

ABSTRACT
Biallelic ENPP1 deficiency in humans induces generalized arterial calcification of infancy (GACI) and/or autosomal recessive hypophosphatemic rickets type 2 (ARHR2). The latter is characterized by markedly increased circulating FGF23 levels and renal phosphate wasting, but aberrant skeletal manifestations associated with heterozygous ENPP1 deficiency are unknown. Here, we report three adult men with early onset osteoporosis who presented with fractures in the thoracic spine and/or left radius, mildly elevated circulating FGF23, and hypophosphatemia. Total hip bone mineral density scans demonstrated osteoporosis (Z‐score < −2.5) and HRpQCT demonstrated microarchitectural defects in trabecular and cortical bone. Next‐generation sequencing revealed heterozygous loss‐of‐function mutations in ENPP1 previously observed as biallelic mutations in infants with GACI. In addition, we present bone mass and structure data as well as plasma pyrophosphate (PPi) data of two siblings suffering from ARHR2 in comparison to their heterozygous and wild‐type family members indicative of an ENPP1 gene dose effect. The skeletal phenotype in murine Enpp1 deficiency yielded nearly identical findings. Ten‐week‐old male Enpp1 asj/asj mice exhibited mild elevations in plasma FGF23 and hypophosphatemia, and micro‐CT analysis revealed microarchitectural defects in trabecular and cortical bone of similar magnitude to HRpQCT defects observed in humans. Histomorphometry revealed mild osteomalacia and osteopenia at both 10 and 23 weeks. The biomechanical relevance of these findings was demonstrated by increased bone fragility and ductility in Enpp1 asj/asj mice. In summary, ENPP1 exerts a gene dose effect such that humans with heterozygous ENPP1 deficiency exhibit intermediate levels of plasma analytes associated with bone mineralization disturbance resulting in early onset osteoporosis. © 2019 The Authors. Journal of Bone and Mineral Research published by American Society for Bone and Mineral Research.
Keywords: DISORDERS OF CALCIUM/PHOSPHATE METABOLISM, OSTEOPOROSIS, OSTEOMALACIA AND RICKETS, FGF23, ARHR2, GACI
Introduction
ENPP1 deficiency in humans is associated with generalized arterial calcification of infancy (GACI), a rare and often fatal neonatal calcification disorder characterized by arterial calcifications in the medium and large arteries. GACI may begin as early as the late second trimester and culminates in death in affected infants around 6 months of age in about 50% to 60% of patients. The calcifications arise as a consequence of the generalized deficiency of pyrophosphate1 resulting from biallelic mutations that inactivate the genes encoding either the ectoenzyme ENPP12 (type 1, about 75% of patients), which hydrolyzes ATP to generate pyrophosphate, or ABCC63, 4 (type 2, about 25% of patients), a transmembrane protein that when mutated results in low plasma pyrophosphate (PPi). If affected individuals reach the age of 6 months they are likely to survive and over time may experience spontaneous reversal of vascular calcifications,5, 6, 7, 8, 9 but severe vascular narrowing may persist along with the development of skeletal deformities described below.
Children with ENPP1 deficiency may manifest a form of hypophosphatemic rickets (autosomal recessive hypophosphatemic rickets type 2 [ARHR2]) whether or not they previously had arterial calcifications. The bone disease in ARHR2 may be severe and symptomatic, leading to repeated fractures of the long bones, rachitic skeletal deformities, and impaired growth and development.10, 11 In ARHR2, elevated serum levels of FGF23 and consequent renal phosphate wasting leading to hypophosphatemia are thought to lead to the rachitic phenotype, but the relationship between ENPP1 deficiency, plasma PPi and FGF23 levels, and their contribution to skeletal pathology is not yet understood.
Systematic studies of ENPP1 deficiency in adults are unavailable, but sporadic case reports of adults with homozygous ENPP1 deficiency document a history of childhood hypophosphatemic rickets and the development of polyarthralgias as adults12 along with progressive calcifications in the anterior and/or posterior spinal ligaments.12, 13, 14 The dual phenotypes of spinal calcifications and hypophosphatemic rickets often lead to presumptive diagnoses of XLH and/or ossification of the posterior longitudinal ligament (OPLL) until genetic analysis confirming ENPP1 deficiency is performed.
Murine models of Enpp1 deficiency exhibit the essential characteristics of human GACI, including arterial calcifications, neointimal hyperplasia, cardiac dysfunction, and myocardial infarctions.15, 16, 17 However, in contrast to the rickets present in human ARHR2, the skeletal phenotype present in Enpp1‐deficient mice is reported to be osteopenic/osteoporotic.18, 19, 20, 21, 22 Osteomalacia, the defining histology of rickets in ARHR2 children, has thus far not been observed in murine models. Conversely, the osteopenia/osteoporosis present in murine Enpp1 deficiency has not been observed in humans with ENPP1 deficiency, findings that question the relevance of the murine model to the human disease. Collectively, the discrepant phenotypes in mouse and man undermine our ability to define the pathogenesis and mechanism of an ultra‐rare bone disease and hampers the development of therapeutics designed to treat the skeletal phenotype by excluding potentially insightful mammalian models.
The discordance in the skeletal phenotype present in mouse and human ENPP1 deficiency is surprising given that murine models of monogenic bone disease have been highly predictive of human skeletal pathophysiology in the past. We, therefore, reexamined the skeletal consequences of ENPP1 deficiency in both man and mouse and surprisingly discovered that human heterozygous ENPP1 deficiency may result in lowered plasma PPi, mildly elevated FGF23, phosphate wasting, and early onset osteoporosis, which are all biochemical and skeletal findings present in murine homozygous Enpp1 deficiency. The combined studies demonstrate that human ENPP1 deficiency results in a gene dose effect and supports the use of plasma FGF23, PPi, and ENPP1 genetic screening in the clinical evaluation of patients with unexplained early onset osteopenia/osteoporosis.
Materials and Methods
Patients
The human study was performed in accordance with the local ethics committee (PV5364) and the Declaration of Helsinki. Between January 1, 2014, and December 31, 2016, deep phenotyping and genotyping for the presence of mutations/rare variants in 386 bone related genes was performed in 172 patients diagnosed with early onset osteoporosis (EOOP) who presented to the specialized osteologic outpatient clinic (The National Bone Board) of the University Medical Center Hamburg‐Eppendorf. EOOP patients were defined as premenopausal women and men aged <50 years suffering from osteoporotic fractures or presenting with dual‐energy X‐ray absorptiometry (DXA) measures within the osteoporotic range (T‐score < −2.5) in the absence of risk factors for secondary osteoporosis, such as glucocorticoid treatment, cancer, other metabolic bone diseases, renal dysfunction (GFR < 60 mL/min), and without a history of severe malnutrition at the time of the study or at any time in the past. Within the same time period, two undiagnosed siblings presented at the National Bone Board with the clinical phenotype of hypophosphatemic rickets.
Human genetic analysis
To test for disease‐causing genetic variants, DNA was isolated from patients’ peripheral blood and analyzed by a custom‐designed SureSelect XT gene panel (Agilent, Santa Clara, CA, USA) comprising 386 genes associated with changes in bone mass, skeletal dysplasias, dysostoses, or connective tissue diseases as described recently.23, 24 The pathogenicity of the prioritized variants was judged using PhenIX,25 which integrates variant scoring by PolyPhen, SIFT, and MutationTaster.
Assessment of bone metabolism and skeletal status in humans
In all patients, the areal bone mineral density (aBMD) was evaluated by DXA (Lunar iDXA, GE Healthcare, Madison, WI, USA). Fracture history and current fracture status were determined by comprehensive assessment of medical history as well as radiography or vertebral morphometry by DXA.
Biochemical bone turnover markers were analyzed from all patients’ serum (calcium, phosphate, osteocalcin, bone‐specific alkaline phosphatase [BAP], PTH) and urine (DPD cross‐links). In patients with detected ENPP1 mutation, serum levels of c‐terminal FGF23 and urinary phosphate clearance in 24‐hour urine were also measured, and tubular phosphate reabsorption (TPR) was calculated. Automated immunoassays were used to measure 25‐OH‐D (Centauer, Siemens, Munich, Germany) and 1,25‐(OH)2‐D (Liaison, Diasorin, Stillwater, MN, USA).
Bone microstructure of the patients with detected ENPP1 mutation was further analyzed using high‐resolution peripheral quantitative computed tomography (HR‐pQCT; XtremeCT, Scanco Medical, Bruttisellen, Switzerland) at the nondominant distal radius and tibia in a standardized procedure as previously described using the in vivo protocol.24, 26 Bone geometric parameters included cortical bone area (Ct. bone area, mm2) and trabecular bone area (Tb. bone area, mm2); volumetric bone mineral density (vBMD) measurements included total vBMD (total BMD, mgHA/cm3), cortical BMD (Ct.BMD, mgHA/cm3), and trabecular BMD (Tb.BMD, mgHA/cm3). Bone microstructure values include bone volume‐to‐total volume ratio (BT/TV), trabecular number (Tb.N, 1/mm), separation (Tb.Sp, mm), and thickness (Tb.Th, mm), as well as cortical thickness (Ct.Th, mm).
Enpp1asj/asj mouse model
Animal care and maintenance were provided through Yale University Animal Resource Center at Yale University (New Haven, CT, USA). All procedures were approved by the Animal Care and Use Committee of Yale University and complied with the US National Institutes of Health guide for the care and use of laboratory animals. Heterozygous Enpp1 asj/WT (genotype C57BL/6J‐Enpp1asj/GrsrJ; Jackson Laboratory, Bar Harbor, ME, USA; stock number 012810) breeding pairs were maintained on normal chow throughout the entire experiment. Food and water were delivered ad libitum. The animal colony was housed in pathogen‐free conditions. All experimental animals were maintained on normal chow in utero through completion of the study. Litters were genotyped on day 8 and weaned at day 21. After weaning, sibling pairs were sequentially divided into cohorts and were consecutively enrolled in the age‐based experimental trials by sex. Enrollment of the 10‐ and 23‐week Enpp1 wt and Enpp1 asj/asj cohorts spanned 4 months. Once the enrollment of an experimental group began, both sexes of the appropriate genotype were consecutively enrolled in an experimental cohort with the exclusion of severely runted animals weighing <5.5 g at 14 days of life. After weaning, all experimental animals were housed with littermates to allow for cooperative grooming and nesting. Experimentalists were not blinded during the study. Study numbers consisted of eight 10‐week, eight 23‐week, and eleven 46‐week Enpp1 wt mice, seven 10‐week and four 23‐week Enpp1 asj/asj mice, and seven 10‐week and seven 46‐week Enpp1 asj/wt mice.
Histology, histomorphometry, and micro‐CT of murine skeleton
Tibias and femora were stripped of soft tissue, fixed in 70% ethanol, dehydrated, and embedded in methyl methacrylate before being sectioned and stained with toluidine blue.27 Histomorphometric measurements were performed on a fixed region just below the growth plate corresponding to the primary spongiosa28 and analyzed by Osteomeasure software (Osteometrics, Atlanta, GA, USA). For μCT, tibias or femora were stripped of soft tissue and stored in 70% ethanol at 4°C. The bones were scanned using a Scanco μCT‐35 (Scanco) and analyzed for numerous structural parameters at both the proximal tibia or distal femur just below the growth plate (trabecular bone) and at the tibial or femoral midshaft (cortical bone).
Murine bone biomechanical testing
All femurs were loaded to failure with four‐point bending. All whole‐bone tests were conducted by loading the femur in the posterior to anterior direction, such that the anterior quadrant was subjected to tensile loads. The widths of the lower and upper supports of the four‐point bending apparatus were 7 mm and 3 mm, respectively. Tests were conducted with a deflection rate of 0.05 mm/s using a servohydraulic testing machine (Instron model 8874; Instron Corp., Norwood, MA, USA). The load and midspan deflection were acquired directly at a sampling frequency of 200 Hz. Load‐deflection curves were analyzed for stiffness, maximum load, and work to fracture. Yield is defined as a 10% reduction in the secant stiffness (load range normalized for deflection range) relative to the initial tangent stiffness. Post‐yield deflection, which is defined as the deflection at failure minus the deflection at yield, was measured also. Femurs were tested at room temperature and kept moist with phosphate‐buffered saline (PBS).
qBEI
Quantitative backscattered electron imaging (qBEI) was performed to analyze the bone mineral density distribution (BMDD) and osteocyte lacunar morphology. As previously described,29 methyl methacrylate embedded murine tibias were ground, polished, and carbon‐coated before being imaged in a scanning electron microscope (LEO 435, LEO Microscopy Ltd., Cambridge, UK) with a backscattered electron detector (Type 202, K.E. Developments Ltd., Cambridge, UK). The electron beam energy was kept constant at 20 keV during measurements, as well as probe current at 680 ± 1 pA and the working distance at 20 mm. Mean calcium weight percentage (CaMean), most frequent calcium weight percentage (CaPeak), and heterogeneity of the calcium distribution (CaWidth) were evaluated based on the generated gray value images representing the calcium content of the tibial cross sections.30 Further, the images were taken to determine the mean osteocyte lacunar area (Ot.Lc.Ar) and number of osteocytes per mineralized matrix (N.Ot.Lc/B.Ar).
Quantification of plasma PPi
All human and murine plasma PPi samples were analyzed in a blinded fashion. Blood plasma was prepared and the PPi assay was performed as previously described31, 32 with minor variations. After plasma isolation and filtration, the plasma samples were diluted in 1:1 with 50 mM Tris‐Acetate pH 8.0 buffer followed by filtration through a 30 kDa membrane (Amicon, MilliporeSigma, Burlington, MA, USA) via ultracentrifugation. The samples were aliquoted for single use and stored frozen at −80°C. The plasma isolation, removal of platelets via filtration, and dilution and freezing of samples were performed within 1 hour of blood collection. The PPi assay was carried out by mixing 5 μL plasma samples with 35 μL mixture composed of 50 mM Hepes pH 7.4, 16 μM adenosine phosphosulfate, 32 mU/mL ATP sulfurylase, and 80 mM MgSO4. Standards were prepared using serial dilutions of a freshly prepared 10 mM PPi stock solution (Acros Organics, Thermo Fisher Scientific, Waltham, MA, USA; A0371139) and run in parallel with the samples. All experimental solutions were prepared in a single 96‐well PCR plate, which is subjected to the following cycle to activate and deactivate ATP sulfurylase: 37°C 10 minutes, 90°C 10 minutes, 4°C for hold. Bactiter‐Glo microbial cell viability assay (Promega, San Luis Obispo, CA, USA; G8231) was used by mixing 20 μL product from the reaction and 20 μL of the Bactiter‐Glo reagent. Luminescence signal was then read by Bio‐Teck synergy HT multi‐detection microplate reader at room temperature. Data analysis was performed via GraphPad Prism 7. Background PPi in the samples and water was subtracted and normalized from the samples, and the values were interpolated using Prism (GraphPad 7, La Jolla, CA, USA).
ELISA assays for PTH1‐84 and FGF‐23
Mouse PTH 1‐84 ELISA kits were purchased from Quidel Corporation (San Diego, CA, USA; catalog number 60‐2305). Mouse/Rat FGF‐23 (intact) ELISA kit was purchased from Quidel Corporation (catalog number 60‐6800). Forty‐five‐microliter plasma samples were used for all the above ELISA experiments. Data analysis was performed via GraphPad Prism 7.
Enzyme kinetic assay
Human polymorphisms were engineered into the human ENPP1‐Fc construct15 using the Quikchange II‐XL Site Directed Mutagenesis from Agilent Technologies (Santa Clara, CA, USA). After sequence verification, constructs were transfected into CHO‐K1 cells using lipofectamine 2000 from Thermo Fisher Scientific. Forty eight hours after transfection, 10 μL of supernatant was mixed with 90 μL of assay buffer containing 250 mM Tris pH 8.0, 500 mM NaCl, 0.05% Triton X‐100, and 1 mM Thymidine 5′‐monophosphate p‐nitrophenyl. The velocity of the p‐nitrophenyl group liberated from the chromogenic substrate was reported as change at OD 405 nM/min, in replicates of at least 5 for each construct, and normalized to % WT.
Results
Patients
Between January 1, 2014, and December 31, 2016, 172 patients with unexplained early onset osteoporosis presenting at a specialized osteologic outpatient clinic for musculoskeletal disorders (The National Bone Board) of the University Medical Center Hamburg‐Eppendorf were analyzed by a custom‐designed SureSelect XT gene panel (Agilent) comprising 386 genes in which mutations have been associated with changes in bone mass, skeletal dysplasias, dysostoses, or connective tissue diseases. In 2 of 172 early onset osteoporosis (EOOP) patients, heterozygous mutations were found in ENPP1 (patient A1 and B2). Segregation analysis revealed one more affected family member (patient B1), which demonstrated that only affected family members carried the respective mutation. In addition, the segregation analysis also demonstrated that the other rare variants observed were clinically silent, especially with respect to phosphate metabolism (Supplemental Table S1).
The mutations detected were c.2330A>G/p.His777Arg in family A (patient A1) and c.1412A>G/p.Tyr471Cys in family B (patient B1 and B2, Fig. 1 A, B). Both mutations have been reported in GACI patients, and we further characterized the effect of these mutations on enzyme activity by performing steady‐state kinetics on cell culture supernatants of CHO cells expressing a soluble form of ENPP1. We found that the Y471C mutation reduced the enzymatic velocity by 70% and the H777R by 95% when compared with WT ENPP1 (Fig. 1C). Our findings concur with other reports demonstrating residual enzymatic activity in these ENPP1 variants.7, 33 In addition to the patients with monoallelic ENPP1 deficiency, three unaffected siblings (A2, A3, and B3) were identified and examined to provide an unaffected sibling pair cohort.
Figure 1.
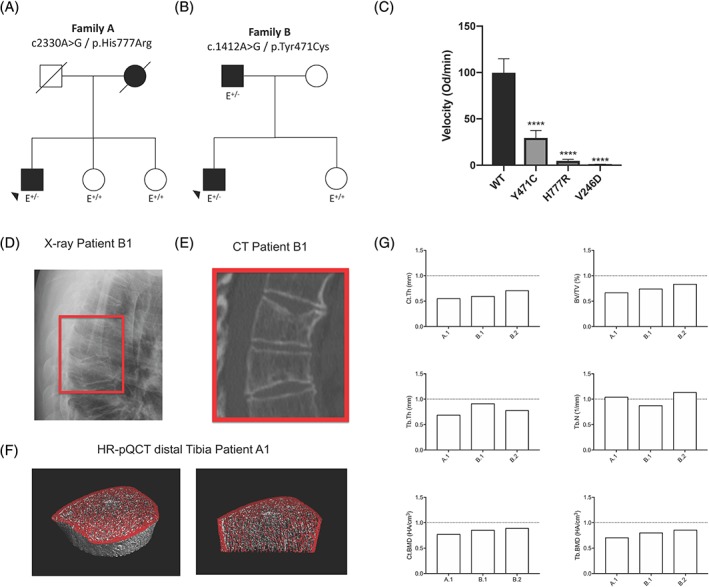
Clinical data of EOOP patients (A) Pedigree and ENPP1 mutation of family A. (B) Pedigree and ENPP1 mutation of family B. (C) Comparison of the enzymatic activity of WT, Y471C, and H777R ENPP1 variants in the human patients with the V246D mutation present in Enpp1 asj mice. The Y471C mutation reduced the velocity of the enzymatic reaction by 70%, and the H777R mutation by 95%, when compared with WT ENPP1. The V246D mutation in Enpp1 asj mice demonstrated nearly absent enzymatic activity. (D) Radiograph of the thoracic spine of patient B1 (lateral view) and (E) the respective scan obtained by computed tomography (CT). (F) HR‐pQCT scans of the distal tibia of patient A1. (G) Bone microstructure parameters of the distal tibia of patients A1, B1, and B2 (carrying heterozygous ENPP1 mutations) obtained by HR‐pQCT in comparison to a healthy sex‐ and age‐matched control group.35
Whereas patients A1 and B1 presented with multiple vertebral fractures, the DXA Z‐score was strongly decreased in patients A1 and B2 (Table 1 and Fig. 1 D–F), in contrast to their unaffected siblings who only suffered from single traumatic fractures and exhibited normal bone mineral density. Biochemical analyses revealed decreased phosphorus and elevated c‐terminal FGF23 serum levels. Corresponding to these findings, urinary phosphate clearance was elevated in affected patients, and tubular phosphate reabsorption was reduced (Table 2). Microstructural bone analysis by HR‐pQCT demonstrated a combined trabecular and cortical bone loss in all three affected individuals at the distal tibia (Fig. 1 G and Supplemental Table S2). Levels of hemoglobin, hematocrit, and ferritin were within the physiological range in all individuals examined.
Table 1.
Sex, Age, Mutation Status, Fracture Assessment, and Bone Mineral Density Measures in EOOP patients and family members
| Parameter | A1+/− | A2+/+ | A3+/+ | B1+/− | B2+/− | B3+/+ |
|---|---|---|---|---|---|---|
| Sex | M | F | F | M | M | F |
| Age (years) | 60 | 44 | 52 | 69 | 44 | 39 |
| ENPP1 mutation | Het. | None | None | Het. | Het. | None |
| DNA change | c2330A>G | n/a | n/a | c.1412A>G | c.1412A>G | n/a |
| Protein change | p.His777Arg | n/a | n/a | p.Tyr471Cys | p.Tyr471Cys | n/a |
| Childhood fractures | No | Yes | No | No | No | No |
| Vertebral fractures | 5 | 0 | 0 | 5 | 0 | 0 |
| Nonvertebral fractures | 0 | 1a | 0 | 1 | 1 | 1b |
| T‐score (lumbar spine) | −4.2 | −0.9 | 0.1 | −1.2 | −0.3 | 0.7 |
| T‐score (total hip) | −3.1 | −1.3 | −0.5 | −0.2 | −3.0 | 0.7 |
| Z‐score (lumbar spine) | −4.1 | −1.0 | 0.5 | −1.2 | −0.2 | 0.7 |
| Z‐score (total hip) | −2.7 | −1.3 | −0.1 | 0.2 | −2.7 | 0.8 |
M = male; F = female; Het. = heterozygous.
Traumatic forearm fracture in childhood.
Traumatic nasal fracture.
Table 2.
Biochemical Measures of Bone and Mineral Metabolism in EOOP patients and family members
| Parameter | Unit | A1+/− | A2+/+ | A3+/+ | B1+/− | B2+/− | B3+/+ | Reference values |
|---|---|---|---|---|---|---|---|---|
| Serum | ||||||||
| Calcium | mmol/L | 2.13 | 2.10 (↓) | 2.21 | 2.10 (↓) | 2.33 | 2.19 | 2.10–2.74 |
| Phosphate | mmol/L | 0.59 (↓) | 0.93 | 0.87 | 0.74 (↓) | 0.68 (↓) | 0.88 | 0.77–1.50 |
| Osteocalcin | μg/L | 6.0 (↓) a | 16.2 | 12.2 | 13.2 | 20.6 | 20.7 | 12.0–52.1 |
| BAP | μg/L | 4.5 (↓) a | 7.4 | 5.7 | 11.3 | 10.2 | 11.1 | 5.5–22.9 |
| 25‐OH‐D | μg/L | 40.6 | 34.5 | 40.3 | 29.5 (↓) | 64.8 | 45.7 | >30.0 |
| 1,25‐(OH)2‐D | ng/L | 41.0 | n/a | n/a | n/a | 65.0 | n/a | 20.0–79.0 |
| PTH | μg/L | 59.1 | 37.1 | 19.6 | 51.7 | 73.9 | 67.6 | 17.0–84.0 |
| Cross‐links | nmol/mmol | 3 | 5 | 9 (↑) | 2 | 3 | 8 (↑) | 2–5/3–7b |
| cFGF23 | kRU/L | 124 (↑) | ‐ | ‐ | 142 (↑) | 104 | 56 | 26–110 |
| Calculated measures or renal phosphate handling | ||||||||
| Phosphate‐clearance | mL/min | 28.0 (↑) | ‐ | ‐ | 24.5 (↑) | 32.0 (↑) | ‐ | 5.5–16.0 |
| TPR | % | 80.9 (↓) | ‐ | ‐ | 72.7 (↓) | 76.2 (↓) | ‐ | 82.0–90.0 |
| TMP/GFR | mg/dL | 2.0 (↓) | ‐ | ‐ | 2.0 (↓) | 1.6 (↓) | ‐ | 2.5–4.2 |
BAP = bone alkaline phosphatase; PTH = parathyroid hormone; TPR = tubular phosphate reabsorption.
Parameters outside age‐ and sex‐specific reference range are in bold.
Low bone turnover state under denosumab treatment.
Sex‐specific reference values for male/female.
In addition, two patients with the clinical phenotype of hypophosphatemic rickets were diagnosed as ARHR2 by verification of homozygous ENPP1 mutations (c.755A>G/p.Tyr252Cys). Segregation analysis in this family (family C) revealed heterozygous mutations in two family members (mother and one sibling) and no mutations in the other two siblings. Plasma PPi level analysis in this family showed a significant relationship between mutational status and PPi level, with the highest PPi levels in unmutated ENPP1, significantly reduced levels in patients with heterozygous loss‐of‐function (LOF) mutations and severely decreased levels in homozygous LOF family members (Fig. 2 A–E and Table 3).
Figure 2.
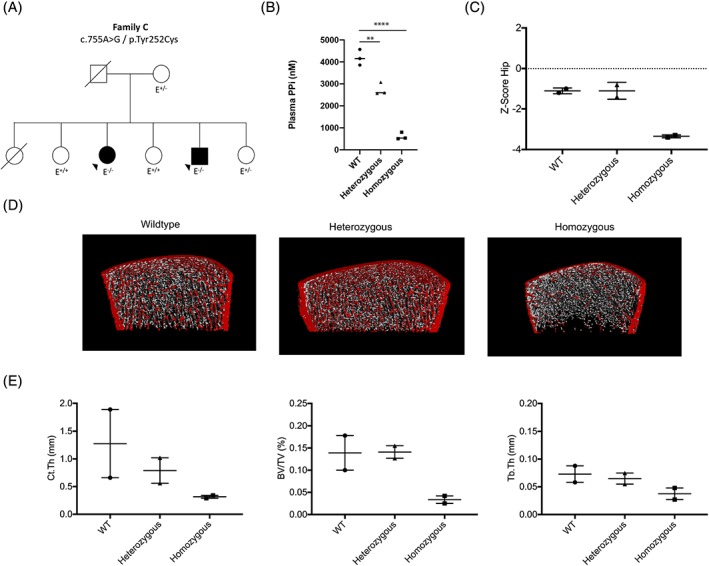
Clinical data of ARHR2 patients and their family members. (A) Pedigree and ENPP1 mutation of family C. (B) PPi plasma levels of the family members in relationship to their mutational status. (C) Hip Z‐scores obtained by DXA. (D) Representative HR‐pQCT scans of the distal tibia, and (E) selected structural parameters. **p < 0.01; ****p < 0.001 (Student's unpaired t test).
Table 3.
Biochemical Measures of Bone and Mineral Metabolism in ARHR2 patients and family members
| Parameter | Unit | C1+/− | C2+/+ | C3−/− | C4+/+ | C5−/− | C6+/− |
|---|---|---|---|---|---|---|---|
| Serum | |||||||
| Calcium | mmol/L | 2.22 | 2.34 | 2.28 | 2.24 | 2.18 | 2.18 |
| Phosphate | mmol/L | 0.83 | 1.39 | 0.63 (↓) | 1.57 | 0.56 (↓) | 1.11 |
| Osteocalcin | μg/L | 12.1 | 46.4 | 40.7 | 70.1 | 41.4 | 30.1 |
| BAP | μg/L | 8.5 | 58.3 | 179.1 | 96.9 | 186.3 | 20.1 |
| 25‐OH‐D | μg/L | 8.6 (↓) | 13.8 (↓) | 10.4 (↓) | 14.9 (↓) | 7.4 (↓) | 8.5 (↓) |
| PTH | μg/L | 75.0 | 62.6 | 59.2 | 48.7 | 82.2 | 59.1 |
| Cross‐links | nmol/mmol | 6 | 28 | 77 (↑) | 22 | 34 (↑) | 12 |
| cFGF23 | kRU/L | 141 (↑) | 46 | 265 (↑) | 44 | 123 (↑) | 130 (↑) |
BAP = bone alkaline phosphatase; PTH = parathyroid hormone.
Parameters outside age‐ and sex‐specific reference range are in bold.
Murine phenotype of Enpp1 deficiency
To understand the pathophysiology of osteopenia noted in the patients above, we studied the bone phenotype of Enpp1 asj mice,16 which possess a V246D point mutation near the Enpp1 catalytic threonine (at position 238), which we demonstrate to almost completely eliminate ENPP1 activity in the human protein (Fig. 1 F). Heterozygous mice with the asj mutation (Enpp1 asj/WT mice) demonstrated plasma PPi comparable to WT sibling pairs (Supplemental Fig. S1), and correspondingly, micro‐CT analysis of their tibias exhibited no significant differences with WT sibling pairs at 10 weeks (Supplemental Tables S3 and S4). The findings demonstrate the lack of a plasma PPi biochemical phenotype in heterozygous murine Enpp1 deficiency at 10 weeks and the lack of a skeletal phenotype in heterozygous murine Enpp1 deficiency at 10 and 46 weeks.
In contrast, homozygous Enpp1 asj/asj mice at 10 weeks exhibited biochemical findings in FGF23 and plasma phosphorous similar to humans with heterozygous ENPP1 deficiency—mildly low plasma phosphorous and slightly elevated intact FGF23 (Table 4). In addition, PTH levels were noted to be elevated in Enpp1 asj/asj mice at 10 weeks. Histology performed on tibias of 10‐week WT and Enpp1 asj/asj sibling pairs demonstrated thinning of the cortex and reduced trabecular bone (Fig. 3 A), and micro‐CT confirmed an osteopenic phenotype in both trabecular and cortical bone compartments. Trabecular BV/TV in 10‐week‐old Enpp1 asj/asj males was 57% of WT, trabecular thickness was 83% of WT, apparent density (of TV) was 55% of WT, and cortical thickness in Enpp1 asj/asj mice was 78% of WT (Fig. 3 B, C and Supplemental Tables S5 and S6). Histomorphometry of tibias from 10‐week‐old Enpp1‐deficient mice confirmed the osteopenia—BV/TV in male Enpp1 asj/asj was 48% of WT, trabecular spacing was increased to 190% of WT, and trabecular number was reduced to 56% of WT (Supplemental Table S7). In addition, histomorphometrical parameters revealed concurrent mild osteomalacia as evidenced by increased osteoid thickness (130% of WT) and osteoid volume/bone volume (238% of WT), as well as a prolonged mineralization lag time in male Enpp1 asj/asj mice, which was 295% of WT (Fig. 3 D and Supplemental Table S7). To verify that the histomorphologic and micro‐CT changes observed in Enpp1 deficiency at 10 weeks resulted in functional changes in bone strength, we compared biomechanical properties by loading femurs to failure using four‐point bending.34 Compared with WT littermates, the bones from Enpp1 asj/asj mice were much less stiff, requiring significantly less total work until fracture and were able to bear significantly less maximal load at either age (Fig. 3 E). To determine if the osteopenia was due to increased bone resorption, we measured the concentration of cross‐linked C‐teleopeptide from type I collagen (CTX) in the plasma of WT and Enpp1 asj/asj mice at 10 weeks and observed no significant differences (Fig. 3 F). From this data, we concluded that Enpp1 deficiency resulted in no changes in osteoclast formation or resorptive activity. We noted that these observations concur with similar findings in Enpp1 null mice.20
Table 4.
Mean Plasma Biochemical Measures of Bone and Mineral Metabolism of 10‐ and 23‐week WT and Enpp1 asj/asj Mice
| Parameter | Unit | 10 weeks: Enpp1wt | 10 weeks: ENPP1asj/asj | 23 weeks: Enpp1wt | 23 weeks: ENPP1asj/asj |
|---|---|---|---|---|---|
| Calcium | mg/dL | 9.36 | 9.03 | 8.28 | 7.23 |
| Phosphate | mg/dL | 6.22 | 4.78** (↓) | 5.02 | 4.65 |
| PTH | pg/mL | 129 | 287** (↑) | 485 | 187** (↓) |
| FGF23 | pg/mL | 115 | 426****(↑) | 219 | 231 |
| PPI | nM | 1900 | 142.9**** (↓) | 2030 | 542.8**** (↓) |
PTH = parathyroid hormone.
p < 0.01.
p < 0.001 (Student's unpaired t test).
Figure 3.
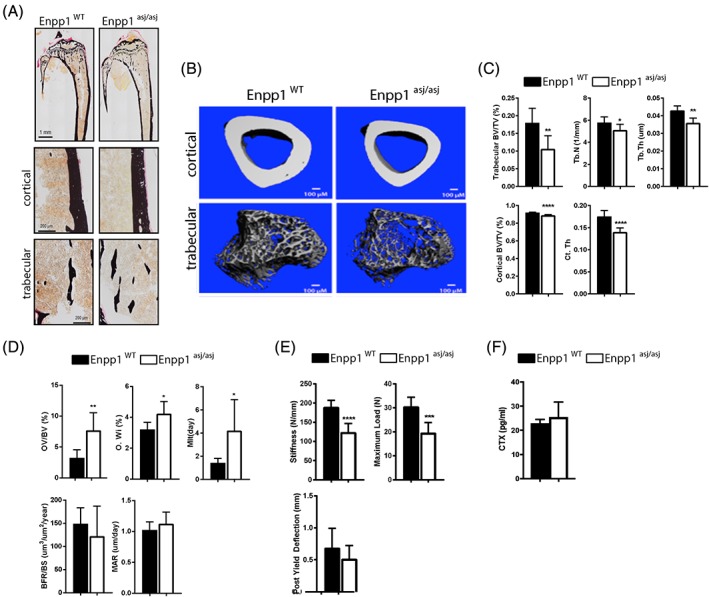
Bone histology of WT and Enpp1 asj/asj mice at 10 weeks. (A) Von Kossa staining of whole tibia (top) and cortical and trabecular bone (bottom) of 10 ‐week WT and Enpp1 asj/asj male mice. (B) Reconstructed images from micro‐CT scans of tibial trabecular and cortical bone. (C) Micro‐CT quantification of trabecular BV/TV, trabecular thickness (Tb.Th), and trabecular number (Tb.N) and cortical BV/TV and cortical thickness (Ct.Th) (D). Histomorphometric quantification of osteoid volume (OV/BV), osteoid width (O.Wi), mineralization lag time (MLT), bone formation rate (BFR/BS), mineral apposition rate (MAR). (E) Biomechanical quantification by four‐point bending of femur bone parameters (stiffness, maximum load, and post yield deflection). (F) Concentration of C‐telopeptide of type 1 collage (CTX) in plasma of 10‐week WT and Enpp1 asj/asj mice. n ≥ 7 (C), n ≥ 6 (D), and n ≥ 7 (E). Data are the means ± SEM. *p < 0.05; **p < 0.01; ***p < 0.005, ****p < 0.001 (Student's unpaired t test).
At 23 weeks, the plasma analytes suggest progression of the bone disease in Enpp1 asj/asj mice. Twenty‐three‐week male Enpp1 asj/asj mice exhibit mild elevations in FGF23 and, as a consequence, mildly reduced serum phosphorous levels, not significantly different from WT, and reduced serum PTH levels (Table 4). Tibial bones of 23‐week male Enpp1 asj/asj mice continued to demonstrate an osteopenic phenotype both micro‐CT (trabecular BV/TV at 54% of WT and cortical thickness at 78% of WT) and histomorphometry (BV/TV at 40% of WT, trabecular thickness was decreased to 69% of WT, and trabecular spacing was increased to 200% of WT; Fig. 4 A‐C and Supplemental Tables S5–S7). Compared with 10‐week mice, the bone formation rate (BFR/BS) was significantly decreased (33% of WT), suggesting the evolution of a low bone turnover state with age, with continued evidence of osteomalacia (MAR 62% of WT, MLT 196% of WT; Fig. 4 D and Supplemental Table S7). Biomechanical findings at 23 weeks continued to exhibit reduced bone stiffness and maximum load but also demonstrated increased ductility, suggesting that the accumulation of unmineralized matrix over time has altered the material properties of bone (Fig. 4E). Finally, plasma concentrations of CTX demonstrated no increase in osteoclast activity at 23 in Enpp1 asj/asj mice (Fig. 4 F).
Figure 4.
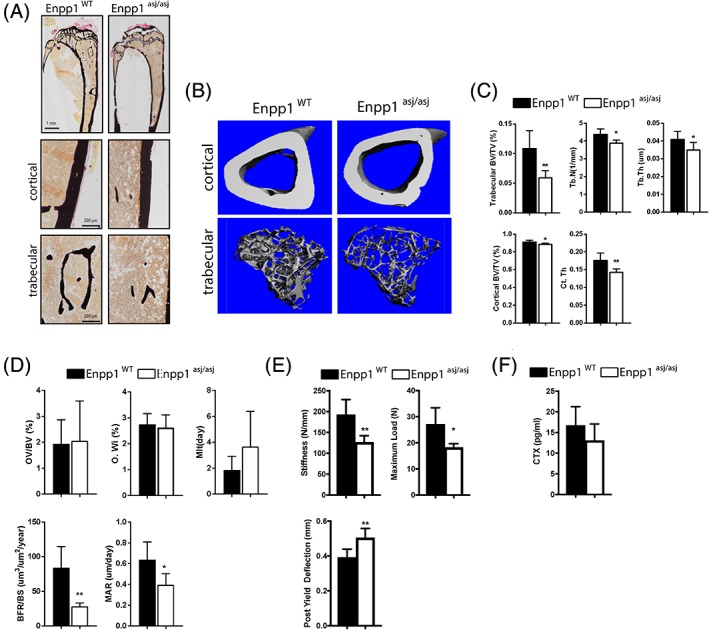
Bone histology of WT and Enpp1 asj/asj mice at 23 weeks. (A) Von Kossa staining of tibia (top) and cortical and trabecular bone (bottom) of 23 ‐week WT and Enpp1 asj/asj male mice. (B) Reconstructed images from micro‐CT scans of tibial trabecular and cortical bone. (C) Micro‐CT quantification of trabecular BV/TV, trabecular thickness (Tb. Th), and trabecular number (Tb.N) and cortical BV/TV and cortical thickness (Ct.Th) (D). Histomorphometric quantification of osteoid volume (OV/BV), osteoid width (O.Wi), mineralization lag time (MLT), bone formation rate (BFR/BS), mineral apposition rate (MAR). (E) Biomechanical quantification by four‐point bending of femur bone parameters (stiffness, maximum load, and post yield deflection). (F) Concentration of C‐telopeptide of type 1 collage (CTX) in plasma of 23‐week WT and Enpp1 asj/asj mice. n ≥ 4 (C–E). Data are the means ± SEM. *p < 0.05; **p < 0.01 (Student's unpaired t test).
Quantitative backscattered electron imaging
To investigate the degree of cortical bone mineralization and its effects on bone quality, we used quantitative backscattered electron imaging (qBEI) to measure bone mineral density distribution (BMDD) and osteocyte lacunar area and density. Calcium weight percentages in cortical bones of 10‐week‐old male Enpp1 asj/asj mice were greater than that of age‐matched WT mice (Fig. 5), which is represented by slightly increased CaMean (+1.4%, p = 0.046) and CaPeak (+1.6%, p = 0.033) values, whereas heterogeneity (CaWidth, p = 0.834) is unchanged. In 23‐week‐old mice, the cortical bone of both Enpp1 asj/asj and WT mice showed a higher degree of mineralization (CaMean) in comparison to the 10‐week‐old mice. However, this maturation effect was more pronounced in the WT mice (WT: +4.4%, asj: +2.7%). When comparing the degree of mineralization of Enpp1 asj/asj and WT mice, it is evident that significant changes toward higher mineralization can only be found in the 10‐week‐old mice, whereas differences in mineralization patterns have leveled off in the older 23‐week‐old mice.
Figure 5.
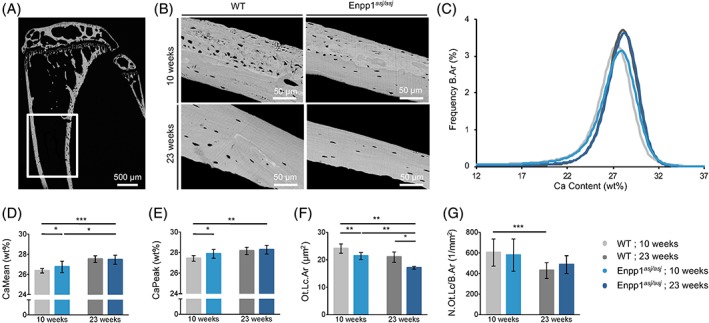
BMDD and osteocyte lacunar morphology in tibias of male mice determined by qBEI. (A) Measurements were performed distal of the proximal trabecular area at the medial and lateral side of 10‐ and 23 ‐week‐old mice. (B) QBEI images show enhanced lamellar bone in 23 ‐week‐old Enpp1 asj/asj and WT mice and (C) BMDD shift (black arrows) to a higher mineralized matrix at 23 weeks, which is represented by increased CaMean and CaPeak (only in WT mice). (D) CaMean (mean ± SD) and (E) CaPeak in 10 ‐week‐old Enpp1 asj/asj mice are increased compared with age‐related WT mice. In 23‐week‐old mice, this difference is no longer apparent. (F) Ot.Lc.Ar in Enpp1 asj/asj mice is decreased at 10 and 23 weeks, and with age the Ot.Lc.Ar decreases in both genotypes. The reduction is osteocyte lacunae area is consistent with the mineralization defect observed in the vasculature and could contribute to the increased osteocytic FGF23 production observed in the Enpp1 deficient animals. (G) N.Ot.Lc/B.Ar is unchanged in Enpp1‐deficient mice compared to WT mice at 10 and 23 weeks, and decreases with age only in WT mice. *p < 0.05; **p < 0.01; ***p < 0.005 (Student's unpaired t test).
Additionally, at 10 weeks of age, the mean osteocyte lacunar area was significantly reduced by 11.2% in Enpp1 asj/asj mice (p = 0.003), wherein the number of osteocyte lacunae per bone surface area (N.Ot.Lc/B.Ar) is similar to WT mice and is still reduced in Enpp1 asj/asj mice (by 18.6%, p = 0.007) at 23 weeks.
Discussion
In humans, homozygous ENPP1 deficiency results in the infantile disorder called GACI and a phosphate wasting rachitic disorder in older children called ARHR2. Furthermore, a SNP variant in ENPP1 (rs1974201) was identified in the Framingham Osteoporosis Study that was associated with unfavorable hip geometry and predictive of femoral neck fractures.36 This study also examined other central regulators of bone mineralization including ANKH and ALPL, but only ENPP1 returned an association with unfavorable skeletal parameters. The study suggests that ENPP1 is a central regulator of the extracellular purinergic metabolic pathway, which is responsible for the generation of essential skeletal mineralization metabolites through the generation of extracellular Pi, PPi, and adenosine.
Although ENPP1 deficiency has not previously been identified as a causative factor of low bone mass in humans, the Department of Osteology and Biomechanics in Hamburg identified a small cohort of middle‐age men with monoallelic ENPP1 deficiency and early onset osteoporosis. The patients exhibited multiple vertebral or radial fractures, elevated c‐terminal FGF23, decreased phosphorous, elevated urinary phosphate clearance, and clinical signs of osteoporosis. HR‐pQCT measurements to determine tibial microstructure confirmed low bone mass (cortical thickness 55% to 70% of WT, trabecular bone volume 70% to 85% of age‐ and sex‐matched controls), and DXA scores of the hip and spine confirmed osteoporosis. The biochemical and bone mineralization findings suggest that heterozygous ENPP1 deficiency induces mildly elevated FGF23 levels and concomitant low levels of plasma phosphate and pyrophosphate that results in low bone mass and a severe, premature osteoporosis.
The skeletal phenotype and plasma biochemistry present in Enpp1 asj/asj mice were remarkably similar to the early onset osteoporosis patients with heterozygous ENPP1 deficiency. Namely, Enpp1 asj/asj mice also exhibit mildly increased FGF23 and mildly low plasma phosphorous. The murine skeletal phenotype exhibits a severe osteopenia and a mild osteomalacia, reflective of the mineralization imbalances present in human ENPP1 deficiency. Mild osteomalacia in Enpp1 asj/asj mice at 10 weeks is evidenced by the prolonged mineralization lag time and the accumulation of osteoid (increased osteoid width and volume; Fig. 3 D). At 23 weeks, features of both osteomalacia and osteoporosis persisted (Fig. 4 D), and biomechanical testing also demonstrated increased bone fragility and increased bone ductility (Fig. 4 E), consistent with the combined presence of osteomalacia and osteoporosis. Notably, defects in bone microarchitecture documented by micro‐CT in both 10‐ and 23‐week Enpp1 asj/asj mice—specifically, reductions in cortical and trabecular thickness and increased trabecular spacing (Figs. 3 C and 4 C and Supplemental Tables S5 and S6)—closely parallel the bone microarchitectural defects documented by HR‐pQCT in humans with heterozygous ENPP1 deficiency, which demonstrate nearly identical deficits in the same parameters (Supplemental Table S2). The findings demonstrate a close correspondence between the skeletal phenotype present in murine and human ENPP1 deficiency and support the use of the murine model to investigate disease pathogenesis.
In humans, heterozygous ENPP1 deficiency induces mild FGF23 elevations, which, in turn, generates mild phosphate wasting that may be clinically overlooked, whereas homozygous ENPP1 deficiency results in overt elevations of FGF23, pronounced phosphate wasting, and clinically apparent rickets during childhood, all supporting the presence of an ENPP1 gene dose effect in humans. We tested this hypothesis by measuring plasma PPi levels in a single pedigree whose family members contained all variations of an ENPP1 gene dose (Fig. 2 B). Our results demonstrated that individuals with heterozygous ENPP1 deficiency had plasma PPi levels intermediate to those of siblings/children without ENPP1 deficiency and siblings with homozygous ENPP1 deficiency (Fig. 2 B). Parallel experiments in Enpp1‐deficient mice demonstrated that the murine gene dose is not comparable with humans—unlike humans with monoallelic ENPP1 deficiency, Enpp1 asj/wt mice do not exhibit decreased plasma PPi but do manifest slight elevations in FGF23 (Supplemental Fig. S1). It is therefore not surprising that micro‐CT data of mice with heterozygous Enpp1 deficiency showed no signs of skeletal abnormality (Supplemental Tables S3 and S4).
ENPP1 generates PPi, a potent endogenous mineralization inhibitor, and therefore ENPP1 deficiency should result in increased and not decreased bone mass. The mechanism by which ENPP1 deficiency induces osteopenia is therefore not readily apparent, but a putative mechanism may be related to changed osteocyte function induced by low plasma PPi. To investigate osteocyte characteristics and tissue mineralization, we used qBEI, which demonstrated higher mean calcium content and lower osteocyte lacunar size in 10‐week Enpp1 asj/asj mice. Although typical lacunar mineralization as described for human bone was not apparent in mice29, the newly obtained data suggest that PPi deficiency is related to lower osteocyte lacunar size. Changes in lacunar size may be associated with increased plasma FGF23 and reduced serum phosphate, while increased osteoid indices were also observed in ENPP1 deficiency. Progressive osteocyte loss has been reported in Enpp1 null mice,20 consistent with the notion that decreased plasma PPi may be harmful to osteocytes, possibly by compromising the vascular channels supplying nutrients or directly impinging on osteocyte lacunae. The aggregate findings suggest an association between plasma PPi, bone mass, and osteocyte function, but further experiments are required before establishing the precise mechanism of low bone mass due to ENPP1 deficiency.
Diseases inducing decreased bone mass and increased vascular calcification are called “paradoxical mineralization disorders,” and the etiology of these disorders is presently unknown. The association of ENPP1 deficiency with human osteoporosis and GACI identifies ENPP1 as a candidate pathogenic mechanism for this poorly understood syndrome. In addition to aging adults, patients with chronic kidney disease bone mineralization disorder (CKD‐MBD) also exhibit paradoxical mineralization whereby vascular calcifications coexist with decreased bone mass and increased bone fragility.37 Our findings suggest that ENPP1 and PPi levels should be evaluated in patients with early onset osteoporosis of unknown etiology and should also be investigated as a pathogenic mechanism in paradoxical mineralization disorders.
Disclosures
DTB is an inventor of patents owned by Yale University that describe therapeutics for ENPP1 deficiency. DTB is an equity holder and receives research and consulting support from Inozyme Pharma. RO received travel reimbursement and honorarium from Inozyme Pharma. TOC received honoraria from Inozyme Pharma and serves on its Clinical Advisory Board. DT is an employee of Inozyme Pharma.
Supporting information
Supplemental Fig. S1. Plasma pyrophosphate and intact FGF23 in 10‐week Enpp1wt and Enpp1asj/+ male mice. Serum concentrations pyrophosphate and intact FGF23 in 10‐week WT and Enpp1 asj/wt male mice. No significant differences between plasma pyrophosphate was observed, but plasma FGF23 was slightly elevated in the heterozygous mice. *p < 0.05 (Student's unpaired t test).
Supplemental Table S1. Mutation status, frequency, phenotype match, inheritance match, and segregation analysis of all rare variants detected by the gene panel analysis.
Supplemental Table S2. Bone microstructure assessed at distal tibia by HR‐pQCT and percentages of sex‐ and age‐specific normative data.(26)
Supplemental Table S3. Trabecular bone architecture and density of 10‐ and 46‐week‐old wild‐type and heterozygous Enpp1 asj/WT male mice.
Supplemental Table S4. Cortical bone architecture and density of 10‐ and 46‐week‐old wild‐type and heterozygous Enpp1 asj/WT male mice.
Supplemental Table S5. Trabecular bone architecture and density of 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.
Supplemental Table S6. Cortical bone architecture and density of 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.
Supplemental Table S7. Skeletal histomorphometry of tibia from 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.
Acknowledgments
Parts of the study were funded by the German Federal Ministry of Education and Research (BMBF) within the project “Detection and Individualized Management of Early Onset Osteoporosis (DIMEOS).” Plasma analytes in mice were performed by the George M O'Brien Kidney Center at Yale, and funded by NIH grant P30 DK079310.
Authors’ roles: Study design: RO, MCH, TMC, and DTB. Study conduct: RO and DTB. Data collection: KZ, RO, JS, UK, PRS, NDM, DK, SvK, SMT, MCH, and DTB. Data analysis: KZ, RO, JS, UK, NDM, DK, SMT, SvK, MCH, DT, BB, TOC, and DTB. Data interpretation: KZ, RO, MA, TS, UK, PRS, SvK, NDM, MCH, TOC, and DTB. Drafting manuscript: KZ, RO, NDM, SMT, TOC, and DTB. Revising manuscript content: KZ, RO, MA, TS, UK, BB, PRS, NDM, DK, SMT, DT, TOC, and DTB. Approving final version of manuscript: KZ, RO, MA, TS, BB, NDM, DK, SMT, MCH, DT, TOC, and DTB. The following individuals take responsibility for the integrity of the data analysis: KZ, RO, and DTB.
References
- 1. Rutsch F, Vaingankar S, Johnson K, et al. PC‐1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158(2):543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rutsch F, Ruf N, Vaingankar S, et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34(4):379–81. [DOI] [PubMed] [Google Scholar]
- 3. Li Q, Brodsky JL, Conlin LK, et al. Mutations in the ABCC6 gene as a cause of generalized arterial calcification of infancy: genotypic overlap with pseudoxanthoma elasticum. J Invest Dermatol. 2014;134(3):658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nitschke Y, Baujat G, Botschen U, et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet. 2012;90(1):25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marrott PK, Newcombe KD, Becroft DM, Friedlander DH. Idiopathic infantile arterial calcification with survival to adult life. Pediatr Cardiol. 1984;5(2):119–22. [DOI] [PubMed] [Google Scholar]
- 6. Ciana G, Trappan A, Bembi B, et al. Generalized arterial calcification of infancy: two siblings with prolonged survival. Eur J Pediatr. 2006;165(4):258–63. [DOI] [PubMed] [Google Scholar]
- 7. Rutsch F, Boyer P, Nitschke Y, et al. Hypophosphatemia, hyperphosphaturia, and bisphosphonate treatment are associated with survival beyond infancy in generalized arterial calcification of infancy. Circ Cardiovasc Genet. 2008;1(2):133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyai K, Ariyasu D, Numakura C, Yoneda K, Nakazato H, Hasegawa Y. Hypophosphatemic rickets developed after treatment with etidronate disodium in patient with generalized arterial calcification in infancy. Bone Rep. 2015;3:57–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sholler GF, Yu JS, Bale PM, Hawker RE, Celermajer JM, Kozlowski K. Generalized arterial calcification of infancy: three case reports, including spontaneous regression with long‐term survival. J Pediatr. 1984;105(2):257–60. [DOI] [PubMed] [Google Scholar]
- 10. Levy‐Litan V, Hershkovitz E, Avizov L, et al. Autosomal‐recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86(2):273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lorenz‐Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss‐of‐function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal‐recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86(2):267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mehta P, Mitchell A, Tysoe C, Caswell R, Owens M, Vincent T. Novel compound heterozygous mutations in ENPP1 cause hypophosphataemic rickets with anterior spinal ligament ossification. Rheumatology (Oxford). 2012;51(10):1919–21. [DOI] [PubMed] [Google Scholar]
- 13. Nakamura I, Ikegawa S, Okawa A, et al. Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum Genet. 1999;104(6):492–7. [DOI] [PubMed] [Google Scholar]
- 14. Saito T, Shimizu Y, Hori M, et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49(4):913–6. [DOI] [PubMed] [Google Scholar]
- 15. Albright RA, Stabach P, Cao W, et al. ENPP1‐Fc prevents mortality and vascular calcifications in rodent model of generalized arterial calcification of infancy. Nat Commun. 2015;6:10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li Q, Guo H, Chou DW, Berndt A, Sundberg JP, Uitto J. Mutant Enpp1asj mice as a model for generalized arterial calcification of infancy. Dis Model Mech. 2013;6(5):1227–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nitschke Y, Yan Y, Buers I, Kintziger K, Askew K, Rutsch F. ENPP1‐Fc prevents neointima formation in generalized arterial calcification of infancy through the generation of AMP. Exp Mol Med. 2018;50(10):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi Y, Goto S, Tanno T, Yamazaki M, Moriya H. Regional variations in the progression of bone loss in two different mouse osteopenia models. Calcif Tissue Int. 1998;62(5):426–36. [DOI] [PubMed] [Google Scholar]
- 19. Okawa A, Goto S, Moriya H. Calcitonin simultaneously regulates both periosteal hyperostosis and trabecular osteopenia in the spinal hyperostotic mouse (twy/twy) in vivo. Calcif Tissue Int. 1999;64(3):239–47. [DOI] [PubMed] [Google Scholar]
- 20. Hajjawi MO, MacRae VE, Huesa C, et al. Mineralisation of collagen rich soft tissues and osteocyte lacunae in Enpp1 ‐/‐ mice. Bone. 2014;69:139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mackenzie NC, Zhu D, Milne EM, et al. Altered bone development and an increase in FGF‐23 expression in Enpp1 ‐/‐ mice. PLoS ONE. 2012;7(2):e32177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Babij P, Roudier M, Graves T, et al. New variants in the Enpp1 and Ptpn6 genes cause low BMD, crystal‐related arthropathy, and vascular calcification. J Bone Miner Res. 2009;24(9):1552–64. [DOI] [PubMed] [Google Scholar]
- 23. Hawellek T, Hubert J, Hischke S, et al. Microcalcification of lumbar spine intervertebral discs and facet joints is associated with cartilage degeneration, but differs in prevalence and its relation to age. J Orthop Res. 2017;35(12):2692–9. [DOI] [PubMed] [Google Scholar]
- 24. Schmidt T, Ebert K, Rolvien T, et al. A retrospective analysis of bone mineral status in patients requiring spinal surgery. BMC Musculoskelet Disord. 2018;19(1):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zemojtel T, Kohler S, Mackenroth L, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease‐associated genome. Sci Transl Med. 2014;6(252):252ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Butscheidt S, Delsmann A, Rolvien T, et al. Mutational analysis uncovers monogenic bone disorders in women with pregnancy‐associated osteoporosis: three novel mutations in LRP5, COL1A1, and COL1A2. Osteoporos Int. 2018;29(7):1643–51. [DOI] [PubMed] [Google Scholar]
- 27. Ware CB, Horowitz MC, Renshaw BR, et al. Targeted disruption of the low‐affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development. 1995;121(5):1283–99. [DOI] [PubMed] [Google Scholar]
- 28. Parfitt AM, Drezner MK, Glorieux FH, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 1987;2(6):595–610. [DOI] [PubMed] [Google Scholar]
- 29. Busse B, Djonic D, Milovanovic P, et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell. 2010;9(6):1065–75. [DOI] [PubMed] [Google Scholar]
- 30. Roschger P, Fratzl P, Eschberger J, Klaushofer K. Validation of quantitative backscattered electron imaging for the measurement of mineral density distribution in human bone biopsies. Bone. 1998;23(4):319–26. [DOI] [PubMed] [Google Scholar]
- 31. Jansen RS, Duijst S, Mahakena S, et al. ABCC6‐mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation‐brief report. Arterioscler Thromb Vasc Biol. 2014;34(9):1985–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jansen RS, Kucukosmanoglu A, de Haas M, et al. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc Natl Acad Sci U S A. 2013;110(50):20206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stella J, Buers I, van de Wetering K, Hohne W, Rutsch F, Nitschke Y. Effects of different variants in the ENPP1 gene on the functional properties of ectonucleotide pyrophosphatase/phosphodiesterase family member 1. Hum Mutat. 2016;37(11):1190–201. [DOI] [PubMed] [Google Scholar]
- 34. Jepsen KJ, Pennington DE, Lee YL, Warman M, Nadeau J. Bone brittleness varies with genetic background in A/J and C57BL/6J inbred mice. J Bone Miner Res. 2001;16(10):1854–62. [DOI] [PubMed] [Google Scholar]
- 35. Burt LA, Liang Z, Sajobi TT, Hanley DA, Boyd SK. Sex‐ and site‐specific normative data curves for HR‐pQCT. J Bone Miner Res. 2016;31(11):2041–7. [DOI] [PubMed] [Google Scholar]
- 36. Cheung CL, Livshits G, Zhou Y, et al. Hip geometry variation is associated with bone mineralization pathway gene variants: the Framingham Study. J Bone Miner Res. 2010;25(7):1564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eddington H, Sinha S, Kalra PA. Vascular calcification in chronic kidney disease: a clinical review. J Ren Care. 2009;35(Suppl 1):45–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1. Plasma pyrophosphate and intact FGF23 in 10‐week Enpp1wt and Enpp1asj/+ male mice. Serum concentrations pyrophosphate and intact FGF23 in 10‐week WT and Enpp1 asj/wt male mice. No significant differences between plasma pyrophosphate was observed, but plasma FGF23 was slightly elevated in the heterozygous mice. *p < 0.05 (Student's unpaired t test).
Supplemental Table S1. Mutation status, frequency, phenotype match, inheritance match, and segregation analysis of all rare variants detected by the gene panel analysis.
Supplemental Table S2. Bone microstructure assessed at distal tibia by HR‐pQCT and percentages of sex‐ and age‐specific normative data.(26)
Supplemental Table S3. Trabecular bone architecture and density of 10‐ and 46‐week‐old wild‐type and heterozygous Enpp1 asj/WT male mice.
Supplemental Table S4. Cortical bone architecture and density of 10‐ and 46‐week‐old wild‐type and heterozygous Enpp1 asj/WT male mice.
Supplemental Table S5. Trabecular bone architecture and density of 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.
Supplemental Table S6. Cortical bone architecture and density of 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.
Supplemental Table S7. Skeletal histomorphometry of tibia from 10‐ and 23‐week‐old wild‐type and homozygous Enpp1 asj/asj male mice.