

Abstract
The incidence of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) is a common health problem in the clinic and is projected to increase in prevalence in the future. Mechanical ventilation is commonly used to provide respiratory support and has become indispensable in emergency and critical medicine. However, ventilator use can result in lung tissue damage, collectively termed ventilator-induced lung injury (VILI). In the present study, phosphoprotein profiling of blood and tissue samples from ventilated and non-ventilated mice was performed and key changes in protein levels and cell signaling during VILI were identified. Activation of the PI3K/AKT and mitogen activated protein kinase signaling pathways, in addition to changes in expression of cancer, inflammatory and cell-death related proteins were detected in response to mechanical ventilation. Focal adhesion-related protein levels and signaling pathways were also significantly altered in an injury model compared with control. VILI can affect patient mortality in ALI and ARDS cases, and no targeted treatment options currently exist. Identifying biomarkers and understanding the signaling pathways associated with VILI is critical for the development of future therapies.
Keywords: ventilation-induced lung injury, PI3K/AKT, mitogen activated protein kinase, focal adhesion-related signaling
Introduction
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are common in critical medicine, affecting as many as 200,000 Americans per year and with an overall mortality rate of approximately 40% (1,2). Acute respiratory failure is the most serious morbidity stemming from these pathologies. Mechanical ventilation is a commonly used modality for respiratory support and has become an indispensable technique for patient rescue and treatment in both emergency and critical medicine. However, ventilator use has advantages and disadvantages. On the one hand, ventilators assist patients in breathing and provide improved oxygenation. On the other hand, ventilation results in varying degrees of lung tissue damage, with a growing body of clinical evidence suggesting that mechanical ventilation duration is positively correlated with patient mortality (3-5). Lung damage induced by mechanical ventilation is referred to as ventilator-induced lung injury (VILI). Although the mechanisms underlying VILI remain unclear (6,7), VILI has been demonstrated to contribute to mortality in patients with ARDS (8,9).
Phosphorylation is an important post-translational protein modification, and it is estimated that more than one-third of all proteins in mammalian cells can be phosphorylated. Phosphorylated proteins play important roles in cellular functions (10-14), including signal transduction, differentiation, proliferation, cycle regulation, metabolism, transcription and translation, degradation and survival. Phosphoproteomic analyses identify, catalog, and characterize phosphorylated proteins (15). Because phosphorylated proteins are involved in numerous biological processes, the current study aimed to further clarify the mechanism of lung injury through the detection of phosphorylated proteins. The present study wish to further elucidate and clarify the mechanisms of VILI in mice using phosphoproteomics.
Materials and methods
Animal models
The Animal Use and Care Committee of the Second Military Medical University of China approved all protocols. Adult male Institute of Cancer Research (ICR) mice (age, 7-9 weeks; weight 20-28 g) were obtained from the Experimental Animal Center of the Second Military Medical University. The ICR mice were randomly divided between control (CON; n=3) and mechanically ventilated (VEN; n=3) groups. All mice were housed in individual cages in a temperature-controlled room (23±1˚C) with 0.05% CO2 and 12-h light/dark cycles and acclimatized for 1 week prior to experimentation. Food and water were available ad libitum until 8 h before the experiment. Eight hours prior to experimentation, access to food was removed, but free access to water was maintained. All mice were anesthetized using ketamine (100 mg/kg; Fujian Gutian Yuanhang Medical Co., Ltd.) and xylazine (10 mg/kg; Sigma-Aldrich; Merck KGaA) via intraperitoneal injection. Endotracheal intubations were then performed. CON group mice were able to breathe spontaneously and VEN group mice were connected to an Inspira ventilator (Harvard Apparatus Ltd.) and ventilated for 4 h at a volume of 30 ml/kg and a rate of 70 breaths/min. At the 4-h time-point, all mice were administered an intra-peritoneal dose of ketamine (100 mg/kg) and xylazine (10 mg/kg) and cardiac puncture was carried out as a terminal procedure to withdraw 0.5-1 ml of blood. The right ventricle was also rapidly perfused with normal saline (at 4˚C) to remove blood from the lungs. While still under anesthesia, cervical dislocation was carried out to euthanize the animals. Lung tissue samples were then harvested and stored at -80˚C prior to analysis.
Hematoxylin and eosin (H&E) staining
The lower left lobes of mouse lungs were collected for examination. Lungs were excised, fixed in 4% paraformaldehyde for 24 h at 4˚C, dehydrated using a series of increasing concentrations of ethanol, cleared in xylene, and embedded in paraffin. Paraffin-embedded lung tissues were cut into 5-µm sections and stained with H&E before observation using light microscopy, as previously described (13).
Detection of lung wet to dry ratio
After euthanasia of the mice, the lower right lobes of lungs were collected and immediately weighed. This initial value was termed wet weight (W). Lung tissues were then dried at 80˚C for 48 h and weighed again, with this second value termed dry weight (D). The W/D ratio was then calculated to evaluate the extent of lung edema.
Western blotting
Western blotting was performed to determine the expression levels of target proteins. Briefly, lung tissues from the middle lobes were homogenized and lysed in ice-cold RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 50 mM NaF, 1% NP-40, 0.1% DOC, 0.1% SDS, 1 mM EDTA, 1 mM PMSF, 1 mg/ml leupeptin). (Beyotime Institute of Biotechnology). After incubation for 5 min at 95˚C the supernatant was harvested and protein concentration quantified using the bicinchoninic acid assay (Bio-Rad Laboratories, Inc.). A total of 30 µg of protein from each sample was loaded into a 10% gel (Bio-Rad Laboratories, Inc.) for SDS-PAGE. The proteins were subsequently transferred onto PVDF membranes, which were blocked in 5% non-fat milk in 0.1% TBST for 2 h at room temperature. All antibodies were used at a dilution of 1:1,000. Membranes were then incubated with primary antibodies against Akt (cat. no. 9272S; Cell Signaling Technology, Inc.), phospho (p)-Akt and GAPDH (cat. nos. sc-377556 and sc-47724, respectively; both Santa Cruz Biotechnology, Inc.) at 4˚C overnight, rinsed in TBST three times (5 min for each rinse), and incubated with a horseradish peroxidase-conjugated secondary antibody (cat. no. ab205718; 1:2,000; Abcam) for 1 h at room temperature. Membranes were then washed with TBST a further 3 times. Visualization was performed using a chemiluminescence kit (SuperSignal™ West Pico substrate; Pierce; Thermo Fisher Scientific, Inc.), and images of protein bands captured Protein densitometry was performed using Quantity One 4.3.0 imaging software (Bio-Rad Laboratories, Inc.).
Protein extraction
Phosphoprotein profiling was performed using the Phospho Explorer Array PEX100 kit (Full Moon BioSystems Inc.) containing 1,318 antibodies. A magnetic serum albumin depletion kit (cat. no. LSKMAGD12) was purchased from EMD Millipore. Antibody arrays and cell lysates were prepared according to manufacturer's protocol. In brief, cell lysates obtained from the upper lobes of the lungs of CON and VEN group mice were biotinylated usingthe aforementioned antibody array assay kit. Antibody microarray slides were treated with a blocking solution for 30 min at room temperature, rinsed with Milli-Q grade water for 3-5 min, and dried with compressed nitrogen. Slides were then incubated with biotin-labeled cell lysates (~100 µg protein per slide) in coupling solution at room temperature for 2 h. The array slides were washed 4-5 times with 1x Wash Solution and rinsed extensively with Milli-Q grade water prior to detection of bound biotinylated protein using Cy3-conjugated streptavidin. The slides were then scanned using a GenePix 4000 scanner (Molecular Devices LLC) and the images analyzed using GenePix Pro 6.0 (Molecular Devices, LLC). The fluorescence signal of each antibody was obtained from antibody spot fluorescence intensity. Protein phosphorylation expression profiling was carried out using a Phospho Explorer protein array. A ratio computation was used to measure the extent of protein phosphorylation and was calculated as follows: Phosphorylation ratio=phosphorylation value/dephosphorylation value.
Bioinformatics analysis
Three independent samples for each experimental group were prepared under the same conditions and used for antibody array experiments. Array-measured protein expression was normalized to the internal positive control β-actin (Sigma-Aldrich; Merck KGaA) and a saline (Sigma-Aldrich; Merck KGaA) negative control. Statistical significance was determined by either Student's t-test or ANOVA using MATLAB 9.1. Omics Bean (http://www.omicsbean.cn/) was used to analyze the data and the resulting data are presented as the mean ± SEM. In cases where protein expression array identified significant fold changes, the roles of the proteins were checked for pathway mapping. For this, Gene Ontology software (http://omicslab.genetics.ac.cn/GOEAST/php/customized_microarray.php) was used to map these proteins which were subsequently checked for protein-protein interaction (PPI) mapping using Cytoscape software (version 3.2.0; https://cytoscape.org/) to identify candidate pathways and inherent hubs formed. GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were used to further understand the signaling pathways involved in VILI.
Statistical analysis
Differences between control (CON; n=3) and mechanically ventilated (VEN; n=3) groups were analyzed using unpaired Student's t-test in SPSS 17.0 (SPSS, Inc.). Only proteins found to differ in expression level by >30% and that returned a P<0.05 were considered statistically significant. Protein phosphorylation expression profiling was carried out using a Phospho Explorer protein array and Agilent GeneSpring GX 9 software (Agilent Technologies Inc.) and Metacore bioinformatics software (https://portal.genego.com/) were used to test the phosphorylation levels of proteins.
Results
Confirmation of the VILI mouse model
In order to determine whether the application of mechanical ventilation induced lung injury in mice, lung morphology was examined in VEN and CON mice post-mortem using H&E staining. While the CON group demonstrated normal lung structure and minimal inflammatory cell presence (Fig. 1), the VEN group showed neutrophil sequestration and infiltration around the pulmonary vessels and the airway, specifically distributed in the alveolar and interstitial spaces (Fig. 1). Edema extent was examined using the W/D weight ratio, and the results indicated that edema extent was significantly elevated in the VEN group compared with the CON group (Fig. 2; P<0.05).
Figure 1.

Lung hematoxylin and eosin staining. CON group mice showed normal lung structure, while VEN group mice lungs presented neutrophil sequestration and infiltration in the alveolar and interstitial spaces. Magnification, x200. CON, control; VEN, mechanically ventilated.
Figure 2.
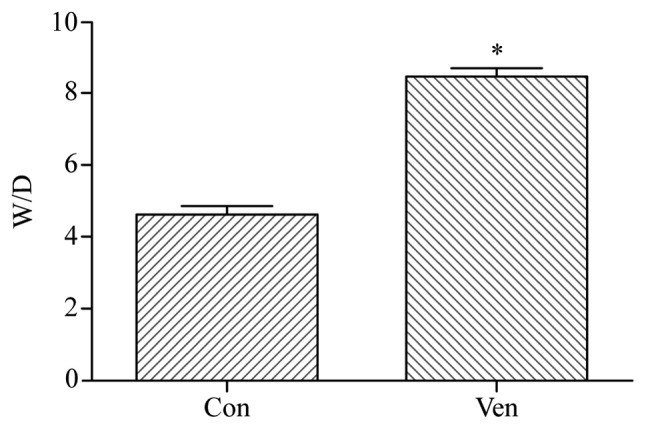
Lung tissue W/D weight ratio. VEN mice showed significantly elevated W/D ratio compared with CON. *P<0.05 vs. CON. CON, control; D, dry; VEN, mechanically ventilated; W, wet.
Proteomic analysis
Raw protein expression values were normalized and then subjected to a fold-change cut-off of >|1.5|. Fold-differences in protein expression with P<0.05 were considered significant. Notable differences in protein expression between CON and VEN group mice were observed in 115 proteins, with 37 increased and 78 decreased protein levels (Table SI). Gene changes are presented as a heatmap (Fig. 3).
Figure 3.

Heatmap of the 6 samples. Proteins are presented in columns while rows represent individual samples. Red represents the proteins with upregulated phosphorylation levels in thr VEN group. Green represents the proteins with downregulated phosphorylation levels in the VEN group. Black represents unaltered protein phosphorylation levels in the VEN group. CON, control; VEN, mechanically ventilated.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis
To further understand the signaling pathways involved in VILI, bioinformatics analysis was performed. Homologous analysis of differentially expressed proteins based on GO was performed, which showed that differences in protein expression levels observed in VEN group mice were primarily in biological processes (Fig. 4). Biological process analysis found that most of these proteins were involved in ‘positive regulation of cellular processes’, ‘response to organic substance’ and ‘positive regulation of metabolic process’.
Figure 4.

Homologous analysis of differentially expressed proteins related to biological processes in mechanically ventilated mice. The top 10 most significantly enriched biological processes are presented.
Based on GO analysis results, potential pathways involved in VILI and control group were investigated using KEGG pathway analysis (Fig. 5). In VEN group mice, the PI3K-AKT signaling pathway was the most markedly altered. Additionally, ‘pathways in cancer’, ‘focal adhesion’, ‘MAPK signaling pathway’ and ‘Ras signaling pathway’ were also significantly enriched (Fig. 5; Table SII; and Figs. S1-S5).
Figure 5.

Top 10 enriched Kyoto Encyclopedia of Genes and Genomes pathways in the VEN group mice.
PPI analysis
To further understand how the identified proteins of interest interact with each other, bioinformatics analysis using Cytoscape was performed to visualize a PPI network (Fig. 6). This analysis revealed that hub proteins were enriched in ‘inflammatory response’ and ‘regulation of response to wounding’ in VILI mice.
Figure 6.

Protein-protein interaction analysis network summary constructed using Cytoscape software. P-values are for GO/Kyoto Encyclopedia of Genes and Genomes analysis; fold change is for protein.
Validation
In order to confirm the proteomics, GO, and KEGG analyses, western blotting was performed to examine AKT phosphorylation levels in VEN and CON mice. AKT phosphorylation was significantly reduced in the VEN group compared with the CON group (Fig. 7; CON group p-AKT/AKT ratio, 0.758±0.055; VEN group, 0.387±0.037; P<0.05).
Figure 7.

p-Akt/Akt western blotting results with densitometry comparisons between groups. (A) Densitometry of p-AKT/AKT ratios of CON and VEN groups. (B) Representative western blotting membrane. *P<0.05 vs. CON. CON, control; p, phosphorylated; VEN, mechanically ventilated.
Discussion
Numerous studies have shown that protein phosphorylation and dephosphorylation play important roles in a range of physiological and biochemical processes, including cell cycle regulation, signal transduction, neuronal growth and memory processes (10,11). Therefore, exploring the molecular mechanisms underlying protein phosphorylation is critical in examining disease mechanisms and identifying potential therapeutic targets. Since approximately 30% of proteins in eukaryotic cells can be phosphorylated, each protein kinase selectively modifies a specific substrate protein to ensure signal transduction within a complex cellular environment (8,10-17). The results of the present study suggest that phosphorylated proteins are involved in VILI, in particular, phosphorylation of proteins involved in PI3K-AKT and MAPK signaling pathways.
VILI is essentially caused by an uncontrolled inflammatory response. Abnormal mechanical tension in the lung, due to artificial ventilation, initiates inflammatory signal transduction in inflammatory cells such as neutrophils and macrophages (18-21), resulting in cell activation and the promotion of phagocytic function, as well as the synthesis and release of inflammatory mediators such as interleukin (IL)-16, IL-10, and tumor necrosis factor-α (1). PI3K activation has been found to regulate neutrophil activation observed in ALI (18), and both pharmacological and induced pluripotent stem cell-mediated inhibition of the PI3K pathway have been documented to ameliorate inflammatory responses and inflammatory cell activation associated with VILI (19,20). PI3K-AKT-mediated reactive oxygen species (ROS) production has also been linked to VILI (20). Moreover, Wnt/β-catenin signaling, which can be induced by PI3K pathway activation, has been shown to be activated by mechanical ventilation and has been found to have a role in the promotion of inflammation and fibrosis in otherwise healthy rat lungs (21).
The MAPK pathway is likewise involved in a myriad of cellular and systemic processes and mechanisms. In the context of VILI, mechanical stress and stretching of the alveolar epithelium activates epidermal growth factor receptor (EGFR)-mediated MAPK signaling, resulting in elevated pulmonary permeability (22,23). Similarly, inhibition of EGFR-mediated MAPK signaling has been shown to reduce lung inflammation (23). Mechanical stress has been shown to induce the release of ROS through MAPK-dependent pathway activation of xanthine oxidoreductase (24). Finally, MAPK pathway signaling has been demonstrated to facilitate microRNA-127-induced inflammation and pulmonary permeability (22-25). In the present study, bioinformatics-based protein phosphorylation analysis suggested that the ‘MAPK signaling pathway’ was activated in VILI. Focal adhesion formation and behavior has been demonstrated to be pivotal to pulmonary epithelial function, and has been linked with modulation of ALI injury severity (15). Focal adhesions (FAs), large macromolecular complexes mediating interactions between cells and the extracellular matrix (ECM), have been linked to the development of pulmonary fibrosis and injury, as FAs play a part in mediating epithelial function, and epithelial dysfunction is a hallmark of pulmonary injury (16). The inhibition of focal adhesion kinase (FAK), a protein that is activated by integrin-ECM interactions and fibroblast-secreted transforming growth factor-β (TGF-β), has been shown to exacerbate bleomycin-induced lung injury in mice (16). Moreover, FA formation, FAK phosphorylation, and FA arrangement and rearrangement have been shown to mediate preservation of pulmonary vascular barrier function (17). In the present study, the phosphorylation of FA-associated proteins was found to be significantly influenced by VILI. In particular, 3-phosphoinositide-dependent protein kinase 1 (PDK1) phosphorylation was elevated in the VEN group. PDK1 is a downstream kinase of PI3K, and has been linked with cell adhesion and the organization of structural fibers and tubules (26,27). PDK1 has also been suggested to play a role in fibroblast-mediated pulmonary fibrosis, acting as a secondary messenger to activate Akt upon TGFβ-mediated PI3K activation (27). However, PDK1 activation has also been demonstrated to be protective, when in conjunction with serum/glucocorticoid-induced protein kinase 1, in an in vitro cellular model (28). Given the complexity of the PI3K-PDK1-AKT pathway and the diverse range of mechanisms that it modulates, more research is required to clarify how this pathway modulates FA formation during health and injury.
The results of the present study suggested that the expression level of RAC1 was upregulated in VEN mice as shown in Table SII. Rac is a protein that mediates epithelial barrier integrity through interactions with microtubules (29). Given that epithelial dysfunction is a principle element of VILI, changes in Rac levels were anticipated as Rac upregulation and activation in lung injury models is typically linked to protective mechanisms (30). As such, the elevated phosphorylation observed in VEN group mice may be a compensatory mechanism in response to physical damage. In-depth pathway analysis and functional investigation is required to fully characterize the role of Rac in VILI.
While the present study was limited by the small sample size and the fact that only AKT phosphorylation was assessed in a validation study, the results of this study suggest several candidate cellular mechanisms that may be involved in VILI. Future studies will focus on determining how other elements of the PI3K-AKT pathway, as well as other potential candidate pathways, may be involved in VILI. Determining the precise roles of specific proteins, as well as establishing proof of principle for these causal relationships, will be a primary focus for future studies to establish potential targets for future therapeutics against VILI.
Supplementary Material
Acknowledgments
The authors would like to thank Mr Ye Changlin from the Shanghai Institute of Physical Education for his technical support.
Funding
This study was funded by the Shanghai Municipal Commission of Health and Family Planning Funding for Key Developing Disciplines (grant no. 2015ZB0101) and the Multi-Center Clinical Study of Pre-operative Pain Facilitation and Post-operative Cognitive Dysfunction, Joint Research Project of Pudong New Area Health and Family Planning Commission (grant no. PW2015D-3).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
RR and WY made substantial contributions to conception and design; ZR, HD and JD were involved in acquisition of data, analysis and interpretation of data; RR and JD were involved in drafting the manuscript; WY were involved in revising it critically for important intellectual content; all authors gave final approval of the version to be published.
Ethics approval and consent to participate
All protocols were approved by the Animal Use and Care Committee of the Second Military Medical University of China. The animal production license number was SYXK (Shanghai) 2012-0003.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Rezoagli E, Fumagalli R, Bellani G. Definition and epidemiology of acute respiratory distress syndrome. Ann Transl Med. 2017;5(282) doi: 10.21037/atm.2017.06.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu KD, Matthay MA. Advances in critical care for the nephrologist: Acute lung injury/ARDS. Clin J Am Soc Nephrol. 2008;3:578–586. doi: 10.2215/CJN.01630407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuipers MT, van der Poll T, Schultz MJ, Wieland CW. Bench-to-bedside review: Damage-associated molecular patterns in the onset of ventilator-induced lung injury. Crit Care. 2011;15(235) doi: 10.1186/cc10437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rocco PR, Dos Santos C, Pelosi P. Pathophysiology of ventilator-associated lung injury. Curr Opin Anaesthesiol. 2012;25:123–130. doi: 10.1097/ACO.0b013e32834f8c7f. [DOI] [PubMed] [Google Scholar]
- 5.Frank JA, Matthay MA. Science review: Mechanisms of ventilator-induced injury. Crit Care. 2003;7:233–241. doi: 10.1186/cc1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silva PL, Negrini D, Rocco PR. Mechanisms of ventilator-induced lung injury in healthy lungs. Best Pract Res Clin Anaesthesiol. 2015;29:301–313. doi: 10.1016/j.bpa.2015.08.004. [DOI] [PubMed] [Google Scholar]
- 7.Han GJ, Li JQ, Pan CG, Sun JX, Shi ZX, Xu JY, Li MQ. Experimental study of airway pressure release ventilation in the treatment of acute respiratory distress syndrome. Exp Ther Med. 2017;14:1941–1946. doi: 10.3892/etm.2017.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 9.Gattinoni L, Tonetti T, Quintel M. Regional physiology of ARDS. Crit Care. 2017;21 (Suppl 3)(S312) doi: 10.1186/s13054-017-1905-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 11.Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 12.Charbonneau H, Tonks NK. 1002 protein phosphatases? Annu Rev Cell Biol. 1992;8:463–493. doi: 10.1146/annurev.cb.08.110192.002335. [DOI] [PubMed] [Google Scholar]
- 13.Yanjun Liu, Xuejun Lv, Wei Zhao, Ming Dong Hu, Yuying Li, Guanchong Wang, Jiancheng Xu, Guisheng Qian. Sirt1 protects against lipopolysaccharide-induced injury in mouse type II alveolar epithelial cells by deacetylating RelA/p65 subunit of nuclear factor-κB. Journal of the Third Military Medical University. 2017;39:1415–1421. [Google Scholar]
- 14.Cohen P. The origins of protein phosphorylation. Nat Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 15.Suwanmajo T, Krishnan J. Mixed mechanisms of multi-site phosphorylation. J R Soc Interface. 2015;12(20141405) doi: 10.1098/rsif.2014.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wheaton AK, Agarwal M, Jia S, Kim KK. Lung epithelial cell focal adhesion kinase signaling inhibits lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2017;312:L722–L730. doi: 10.1152/ajplung.00478.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Bittman R, Garcia JG, Dudek SM. Junctional complex and focal adhesion rearrangement mediates pulmonary endothelial barrier enhancement by FTY720 S-phosphonate. Microvasc Res. 2015;99:102–109. doi: 10.1016/j.mvr.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li LF, Liu YY, Yang CT, Chien Y, Twu NF, Wang ML, Wang CY, Huang CC, Kao KC, Hsu HS, et al. Improvement of ventilator-induced lung injury by IPS cell-derived conditioned medium via inhibition of PI3K/Akt pathway and IP-10-dependent paracrine regulation. Biomaterials. 2013;34:78–91. doi: 10.1016/j.biomaterials.2012.09.042. [DOI] [PubMed] [Google Scholar]
- 19.Uhlig U, Fehrenbach H, Lachmann RA, Goldmann T, Lachmann B, Vollmer E, Uhlig S. Phosphoinositide 3-OH kinase inhibition prevents ventilation-induced lung cell activation. Am J Respir Crit Care Med. 2004;169:201–208. doi: 10.1164/rccm.200303-343OC. [DOI] [PubMed] [Google Scholar]
- 20.Spassov SG, Donus R, Ihle PM, Engelstaedter H, Hoetzel A, Faller S. Hydrogen sulfide prevents formation of reactive oxygen species through PI3K/Akt signaling and limits ventilator-induced lung injury. Oxid Med Cell Longev. 2017;2017(3715037) doi: 10.1155/2017/3715037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villar J, Cabrera NE, Valladares F, Casula M, Flores C, Blanch L, Quilez ME, Santana-Rodríguez N, Kacmarek RM, Slutsky AS. Activation of the Wnt/β-catenin signaling pathway by mechanical ventilation is associated with ventilator-induced pulmonary fibrosis in healthy lungs. PLoS One. 2011;6(e23914) doi: 10.1371/journal.pone.0023914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otulakowski G, Engelberts D, Gusarova GA, Bhattacharya J, Post M, Kavanagh BP. Hypercapnia attenuates ventilator-induced lung injury via a disintegrin and metalloprotease-17. J Physiol. 2014;592:4507–4521. doi: 10.1113/jphysiol.2014.277616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bierman A, Yerrapureddy A, Reddy NM, Hassoun PM, Reddy SP. Epidermal growth factor receptor (EGFR) regulates mechanical ventilation-induced lung injury in mice. Transl Res. 2008;152:265–272. doi: 10.1016/j.trsl.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdulnour RE, Peng X, Finigan JH, Han EJ, Hasan EJ, Birukov KG, Reddy SP, Watkins JE III, Kayyali US, Garcia JG, et al. Mechanical stress activates xanthine oxidoreductase through MAP kinase-dependent pathways. Am J Physiol Lung Cell Mol Physiol. 2006;291:L345–L353. doi: 10.1152/ajplung.00453.2005. [DOI] [PubMed] [Google Scholar]
- 25.Li Q, Ge YL, Li M, Fang XZ, Yuan YP, Liang L, Huang SQ. miR-127 contributes to ventilator-induced lung injury. Mol Med Rep. 2017;16:4119–4126. doi: 10.3892/mmr.2017.7109. [DOI] [PubMed] [Google Scholar]
- 26.Medina-Tato DA, Ward SG, Watson ML. Phosphoinositide 3-kinase signalling in lung disease: Leucocytes and beyond. Immunology. 2007;121:448–461. doi: 10.1111/j.1365-2567.2007.02663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang W, Wei K, Ho L, Berry GJ, Jacobs SS, Chang CH, Rosen GD. A critical role for the mTORC2 pathway in lung fibrosis. PLoS One. 2014;9(e106155) doi: 10.1371/journal.pone.0106155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caohuy H, Yang Q, Eudy Y, Ha TA, Xu AE, Glover M, Frizzell RA, Jozwik C, Pollard HB. Activation of 3-phosphoinositide-dependent kinase 1 (PDK1) and serum- and glucocorticoid-induced protein kinase 1 (SGK1) by short-chain sphingolipid C4-ceramide rescues the trafficking defect of ΔF508-cystic fibrosis transmembrane conductance regulator (ΔF508-CFTR) J Biol Chem. 2014;289:35953–35968. doi: 10.1074/jbc.M114.598649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karki P, Birukova AA. Microtubules-associated Rac regulation of endothelial barrier: A role of Asef in acute lung injury. J Investig Med. 2017;65:1089–1092. doi: 10.1136/jim-2017-000571. [DOI] [PubMed] [Google Scholar]
- 30.Birukova AA, Wu T, Tian Y, Meliton A, Sarich N, Tian X, Leff A, Birukov KG. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J. 2013;41:165–176. doi: 10.1183/09031936.00148311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.