

Abstract
Nickel enzymes, present in archaea, bacteria, plants, and primitive eukaryotes are divided into redox and nonredox enzymes and play key functions in diverse metabolic processes, such as energy metabolism and virulence. They catalyze various reactions by using active sites of diverse complexities, such as mononuclear nickel in Ni‐superoxide dismutase, glyoxylase I and acireductone dioxygenase, dinuclear nickel in urease, heteronuclear metalloclusters in [NiFe]‐carbon monoxide dehydrogenase, acetyl‐CoA decarbonylase/synthase and [NiFe]‐hydrogenase, and even more complex cofactors in methyl‐CoM reductase and lactate racemase. The presence of metalloenzymes in a cell necessitates a tight regulation of metal homeostasis, in order to maintain the appropriate intracellular concentration of nickel while avoiding its toxicity. As well, the biosynthesis and insertion of nickel active sites often require specific and elaborated maturation pathways, allowing the correct metal to be delivered and incorporated into the target enzyme. In this review, the phylogenetic distribution of nickel enzymes will be briefly described. Their tridimensional structures as well as the complexity of their active sites will be discussed. In view of the latest findings on these enzymes, a special focus will be put on the biosynthesis of their active sites and nickel activation of apo‐enzymes.
Keywords: enzyme maturation, metallocluster, metalloenzymes, nickel active site, redox enzymes
1. INTRODUCTION
At the beginning of life, the environment was highly reducing, with an anoxic atmosphere rich in gases like H2, CO, and CO2 and hot oceans well‐supplied with transition metals such as Fe(II) and Ni(II).1 In order to accommodate these early living conditions, nickel is most likely featured as a key player in catalyst development. Notably, its role was essential as a metal cofactor in the metabolism of methanogenic archaea.2 The “nickel famine” theory has been proposed to play a central role in evolution:3 it correlates the great oxidation event (GEO) with a drastic reduction of nickel flux in archean oceans, consequent to the cooling of the upper‐mantle and the decreased eruption of nickel‐rich ultramafic rocks, which would have starved the major oceanic microbial community, the methanogens.4 Their decline left space for the proliferation of other microorganisms, especially oceanic cyanobacteria, that did not require as much nickel. This lead to an increased production of O2 via photosynthesis, concomitant with a decrease in methane concentration. The appearance of oxygen in the atmosphere 2.4 billion years ago had a dramatic impact on metabolic evolution and many enzymatic reaction pathways used by anaerobic metabolism under reductive conditions were replaced by aerobic ones.5 The result was the segregation of the microorganisms relying on H2 and/or CO oxidation and CO2 reduction pathways to anoxic niches, such as ocean or lake bottoms, animal digestive tracts, and volcanic mud.6 To date, only nine nickel‐dependent enzymes have been identified in archaea, bacteria, plants, and primitive eukaryotes, including [NiFe]‐carbon monoxide dehydrogenase (CODH), acetyl‐CoA decarbonylase/synthase (ACS), [NiFe]‐hydrogenase methyl‐CoM reductase (MCR), urease, Ni‐superoxide dismutase (NiSOD), lactate racemase (LarA), glyoxylase I (GlxI), and acireductone dioxygenase (ARD; Figure 1).7 In contrast, no nickel enzyme has been found in mammalian species.8 Despite their scarcity, they are often essential, playing key functions in diverse metabolic processes, such as energy metabolism and virulence, and functioning as either redox or nonredox enzymes (Table 1). The advantage of nickel as a catalytic center is based on its flexible coordination geometry, allowing diverse biological functions. Moreover, in redox enzymes, the metal environment is critical in adjusting its redox potential and nickel is thus able to cycle through several redox states and to catalyze reactions spanning over 1.5 V, while in nonredox enzymes, Ni(II) is used as a Lewis acid.
Figure 1.

Schematic amino acid sequences and metal‐binding motifs of Ni‐enzymes. Residues involved in Ni or Fe binding are highlighted in green and orange, respectively. Residues involved in both Ni and Fe binding are highlighted in magenta. Residues in interaction with cofactors are highlighted in blue. Accession number in UniProtKB is in italics. Residues constituting the active site are annotated. RrCODH: monomer of CODH from R. rubrum; MtACS: α susbunit of CODH/ACS from Moorella thermoacetica; DgH2ase: small (HydA) and large (HydB) subunit of [NiFe]‐H2ase from Desulfovibrio gigas, sequence signal of HydA (1–23) for periplasmic translocation and propeptide (537–551) processed in HydA during maturation are in barred; MmMCR: α (McrA), ß (McrB), and ɤ (McrG) subunits of MCR from Methanothermobacter marbugensis; SsNi‐SOD: monomer of Ni‐SOD from Streptomyces seoulensis, the processed propeptide (1–14) is in barred; SpUrease: α, ß, and ɤ subunits of urease from Sporosarcina pasteurii; LpLarA: LarA from Lactobacillus planetarium; EcGlxI: monomer of Glx I from E. coli; KoARD: Ni‐ARD from Klebsellia oxytoca
Table 1.
Occurrence of Ni‐enzymes and compilation of the various catalytic reactions
| Occurrence | Enzyme reaction | |
|---|---|---|
| Redox enzymes | ||
| [NiFe]‐hydrogenase | Bacteria, Archaea | H 2 ↔ 2H+ + 2e− |
| CO‐dehydrogenase | Bacteria, Archaea | CO + H2O ↔ CO 2 + 2H+ + 2e− |
| Acetyl‐SCoA synthase | Bacteria, Archaea | CO + CoA‐S− + CH3‐CO3+FeSP ↔ CH 3 C(O)‐S‐CoA + co+FeSP |
| Methyl‐SCoM reductase | Archaea | CH3‐SCoM + CoBSH ↔ CH 4 + CoBS‐SCoM |
| Superoxide dismutase | Bacteria | |
| Marine Eukaryota | 2 O 2 −° + 2H+ ➔ H2O2 + O2 | |
| Nonredox enzymes | ||
| Urease |
Bacteria, Archaea Eukaryota (plants, fungi, algae) |
(NH 2 )CO 2 + H2O ➔ CO2 + 2 NH 3 |
| Lactate racemase |
Bacteria Archaea? |
l‐lactate ↔ d‐lactate |
| Glyoxalase I |
Bacteria Eukaryota (protists, plants) |
CH3‐CO‐C(OH)‐SG ➔ CH 3 ‐CH(OH)‐CO‐SG |
| Acireductone dioxygenase |
Bacteria Eukaryota? |
1,2‐dihydroxy‐3‐keto‐5‐methylhtiopentane + O2 ↔ 3‐methylthiopropanoate + formate + CO |
Note: The most representative kingdoms for each enzyme and the main products or substrates are in bold.
The presence of metalloenzymes in organisms requires a tight regulation of metal homeostasis absolutely critical for cells in order to maintain the appropriate intracellular concentration of essential metals while avoiding toxicity caused by excess amounts.9 On the one hand, if the concentration becomes too low, the cell will suffer from the inactivation of essential enzymes. On the other hand, a nonphysiological metal present at high intracellular concentrations might lead to the replacement of the native metal which in turn might result in inactivation, or the catalytic formation of highly toxic reactive oxygen species. In the case of nickel, the current levels in natural environments are generally in the nanomolar range with the exception of specific niches. Therefore, its scavenging is a critical step and requires tightly controlled and highly efficient import systems.10 Once acquired, the correct metal will be delivered and incorporated into the target enzymes through dedicated protein complexes comprising nickel chaperones and accessory proteins. These proteins participate in nickel delivery, metallocenter assembly, or cofactor synthesis.
In this review, the phylogenetic distribution of Ni‐enzymes will first be presented. Their tridimensional structures as well as the complexity of their active sites will be discussed and a special focus will be put on the biosynthesis of active sites and nickel activation of apo‐enzymes. The details of reaction mechanisms will be excluded, as well as the description of nickel uptake, nickel efflux, and regulatory systems.
2. OCCURRENCE AND BIOLOGICAL RELEVANCE
Most of nickel enzymes are involved in gas processing, either as substrates or as final products of the reaction (CO, CO2, H2, ammonia, O2 and CH4), except GlxI I and LarA. They are divided into anaerobic and aerobic enzymes, depending on their metabolic implication. Ni‐utilizing organisms regroup archaea, bacteria, and some eukaryotes and possess one or several nickel enzymes (Table 1). All enzymes except MCR are found in bacteria, [NiFe]‐H2ase and urease being the most widespread, while the other ones show a limited and mosaic distribution. [NiFe]‐H2ase is also the most widespread Ni‐enzyme in Archaea.
2.1. Redox enzymes
H2ases have been classified into three phylogenetically unrelated classes, based on the metal composition of their active site: [Fe]‐, [FeFe]‐, and [NiFe]‐H2ases.6 The two latter types reversibly catalyze the oxidation of molecular hydrogen into protons and electrons. Hydrogen can have a double role, either as an energy source or as a final product to remove a possible excess of reducing equivalents. [NiFe]‐H2ases, found in bacteria and archaea, have been classified in four main groups depending on their function. Group 1 contains membrane‐associated H2 uptake hydrogenases; soluble uptake hydrogenases and sensory hydrogenases form the Group 2; heteromultimeric cytoplasmic hydrogenases harboring a reducible cofactor (F420 or NAD(P)) are found in Group 3 and the last group consists of energy‐conserving hydrogenases.11
Carbon monoxide dehydrogenase (CODH) plays a central role in carbon metabolism in anaerobic microorganisms, by reversibly catalyzing the oxidation of CO into CO2 for a variety of metabolic purposes.12 In the direction of CO oxidation, monofunctional CODH is used by various carboxydotrophic archaea and bacteria for energy production. In the direction of CO2 reduction, CODH can be coupled with acetyl‐SCoA synthase to form either the CODH/ACS complex in anaerobic bacteria or the acetyl‐CoA decarbonylase/synthase (ACDS) multienzyme complex in Archaea.13 The complex catalyzes the synthesis of acetyl‐CoA from coenzyme A condensed with CO (derived from CO2 reduction catalyzed by CODH) and a methyl group, derived from a corrinoid/ Fe–S protein Co(III)‐FeSP. CODH and ACS play an important role in the Wood–Ljungdahl (WL) pathway, one of the six carbon fixation pathways known on Earth. This pathway is found in acetogens for energy conservation and autotrophic carbon assimilation, with acetate as the end product. In methanogenic archaea, it is only used for CO2 fixation, while energy conservation is achieved by methanogenesis, which is thermodynamically more favorable. The WL pathway can also act in reverse (in the oxidative direction) to produce reducing power from the oxidation of organic compounds, illustrated by the coupling of the oxidation of acetate to H2 and CO2, to the reduction of sulfate in sulfate‐reducing bacteria. The WL pathway consists of two branches, defined as the methyl (or eastern) and the carbonyl (or western) branches. Although its overall scheme is conserved, the bacterial and archaeal methyl branches involve different cofactors, C1‐carriers and enzymes, while the carbonyl branch is common in all microorganisms. In the methyl branch, one molecule of CO2 undergoes a six‐electron reduction to yield a methyl group, condensed with CoA and CO coming from the reduction of CO2 in the carbonyl branch, to make acetyl‐CoA, a key molecule in protein, carbohydrate, and lipid metabolism.13 ACS can also be found as an independent monofunctional enzyme in carboxydotrophs.
MCR is strictly restricted to Archaea and plays an important role in carbon metabolism, being the key enzyme in biological methane formation.14 This enzyme catalyzes the reduction of methyl‐S‐coenzyme M (CH3‐S‐CoM) by coenzyme B (CoB) to yield methane, which is the final step of methanogenic Archaea metabolism. These strictly anaerobic microorganisms, able to grow on acetate, methanol, formate, or CO2 and H2, are responsible for more than 90% of the methane present on earth.15
SOD emerged with the rise in O2 levels in the atmosphere about 2 billion years ago. Its role is to protect biological systems from the oxidative damage caused by superoxide radical anions (O2 −°), a byproduct of oxygen metabolism.16 The enzyme catalyzes the dismutation of superoxide anions to hydrogen peroxide and oxygen (Table 1). Its active site can be composed of different metals. The most common ones are constituted either by a di‐nuclear Cu–Zn center or by a mononuclear Fe or Mn atom. The discovery of NiSOD in 1996 in Streptomyces species came as a surprise to the community because Ni(II) is the only stable oxidation state in aqueous media and cannot catalyze the superoxide disproportionation in aqueous solution.17 The adaptations required to use this metal as catalytic center resulted in a unique emergence of this enzyme which has no sequence homology with the other SODs. NiSOD is commonly found in cyanobacteria, which were the first to produce oxygen, and is also encoutered in some marine eukaryotes, making NiSOD the most abundant SOD in modern oceans.18
2.2. Nonredox enzymes
Urease was the first example that nickel could be used as an enzyme cofactor.19 This enzyme is found in a plethora of organisms including plants, fungi, algae, archaea, and bacteria and catalyzes the hydrolysis of urea to produce ammonia and carbamate. The latter decomposes spontaneously to ammonia and carbonic acid. The widespread occurrence of urease is related to the profusion of urea in diverse ecological niches, where the enzyme is involved in different biological processes and is an active player in the global nitrogen cycle.20 Interestingly, urease is also established as a virulence factor in several pathogenic bacteria. In this case, bicarbonate and ammonia are used as buffering compounds to maintain a neutral cytosolic pH, allowing the microorganisms to resist acidic environments, such as the digestive or the urinary tract, enhancing their survival during host colonization. One typical example is the gastric pathogen Helicobacter pylori for which urease constitutes a major virulence determinant to ensure its survival in the acidic human stomach.21
Glyoxalase I (Glo1 or GlxI) employs intracellular thiols to convert α‐ketoaldehydes such as methylglyoxal, a highly cytotoxic metabolic by‐product, into nontoxic d‐hydroxyacids.7 The enzyme works in concert with glyoxalase II, and converts the hemitioacetal, formed by the reaction of methylglyoxal with reduced glutathione to S‐d‐lactoylglutathione, used in turn by GlxII as a substrate to re‐generate reduced glutathione and produce d‐lactate. GlxI was described as a zinc‐dependent enzyme until 1998, when the activity of GlxI from Escherichia coli was shown to be maximized by Ni(II), while Zn(II) substitution resulted in enzyme inactivation.22 NiGlx1 have been described in bacteria, in protists such as Trypanosoma and Leishmania and in plants.
Acireductone dioxygenase participates to the methionine salvage pathway. During this pathway, 5′‐methylthioadenosin is converted into acireductone which can be used as a substrate to generate two different products.23 This enzyme is unique in exhibiting two distinct reactivities depending on the nature of the metal present in the active site. 7 When loaded with Fe, ARD produces a ketoacid precursor that can be recycled back to methionine. If instead Ni is the active center, ARD catalyzes an off‐ pathway which converts the acireductone into formate, carbon monoxide, and methylthiobutyric acid. In the latter case, the enzyme is presumably implicated in regulatory and/or signaling functions. This dual chemistry of ARD was originally discovered in the bacterium Klebsiella oxytoca.24 This activity has been confirmed in vitro for mammalian Mus musculus 25 and human ARD. However, it is unlikely that NiARD plays a biologically relevant role in eukaryotes.26 While the Fe‐containing form uses Fe(II) to activate dioxygen for the oxidation of the substrate via a redox chemistry (oxidative cleavage of C1—C2 bond), Ni(II) is used as a Lewis acid to activate the substrate toward reaction with O2 (cleavage of C1—C2 and C2—C3 bonds). The phylogenetic distribution of Ni‐dependent ARD is difficult to estimate since the nature of the physiological metal is not easily determined.
LarA, recently identified as the ninth known nickel‐dependent enzyme, catalyzes the interconversion between l‐ and d‐lactic acid isomers. The only characterized enzyme is the one found in Lactobacillus species, where it may act as a rescue enzyme under stress conditions to ensure d‐lactate production.27 d‐lactate is generally produced by the d‐lactate dehydrogenase and is an essential component of the cell wall peptidoglycan in Lactobacillus. Since then, larA genes have been identified in other bacteria and some archaea, although they have not been clearly identified as Ni‐dependent enzymes.
3. OVERALL STRUCTURES
X‐ray structures of the nine Ni‐enzymes described so far have been solved (Figure 2).
Figure 2.

Overall structures of Ni‐enzymes. (a). CODH dimer from Rhodospirillum rubrum (PDB code: 1JQK). The N‐terminal domain is in magenta and the two Rossman‐like domains are in green and blue. C‐clusters are depicted in spheres B. α2ß2 tetramer of CODH/ACS from Moorella thermoacetica (PDB code: 1OAO). The two ß subunits of CODH are colored in blue and cyan. The two ACS subunits are in open or closed conformation. A‐ and C‐clusters are depicted in spheres. C. Archetypical structure of [NiFe]‐hydrogenase from Desulfovibrio gigas (PDB code: 1FRV). The large subunit is in magenta and the small subunit in green. The buried active site is depicted in spheres. D. Dimer of αßɤ MCR from Methanothermobacter marbugensis (PDB code: 3POT). The α, ß, and ɤ subunits are in blue, cyan, and magenta, respectively. Coenzyme F430 is depicted in orange and Coenzyme B in green. E. Ni‐SOD hexamer from Streptomyces seoulensis (PDB code: 1Q0D). (f). Trimer of αßɤ urease from Sporosarcina pasteurii (PDB code: 4CEU). The α, ß, and ɤ subunits are in magenta, blue, and cyan, respectively. Helix‐turn‐helix motif (residues 375–405) belonging to subunit α is highlighted in orange. (g). Closed conformation of LarA from Lactobacillus plantarium (PDB code: 5HUQ). The two domains are in orange and blue and the pincer nucleotide is in magenta. (h). Glyoxalase I dimer from E. coli (PDB code: 1F9Z). (i). Ni‐ARD monomer from Klebsellia oxytoca (PDB code: 1ZRR) α‐helices are in cyan and ß‐strands in magenta. In all structures, nickel ions are depicted as green spheres and FeS clusters are depicted in red and yellow spheres for iron and sulfur atoms, respectively
The first structures of two CODH were solved in 2001,28, 29 revealing that monofunctional enzymes are homodimers (Figure 2a). Each monomer is composed of three different domains known as the N‐terminal helical domain, and two central and C‐terminal α/β (Rossmann‐like) domains. The homodimer contains five metalloclusters: two unique [Ni‐4Fe‐4S] clusters forming the active sites (namely C‐cluster), two [4Fe‐4S] clusters (namely B‐cluster), and one intermolecular [4Fe‐4S] cluster (namely d‐cluster). Later, the X‐ray structure of the bifunctional ACS/CODH enzyme from Moorella thermoacetica was solved.30, 31 In this case, the α2β2 tetrameric protein is formed by two CODH β‐subunits in the center and one ACS α‐subunit on each extremity. The ACS subunit contains three different domains: the N‐terminal domain, responsible for the interaction of ACS with CODH, the central domain and the C‐terminal domain, which contains the active site, called A‐cluster. The whole ACS‐CODH complex can adopt an open or closed state exhibiting significant conformational changes (Figure 2b). Only the open form contains an active dinuclear Ni site.30, 32 Moreover, the bifunctional enzyme contains an extensive cavity network, connecting the active sites by hydrophobic channels to allow diffusion of CO produced by CODH to the ACS active site. On this basis, a finely controlled tunnel gating mechanism has been proposed, regulating in this way the efficient CO diffusion inside the enzyme, avoiding at the same time the release of this toxic gas into the cell.
Standard [NiFe]‐hydrogenase are heterodimers consisting of a large subunit hosting the active site and a small subunit that binds FeS clusters needed for electron transfer. A common feature among all the available X‐ray structures is the highly buried position of the active site, which implies the presence of hydrophobic tunnels for dihydrogen access/release, as well as proton pathways in addition to electron relays.11 The archetypical structure of [NiFe]‐hydrogenase from Desulfovibrio gigas (Figure 2c) revealed the presence of three FeS clusters with one proximal and one distal [Fe4S4] clusters and a median [Fe3S4] cluster.33 The small subunit is composed of two domains: Domain I has a flavodoxin‐like fold containing the proximal cluster and domain II binds the median and distal FeS clusters. Domain II is absent from some NiFe‐hydrogenases. The large subunit possesses five domains, with the active sites between the two α/ß Domains I and II. The two subunits possess an extensive contact interface of about 3,500 Å2. Variations are found in [NiFeSe]‐hydrogenase that possesses a median [Fe4S4] instead of [Fe3S4] cluster34 and in O2‐tolerant enzymes which have a modified proximal cluster corresponding to a [Fe4S3] center.35 This heterodimeric prototype is often involved in protein complexes and interacts with various redox partners.
The crystal structure of MCR from Methanothermobacter marburgensis, reveals a hexamer arranged as a dimer of heterotrimers (αβϒ)2 (Figure 2d).36 The enzyme contains two spatially separated active sites, namely coenzyme F430, one in each α subunit. The X‐ray structures revealed that coenzymes F430, M, and B and the heterodisulfide CoM—S—S—CoB are embedded inside a narrow channel of 50 Å, from the protein surface to the buried active site.37 Residues from different subunits form the channel showing that one αβϒ trimer cannot constitute an active catalytic unit. This enzyme presents the ability to undergo several unusual posttranslational amino acid modifications located near the active site in the McrA subunit, mainly methylation of cysteine, histidine, arginine and glutamine residues catalyzed by S‐adenosyl‐l‐methionine (SAM) dependent N‐ and S‐methyltransferases.38 These posttranslational modifications could serve as important factors to increase enzyme stability.
The quaternary structure of NiSOD are exclusive to the SOD family.39 It forms a homo‐hexamer with each subunit adopting a four‐helix bundle fold (Figure 2e). The 7/8 N‐terminal residues of the mature protein form a “hook” protruding from the 4‐helix bundle to chelate the Ni active center. The topology of the biological unit possesses six independent active sites which do not interact with each other. Interestingly, the 14 amino acids at the N‐terminus are processed into a mature and active form of 117 amino acids leaving the Ni‐coordinating histidine as the first residue (Figure 1).
Currently, about 50 different structures of ureases have been deposited in the Protein Data Bank, most from bacteria.40 Among them, the most representative protein architecture corresponds to a trimer of trimers (αβϒ)3 in which the α subunit harbors the catalytic center, making a total count of three active sites per biological unit (Figure 2f).20 In other bacterial ureases, the quaternary structure corresponds to an (αβ)3 trimer in which the β subunit is a fusion of the β and ϒ subunits. In H. pylori, the enzyme is a tetramer of (αβ)3 trimers containing 12 active sites. In plants, the fusion of the three α, β, and ϒ subunits forms a single α subunit, organized as a dimer of homotrimers (α3)2. Despite variable quaternary structures, the secondary and tertiary structures of all ureases are very similar. The α subunits consist of a TIM barrel domain and a β‐sheet domain, the β subunits are located on the external surface of the trimer and are mainly composed of β‐sheets, and the γ subunits consist of domains containing both α‐helices and β‐sheets. Importantly, the α subunits possess a highly conserved flexible helix‐turn‐helix motif, flanking the active site, proposed to be involved in the modulation of substrates and products diffusion to and from the active site41 (Figures 1 and 2).
The intrinsic instability of LarA has limited the available knowledge of enzyme structure and function. Since 2014, studies conducted on lactate racemization in L. plantarum 27 and notably the determination of the crystal structure of holo‐LarA in 2015,42 have brought significantly new information on this enzyme, particularly its identification as a nickel enzyme. Surprisingly, although nickel is absolutely required for the activity, the metal easily dissociates from the enzyme. The determination of LarA structure in complex with its physiological metal was only possible thanks to the addition of sulfite, shown to stabilize LarA and delay spontaneous Ni leakage. Ni(II) is bound to the protein via a Ni‐pincer nucleotide (NPN), covalently bound to the protein by forming a thioamide with a lysine residue. LarA possesses 18 ß‐strands and 16 α‐helices arranged in a novel fold composed of two domains connected by two hinges (Figure 2g). The two molecules constituting the asymmetric unit adopt an open or closed form, with different degrees of closure upon the catalytic site. The closed conformation shields the nickel from the solvent thanks to the close proximity of the N‐ and C‐termini. The open conformation results in a loss of the nickel ion due to the easy accessibility of the active site to the solvent. This implies that the access of the substrate to the catalytic center has to be rapid to avoid enzyme deactivation and that LarA is most likely able to switch dynamically between open and closed conformations.
The structure of Glx I from E. coli is a homodimer with an overall fold similar to that of the human enzyme. Each subunit is made‐up of two mixed‐ß sheet domains. The dimer comprises two antiparallel monomers, with two independent metal active sites within curved‐ß sheets at the dimer interface (Figure 2h). The X‐ray structures of the enzyme in its apo‐form or in a complex with a series of divalent metals brought key information about the importance of the metal coordination geometry in the catalytic mechanism.43 Notably, the active forms of E. coli and human GlxI bind Ni(II) and Zn(II), respectively, adopting an octahedral geometry. In contrast, the E. coli enzyme binds Zn(II) in a trigonal bipyramidal geometry leading to an inactive form, supporting the need for an octahedral environment to promote the catalysis independently from the metal nature. The main structural differences between the two classes of Glyoxalases is the absence of a long N‐terminal arm in the Ni‐enzymes and deletions in three other regions. In particular, the absence of residues 73–87 has a dramatic impact on the metal activation profile.44
The first molecular structure of ARD was solved from NMR data on the enzyme from K. oxytoca.45 This enzyme belongs to the cupin structural superfamily, regrouping functionally diverse proteins possessing a conserved ß‐barrel fold (known as cupin motif; Figure 2i). Many other nonheme iron‐dependent oxygenases share the cupin motif. The Ni(II)‐ and Fe(II)‐forms of ARD have been shown to be separated chromatographically indicating that depending on the bound metal, the enzyme can adopt different conformations.23 The most readily observable structural difference between the two forms is the presence of a more disordered helix on top of the ß‐barrel in the Fe(II)‐ bound form, rendering the active site significantly more solvent accessible. On the opposite face, the movement of two helices induces a drastic re‐arrangement. This phenomenon is referred to as a structural entropy switch, with a disordered C‐terminus and an ordered N‐terminus in the Fe‐form while the opposite state is observed in the Ni‐form.45 Although the origin of these conformational changes are not obvious, the metal nature is likely to induce different selectivity to catalyze the two distinct reactions.
4. ACTIVE SITES
Protein environment has a great impact on the transition metals present in metalloenzyme active sites. Particularly, the solvent exposure as well as the nature of the donor ligands involved in metal coordination strongly determine the chemical properties of metallic active centers. The protein scaffold has a protective effect toward oxidative damage in anaerobic enzymes, where the active site is often localized in a buried position with low access to the solvent. This implicates the existence of dedicated channels controlling the diffusion of products and substrates, specific proton channels, and FeS clusters as electron relays between the active site and the physiological partner(s) (as in the case of CODH, ACS, hydrogenases, MCR; Figure 3). Locally, the nature of the coordinating ligands drives the redox properties of the metal, by adjusting the redox potentials required for biological processes and by giving access to the different redox states involved during the catalysis. In order to meet this demand, S‐donor rich ligand environments are favored in redox enzymes. In contrast, in nonredox enzymes where Ni(II) acts as a Lewis acid catalyst, O/N‐donor ligands are privileged. Protein crystallography has played a decisive role in the characterization of the active center of Ni‐enzymes.
Figure 3.
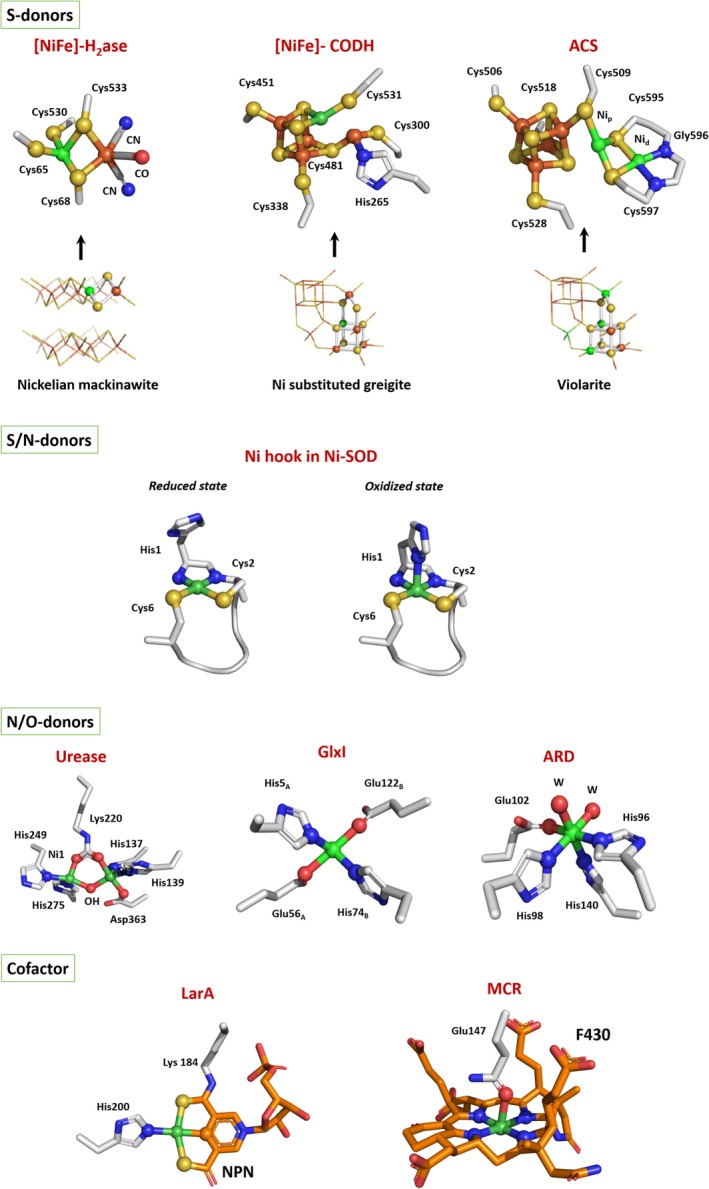
Active sites of Ni‐enzymes. S‐donors: comparison between natural sulfide nickelian mackinawite, greigite, and violarite and early nickel enzymes (adapted from Reference 46). Nickel and active site coordinating atoms are in spheres. Ni is in green, nitrogen in blue, sulfur in yellow, and oxygen in red. W, water molecule
4.1. Cysteine environment
CODH, ACS, and [NiFe]‐H2ase have in common a dinuclear Ni–Ni/Fe active site coordinated to the protein via one or several cysteine residues with a bridging position for substrate/product/intermediate binding in CODH and [NiFe]‐H2ase (Figure 3). These three enzymes are defined as pre‐Last Universal Common Ancestor (LUCA) enzymes whose active sites display structural affinities with natural metal‐sulfides catalysts.46
The determination of the first crystal structure of a [NiFe]‐H2ase and the characterization of its active site in 1995 had a considerable impact in the scientific community.33 First, the structure revealed the nature of the dinuclear NiFe active site. Second, the electron density map revealed the existence of three terminally bound diatomic ligands to Fe. From FTIR studies, they were assigned to one CO and two CN− ligands.47 Ni is coordinated to the protein via two CXXC motifs. Two cysteine thiolates (one from each motif) bridge Fe and Ni while the two other cysteine thiolates terminally binds Ni. In [NiFeSe]‐hydrogenases, one of the terminal cysteines is replaced by a seleno‐cysteine. The coordination sphere of Fe is completed by the three diatomic ligands, leaving an available Ni–Fe bridging site for substrate binding. The binding of the active site Fe to CO and CN− ligands promotes a low‐redox state of the metal ion, making it a relatively soft Lewis acid, favoring its binding to the soft Lewis base H−, a probable step in catalysis. During catalysis, nickel undergoes a redox transition from Ni(II) to Ni(III).11
X‐ray structure of CODH revealed the unique nature of the active site, a distorted [NiFe3S4] cubane coordinated to a mononuclear Fe site (Fe1) (Figure 3).29 The protein provides a terminal cysteine thiolate ligand to both Ni and Fe1 and a histidine ligand to Fe1. Ni coordination is completed by two inorganic S from the cluster and a labile position where the substrate/product (CO or CO2) binds while a water molecule binds to Fe1.28 Crystal structures of ACS/CODH revealed the nature of the active site (A‐cluster) of ACS.30 This dinuclear site consists of a proximal Ni (Nip), which is the catalytic center, linked to a [4Fe‐4S] cluster via a bridging cysteine thiolate. Nip is bridged to a distal Ni (Nid) via two additional cysteine thiolates. A labile position is present for substrate and/or intermediates binding. Concerning the Nid, its coordination is held by two thiolates and two backbone amide groups (one from Gly and one from Cys) from an unusual Cys‐Gly‐Cys motif, in a S2N2 square planar geometry. Nid is postulated not to be involved in redox chemistry and to remain in the Ni(II) state. It has been proposed to have a role either as a supporting ligand to stabilize the proximal nickel in a low‐valent redox level or for the generation of an open site on the catalytic metal thanks to its hemilabile ring‐opening property.48, 49 Nip, on the other hand, presents different oxidation states during the catalysis.
The identification of NiSOD as a redox enzyme highlighted the role of the protein environment of the Ni center to both support the Ni(III) oxidation state and lower the potential of the Ni(III)/(II) couple by over 2 V. The metal binding site is in fact formed by the N‐terminal “Ni‐hook,” which coordinates a Ni ion via the thiolate and amidate groups of Cysteine 2, the thiolate group of Cysteine 6 and the amine and imidazole groups of histidine 1.16 The binding coordination of nickel changes according to the metal oxidation state, switching from a pyramidal geometry for the oxidized Ni(III) state to a N2S2 square planar geometry for the reduced Ni(II) one, losing the apical imidazole donor (Figure 3). While the thiolate and imidazole ligands are critical to maintain the nickel ion redox state, the role of the mixed amine‐amidate coordination is the subject of different studies.50, 51, 52 Several NiSOD model peptides and complexes have been synthesized in which the N‐donor ligands were varied from the natural mixed amine/amidate environment to a bis‐amidate one, leading exclusively to catalytically inactive species. One reason would be that bis‐amidate complexes are considered to be more reactive toward O2. Thus, the mixed amine/amidate coordination would be necessary to increase the resistance toward substrates and products of the reaction.
4.2. Histidine‐rich environment
Histidine‐rich environments are found in three nonredox enzymes, namely, urease, ARD, and GlxI where nickel is in a Ni(II) oxidation state (Figure 3).
In the case of urease, the active site is a di‐nuclear center, with the two nickel ions spatially separated by 3.5–3.7 Å, bridged by a carbamylated lysine residue and coordinated by two histidine residues.20 Water molecules fill the remaining coordination positions, yielding a pentacoordinated distorted square‐pyramidal based geometry (Ni1), while the second nickel (Ni2) is additionally bound to an aspartate resulting in a hexacoordinated distorted octahedral geometry. Recently, the X‐ray structure of urease in complex with its substrate has been solved, showing that urea binds in a bridging position between the two Ni sites.53
In GlxI, Nickel coordination includes a histidine and a glutamic acid of each monomer as well as two water molecules to complete the octahedral geometry required for catalysis. In ARD, the ligand characterization by spectroscopic and mutagenesis studies, suggested an octahedral coordination geometry for both metals involving three histidines and one glutamic acid as possible donors supplemented by two additional waters.43
4.3. Ni‐containing cofactors
In methanogens, unique cofactors have evolved which are restricted to this group of archaea. In MCR, nickel was shown to be a constituent of a low molecular weight cofactor (F430) identified as a tetrapyrrole derivative, also known as corphin (Figure 3).37 This is the most reduced tetrapyrrole‐metal complex found in nature. The name of the cofactor derives from its absorption band, precisely at 430 nm, in the Ni (II) oxidation state. A Ni(I) state is also involved during catalysis, implying that a tight control of the metal redox state is required for its functionality. The F430 is not covalently bound to the protein. The ring of the F430 cofactor chelates the nickel ion and its coordination is completed via an axial glutamine 147 on the opposite side compared to the channel. Trans to the latter, there is vacant site available for substrate and product binding.
In LpLarA, a nicotinic acid mononucleotide derivate with two added thiocarboxylate groups, one of which forms a thioamide with the lysine 184, is present in the active site and is called nickel‐pincer nucleotide (NPN)42 (Figure 3). Pincer complexes are not new in chemistry but notably this is the only example of such a molecule in an enzyme. The nickel is bound in a distorted planar arrangement to the NPN cofactor via a SCS ligand environment based on a pyridinium‐3,5‐bisthiocarboxylic acid mononucleotide. The cofactor is covalently bound to the protein via a thioamide bond with a lysine residue and the nickel coordination sphere is completed by histidine 200. The stable Ni—C bond in the resting/stable state of this enzyme is unexpected. Indeed, in other Ni‐containing enzymes, Ni—C bonds have only been observed as reaction intermediates.18 This coordination is essential to guarantee the stability of the metal in the complex. In fact, it appears that the switch between the open and closed conformations is due to a distortion of the NPN pincer coordination and displacement of the His200.
5. METALLOCENTER BIOSYNTHESIS
In the cell, nickel has to be correctly delivered and incorporated into the desired enzyme active sites. These processes often require an intricate team of accessory proteins, working in concert for the biosynthesis of these complex metalloclusters enzymes. The addition of Ni(II) excess to microbial cultures can often compensate for the lack of their dedicated accessory proteins, leading to partly activated enzymes. It suggests that the maturation process can operate spontaneously, but not through a fully effective process. Moreover, considering the low nickel levels present in a cell, the maturation machinery is necessary and works in a well‐organized manner in space and time. While nickel chaperones involved in the biogenesis of several active sites have been thoroughly characterized, understanding their role at the molecular level in the nickel delivery steps remains to be clarified. In fact, these studies are made highly complex by the modularity of the nickel‐binding properties and allosteric regulations induced by protein/protein interactions or protein/metal interactions. In the next section, the maturation pathways of urease, H2ase, LarA, MCR, CODH, and ACS are discussed. Concerning the other enzymes, little is known about the biosynthesis of their active sites. All the described maturation machineries have in common the presence of an NTP‐hydrolyzing maturation factor, belonging to the SIMIBI‐class of NTPases,54 a histidine‐rich protein (HRP) and eventually additional nickel chaperones.18 NTPases potentially play a regulatory role by affecting interaction of partners in protein complexes or nickel ion affinity while HRP are often described as storage proteins.
5.1. Urease
Urease is first produced in its apo‐form, which then needs to be activated through two main steps: lysine carbamylation and nickel insertion into the active site.20 Considering the buried position of this site in the apo‐protein, the occurrence of a protein folding modification is predicted to allow nickel insertion. In addition to the three enzyme subunits, the urease gene cluster encodes four accessory proteins: UreD/H, UreE, UreF, and UreG, involved in nickel delivery and urease maturation. The urease maturation is dependent on GTP hydrolysis catalyzed by the nickel‐dependent GTPase UreG,80 and requires the formation of an UreFGH‐Urease complex. On the other hand, UreE is described as the metallochaperone that provides Ni(II) to the enzyme,81, 82 via its interaction with UreG. Interestingly, UreG has been identified as an intrinsically disordered protein (IDP), with a very low GTPase activity that is greatly enhanced by the complex formation.80 The UreH2F2G2 complex has been isolated and crystallized,83 revealing the existence of a water tunnel originating from the Ni‐binding site of UreG located at the dimer interface, passing through UreF and exiting UreD/H84 to finally provide Ni(II) to urease active site, as supported by structural models.
In view of the most recent results, a model was postulated for the overall maturation pathway of urease, in which the role of conformational changes in UreG during the GTP hydrolyzing/partner binding is highlighted (Figure 4).85 In this scenario, the UreH2F2G2 complex is destabilized by GTP‐binding, causing the release of UreG2 and allowing its interaction with Ni‐UreE2 to form the Ni‐(UreEG)2 complex. The Ni(II) transfer from UreE to UreG is followed by the complex dissociation and the urease‐bound (UreHF)2 complex will then interact with Ni‐(UreG)2. The subsequent GTP hydrolysis induces conformational changes promoting the nickel release from UreG to apo‐urease, via UreH and UreF. TheGDP‐UreG2 prefers then to bind to (UreHF)2, regenerating the UreH2F2G2.
Figure 4.

Maturation of urease. In this mechanism, the nickel‐charged UreG dimer is recruited to form the activation complex with apo‐urease and UreF2H2. GTP hydrolysis promotes the nickel release for urease maturation (adapted from Reference 85)
5.2. [NiFe]‐hydrogenase
The biosynthesis of [Ni—Fe]‐hydrogenase dinuclear active site is a multistep process, with three consecutive events: (i) Fe(CN)2CO center biosynthesis and insertion followed by (ii) nickel delivery and finally (iii) the removal of the C‐terminal tail (CTT) of the large subunit (LSU) to lock the active site in place (Figure 5).55 Once processed, the mature LSU associates to the small subunit (SSU) and in the case of membrane‐bound and periplasmic enzymes, the mature dimeric form is exported via the Twin‐Arginine Translocation (TAT) system. At least six strictly conserved accessory proteins are required, namely HypABCDEF,56 for the active site biosynthesis, in addition to the nickel chaperone SlyD found in some microorganisms57 and the specific protease cleaving the CTT (HycI in E. coli). In detail, the LSU is maturated according to the following steps. At first, HypEF mediates the formation of the Fe(CN)2CO center in concert with HypCD. HypC is assumed to coordinate the Fe(CN)2CO moiety via Cys2 together with the Cys41 of HypD, and to interact directly with the LSU precursor. This step is followed by the insertion of this Fe center into the protein.
Figure 5.

Maturation of [NiFe]‐hydrogenase. The proposed mechanism for the enzyme biosynthesis is divided into three successive steps. 1. The Fe(CN)2CO moiety is synthesized prior to its insertion into the enzyme. 2. Nickel is inserted directly into the enzyme (adapted from Reference 60). 3. The proteolysis of the C‐terminal tail is necessary for the enzyme activation step (adapted from Reference 64)
The insertion of Ni occurs via a NTP‐dependent mechanism mediated by the Ni/Zn‐dependent NTPase HypB,58 the nickel chaperone HypA,59 and in some case SlyD. In H. pylori, HypA and HypB are also recruited for the maturation of urease, through their respective interaction with UreE and UreG.21 In E. coli, HypB clearly interacts individually with HypA and SlyD, but does not interact directly with the LSU.60 The role of SlyD is not well understood despite its high affinity (in the nM range) for HypB: it is thought to facilitate Ni(II) release from HypB toward other partners,61 but also prevents HypB‐HypA interaction in competition experiments, suggesting a stepwise process with HypB as a central component.60 HypA contains two metal site, a structural Zn(II) site and a unique Ni(II) binding site at the N‐terminus. Recently, a dual NMR and computational approach revealed that HypA is an elongated protein with two metal‐binding domains connected by a flexible linker, with Ni(II)‐binding stabilizing the N‐terminal helix and probably impacting its interaction with its partner.59 In the current model, HypA serves as a nickel relay from HypB to LSU precursor. HypB possesses a strictly conserved Ni/Zn‐binding site embedded in the NTPase domain (G‐domain), while additional Histidine‐rich clusters and/or a N‐terminal high affinity site are present in some cases, whose function is not well understood.62 Recent studies proposed that GTP hydrolysis governs the interaction between HypA and HypB via the formation of a Ni‐HypB2A complex with Ni(II) being rapidly transferred to HypA from GDP‐loaded HypB2 induced by a drastic conformational change and an increased affinity of HypA for Ni(II) once in complex with HypB, followed by the dissociation of the HypAB2 complex and the formation of Ni‐HypA/LSU complex.63
The proteolytic cleavage of the C‐terminal tail of LSU is a nickel‐promoted key step in the biosynthetic process of [NiFe]‐H2ases, highlighting the pivotal role of Ni in endopeptidase recognition during the maturation process.64 The role of the CTT is to allow the interaction with the Hyp machinery while avoiding the premature interaction with the SSU. The recent crystal structure of the premature LSU in complex with HypA revealed that Ni insertion into the active site induces spatial rearrangements in LSU. In this scenario, the CTT adopts a ß‐strand conformation and forms a ß‐sheet with the N‐terminal ß‐strands, preventing the endopeptidase accessing its recognition site. Ni‐loaded HypA interacts with both the N‐terminus and the C‐terminal extension, putting in close proximity the Ni‐binding site of HypA to the active site of H2ase likely allowing a direct metal transfer. Ni interaction with one of the cysteine‐coordinating ligand within the active site induces the conformational change and the release and solvent exposure of the CTT. Additional molecular dynamical studies revealed that the CTT is an intrinsically disordered region (IDR), facilitating its recognition by the protease and the subsequent formation of the mature enzyme.65
Despite extensive studies on this enzyme, all the data presented here highlight the difficulty to identify clearly a precise mechanism for the active site biosynthesis and to propose a universal model for H2ase maturation.
5.3. Carbon monoxide dehydrogenase
As for hydrogenase, the maturation of [NiFe]‐CODH is a multistep process involving two consecutive events: an initial FeS cluster insertion followed by the nickel delivery. The biosynthesis process of C‐cluster starts with the insertion of Fe and S atoms, to form a Fe4S4 cluster, likely performed by the classical ISC/SUF machineries responsible for FeS cluster biogenesis, as shown by the production of a fully FeS‐loaded CODH coproduced in E. coli with the ISC machinery.66 The following step is the insertion of the Ni atom thanks to three nickel‐binding proteins: CooC, CooT, and CooJ.67 Phylogenetic analyses showed that CooC is widely distributed in microorganisms possessing a [NiFe]‐CODH, generally in operon with the enzyme, and is essential for CODH maturation under physiological conditions.68 On the contrary, CooT and CooJ are more difficult to identify in genomes. Although CooC is described as the major player in CODH maturation, CooJ and CooT are nevertheless relevant for nickel delivery to CODH in R. rubrum in vivo.67 Moreover, recent phylogenetic analyses highlighted the existence of at least 111 CooT homologues in anaerobic archaea and bacteria69 and at least 46 CooJ homologues in bacteria,70 suggesting different maturation pathways for CODH with CooC +/− CooT and/or CooJ. CooC is a nickel‐dependent ATPase belonging to the same class as UreG and HypB and displaying a similar cross‐correlation between nucleotide hydrolysis and nickel binding.71 The hypothesis is that the enzyme cycles dynamically between an open Ni(II) state and a closed ATP‐dependent state that drives the metal release.72 The recent characterization of CooJ from R. rubrum revealed that the protein is a homodimer comprising a coiled‐coil flanked by two distinct histidine tails.70 All CooJ homologs possess two spatially separated nickel‐binding motifs: a variable C‐terminal histidine tail and a strictly conserved “H‐(W/F)‐X2‐H‐X3‐H” motif, binding Ni(II) in the nanomolar range, suggesting a dual function for CooJ both as a Ni chaperone and as a Ni storage protein.73 CooT is an additional nickel chaperone displaying specific features. The 111 CooT homologues show a remarkably large variability, with Cys2 being the only strictly conserved residue. The evolutionary conservation of the solvent‐exposed cysteine in CooT protein suggests its importance to meet a specific and essential functional role that remains to be discovered. In the CooT family, two different Ni(II)‐binding motifs, namely the “Cys2” (about 30% of CooT proteins) and the “Cys2His55” motifs were identified.74 A recent study on the “Cys2” motif group suggests that the diamagnetic Ni(II) adopts a square‐planar geometry with bis‐amidate/bis‐thiolate ligands, coordinated by a single cysteine motif. This would be unprecedented in biology and raises the question of its role in the activation of CODH at the molecular level.
Mössbauer and EPR studies showed that the mononuclear Fe site (Fe1) present in the C‐cluster in RrCODH is only observed in the presence of nickel. Therefore, nickel incorporation into C‐cluster likely alters the coordination environment of one iron atom.75 Site‐directed mutagenesis of residues coordinating C‐cluster in RrCODH highlighted that the only critical residue required for Ni binding is His265.76 Surprisingly, all cysteine mutants, even the one affecting the residue directly involved in Ni coordination, contain one Ni equivalent per monomer, suggesting that the Ni(II) ion is bound to another position in the protein, forming a putative intermediate state. Conversely, His265 that coordinates the mononuclear Fe1 ion, is an essential residue in the C‐cluster biogenesis. If the C‐cluster precursor is a classical Fe4S4 cubane cluster, as shown by spectroscopic studies, nickel insertion would imply bond breaking, involving His265 and Cys300. Very recent structural studies on CODH from Desulfovibrio vulgaris (DvCODH) revealed the great plasticity of the C‐cluster and are in agreement with the “bond breaking” theory, with His266 (His265 in RrCODH) and Cys302 (Cys300 in RrCODH) serving as the nickel binding site before its transfer to its stable active site (Figure 6a.77, 78 In DvCODH, the d‐cluster is a Fe2S2 center that would be involved in nickel insertion by mediating electron transfer, working in concert with CooC to control the redox state of the active site, ensuring thus the right metalation of the cluster. The necessity of a reduced cluster intermediate in the mechanism is reinforced by the requirement of reducing conditions to activate Ni‐deficient CODH in vitro by addition of nickel salts in solution. Although never observed spectroscopically, a [Fe3S4]‐Fe1 precluster would be a key intermediate in the C‐cluster biosynthesis, just before nickel insertion. To date, many uncertainties exist regarding nickel insertion into the C‐cluster, a mechanism that appears to be nonstraightforward and likely complicated by the presence of the Fe1 site.
Figure 6.

Maturation of ACS/CODH. (a) Proposed mechanism for CODH maturation: Fe and S atoms are first inserted to form a classical [Fe4S4] cluster. The following steps remain unclear to yield the active C‐cluster (adapted from Reference 77). (b) Proposed mechanism for ACS maturation (adapted from Reference 79)
5.4. Acetyl coenzyme A synthase
Little is known about the maturation of ACS and how the peculiar A‐cluster is formed and inserted. As for CODH and [NiFe]‐H2ase, nickel insertion seems to be the second step in the enzyme biosynthesis. Recently, the ATPase AcsF was shown to be involved in the active site biogenesis.79 Initially, two major groups of CooC proteins have been identified depending on their genomic context: the “CooC‐like” group, positioned in the monofunctional CODH operons and the “AcsF‐like” group, found in the ACS/CODH operons (or gene clusters). The study of AcsF from C. hydrogenoformans revealed that the enzyme is actually specifically required for the activation of ACS and cannot be replaced by CooC proteins. In light of these results, the division in two distinct classes of CooC and AcsF involved in CODH and ACS maturation, respectively, should be considered. AcsF/ACS complex formation was proposed to induce the creation of at least one high‐affinity Ni‐binding site (Figure 6b).
5.5. Lactate racemase
Among the lar gene cluster, LarB is responsible for the biosynthesis of pyridinium 3,5‐biscarboxylic acid mononucleotide (P2CMN) by catalyzing the carboxylation of nicotinic acid adenine dinucleotide (NaAD).86 Subsequently, LarE converts P2CMN into pyridinium 3,5‐biscarboxylic acid mononucleotide (P2TMN) via two sulfur transfer reactions and LarC provides the nickel to generate the active form of the pincer cofactor that will bind to LarA.87, 88 LarE belongs to the PP‐loop pyrophosphatase family and possesses two independent domains: a N‐terminal domain containing the conserved PP‐loop SGGXDS motif and a C‐terminal domain which is not common to any member of this N‐type ATPase family, revealing a double function for LarE. The enzyme catalyzes the AMPylation of the pyridinium carboxyl groups followed by a sulfur transfer, via the conserved cysteine 176 present in the C‐terminal part of the protein as a sulfur donor to convert P2CMN to P2TMN. This hypothesis has been recently proven thanks to the determination of the crystal structures of LarE from L. plantarum and its C176A mutant, in their apo‐form and in complex with coenzyme A (CoA).87 This in vitro study revealed the essential role of Cys176 as the catalytic site for the sulfur insertion, as well as the role of CoA persulfilde in the regeneration of active LarE, although the physiological relevance of this cycling mechanism remains unclear. Finally, LarC would directly insert nickel into the incomplete pincer ligand, still bound to LarE, promoting the formation of the nickel‐pincer nucleotide, which then would be transferred to LarA (Figure 7a).89 LarC forms two S—Ni bonds and a peculiar stable C—Ni bond. In contrast with the other nickel accessory proteins, it is the only example able to catalyze the formation of a C—Ni bond by insertion of the metal into an organic molecule, via a CTP‐dependent cyclometalation, inducing a possible conformational modification. From LarC sequence, a His‐rich region, not strictly required for LarC activity, was identified as a possible nickel storage. The structure of the C‐terminal domain was solved in the presence or absence of CTP, allowing the identification of a peculiar binding site that does not correspond to any known nucleotide‐binding motif.90 However, how the CTP hydrolysis is related to the nickel insertion still remains to be investigated.
Figure 7.

Maturation of LarA and MCR. (a). Proposed mechanism for the biosynthesis of the nickel‐pincer nucleotide, followed by its incorporation into LarA (adapted from Reference 89). (b). Biosynthetic pathway of coenzyme F430 by the Cfb machinery (adapted from Reference 91) and proposed working model for MCR assembly (adapted from Reference 93)
5.6. MCR
The presence of the coenzyme F430 in MCR led to the search for specific chelatases, known to catalyze the metalation of naturally occurring tetrapyrolles. A few years ago, five genes designated cfbABCDE, conserved in all methanogens, were identified as responsible for the biosynthesis of this specific cofactor.91, 92 Four enzymatic steps are required to transform the sirohydrochlorin precursor into coenzyme F430 (Figure 7b). The first step is performed by the CfbA protein shown to be the specific nickel chelatase (CfbA) promoting the insertion of Ni into sirohydrochlorin; Then, two mechanisms are proposed with CfbB or CfbE catalyzing the amidation of two carboxylic moieties to yield the Ni‐sirohydrochlorin a, c amide intermediate. Following a reducing step thanks to a Fe protein CfbC and a MoFe protein CfbD, both homologs to NifH and NifD proteins, respectively, implicated in nitrogenase biosynthesis. Finally, CfbE or CfbF could act as an activator of the g‐propionate side chain for intramolecular C—C bond formation to produce the carboxylic F‐ring and thus act as a coenzyme F430 synthase. At this point, the assembly of the active MCR is not fully understood. The mcrBDCGA gene cluster contains two conserved genes, mcrC and mcrD whose function is unknown. A working model has been proposed where McrB would serve as a scaffold for the assembly of the other subunits (Figure 7b).93 The McrBDGA complex would be formed first. McrD would both prevent the folding of the McrA subunit to ensure the correct posttranslational modifications and facilitate the insertion of the coenzyme F430 that would lead to the release of McrD before a final activation step in which McrC could belong to a reductase complex allowing the formation and stabilization of the Ni(I) active state.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest with the contents of this article.
ACKNOWLEDGMENTS
This work was supported by “the ITERLIS PhD program, CEA Life sciences” for MA's PhD funding. This work has been partially supported by Labex ARCANE and CBH‐EUR‐GS (ANR‐17‐EURE‐0003).
Alfano M, Cavazza C. Structure, function, and biosynthesis of nickel‐dependent enzymes. Protein Science. 2020;29:1071–1089. 10.1002/pro.3836
Funding information Commissariat à l'Énergie Atomique et aux Énergies Alternatives
REFERENCES
- 1. Konhauser KO, Pecoits E, Lalonde SV, et al. Oceanic nickel depletion and a methanogen famine before the great oxidation event. Nature. 2009;458:750–753. [DOI] [PubMed] [Google Scholar]
- 2. Thauer RK. My lifelong passion for biochemistry and anaerobic microorganisms. Annu Rev Microbiol. 2015;69:1–30. [DOI] [PubMed] [Google Scholar]
- 3. Konhauser KO, Robbins LJ, Pecoits E, Peacock C, Kappler A, Lalonde SV. The archean nickel famine revisited. Astrobiology. 2015;15:804–815. [DOI] [PubMed] [Google Scholar]
- 4. Wang SJ, Rudnick RL, Gaschnig RM, Wang H, Wasylenki LE. Methanogenesis sustained by sulfide weathering during the great oxidation event. Nat Geosci. 2019;12:296–300. [Google Scholar]
- 5. Raymond J, Segrè D. The effect of oxygen on biochemical networks and the evolution of complex life. Science. 2006;311:1764–1767. [DOI] [PubMed] [Google Scholar]
- 6. Fontecilla‐Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A. Structure‐function relationships of anaerobic gas‐processing metalloenzymes. Nature. 2009;460:814–822. [DOI] [PubMed] [Google Scholar]
- 7. Boer JL, Mulrooney SB, Hausinger RP. Nickel‐dependent metalloenzymes. Arch Biochem Biophys. 2014;544:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zambelli B, Uversky VN, Ciurli S. Nickel impact on human health: An intrinsic disorder perspective. Biochim Biophys Acta Proteins Proteom. 2016;1864:1714–1731. [DOI] [PubMed] [Google Scholar]
- 9. MacOmber L, Hausinger RP. Mechanisms of nickel toxicity in microorganisms. Metallomics. 2011;3:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li Y, Zamble DB. Nickel homeostasis and nickel regulation: An overview. Chem Rev. 2009;109:4617–4643. [DOI] [PubMed] [Google Scholar]
- 11. Fontecilla‐Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/function relationships of [NiFe]‐ and [FeFe]‐hydrogenases. Chem Rev. 2007;107:4273–4303. [DOI] [PubMed] [Google Scholar]
- 12. Robb FT, Techtmann SM. Life on the fringe: Microbial adaptation to growth on carbon monoxide. F1000Research. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Adam PS, Borrel G, Gribaldo S. Evolutionary history of carbon monoxide dehydrogenase/acetyl‐CoA synthase, one of the oldest enzymatic complexes. Proc Natl Acad Sci U S A. 2018;115:E1166–E1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Evans PN, Boyd JA, Leu AO, et al. An evolving view of methane metabolism in the Archaea. Nat Rev Microbiol. 2019;17:219–232. [DOI] [PubMed] [Google Scholar]
- 15. Kietäväinen R, Purkamo L. The origin, source, and cycling of methane in deep crystalline rock biosphere. Front Microbiol. 2015;6:725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sheng Y, Abreu IA, Cabelli DE, et al. Superoxide dismutases and superoxide reductases. Chem Rev. 2014;114:3854–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Youn HD, Kim EJ, Roe JH, Hah YC, Kang SO. A novel nickel‐containing superoxide dismutase from Streptomyces spp. Biochem J. 1996;318:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zamble D. Introduction to the Biological Chemistry of Nickel. RSC Metallobiology 2017;2017:1–11. [Google Scholar]
- 19. Dixon NE, Gazzola C, Blakeley RL, Zerner B. Jack bean urease (EC 3.5.1.5). A metalloenzyme. A simple biological role for nickel? J Am Chem Soc. 1975;97:4131–4133. [DOI] [PubMed] [Google Scholar]
- 20. Mazzei L, Musiani F, Ciurli S. Urease. RSC Metallobiology. Vol. 2017. pp. 60–97; 2017.
- 21. De Reuse H, Vinella D, Cavazza C. Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of helicobacter pylori. Front Cell Infect Microbiol. 2013;3:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Clugston SL, Barnard JFJ, Kinach R, et al. Overproduction and characterization of a dimeric non‐zinc glyoxalase I from Escherichia coli: Evidence for optimal activation by nickel ions. Biochemistry. 1998;37:8754–8763. [DOI] [PubMed] [Google Scholar]
- 23. Dai Y, Wensink PC, Abeles RH. One protein, two enzymes. J Biol Chem. 1999;274:1193–1195. [DOI] [PubMed] [Google Scholar]
- 24. Chai SC, Ju T, Dang M, Goldsmith RB, Maroney MJ, Pochapsky TC. Characterization of metal binding in the active sites of acireductone dioxygenase isoforms from Klebsiella ATCC 8724. Biochemistry. 2008;47:2428–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Deshpande AR, Wagenpfeil K, Pochapsky TC, Petsko GA, Ringe D. Metal‐dependent function of a mammalian acireductone dioxygenase. Biochemistry. 2016;55:1398–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Miłaczewska A, Kot E, Amaya JA, et al. On the structure and reaction mechanism of human acireductone dioxygenase. Chemistry. 2018;24:5225–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Desguin B, Goffin P, Viaene E, et al. Lactate racemase is a nickel‐dependent enzyme activated by a widespread maturation system. Nat Commun. 2014;5:3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Drennan CL, Heo J, Sintchak MD, Schreiter E, Ludden PW. Life on carbon monoxide: X‐ray structure of Rhodospirillum rubrum Ni‐Fe‐S carbon monoxide dehydrogenase. Proc Natl Acad Sci U S A. 2001;98:11973–11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dobbek H, Svetlitchnyi V, Gremer L, Huber R, Meyer O. Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni‐4Fe‐5S] cluster. Science. 2001;293:1281–1285. [DOI] [PubMed] [Google Scholar]
- 30. Darnault C, Volbeda A, Kim EJ, et al. Ni‐Zn‐[Fe4‐S4] and Ni‐Ni‐[Fe4‐S4] clusters in closed and open α subunits of acetyl‐CoA synthase/carbon monoxide dehydrogenase. Nat Struct Biol. 2003;10:271–279. [DOI] [PubMed] [Google Scholar]
- 31. Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. A Ni‐Fe‐cu center in a bifunctional carbon monoxide dehydrogenase/acetyl‐CoA synthase. Science. 2002;298:567–572. [DOI] [PubMed] [Google Scholar]
- 32. Gencic S, Grahame DA. Nickel in subunit β of the acetyl‐CoA decarbonylase/synthase multienzyme complex in methanogens: Catalytic properties and evidence for a binuclear Ni‐Ni site. J Biol Chem. 2003;278:6101–6110. [DOI] [PubMed] [Google Scholar]
- 33. Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla‐Camps JC. Crystal structure of the nickel‐iron hydrogenase from Desulfovibrio gigas . Nature. 1995;373:580–587. [DOI] [PubMed] [Google Scholar]
- 34. Garcin E, Vernede X, Hatchikian EC, Volbeda A, Frey M, Fontecilla‐Camps JC. The crystal structure of a reduced [NiFeSe] hydrogenase provides an image of the activated catalytic center. Structure. 1999;7:557–566. [DOI] [PubMed] [Google Scholar]
- 35. Volbeda A, Amara P, Darnault C, et al. X‐ray crystallographic and computational studies of the O 2‐tolerant [NiFe]‐hydrogenase 1 from Escherichia coli . Proc Natl Acad Sci U S A. 2012;109:5305–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ermler U, Grabarse W, Shima S, Goubeaud M, Thauer RK. Crystal structure of methyl‐coenzyme M reductase: The key enzyme of biological methane formation. Science. 1997;278:1457–1462. [DOI] [PubMed] [Google Scholar]
- 37. Cedervall PE, Dey M, Li X, et al. Structural analysis of a Ni‐methyl species in methyl‐coenzyme M reductase from methanothermobacter marburgensis. J Am Chem Soc. 2011;133:5626–5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deobald D, Adrian L, Schöne C, Rother M, Layer G. Identification of a unique radical SAM methyltransferase required for the sp3‐C‐methylation of an arginine residue of methyl‐coenzyme M reductase. Sci Rep. 2018;8:7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wuerges J, Lee JW, Yim YI, Yim HS, Kang SO, Carugo KD. Crystal structure of nickel‐containing superoxide dismutase reveals another type of active site. Proc Natl Acad Sci U S A. 2004;101:8569–8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kappaun K, Piovesan AR, Carlini CR, Ligabue‐Braun R. Ureases: Historical aspects, catalytic, and non‐catalytic properties—A review. J Adv Res. 2018;13:3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ciurli S, Benini S, Rypniewski WR, Wilson KS, Miletti S, Mangani S. Structural properties of the nickel ions in urease: Novel insights into the catalytic and inhibition mechanisms. Coordination Chemistry Reviews. 1999;190–192:331–355. [Google Scholar]
- 42. Desguin B, Zhang T, Soumillion P, Hols P, Hu J, Hausinger RP. A tethered niacin‐derived pincer complex with a nickel‐carbon bond in lactate racemase. Science. 2015;349:66–69. [DOI] [PubMed] [Google Scholar]
- 43. He MM, Clugston SL, Honek JF, Matthews BW. Determination of the structure of Escherichia coli glyoxalase I suggests a structural basis for differential metal activation. Biochemistry. 2000;39:8719–8727. [DOI] [PubMed] [Google Scholar]
- 44. Suttisansanee U, Ran Y, Mullings KY, Sukdeo N, Honek JF. Modulating glyoxalase i metal selectivity by deletional mutagenesis: Underlying structural factors contributing to nickel activation profiles. Metallomics. 2015;7:605–612. [DOI] [PubMed] [Google Scholar]
- 45. Pochapsky TC, Pochapsky SS, Ju T, Hoefler C, Liang J. A refined model for the structure of acireductone dioxygenase from Klebsiella ATCC 8724 incorporating residual dipolar couplings. J Biomol NMR. 2006;34:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nitschke W, McGlynn SE, Milner‐White EJ, Russell MJ. On the antiquity of metalloenzymes and their substrates in bioenergetics. Biochim Biophys Acta Bioenerg. 2013;1827:871–881. [DOI] [PubMed] [Google Scholar]
- 47. Pierik AJ, Roseboom W, Happe RP, Bagley KA, Albracht SPJ. Carbon monoxide and cyanide as intrinsic ligands to iron in the active site of [NiFe]‐hydrogenases. NiFe(CN)2CO, biology's way to activate H2. J Biol Chem. 1999;274:3331–3337. [DOI] [PubMed] [Google Scholar]
- 48. Krishnan R, Riordan CG. Cys‐Gly‐Cys tripeptide complexes of nickel: Binuclear analogues for the catalytic site in acetyl coenzyme a synthase. J Am Chem Soc. 2004;126:4484–4485. [DOI] [PubMed] [Google Scholar]
- 49. Golden ML, Reibenspies JH, Darensbourg MY. Capture of NiII, CuI and ZnII by thiolate sulfurs of an N2S2Ni complex: A role for a metallothiolate ligand in the acetyl‐coenzyme a synthase active site. Chem Commun. 2003;3:1824–1825. [DOI] [PubMed] [Google Scholar]
- 50. Huang HT, Dillon S, Ryan KC, et al. The role of mixed amine/amide ligation in nickel superoxide dismutase. Inorg Chem. 2018;57:12521–12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mathrubootham V, Thomas J, Staples R, McCraken J, Shearer J, Hegg EL. Bisamidate and mixed amine/amidate NiN2S2 complexes as models for nickel‐containing acetyl coenzyme a synthase and superoxide dismutase: An experimental and computational study. Inorg Chem. 2010;49:5393–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tietze D, Sartorius J, Koley Seth B, et al. New insights into the mechanism of nickel superoxide degradation from studies of model peptides. Sci Rep. 2017;7:17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mazzei L, Cianci M, Benini S, Ciurli S. The structure of the elusive urease–urea complex unveils the mechanism of a paradigmatic nickel‐dependent enzyme. Angew Chem Int Ed. 2019;58:7415–7419. [DOI] [PubMed] [Google Scholar]
- 54. Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P‐loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. [DOI] [PubMed] [Google Scholar]
- 55. Lacasse MJ, Zamble DB. [NiFe]‐hydrogenase maturation. Biochemistry. 2016;55:1689–1701. [DOI] [PubMed] [Google Scholar]
- 56. Böck A, King PW, Blokesch M, Posewitz MC. Maturation of hydrogenases. Adv Microb Physiol. 2006;51:1–71. [DOI] [PubMed] [Google Scholar]
- 57. Kaluarachchi H, Sutherland DEK, Young A, Pickering IJ, Stillman MJ, Zamble DB. The Ni(II)‐binding properties of the metallochaperone SlyD. J Am Chem Soc. 2009;131:18489–18500. [DOI] [PubMed] [Google Scholar]
- 58. Sydor AM, Lebrette H, Ariyakumaran R, Cavazza C, Zamble DB. Relationship between Ni(II) and Zn(II) coordination and nucleotide binding by the helicobacter pylori [NiFe]‐hydrogenase and urease maturation factor HypB. J Biol Chem. 2014;289:3828–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Spronk CAEM, Żerko S, Górka M, et al. Structure and dynamics of helicobacter pylori nickel‐chaperone hypa: An integrated approach using nmr spectroscopy, functional assays and computational tools. J Biol Inorg Chem. 2018;23:1309–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Khorasani‐Motlagh M, Noroozifar M, Kerman K, Zamble DB. Complex formation between the Escherichia coli [NiFe]‐hydrogenase nickel maturation factors. Biometals. 2019;32:521–532. [DOI] [PubMed] [Google Scholar]
- 61. Kaluarachchi H, Zhang JW, Zamble DB. Escherichia coli SlyD, more than a Ni(II) reservoir. Biochemistry. 2011;50:10761–10763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Khorasani‐Motlagh M, Lacasse MJ, Zamble DB. High‐affinity metal binding by the Escherichia coli [NiFe]‐hydrogenase accessory protein HypB is selectively modulated by SlyD. Metallomics. 2017;9:482–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lacasse MJ, Summers KL, Khorasani‐Motlagh M, George GN, Zamble DB. Bimodal nickel‐binding site on Escherichia coli [NiFe]‐hydrogenase metallochaperone HypA. Inorg Chem. 2019;58:13604–13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kwon S, Watanabe S, Nishitani Y, et al. Crystal structures of a [NiFe] hydrogenase large subunit HyhL in an immature state in complex with a Ni chaperone HypA. Proc Natl Acad Sci U S A. 2018;115:7045–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Albareda M, Pacios LF, Palacios JM. Computational analyses, molecular dynamics, and mutagenesis studies of unprocessed form of [NiFe] hydrogenase reveal the role of disorder for efficient enzyme maturation. Biochim Biophys Acta Bioenerg. 2019;1860:325–340. [DOI] [PubMed] [Google Scholar]
- 66. Jeoung JH, Dobbek H. Carbon dioxide activation at the Ni,Fe‐cluster of anaerobic carbon monoxide dehydrogenase. Science. 2007;318:1461–1464. [DOI] [PubMed] [Google Scholar]
- 67. Kerby RL, Ludden PW, Roberts GP. In vivo nickel insertion into the carbon monoxide dehydrogenase of Rhodospirillum rubrum: Molecular and physiological characterization of cooCTJ. J Bacteriol. 1997;179:2259–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Alfano M, Cavazza C. The biologically mediated water‐gas shift reaction: Structure, function and biosynthesis of monofunctional [NiFe]‐carbon monoxide dehydrogenases. Sustain Energy Fuels. 2018;2:1653–1670. [Google Scholar]
- 69. Timm J, Brochier‐Armanet C, Perard J, et al. The CO dehydrogenase accessory protein CooT is a novel nickel‐binding protein. Metallomics. 2017;9:575–583. [DOI] [PubMed] [Google Scholar]
- 70. Alfano M, Pérard J, Carpentier P, et al. The carbon monoxide dehydrogenase accessory protein CooJ is a histidine‐rich multidomain dimer containing an unexpected Ni(II)‐binding site. J Biol Chem. 2019;294:7601–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jeoung JH, Giese T, Grünwald M, Dobbek H. CooC1 from Carboxydothermus hydrogenoformans is a nickel‐binding ATPase. Biochemistry. 2009;48:11505–11513. [DOI] [PubMed] [Google Scholar]
- 72. Jeoung JH, Giese T, Grünwald M, Dobbek H. Crystal structure of the ATP‐dependent maturation factor of Ni,Fe‐containing carbon monoxide dehydrogenases. J Mol Biol. 2010;396:1165–1179. [DOI] [PubMed] [Google Scholar]
- 73. Alfano M, Pérard J, Cavazza C. Nickel‐induced oligomerization of the histidine‐rich metallochaperone CooJ from Rhodospirillum rubrum. Inorganics. 2019;7:84. [Google Scholar]
- 74. Alfano M, Veronesi G, Musiani F, et al. A solvent‐exposed cysteine forms a peculiar NiII‐binding site in the metallochaperone CooT from Rhodospirillum rubrum. Chemistry. 2019;25:15351–15360. [DOI] [PubMed] [Google Scholar]
- 75. Spangler NJ, Lindahl PA, Bandarian V, Ludden PW. Spectroelectrochemical characterization of the metal centers in carbon monoxide dehydrogenase (CODH) and nickel‐deficient CODH from Rhodospirillum rubrum. J Biol Chem. 1996;271:7973–7977. [DOI] [PubMed] [Google Scholar]
- 76. Spangler NJ, Meyers MR, Gierke KL, Kerby RL, Roberts GP, Ludden PW. Substitution of valine for histidine 265 in carbon monoxide dehydrogenase from Rhodospirillum rubrum affects activity and spectroscopic states. J Biol Chem. 1998;273:4059–4064. [DOI] [PubMed] [Google Scholar]
- 77. Wittenborn EC, Cohen SE, Merrouch M, et al. Structural insight into metallocofactor maturation in carbon monoxide dehydrogenase. J Biol Chem. 2019;294:13017–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wittenborn EC, Merrouch M, Ueda C, et al. Redox‐dependent rearrangements of the nifes cluster of carbon monoxide dehydrogenase. Elife. 2018;7:e39451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gregg CM, Goetzl S, Jeoung JH, Dobbek H. AcsF catalyzes the ATP‐dependent insertion of nickel into the Ni, Ni‐[4Fe4S] cluster of acetyl‐CoA synthase. J Biol Chem. 2016;291:18129–18138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zambelli B, Stola M, Musiani F, et al. UreG, a chaperone in the urease assembly process, is an intrinsically unstructured GTPase that specifically binds Zn2+. J Biol Chem. 2005;280:4684–4695. [DOI] [PubMed] [Google Scholar]
- 81. Mulrooney SB, Ward SK, Hausinger RP. Purification and properties of the Klebsiella aerogenes UreE metal‐binding domain, a functional metallochaperone of urease. J Bacteriol. 2005;187:3581–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Song HK, Mulrooney SB, Huber R, Hausinger RP. Crystal structure of Klebsiella aerogenes UreE, a nickel‐binding metallochaperone for urease activation. J Biol Chem. 2001;276:49359–49364. [DOI] [PubMed] [Google Scholar]
- 83. Fong YH, Wong HC, Yuen MH, Lau PH, Chen YW, Wong KB. Structure of UreG/UreF/UreH complex reveals how urease accessory proteins facilitate maturation of helicobacter pylori urease. PLoS Biol. 2013;11:e1001678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Farrugia MA, Wang B, Feig M, Hausinger RP. Mutational and computational evidence that a nickel‐transfer tunnel in UreD is used for activation of Klebsiella aerogenes urease. Biochemistry. 2015;54:6392–6401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yuen MH, Fong YH, Nim YS, Lau PH, Wong KB, Fletterick RJ. Structural insights into how GTP‐dependent conformational changes in a metallochaperone UreG facilitate urease maturation. Proc Natl Acad Sci U S A. 2017;114:E10890–E10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Desguin B, Soumillion P, Hols P, Hausinger RP. Nickel‐pincer cofactor biosynthesis involves LarB‐catalyzed pyridinium carboxylation and LarE‐dependent sacrificial sulfur insertion. Proc Natl Acad Sci U S A. 2016;113:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Fellner M, Desguin B, Hausinger RP, Hu J. Structural insights into the catalytic mechanism of a sacrificial sulfur insertase of the N‐type ATP pyrophosphatase family, LarE. Proc Natl Acad Sci U S A. 2017;114:9074–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fellner M, Rankin JA, Desguin B, Hu J, Hausinger RP. Analysis of the active site cysteine residue of the sacrificial sulfur insertase LarE from lactobacillus plantarum. Biochemistry. 2018;57:5513–5523. [DOI] [PubMed] [Google Scholar]
- 89. Hausinger RP. New metal cofactors and recent metallocofactor insights. Curr Opin Struct Biol. 2019;59:1–8. [DOI] [PubMed] [Google Scholar]
- 90. Desguin B, Fellner M, Riant O, et al. Biosynthesis of the nickel‐pincer nucleotide cofactor of lactate racemase requires a CTP‐dependent cyclometallase. J Biol Chem. 2018;293:12303–12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zheng K, Ngo PD, Owens VL, Yang XP, Mansoorabadi SO. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science. 2016;354:339–342. [DOI] [PubMed] [Google Scholar]
- 92. Moore SJ, Sowa ST, Schuchardt C, et al. Elucidation of the biosynthesis of the methane catalyst coenzyme F 430. Nature. 2017;543:78–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Lyu Z, Chou CW, Shi H, et al. Assembly of methyl coenzyme M reductase in the methanogenic archaeon Methanococcus maripaludis. J Bacteriol. 2018;200:00717"–00746. [DOI] [PMC free article] [PubMed] [Google Scholar]