZC3H15 (Table 2). ZnF domain proteins were distributed across interconnected and isolated nodes of the network, as annotated by STRING (Figure 4d). Overlap between functional cluster annotation is shown in Figure 4e. According to available literature and protein interaction database BioGRID, 18.9% of the network is comprised of previously validated p38α interaction partners (Figure 4f, Table S9). 10.8% are known p38α phosphorylation targets (Figure 4g). Based on kinase substrate prediction by KEA2 36 and GPS3.0, 37 21.6 or 75.7% of the biotinylated network proteins respectively, are putative p38α substrates (Figure 4g, Table S10). Taken together, our BioID2 results identify interactions of p38α in cultured cells with proteins of heterogeneous functions, including stress‐ and cytokine‐signal transduction, DNA damage response, and regulation of transcription and translation, thus explaining the pleiotropic role of p38α in cells.
3. p38α COLOCALIZES WITH XPA IN HEK 293T CELLS
Our biotinylation approach identified previously established as well as novel interactions with ZnF domain proteins. The ZnF domain protein XPA, which was found to be biotinylated protein when expressing BioID2‐p38α, is a component of the UV‐induced DNA damage response. 38 XPA is a target of MK2 downstream of p38α when cells are exposed to UV. 39 Our results indicate that p38α engages in a complex with XPA because we find biotinylated XPA peptides solely in cells expressing BioID2‐p38α (Table S6). We addressed whether the interaction of p38α with XPA in HEK293T cells identified by our biotinylation approach could be confirmed by immunofluorescence as an alternative method. We expressed HA‐tagged XPA in HEK293T cells at subconfluency either with or without FLAG‐tagged p38α. Expression of both XPA and p38α was confirmed by immunoblot (Figure 5a). We treated synchronized cells at subconfluency with UV light or mock irradiation (Figure 5b). We then fixed and immunostained cells with antibodies to both HA and FLAG tags to visualize overexpressed XPA and p38α, respectively, by epifluorescence (Figure 5b). Both FLAG‐p38α and HA‐XPA were detectable in cells at subconfluency and showed mainly cytoplasmic localization in mock‐treated cells. More cells showed nuclear FLAG‐p38α signal upon UV irradiation then upon mock irradiation. XPA translocated into the nucleus upon UV irradiation (Figure 5b). These results indicate a potential colocalization of FLAG‐p38α and HA‐XPA in 293T cells.
Figure 5.
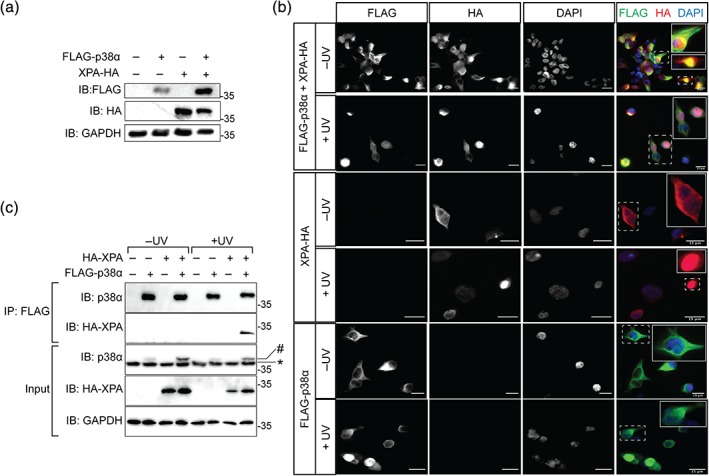
UV irradiation‐induced colocalization and protein complex between p38α and XPA in cultured cells. (a) Immunoblot of 293T cells expressing enhanced green fluorescence protein (eGFP) (n = 1), FLAG‐p38α (n = 1), XPA‐HA (n = 1), or FLAG‐p38α and XPA‐HA (n = 1). Protein expression was detected by immunoblotting using an antibody for FLAG, HA, or glyceraldehyde 3‐phosphate dehydrogenase (GAPDH). GAPDH signal confirmed equal loading. (b) Immunofluorescence of FLAG‐p38α and HA‐XPA in 293T cells transfected with constructs encoding either FLAG‐p38α, XPA‐HA, or both FLAG‐p38α and XPA‐HA with or without treatment with UV radiation (350 J/m2). Cells were labeled with antibodies for FLAG and HA, and with DAPI to visualize nuclei. Arrows, areas of colocalization of FLAG and HA signals. Scale bare, 15 μm. (c) Immunoprecipitation (IP) from lysates from cells expressing FLAG‐p38α and HA‐XPA 1 hr after mock or UV irradiation. Protein complexes were precipitated with anti‐FLAG antibody. Immunoblots (IB) were probed for p38α, HA tag, and GAPDH as loading control. Representative results for two independent experiments are shown
We addressed whether XPA and p38α can be found in a complex using coimmunoprecipitation. Lysates from UV‐ or mock‐treated cells expressing HA‐XPA and FLAG‐p38α were incubated with anti‐FLAG antibody and immunocomplexes were isolated and analyzed by immunoblot. HA‐XPA was detected together with immunoprecipitated FLAG‐p38α only upon UV treatment, yet not after mock treatment (Figure 5c). These results suggest that p38α can engage in a complex with XPA upon UV irradiation in cells.
XPA has previously been shown to enter the nucleus upon exposure of cells to UV light. 39 XPA interacts with proliferating cell nuclear antigen (PCNA) upon UV irradiation. 40 PCNA is critical for DNA replication and repair 41 and localizes to replication foci during DNA synthesis. 42 To address an interaction of p38α with both XPA and PCNA in cells upon UV irradiation, we performed proximity ligation assay (PLA) that results in fluorescence signal upon complex detection of two antigens with high sensitivity. 43 Cells expressing FLAG‐p38α and HA‐XPA showed PLA signal, which was markedly more intense upon UV irradiation (Figure 6a,b). Notably, nuclear PLA signal appeared when cells were exposed to UV, suggesting partial localization of XPA‐p38α complex to the nucleus (Figure 6a). Control conditions with single transfection showed no PLA signal (Figure 6a). These results support the presence of a UV‐induced interaction between p38α and XPA. We performed PLA for p38α and PCNA in cells that were transfected with FLAG‐p38α and either mock or UV irradiated. PLA signal was detectable in individual mock‐irradiated cells expressing FLAG‐p38α (Figure 6c). However, UV‐irradiated cells showed pronounced PLA for p38α in and surrounding nuclei (Figure 6c,d). Control cells that were not transfected with FLAG‐p38α showed minimal PLA signal upon UV irradiation (Figure 6c). In summary, these results support that p38α can colocalize with DNA repair factors XPA and PCNA upon UV irradiation.
Figure 6.
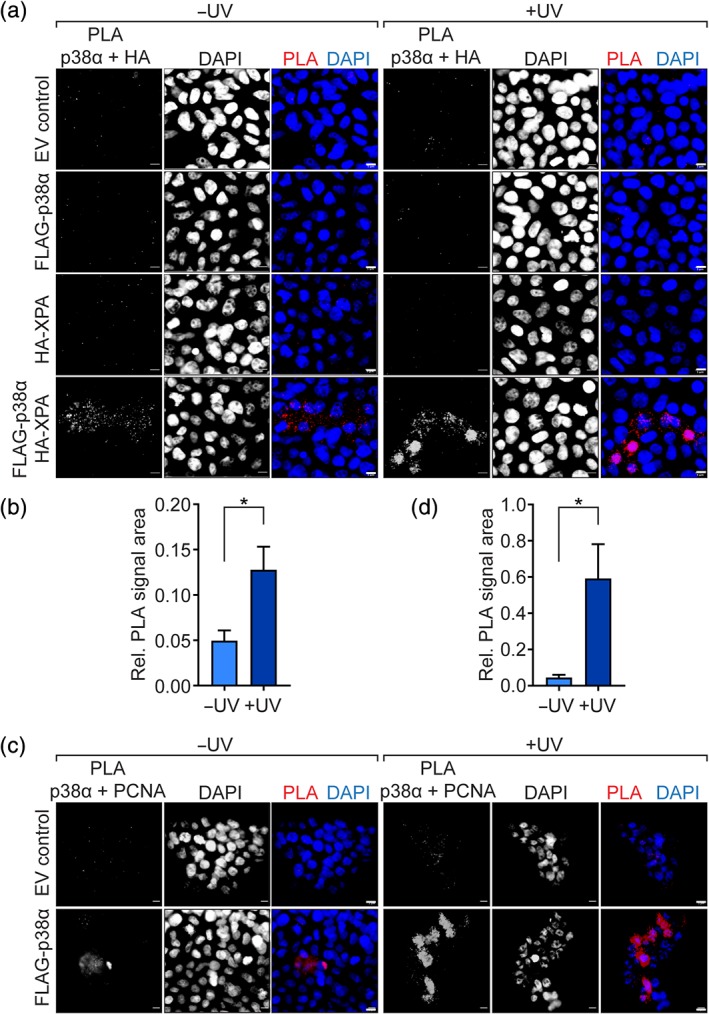
Proximity ligation assay (PLA) confirms interaction of p38α with XPA and proliferating cell nuclear antigen (PCNA) upon UV irradiation. (a) PLA signals for combination of anti‐HA and anti‐p38α in 293T cells expressing FLAG‐p38α or with empty vector (EV) transfection. Cells were UV irradiated (350 J/m2) or mock treated as indicated. DAPI, nuclei. Scale bar, 5 μm. (b) Quantification of HA‐p38α PLA signal in cells expressing FLAG‐p38α and HA‐XPA with UV or mock treatment (n = 64–211 cells per condition). * p < .05 (Student's t test). (c) PLA signals for combination of anti‐PCNA and anti‐p38α in 293T cells expressing FLAG‐p38α and/or HA‐XPA. EC control, EV control. Cells were UV irradiated (350 J/m2) or mock treated as indicated. DAPI, nuclei. Scale bar, 5 μm. (d) Quantification of PCNA‐p38α PLA signal in cells expressing FLAG‐p38α with UV or mock treatment (n = 29–35 cells per condition). * p < .05 (Student's t test)
Taken together, these results corroborate an interaction between the p38α MAP kinase cascade and the UV‐damage response factor XPA as suggested from our biotinylation proximity labeling with BioID2.
4. DISCUSSION
This study identified putative p38α binding partners using proximity labeling using the modified biotin ligase BioID2. Studies to date have identified interactors with affinities strong enough to withstand conventional copurification procedures,32, 33 potentially overrepresenting more stable interactions relative to transient ones. Proximity labeling using BioID2 circumvents this limitation by labeling a putative interacting protein within distinct spatial vicinity of the bait protein. In our experiments with BioID2‐p38α, this radius is approximately 3.5 nm surrounding p38α given previous experiments with BioID2 and the linker length between the domains of the BioID2‐p38α fusion protein.35, 44 The ability of BioID2 to label protein interactions as they occur, “on‐the‐go” as a manner of speaking, within living cells without the need for cell lysis and exchange of natural milieu surrounding protein complexes allowed us to detect interactions of lower affinity, not identified previously by other methods.35, 44 Covalent modification of lysine with biotin is not readily reversible in mammalian cells. 44 Thus, recovered biotinylated peptides reflect protein interactions even after they occurred and complexes have dissociated, conserving interaction events in biotinylated peptide signatures, which are, however, dependent on protein stability and detection limits of MS.34, 44
The GO profile of background control (BioID2) biotinylated proteins was comparable to that of the negative control (eGFP) HEK293T total proteome. Top annotated clusters from each condition had enriched pathways associated with cell adhesion molecules, translation, and mRNA processing (Table S4). We found a statistically significant between‐groups difference in biotinylation profiles (p < 1.0 × 10−4; Figure 2c). Biotinylation profiles were based on protein identities and their abundance (SC) in BioID2 and BioID2‐p38α samples as well as a rigorous background subtraction that eliminated proteins identified in both data sets. Differences between our experimental groups were therefore attributable to the presence of p38α in BioID2‐p38α samples. To quantify a relationship between biotin ligase constructs and intracellular proteins, we compared mean biotin‐modified lysine residues per protein identity (unique peptides assigned to one protein identity were disregarded). Biotin ligations per protein in BioID2 samples were ∼3.5 compared to ∼2.3 in BioID2‐p38α (Table 1). This suggests that soluble nonfusion protein BioID2 was potentially more efficient in biotinylating proximal proteins than BioID2 in a fusion protein with p38α, resulting in a larger number of unique biotinylated peptides identified in the BioID2 control samples (Table 1). This may reflect steric effects of bait proteins on BioID2 35 or the nature of the proximal milieu surrounding BioID2 when fused to a bait protein. 44 Marked overexpression of BioID2‐p38α compared to endogenous p38α levels could affect proximity biotinylation results. Based on our comparisons of endogenous p38α with expressed BioID2‐p38α was (Figure 3), biotinylation interactomes may likely be impacted by less than stoichiometric competition of BioID2‐p38α with endogenous p38α. Thus, biotinylation of interactions by BioID2‐p38α is not likely impacted by nonphysiologic abundance of BioID2‐p38α fusion protein. Future studies may wish to perform BioID experiments in genetic knockout backgrounds, however, keeping in mind that genetic ablation (e.g., in the case of p38α) may have impacts on cell division, physiology, and signaling networks.17, 45, 46
We used relative abundance thresholds in the bait condition to discriminate overrepresented proteins from background as done in previous studies of biotinylation labeling of protein interactions. 47 The resulting list was further restricted by eliminating overlap between BioID and BioID2‐p38α, leading to a set to 37 interactome proteins. Comparison with data from interaction database BioGRID confirmed a substantial overlap with known associations with p38α. The list contained upstream and downstream factors in p38α signaling (e.g., MAP2K4, MAPKAPK3) and included both stable and transient p38α interaction substrates.5, 32, 48 Prediction and annotation of known p38α phosphorylation substrates indicated a considerable potential of phosphotarget identification through our approach. Thus, our approach delineated entire p38α signaling pathways in cultured cells, confirming results obtain from conventional methods over the last 20 years as well as of other proteomic approaches.32, 33 Therefore, BioID2 is a fast and comprehensive way to describe functional pathways upstream and downstream of protein kinases in living cells.
BioID2‐p38α labeled well‐established factors associated with p38 MAP kinase signaling in HEK293T cells at subconfluent culture conditions (Figure 3a,b). Previously reported factors upstream of p38α confirmed in our approach were HGK (MAP4K4), TAK1 (MAP3K7), MAKK4 (MAP2K4), and TAB2, and downstream factors were MAPKAPK3 (MK3) and RelA, a component of the NFκ‐β transcription factor complex. 49 These proteins were part of a densely interconnected 10‐node cluster generated by STRING (Figure 3a) and confirm molecular events of (a) classical p38α activation through MAP kinase kinase MKK4 3 and (b) alternative p38α activation through interaction with TAB2 and subsequent autophosphorylation. 15 Our results may indicate that the TAB2‐dependent activation mechanism of p38α previously reported in lymphocytes and epithelial cells is also employed in cultured HEK293T cells.13, 15 Further investigation is required to elucidate which stimuli engage alternative p38α activation in HEK293T cells. p38α shares some overlap in upstream factors with the JNK signaling pathway (MKK4, TAK1) in this cell type. p38α was shown to inhibit JNK pathway activity in multiple cell types, including HEK293T cells.16, 17 Putative interactions of p38α with factors involved in JNK activation in our data suggests that part of this inhibitory role of p38α on JNK may be mediated through association of p38α with upstream factors of JNK. Consistent with this idea and our data, RNA interference and inhibitor experiments suggest that such a mechanism of MAP pathway cross‐regulation is mediated by impact of p38α on TAK1 and MKK4. 50
Gene ontology classification of the list of 37 p38α interactors from HEK293T cells showed a significant enrichment of transcription regulation, that included factors of p38α MAP kinase signaling factors including TAK1, TAB2, RELA, and p38α itself (p = 1.2 × 10−6; Figure 3c). This also identified transcription factor TEAD1 as a biotinylation target of p38α. TEAD1 is part of the Hippo pathway protein in response to osmotic stress and recently was shown to physically interact with p38α. 51 Notably, this known interaction was not registered in STRING database, showing that our approach can accurately find the most recent interactions identified.
The BioID2‐p38α interactome showed a significantly overrepresented cluster consisting of ZnF domain containing proteins according to DAVID INTERPRO domain ontology analysis (p = 4.9 × 10−3; Figure 3d). Though some ZnF domain proteins were present among background biotinylation, certain ZnF domain proteins were significantly higher in abundance across BioID2‐p38α samples as compared with BioID2 only controls (Tables 2 and S4). ZnF domains share common primary sequence motifs that facilitate zinc ion coordination, support a domain fold for protein–protein/protein–DNA interactions, and can undergo phosphorylation. 52
Among the ZnF domain proteins identified by BioID‐p38α was Xeroderma pigmentosum group A‐complementing protein (XPA), which has a role in the UV‐induced DNA damage response and mutations of which are associated with UV hypersensitivity and cancer.53, 54 A recent study delineated a pathway through p38α and MAPKAP kinase 2 (MK2) in the cellular response to UV damage that targets downstream phosphorylation of XPA. 39 MK2, rather than p38α, was shown to interact with XPA in this study. 39 Our results indicate that either biotinylation was possible in a complex of BioID‐p38α, MK2, and XPA or that a more direct, yet transient interaction between BioID‐p38α and XPA can occur in HEK293T cells. Interestingly, XPA likely engages in transient protein interactions during the DNA damage response. 55 Epifluorescence microscopy indicated colocalization of XPA and p38α when expressed in HEK293T cells (Figure 4c). Our results from fluorescence imaging, coimmunoprecipitation, and PLA imaging corroborate a UV‐induced physical association, whether direct or indirect, between p38α and XPA in HEK293T cells, consistent with a known role of this pathway in UV‐induced DNA damage control.39, 45, 46
We found ZnF protein transcription factor MTA1 biotinylated by BioID‐p38α (Figure 3c,d), suggesting that p38α can impact on gene regulation through MTA1. MTA1 acts in response to UV and osmotic stress and reduces the expression of tumor suppressor phosphatase and tensin homolog on Chromosome 10 (PTEN). 56 Notably, the MTA1 ZnF domain may mediate interactions with both proteins or DN. 57 The functional consequence of a physical interaction of p38α with MTA1 remains to be determined, yet may be part of a UV‐induced DNA damage response network downstream of p38α involving at least XPA1, NFκB, and MTA1. 58
In summary, we show that BioID2 can be used to map entire upstream and downstream pathways and functional protein networks surrounding protein kinases with the example of the pivotal MAP kinase p38α. Interactomes generated with proximity labeling by BioID2 may facilitate identification of kinase regulators and targets that explain physiologic responses of signaling networks in living cells and organisms.
5. EXPERIMENTAL PROCEDURES
5.1. Materials and reagents
Antibodies against the following epitopes were used: myc proto‐oncogene protein (MYC; cat. #986565, 1/2,000, Invitrogen, ThermoFisher, Sydney, Australia), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH; clone 6C5, 1/5,000, Invitrogen, ThermoFisher), p38α (cat #9218S, 1/1,000, Cell Signaling Technologies [CST], Arundel, Australia), flag tag (FLAG; clone M2, cat. #F3165, Sigma, St. Louis, MI), hemagglutinin tag (HA; clone HA‐7, cat. #H3663, 1/5,000, Sigma, Sydney, Australia), PCNA (Abcam), and (HA; clone C29F4, cat. #3724, CST). Horseradish peroxidase (HRP) coupled secondary antibodies used were donkey anti‐mouse (cat. #A16011, ThermoFisher) and goat anti‐rabbit (cat. #ab205718, 1/5,000, Melbourne, Australia). SA conjugated to HRP was used for biotin detection (SA; cat. #3999, CST).
5.2. Plasmid constructs
Plasmid myc‐BioID2‐MCS was a kind gift from Kyle Roux (AddGene plasmid #74223). Coding sequence of p38α was amplified with Q5 PCR (NEB, Ipswich, MA) and cloned into myc‐BioID2‐MCS by HiFi assembly (NEB). For coimmunoprecipitation experiments, amplified p38α‐encoding DNA was cloned into pFLAG‐C1, which was generated by replacing the eGFP coding sequence in peGFP‐c1 with the coding sequence for FLAG tag by oligocloning as previously described. 59 All constructs were confirmed by sequencing.
5.3. Cell culture
Human embryonic kidney 293T (HEK293T) cells were maintained in Dulbecco's Modified Eagle Medium (Gibco, ThermoFisher, Waltham, MA) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 2 mM l‐glutamine at 37°C, 5% CO2, and 55% humidity. Cells were seeded at a density of 5.0 × 105 cells per 60 mm plates 24 hr before transfection. Culture medium was changed 1.5 hr prior to transfection at 70% cell confluency. Plasmid DNA was transfected using polyethyleneimine (PEI) as previously described. 59 Briefly, 4 μg of plasmid DNA and 12 μl of PEI (>98%) were added to saline to a final volume of 420 μl. DNA complexed with PEI was incubated for 15 min, then added dropwise to each cell culture plate.
5.4. UV irradiation
HEK293T cells expressing FLAG‐p38α, HA‐XPA, and/or eGFP cultured on sterilized glass coverslips were irradiated with UV‐B light (350 J/m2) for 1 min in a UV crosslinker (UVP). Cells were fixed 1 hr postirradiation with 4% paraformaldehyde (PFA) in phosphate‐buffered saline (PBS; pH 7.4) for 10 min at room temperature.
5.5. Proximity‐dependent biotin labeling
Eighteen hours posttransfection, culture medium was exchanged with 200 μM biotin‐containing medium and incubated for 48 hr. Cells were lysed and sonicated in RIPA buffer (1 M tris(hydroxymethyl)aminomethane pH 8.0, 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid [EDTA], 1% Nonidet‐P40, 0.1% sodium dodecyl sulfate [SDS], 1 mM sodium vanadate [Na3VO4], 10 mM sodium fluoride [NaF], 0.1% glycerophosphate, 1 mM sodium pyrophosphate [NaPP], 1× Complete Mini protease inhibitor [Roche Applied Science, Sydney, Australia]) and centrifuged (16,000g, 10 min, 4°C). Protein concentration was determined using a Bradford assay (BioRad, Sydney, Australia). Aliquots of 20 μg from each sample were reserved for Western blotting and lysates of 1,000 μg protein per sample were further processed for MS analysis.
5.6. Tryptic digest
Protein precipitation and trypsin digest were performed as previously described. 60 Briefly, three volumes of methanol and one volume each of chloroform and water were added to each sample, vortexed and centrifuged (15,000g, 2 min). Upon removing the solvents, three volumes of methanol were used to resuspend the protein pellet for centrifugation (15,000g, 2 min), and then removed to air‐dry the remaining pellet (15 min). Protein from each sample was resuspended in 200 μl buffer (4 M urea, 0.1% ProteaseMAX trypsin buffer [Promega, Madison, WI, USA]; 50 mM NH4HCO3) and sonicated. Dithiothreitol (Sigma) was added to a 5 mM final concentration and samples were incubated in a thermomixer (800 rpm; 1 hr, 55°C). Iodoacetamide was added to a final 10 mM concentration and reincubated (800 rpm, 20 min, in the dark; 20 min). Trypsin buffer (NH4HCO3 [150 μl, 50 mM], ProteaseMAX [2.5 μl, 1%, in 50 mM NH4HCO3), trypsin (cat. #V5111, Promega) was added to each sample and incubated (300 rpm; 4 hr, 37°C). Trifluoroacetic acid (TFA) was added to 0.1% final concentration to inhibit trypsin, then samples were centrifuged (20,000g; 20 min).
5.7. Buffer exchange and peptide resuspension
Supernatant transferred to solid‐phase extraction tC18 cartridges (cat. #WAT036810, Waters, Milford, MA) as previously described. 61 Briefly, cartridges were sequentially washed with acetonitrile, 0.5% acetic acid in 50% acetonitrile, and 0.1% TFA in dH2O. After adding the peptide samples, cartridges were washed with 3 ml 0.1% TFA in dH2O, followed by 250 μl 0.5% acetic acid in dH2O. Peptides were eluted with a solution of 0.5% acetic acid in 80% acetonitrile and dried overnight in a vacuum concentrator. We used a modified biotinylated peptide‐enrichment protocol. 60 Briefly, peptides were resuspended in 250 μl PBS, 55 μl buffer‐equilibrated Sera‐Mag neutravidin (NA) magnetic bead slurry (cat. #09‐981‐155, GE, ThermoFisher) was added, and mixture was incubated (rotating, 2 hr at RT). NA‐bound peptides were immobilized on a magnetic stand and washed sequentially with 1.5 ml PBS, and then 1.5 ml PBS with sequentially increasing acetonitrile concentrations (2.5, 5,and 10% acetonitrile), rotating for 5 min at each wash step. Bead‐bound peptides were resuspended in 250 μl dissociation buffer (0.2% TFA; 0.1% formic acid; 80% acetonitrile) and incubated (rotating, 15 min) before magnet immobilization and collection of the first supernatant (SN1). To maximize peptide recovery, beads were resuspended and reincubated in 250 μl dissociation buffer under heat (700 rpm, 5 min, 95°C), followed by collection and addition of the second supernatant (SN2) to SN1. Eluates were dried under vacuum and resuspended in 0.2% heptafluorobutyric acid with 1% formic acid for MS analysis.
5.8. MS (LCMSMS), MASCOT database searching, and scaffold data analysis
Samples were run in triplicate (3 μg total protein injected per run) and captured on a C18 cartridge (Acclaim PepMap 100, 5 μm 100 Å, Thermo Scientific Dionex, Waltham, MA) before switching to a capillary column (20 cm) containing C18 reverse phase packing (Reprosil‐Pur, 1.9 μm, 200 Å, Dr Maisch GmbH, Ammerbuch‐Entringen, Germany), and eluted with a 40 min gradient of buffer A (H2O:CH3CN of 98:2 with 0.1% formic acid) to buffer B (H2O:CH3CN of 20:80 with 0.1% formic acid) at 200 nl/min. Data‐dependent acquisition was performed on a QExactive mass spectrometer (ThermoFisher Scientific), run in positive ion mode. Mass spectrometer settings were: ion spray voltage 2000 V, capillary temperature 275–300°C, a survey scan acquired (m/z 375–1,750) and up to 10 multiply charged ions (charge state ≥2+) isolated for MSMS fragmentation (counts >2,500), with nitrogen used as the CID collision gas. Raw datafiles were processed to generate peak lists using MASCOT Distiller (Matrix Science, London, UK) and these searched using the MASCOT search engine (v2.6.2, Matrix Science). Peak lists were matched to amino acid sequences from the SwissProt database (downloaded 14‐12‐18; Mammalia taxonomy, 66,946 entries). Search parameter settings were: peptide and MS/MS tolerances of ±5 ppm and ±0.05 Da, respectively, variable modifications included biotin (K), carbamidomethyl (C), and oxidation (M), peptide charge of 2+ and 3+ and enzyme specificity semitrypsin with up to two missed cleavages allowed. Comparison of data across sample types was performed using Scaffold Q+ software, which uses the ProteinProphet algorithm, 62 and in the current work, identified peptides with FDR = 1.3% and proteins with FDR = 0.5%. Protein identification was based on identification ≥1 unique peptide(s) with peptide score(s) ≥40 and protein score(s) ≥30. All identified biotinylated proteins for triplicates of background and experimental conditions are listed in Table S2.
5.9. Data analysis and gene ontology
BioID2 and BioID2‐p38α biotinylated protein lists were analyzed using criteria adapted from a previously published protocol. 47 Biotinylated protein abundance was inferred from assigned SC, summated from triplicates of each condition. Relative abundance was expressed as fold change in SC (FΔSC) of biotinylated protein abundance for BioID2‐p38α relative to BioID2. Proteins constituting the p38α interactome fulfilled at least one of the following criteria: a significantly higher FΔSC, or if exclusively identified in BioID2‐p38α samples, SC was ≥2, or simply having a FΔSC ≥3. Protein–protein interaction networks were generated using STRING (v10.5). Protein ontology was analyzed using DAVID gene ontology database (v6.8) using gene ontology annotations for the entire human genome. Interactomes and network properties were visually represented using CytoScape (v3.7.1). Existing evidence for protein–protein interactions of p38α was derived from BioGRID (v3.1.180) and STRING (v10.5) databases and through literature search. Putative p38α phosphorylation targets were predicted with KEA2 database 36 or GPS3.0 kinase consensus site prediction software with highest prediction threshold. 37
5.10. RNA isolation, reverse transcription, and cloning of XPA
Human coding DNA (cDNA) was reverse transcribed using total RNA isolated from HEK293T cells using Trizol (Sigma) with a first strand cDNA synthesis kit and poly dT oligonucleotide primers (NEB, MA). Coding sequence of human XPA was amplified and subcloned into pBluescript before cloning with a C‐terminal HA tag into mammalian expression destination vector pcDNA3.1 by HiFi assembly (NEB). All constructs where confirmed by sequencing. After transfecting HEK293T cells, harvested cell lysates were used to confirm expression of proteins by immunoblotting.
5.11. Immunofluorescence imaging
293T cells were plated at a density of 1 × 105 cells per cm2 onto sterilized glass coverslips and transfected the next day. On Day 2, the cell culture medium was supplemented with biotin (200 μM) for 30 hr. Cells were fixed in PBS with 4% PFA at room temperature for 10 min. Cells were washed once with PBS, permeabilize with 0.05% NP‐40 in PBS for 5 min, and washed twice more with PBS before addition of blocking buffer (BB) (3% goat serum; Vector Laboratories, Sydney, Australia) for 45 min. Cells were incubated with mouse anti‐myc (1/500) and rabbit anti‐p38α antibodies (1/500) for 1 hr at room temperature. Following three PBS washes, all samples were incubated with donkey anti‐mouse antibody conjugated to Alexa‐488 (1/500); donkey anti‐rabbit Alexa‐647 (1/500); DAPI (1/1,000); and SA‐Cy3 (1/500). All antibodies, dyes, and molecular were diluted in 3% BB. Coverslips were mounted (FluoroMount) after three more PBS washes. Micrographs were acquired using an epifluorescence microscope (BX51, Olympus) at the same exposure times settings for each channel across samples with CellSens software (Olympus).
5.12. Immunoprecipitation
Immunoprecipitation was performed from cell lysates as previously described. 59 Briefly, cells were lysed in RIPA buffer (20 mM Tris–HCl pH 7.5, 150 mM sodium chloride, 1 mM sodium EDTA, 1 mM EGTA, 1% NP‐40, 1% sodium deoxycholate, 0.1% SDS, 2.5 mM NaPP, 1 mM β‐glycerophosphate, 1 mM NaF, 1 mM Na3VO4, Complete protease inhibitors (Roche)) on ice. Lysates were cleared by centrifugation (16,000g, 10 min, 4°C). Protein concentration was determined by Bradford assay (BioRad). Protein lysate (550 μg) was incubated with 1.2 μg anti‐FLAG antibody (FLAG M2, Sigma) for 2 hr with rotation at 4°C. Buffer‐equilibrated, blocked Protein G magnetic beads (S1430; New England Biolabs) were added, followed by 1 hr incubation at 4°C with rotation. Beads were rinsed once and washed three times with RIPA buffer. Subsequently, beads were resuspended in 40 μl of sample buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 5% β‐mercaptoethanol, 0.01% bromophenol blue, 15% glycerol) and incubated at 90°C for 2 min. Proteins were separated by 10% SDS‐PAGE and transferred onto nitrocellulose membranes (Immobilon, Millipore). Membranes were blocked with 5% skim milk powder in Tris‐buffered saline with 0.1% Tween‐20 (TBS‐T) before probing with primary antibodies for 1 hr at room temperature. After three washes in TBS‐T, secondary antibody HRP conjugates (Jackson) were incubate for 1 hr at room temperature. After additional three washes in TBS‐T, membranes were incubated with chemiluminescence substrate (Immobilon Crescendo, Millipore) and signals were acquired on digital imaging system (Chemidoc, BioRad). Immunoblot signals were quantified in ImageJ/Fiji. 63
5.13. Proximity ligation assay
Cells on sterilized glass coverslips were washed in PBS and fixed using 4% PFA in PBS for 10 min at room temperature. Cells were washed in PBS and permeabilized by incubation for 2 min in PBS containing 0.05% NP‐40. After two PBS washes, cells were incubated with BB provided in the Duolink PLA starter Kit (#DUO92101; Sigma‐Aldrich, Sydney, Australia) and manufacturer instructions were followed to the final step of amplification. Primary antibodies used were rabbit anti‐p38α (1/500) and/or mouse anti‐PCNA (1/500) or mouse anti‐HA (1/500). Cells were counterstained with DAPI (1/1,000). Micrographs were acquired on an epifluorescence microscope (BX51, Olympus) at the same exposure times settings for each channel across samples with CellSens software (Olympus). PLA signal area was quantified by particle analysis and expressed relative to area of nuclei. Image analysis was performed in ImageJ/Fiji. 63
5.14. Statistical analysis
Statistical analyses were performed using Prism (v8.2.0) (GraphPad). Overall intersample condition comparison of protein SC medians was performed using the nonparametric Mann–Whitney test. Intersample condition comparisons of individual biotinylated protein SCs were performed using Fisher's exact test. Comparisons of quantified immunoblot signals were performed with analysis of variance (ANOVA) including Bonferroni posttest. Comparison of PLA signals was performed with unpaired parametric Student's t test. Statistical significance was set at p < .05 (*), p < .01 (**), p < .001 (***), p < .0001 (****). Data are expressed as means ± SEM.
AUTHOR CONTRIBUTIONS
Emmanuel Prikas: Conceptualization; data curation; formal analysis; investigation; methodology; validation; visualization; writing‐original draft; writing‐review and editing. Anne Poljak: Formal analysis; investigation; methodology; validation; writing‐original draft; writing‐review and editing. Arne Ittner: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; resources; supervision; validation; visualization; writing‐original draft; writing‐review and editing.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the contents of this article.
Supporting information
Table S1 BioID2‐Peptides & Sequence.
Table S2 BioID2p38alpha‐Peptides & Sequence.
Table S3 Total Unique Biotinylated Proteins Detected.
Table S4 GO of 719 biotinylated proteins identified in BioID2 samples only.
Table S5 Abundance of biotinylated proteins: BioID2 vs. BioID2‐p38a.
Table S6 Biotinylated proteins found in higher abundance/only in BioID2‐p38a samples vs. BioID2 samples.
Table S7 Annotation Cluster 1 Enrichment Score: 2.3313793326308203.
Table S8 The most biotinylated proteins in experimental samples (BioID2‐p38a) relative to background condition samples (BioID2) were the most abundant proteins used for generating the p38a interactome The abundance of a biotinylated protein was quantified as the number of spectral counts of biotinylated peptide fragments, attributable to a given single protein, present in BioID2‐p38a vs. BioID2 samples Relative protein abundance, or fold change increase, used as the general p38a interactome inclusion criterion, was expressed as the abundance of a biotinylated protein in BioID2‐p38a relative to BioID2 All interactome proteins were non‐redundant proteins; those most biotinylated in BioID2‐p38a relative to BioID2 samples.
Table S9 BioID2‐p38alpha interactome analysis by interaction databases.
Table S10 Known and predicted p38alpha phosphorylation substrates among BioID2‐p38alpha interactome.
ACKNOWLEDGMENTS
This work was supported by the Australian Research Council (ARC) grant DP170100843 to A. I., and the National Health and Medical Research Council (NHMRC) grants APP1143978 to A. I., and by Macquarie University.
Prikas E, Poljak A, Ittner A. Mapping p38α mitogen‐activated protein kinase signaling by proximity‐dependent labeling. Protein Science. 2020;29:1196–1210. 10.1002/pro.3854
Funding information Australian Research Council, Grant/Award Number: DP170100843; National Health and Medical Research Council, Grant/Award Number: APP1143978; Macquarie University
REFERENCES
- 1. Davis RJ. The mitogen‐activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 2. Roux PP, Blenis J. ERK and p38 MAPK‐activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev. 2004;68:320–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. [DOI] [PubMed] [Google Scholar]
- 4. Morrison DK. MAP kinase pathways. Cold Spring Harb Perspect Biol. 2012;4:a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hotamisligil GS, Davis RJ. Cell signaling and stress responses. Cold Spring Harb Perspect Biol. 2016;8:a006072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Han J, Lee JD, Bibbs L, Ulevitch RJ. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science. 1994;265:808–811. [DOI] [PubMed] [Google Scholar]
- 7. Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. [DOI] [PubMed] [Google Scholar]
- 8. Kim C, Sano Y, Todorova K, et al. The kinase p38 alpha serves cell type‐specific inflammatory functions in skin injury and coordinates pro‐ and anti‐inflammatory gene expression. Nat Immunol. 2008;9:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. de Nadal E, Ammerer G, Posas F. Controlling gene expression in response to stress. Nat Rev Genet. 2011;12:833–845. [DOI] [PubMed] [Google Scholar]
- 10. Lo U, Selvaraj V, Plane JM, Chechneva OV, Otsu K, Deng W. p38alpha (MAPK14) critically regulates the immunological response and the production of specific cytokines and chemokines in astrocytes. Sci Rep. 2014;4:7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen P. The search for physiological substrates of MAP and SAP kinases in mammalian cells. Trends Cell Biol. 1997;7:353–361. [DOI] [PubMed] [Google Scholar]
- 12. Kyriakis JM, Avruch J. Mammalian mitogen‐activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev. 2001;81:807–869. [DOI] [PubMed] [Google Scholar]
- 13. Ge B, Gram H, Di Padova F, et al. MAPKK‐independent activation of p38alpha mediated by TAB1‐dependent autophosphorylation of p38alpha. Science. 2002;295:1291–1294. [DOI] [PubMed] [Google Scholar]
- 14. Grimsey NJ, Lin Y, Narala R, Rada CC, Mejia‐Pena H, Trejo J. G protein‐coupled receptors activate p38 MAPK via a non‐canonical TAB1‐TAB2‐ and TAB1‐TAB3‐dependent pathway in endothelial cells. J Biol Chem. 2019;294:5867–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ashwell JD. The many paths to p38 mitogen‐activated protein kinase activation in the immune system. Nat Rev Immunol. 2006;6:532–540. [DOI] [PubMed] [Google Scholar]
- 16. Heinrichsdorff J, Luedde T, Perdiguero E, Nebreda AR, Pasparakis M. p38 alpha MAPK inhibits JNK activation and collaborates with IkappaB kinase 2 to prevent endotoxin‐induced liver failure. EMBO Rep. 2008;9:1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stefanoska K, Bertz J, Volkerling AM, van der Hoven J, Ittner LM, Ittner A. Neuronal MAP kinase p38alpha inhibits c‐Jun N‐terminal kinase to modulate anxiety‐related behaviour. Sci Rep. 2018;8:14296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Staples CJ, Owens DM, Maier JV, Cato AC, Keyse SM. Cross‐talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase‐1 determines cellular sensitivity to UV radiation. J Biol Chem. 2010;285:25928–25940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miura H, Kondo Y, Matsuda M, Aoki K. Cell‐to‐cell heterogeneity in p38‐mediated cross‐inhibition of JNK causes stochastic cell death. Cell Rep. 2018;24:2658–2668. [DOI] [PubMed] [Google Scholar]
- 20. Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. [DOI] [PubMed] [Google Scholar]
- 21. Ben‐Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. Nuclear export of the stress‐activated protein kinase p38 mediated by its substrate MAPKAP kinase‐2. Curr Biol. 1998;8:1049–1057. [DOI] [PubMed] [Google Scholar]
- 22. Pomerance M, Quillard J, Chantoux F, Young J, Blondeau JP. High‐level expression, activation, and subcellular localization of p38‐MAP kinase in thyroid neoplasms. J Pathol. 2006;209:298–306. [DOI] [PubMed] [Google Scholar]
- 23. Gong X, Ming X, Deng P, Jiang Y. Mechanisms regulating the nuclear translocation of p38 MAP kinase. J Cell Biochem. 2010;110:1420–1429. [DOI] [PubMed] [Google Scholar]
- 24. Kotlyarov A, Yannoni Y, Fritz S, et al. Distinct cellular functions of MK2. Mol Cell Biol. 2002;22:4827–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lukas SM, Kroe RR, Wildeson J, et al. Catalysis and function of the p38 alpha.MK2a signaling complex. Biochemistry. 2004;43:9950–9960. [DOI] [PubMed] [Google Scholar]
- 26. Ouwens DM, de Ruiter ND, van der Zon GC, et al. Growth factors can activate ATF2 via a two‐step mechanism: Phosphorylation of Thr71 through the Ras‐MEK‐ERK pathway and of Thr69 through RalGDS‐Src‐p38. EMBO J. 2002;21:3782–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanos T, Marinissen MJ, Leskow FC, et al. Phosphorylation of c‐Fos by members of the p38 MAPK family. Role in the AP‐1 response to UV light. J Biol Chem. 2005;280:18842–18852. [DOI] [PubMed] [Google Scholar]
- 28. Zhao M, New L, Kravchenko VV, et al. Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999;19:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y, Cho YY, Petersen BL, Zhu F, Dong Z. Evidence of STAT1 phosphorylation modulated by MAPKs, MEK1 and MSK1. Carcinogenesis. 2004;25:1165–1175. [DOI] [PubMed] [Google Scholar]
- 30. Puigserver P, Rhee J, Lin J, et al. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator‐1. Mol Cell. 2001;8:971–982. [DOI] [PubMed] [Google Scholar]
- 31. Shimada N, Rios I, Moran H, Sayers B, Hubbard K. p38 MAP kinase‐dependent regulation of the expression level and subcellular distribution of heterogeneous nuclear ribonucleoprotein A1 and its involvement in cellular senescence in normal human fibroblasts. RNA Biol. 2009;6:293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belozerov VE, Lin ZY, Gingras AC, McDermott JC, Michael Siu KW. High‐resolution protein interaction map of the Drosophila melanogaster p38 mitogen‐activated protein kinases reveals limited functional redundancy. Mol Cell Biol. 2012;32:3695–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Varjosalo M, Keskitalo S, Van Drogen A, et al. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013;3:1306–1320. [DOI] [PubMed] [Google Scholar]
- 34. Kim DI, Jensen SC, Noble KA, et al. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell. 2016;27:1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol. 2012;196:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lachmann A, Ma'ayan A. KEA: Kinase enrichment analysis. Bioinformatics. 2009;25:684–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xue Y, Zhou F, Zhu M, Ahmed K, Chen G, Yao X. GPS: A comprehensive www server for phosphorylation sites prediction. Nucleic Acids Res. 2005;33:W184–W187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sugitani N, Sivley RM, Perry KE, Capra JA, Chazin WJ. XPA: A key scaffold for human nucleotide excision repair. DNA Repair. 2016;44:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Borisova ME, Voigt A, Tollenaere MAX, et al. p38‐MK2 signaling axis regulates RNA metabolism after UV‐light‐induced DNA damage. Nat Commun. 2018;9:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gilljam KM, Muller R, Liabakk NB, Otterlei M. Nucleotide excision repair is associated with the replisome and its efficiency depends on a direct interaction between XPA and PCNA. PLoS One. 2012;7:e49199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell. 2007;129:665–679. [DOI] [PubMed] [Google Scholar]
- 42. Aboussekhra A, Wood RD. Detection of nucleotide excision repair incisions in human fibroblasts by immunostaining for PCNA. Exp Cell Res. 1995;221:326–332. [DOI] [PubMed] [Google Scholar]
- 43. Alam MS. Proximity ligation assay (PLA). Curr Protoc Immunol. 2018;123:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roux KJ, Kim DI, Burke B, May DG. BioID: A screen for protein‐protein interactions. Curr Protoc Protein Sci. 2018;91:19.23.11–19.23.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wood CD, Thornton TM, Sabio G, Davis RA, Rincon M. Nuclear localization of p38 MAPK in response to DNA damage. Int J Biol Sci. 2009;5:428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Canovas B, Igea A, Sartori AA, et al. Targeting p38alpha increases DNA damage, chromosome instability, and the anti‐tumoral response to taxanes in breast cancer cells. Cancer Cell. 2018;33:1094–1110. [DOI] [PubMed] [Google Scholar]
- 47. Uezu A, Kanak DJ, Bradshaw TW, et al. Identification of an elaborate complex mediating postsynaptic inhibition. Science. 2016;353:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singhirunnusorn P, Suzuki S, Kawasaki N, Saiki I, Sakurai H. Critical roles of threonine 187 phosphorylation in cellular stress‐induced rapid and transient activation of transforming growth factor‐beta‐activated kinase 1 (TAK1) in a signaling complex containing TAK1‐binding protein TAB1 and TAB2. J Biol Chem. 2005;280:7359–7368. [DOI] [PubMed] [Google Scholar]
- 49. Olson CM, Hedrick MN, Izadi H, Bates TC, Olivera ER, Anguita J. p38 mitogen‐activated protein kinase controls NF‐kappaB transcriptional activation and tumor necrosis factor alpha production through RelA phosphorylation mediated by mitogen‐ and stress‐activated protein kinase 1 in response to Borrelia burgdorferi antigens. Infect Immun. 2007;75:270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cheung PC, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38alpha. EMBO J. 2003;22:5793–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lin KC, Moroishi T, Meng Z, et al. Regulation of Hippo pathway transcription factor TEAD by p38 MAPK‐induced cytoplasmic translocation. Nat Cell Biol. 2017;19:996–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. LaRochelle O, Gagne V, Charron J, Soh JW, Seguin C. Phosphorylation is involved in the activation of metal‐regulatory transcription factor 1 in response to metal ions. J Biol Chem. 2001;276:41879–41888. [DOI] [PubMed] [Google Scholar]
- 53. Cleaver JE, States JC. The DNA damage‐recognition problem in human and other eukaryotic cells: The XPA damage binding protein. Biochem J. 1997;328:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis. 2011;6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fadda E. Role of the XPA protein in the NER pathway: A perspective on the function of structural disorder in macromolecular assembly. Comput Struct Biotechnol J. 2016;14:78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Reddy SD, Pakala SB, Molli PR, et al. Metastasis‐associated protein 1/histone deacetylase 4‐nucleosome remodeling and deacetylase complex regulates phosphatase and tensin homolog gene expression and function. J Biol Chem. 2012;287:27843–27850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Manavathi B, Kumar R. Metastasis tumor antigens, an emerging family of multifaceted master coregulators. J Biol Chem. 2007;282:1529–1533. [DOI] [PubMed] [Google Scholar]
- 58. Mangerich A, Burkle A. Pleiotropic cellular functions of PARP1 in longevity and aging: Genome maintenance meets inflammation. Oxid Med Cell Longev. 2012;2012:321653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ittner A, Chua SW, Bertz J, et al. Site‐specific phosphorylation of tau inhibits amyloid‐beta toxicity in Alzheimer's mice. Science. 2016;354:904–908. [DOI] [PubMed] [Google Scholar]
- 60. Schiapparelli LM, McClatchy DB, Liu HH, Sharma P, Yates JR III, Cline HT. Direct detection of biotinylated proteins by mass spectrometry. J Proteome Res. 2014;13:3966–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Villen J, Gygi SP. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3:1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. [DOI] [PubMed] [Google Scholar]
- 63. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: An open‐source platform for biological‐image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 BioID2‐Peptides & Sequence.
Table S2 BioID2p38alpha‐Peptides & Sequence.
Table S3 Total Unique Biotinylated Proteins Detected.
Table S4 GO of 719 biotinylated proteins identified in BioID2 samples only.
Table S5 Abundance of biotinylated proteins: BioID2 vs. BioID2‐p38a.
Table S6 Biotinylated proteins found in higher abundance/only in BioID2‐p38a samples vs. BioID2 samples.
Table S7 Annotation Cluster 1 Enrichment Score: 2.3313793326308203.
Table S8 The most biotinylated proteins in experimental samples (BioID2‐p38a) relative to background condition samples (BioID2) were the most abundant proteins used for generating the p38a interactome The abundance of a biotinylated protein was quantified as the number of spectral counts of biotinylated peptide fragments, attributable to a given single protein, present in BioID2‐p38a vs. BioID2 samples Relative protein abundance, or fold change increase, used as the general p38a interactome inclusion criterion, was expressed as the abundance of a biotinylated protein in BioID2‐p38a relative to BioID2 All interactome proteins were non‐redundant proteins; those most biotinylated in BioID2‐p38a relative to BioID2 samples.
Table S9 BioID2‐p38alpha interactome analysis by interaction databases.
Table S10 Known and predicted p38alpha phosphorylation substrates among BioID2‐p38alpha interactome.