

Abstract
DNA damage response (DDR) genes orchestrating the network of DNA repair, cell cycle control, are essential for the rapid proliferation of neural progenitor cells. To date, the potential association between specific DDR genes and the risk of human neural tube defects (NTDs) has not been investigated. Using whole-genome sequencing and targeted sequencing, we identified significant enrichment of rare deleterious RAD9B variants in spina bifida cases compared to controls (8/409 vs. 0/298; p = .0241). Among the eight identified variants, the two frameshift mutants and p.Gln146Glu affected RAD9B nuclear localization. The two frameshift mutants also decreased the protein level of RAD9B. p.Ser354Gly, as well as the two frameshifts, affected the cell proliferation rate. Finally, p.Ser354Gly, p.Ser10Gly, p.Ile112Met, p.Gln146Glu, and the two frameshift variants showed a decreased ability for activating JNK phosphorylation. RAD9B knockdowns in human embryonic stem cells profoundly affected early differentiation through impairing PAX6 and OCT4 expression. RAD9B deficiency impeded in vitro formation of neural organoids, a 3D cell culture model for human neural development. Furthermore, the RNA-seq data revealed that loss of RAD9B dysregulates cell adhesion genes during organoid formation. These results represent the first demonstration of a DDR gene as an NTD risk factor in humans.
Keywords: DNA damage response, RAD9B, spina bifida, stem cell
1 |. INTRODUCTION
Neural tube defects (NTDs) result from the failure of neural tube closure (NTC) during the first 4 weeks of human embryonic development (Avagliano et al., 2018; Wallingford, Niswander, Shaw, & Finnell, 2013). They are among the most severe of all human structural malformations, with an average global prevalence ranging from 0.69 to 2.19 per 1,000 newborns (Zaganjor et al., 2016). Although there are multiple types of NTDs, spina bifida (SB) represents the most prevalent form of caudal NTDs, while anencephaly is the most frequent failure of rostral NTC (Avagliano et al., 2018). Numerous studies over decades indicate that maternal periconceptional supplementation with folic acid (FA) can significantly reduce the risk of having an NTD affected pregnancy (Copp, Stanier, & Greene, 2013). Nonetheless, NTDs remain a significant clinical problem, as more than 30% of NTD affected pregnancies may be FA resistant, accounting for a large number (>400,000 pregnancies per year) of affected infants even in countries that employ mandatory folate fortification of the food supply (Copp et al., 2013). Recently, FA-resistant NTDs were reported to be associated with multiple developmental signaling pathways, including the Wnt/PCP (planar cell polarity; Chen, Lei, Cao et al., 2018; Kharfallah et al., 2017; Kibar et al., 2009; Lei et al., 2010, 2019), sonic hedgehog (Murdoch & Copp, 2010), and mitochondrial folate metabolic pathways (Kim et al., 2018). However, mutations in those pathways have been identified in only a small fraction of NTD cases (Greene & Copp, 2014). As a result, the underlying mechanisms and etiologies for most FA-resistant NTDs remain unclear.
The DNA damage response (DDR) elements perform an essential role in governing neural progenitor genome integrity (Enriquez-Rios et al., 2017). Once activated by ATR (ataxia telangiectasia and Rad3-related protein) signaling, RAD9 will form the 9-1-1 complex (RAD9-HUS1-RAD1) to protect genome integrity by activating Chk1, thus establishing a DNA damage checkpoint and preventing potential double-strand breaks (Dufault, Oestreich, Vroman, & Karnitz, 2003; Perez-Castro & Freire, 2012; Sierant, Archer, & Davey, 2010). Functional divergence in the RAD9 protein family is indicated by expression of RAD9B predominantly in the testis, with translocation of RAD9B to the nucleoli under nucleolar stress, yet RAD9B still interacts with components of the 9-1-1 complex. Gene inactivation of the three 9-1-1 genes, Hus1, Rad9a, and Rad9b in mice all resulted in abnormal NTC and increased embryonic lethality (Weiss, Enoch, & Leder, 2000). However, only the Rad9b heterozygous knockout (MRad9B+/−) embryos exhibited NTC defects (exencephaly), which occurred in 30% of these embryos (Leloup et al., 2010). This suggests that Rad9b has a unique and crucial role in NTC. Despite the importance of DDR, 9-1-1 components and RAD9B specifically during murine NTC, there is a significant data gap with respect to whether RAD9B mutations are enriched in human SB patients, nor are the molecular mechanisms by which RAD9B may impact early neurodevelopment well understood.
In this study, we initially performed data mining on 129 SB whole-genome sequencing (WGS) datasets and identified three, predicted to be damaging variants in the RAD9B gene. We further confirmed the association between RAD9B rare deleterious variants and the risk for NTDs by resequencing a validation cohort with 280 SB cases. Functional assays were performed to determine whether any of the variants detected adversely affected RAD9B function. After overexpression in HeLa cells, two of these variants were found to affect protein stability due to truncation, while three of these variants failed to translocate to the nucleus and form DNA damage foci under conditions of oxidative stress. Overexpression of RAD9B mutant plasmids also influenced RAD9B ability to activate JNK signaling and altered the proliferation rate of these cells, which is consistent with previous findings. Knockdown (KD) of RAD9B in embryoid bodies (EBs) derived from human embryonic stem cells (hESCs) demonstrated dysregulation of OCT4 and PAX6 transcriptional circuitry, indicating irregular specification of neural ectoderm. Conversely, loss of RAD9B resulted in impairment of neural differentiation and neural-organoid formation via dysregulating cell adhesion and extracellular matrix (ECM) interaction pathways. These data are consistent with the findings of neurodevelopmental defects observed in mouse models and support the hypothesis that these RAD9B variants may contribute to the etiology of human SB through impairment of early NTC.
2 |. MATERIALS AND METHODS
2.1 |. Human subjects and sequencing
A total of 129 SB infant samples for WGS were collected from the California Birth Defects Monitoring Program and from the national Spina Bifida Clinic at Hamad Medical Corporation in Doha, Qatar, as previously reported (Chen, Lei, Zheng et al., 2018). Sixty samples were collected in California during 1983–1999. All 60 samples were from non-Hispanic white infants with 43% of the samples being male. DNA samples of 108 unaffected healthy individuals with matched ancestry were collected as controls for the Middle Eastern cohort of NTDs (2). Genomic DNA was extracted either from newborn screening bloodspots or infant/child venipuncture samples using the Puregene DNA Extraction Kit (Qiagen, Valencia, CA). Input amounts of DNA from infant bloodspots were in the range of 200–500 ng, while inputs from venipuncture samples were 2–3 μg. All DNA samples were whole-genome sequenced using Illumina chemistries (v3) on HiSeq2500 instruments to yield short insert paired-end reads of 2 × 100 bp. The average depth of coverage was approximately 30× for all samples. The use of specimens, including the collection and inclusion of archived newborn bloodspots, was approved by the California State Committee for the Protection of Human Subjects as well as the Institutional Review Board at Stanford University.
For the Phase II validation study, 280 SB samples were provided by Dr. Nicholas J. Marini from the University of California at Berkeley. For this cohort, mothers with a history of SB-affected pregnancies were recruited from August 2009 to November 2012 through a web-based outreach program supported by 32 Spina Bifida Associations and 61 SB specialty practices within the USA These data collection procedures were approved by the Institutional Review Board at the Children’s Hospital and Research Center Oakland (PI Edward J. Lammer). Mothers were considered fully enrolled in the study if (a) mother confirmed a SB-affected pregnancy, including the phenotype (thoracic or lumbar), (b) mother provided demographic data by completing an online survey, and (c) mother and/or affected offspring provided consent and a saliva sample for DNA extraction. Offspring of Caucasian mothers with reported non-Caucasian fathers were excluded. Genomic DNA was extracted from 500 ul–3 ml saliva samples following the manufacturer’s instruction (DNA Genotek, Kanata, ON, Canada). Total DNA yield was determined using the Quant-iT DNA High Sensitivity Assay Kit (Life Technologies, Carlsbad, CA). RAD9B Sanger resequencing was performed using the ABI3730 DNA Analyzer. Primers for both polymerase chain reaction (PCR) and sequencing are attached in Table S1. This study was approved by the Institutional Review Board (IRB) at the University of Texas at Austin. Written informed consent was obtained from the parents or guardians of all subjects. Recruitment protocols were approved by IRBs in the United States (state of California and Stanford University, the University of Texas at Austin, Weill Cornell Medical College-NY), and the Middle Eastern population receiving their healthcare in Qatar (Hamad Medical Corporation and Weill Cornell Medical College-Qatar), including informed consent documentation provided in both English and Arabic.
2.2 |. Plasmids and siRNAs
pcDNA-3.1(+)-N-eGFP-RAD9B (RAD9B-GFP, clone ID: Z26522) was purchased from Genescript. RAD9B p. Ser318FS, p. Arg401FS, and p. Ser354Gly were introduced into pcDNA-3.1(+)-N-eGFP-Rad9B plasmids by the GeneArt® Site-Directed Mutagenesis System (Life Technologies). Small interfering RNAs (siRNAs) targeting RAD9B Exons 9 and 10 were purchased from Thermo’s (Life Technologies) predesigned RNAi assay pool, containing siRNA-id: 38957, 39050. The scrambled siRNA negative control was purchased from Sigma–Aldrich, product ID: SIC003, MISSION® siRNA Fluorescent Universal Negative Control #1.
2.3 |. Cell culture and transfection
HeLa cells (human cervical cancer) and HEK293T were grown in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. Cells were grown to 50–70% confluent before transfection. Cell transfection was carried out by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
2.4 |. hESC cell culture and transfection
BG01V/hOG (hESC hOct4-GFP reporter) cells were purchased from Invitrogen. hESCs were maintained on a mouse embryonic fibroblast (MEF) feeder layer with hESC media (DMEM/F12 with GLUTAMAX, 20% Knockout Serum Replacement, 0.1 mM NEAA, 55 μM 2-Mercaptoethanol, and bFGF 10 μg/ml). MEF feeder cells were grown in MEF media (DMEM, 10% FBS, NEAA) before inactivation with mitomycin C (10 μg/ml for 3 hr at 37°C). A 10-μM Rock Inhibitor (Y27632) was replenished in hESC media during cell passages. For transfection, 0.5–1 × 106 cells were pelleted and resuspended in 100 μl of transfection solution as performed in Ma et al. (2010). The total transfected siRNA amount used in both embryoid body and neural differentiation was 15 pmol for 1x, 45 pmol for 3x for both embryoid body and neural differentiation.
2.5 |. EB formation and neural differentiation
hESC colonies were treated with collagenase and subsequently split using accutase for 5 min at 37°C. The cells were replated into a round-bottomed 96 well ultra-low attachment plate with EB medium (80% DMEM/F12, 20% HI-FBS, 1% NEAA, and 1% GLUTAMAX) and centrifuged for 2 min at 3,000 rpm. After 2–3 days, EBs were individualized by applying protocol from Vichier-Guerre, Parker, Pomerantz, Finnell, and Cabrera (2017), and replated onto matrigel (9–12 mg/ml; cat# 354230; BD bioscience) coated wells in neural differentiation media (DMEM/F12, Neural Basal [Gibco] 1:1; 1:100 N2 [Invitrogen]; 300 mg/ml bovine serum albumin fraction V; 1: 50 B27 [Invitrogen]; bFGF 1:1000, EGF 1:1000 [Invitrogen growth factor], insulin 20 mg/ml and Anti-anti 1:100). After 21 days, the cells were lifted with accutase and replated on matrigel for neural-organoid formation.
2.6 |. Western blot assay
Cells were transfected with constructs using Lipofectamine 2000 (Invitrogen, Waltham, MA). The cells were lysed with 1% NP40 buffer and were immunoblotted with anti-GFP (Santa Cruz), anti-JNK (CST; Cell Signaling, Danvers, MA), anti-GAPDH(CST), anti-p-JNK(CST), 1RDye® 800CW goat anti-rabbit IgG secondary antibodies (LI-COR, Cambridge, UK) and 1RDye® 680CW goat anti-mouse IgG secondary antibodies (LI-COR). Images were captured by the Odyssey® system (LI-COR). Quantification analysis is performed by using Image J.
2.7 |. Immunocytochemistry and flow cytometry
Cells from various treatment groups were washed in PBS, fixed in 4% (wt/vol) formaldehyde, immunostained overnight with indicated antibodies TUBB3, POU5F1 (1:200; Cell Signal); Nestin, Tuj1, Sox2 (1:50; DSHB); Peripherin (1:200; Abcam); GFP (Santa Cruz), ɤ-H2AX (CST) with 1% BSA (Sigma–Aldrich) and washed three times with cold PBS followed by a 1-hr incubation with the indicated secondary antibody (Cell Signal, Alexa fluor 488, 555, and 647). Images were taken by using a deconvolution microscope (Nikon T2). Canto II Yellow Green Carrousel is used for cell flow cytometry experiments after the individualization of the EB cells.
2.8 |. RT and qRT-PCR
For reverse transcription polymerase chain reaction (RT-PCR) analysis, total messenger RNA (mRNA) was extracted using an RNA Extraction Kit (Zymo Research). First-Strand cDNA was synthesized by a cDNA Reverse Transcription Kit (Applied Biosystem). qPCR was carried out with designed primers: (PAX6: F-5′ GAAGTGGTGCCCGAGGT 3′; R-5′ AGTCCCCAGCCAGACCT 3′; Rad9B: F-5′ AATTT TGCCCATCTTTAGATGTCTGAA 3′; R-5′ ATGTCTGTAGAAGAATTGAATAACTACT 3′; GAPDH: F-5′ GTATTGGGCGCCTGGTCAC 3′; R-5′ CATGTAAACCTGGGGGAATACG 3′). mRNA levels were quantitatively measured by using Applied Biosystem SYBR 2X master Mix on an ABI QuantStudio Flex 7.
2.9 |. Cell proliferation assay
The proliferation assays were performed using the Click-it Alex fluor 647 Edu Assay Kit (Invitrogen) following the protocol provided by the manufacturer with a 12-hr incubation. Quantification analysis was performed by using Image J.
2.10 |. Bioinformatics
The WGS data were processed through, using the CASAVA pipeline (1.9.0a1; Illumina) with build 37 (hg19) of the human reference genome. Variants in each sample were further filtered for high genotype quality (≥20), and annotated according to Human Genome Variation Society (HGVS) nomenclature (http://www.hgvs.org/mutnomen/). A variant was designated as a novel if it was not found in the ExAC (Exome Aggregation Consortium) database. The potential pathogenic effect of the missense variants on protein function was predicted using online programs: PolyPhen V2 (Polymorphism Phenotyping; http://genetics.bwh.harvard.edu/pph2/), SIFT (Sorting Intolerant From Tolerant; https://sift.bii.a-star.edu.sg), and PANTHER (Protein Analysis Through Evolutionary Relationships; http://www.pantherdb.org/). All parameters were set as per the website’s recommendation. The localization of the variants in their protein domains was assessed by Uniprot (http://www.uniprot.org/). Gene scheme structure was generated by using Lollipops (Jay & Brouwer, 2016). RNA-seq data analysis was performed by using kallisto and DEseq2 (Bray, Pimentel, Melsted, & Pachter, 2016). Gene set enrichment analysis (GSEA) is carried out by using javaGSEA2–3.0 (Subramanian et al., 2005).
2.11 |. Statistical analysis
All data (mean ± standard error) were analyzed with a Student’s t test, and p values <.05 were considered statistically significant. Rad9B mutation enrichment was detected by Fisher’s Exact test compared to the ExAC database (variants with MAF <0.001 were counted).
3 |. RESULTS
3.1 |. Rare and novel variants in RAD9B are associated with an increased risk for human SB
Interrogating the WGS data from 129 SB cases, two predicted loss of function (LoF) variants (NM_152442.4:g.110960044 delA, p.S318SfsX28; NM_152442.4:g.110968402 insC, p.A401Rfs4X) and one predicted-to-be-damaging rare missense variant (NM_152442.4:g.110960151A>G, p.Ser354Gly) were identified in the RAD9B gene (MAF < 1%, gnomAD frequency (AF) < 0.001, SIFT < 0.1 or PolyPhen > 0.9). Using the same criteria (Table 1), there were no LoF or predicted-to-be-damaging rare variants identified in 108 unaffected controls. The p.Ser354Gly and p.S318SfsX28 variants were both absent from either the gnomAD (http://gnomad.broadinstitute.org) or the ExAC database (http://exac.broadinstitute.org/; Lek et al., 2016; Table 1). The two RAD9B frameshift variants are both predicted to affect amino acids located in RAD9B’s C-terminal tail motif (Figure 1a), which contains a nuclear localization signal (Perez-Castro & Freire, 2012). The novel missense variant, p.Ser354Gly is shown to affect RAD9B phosphorylation modifications according to the mass spectrometry data from Uniprot (https://www.uniprot.org/). It is worth noting that amino acid 354Ser is located within the low complexity region, which represents a potential transcriptional activation domain of the protein. All three variants were heterozygous in the three different SB patients (Figure 1b).
TABLE 1.
Table for the detected Rad9B mutations in two SB cohorts
| Locus (GRCh37) | Case/Control | Predicted DNA change | Predicted Protein Variants | Function | SNP ID | SIFT | POLYPhen V2 Score | ExAC Frequency | gnomAD Frequency |
|---|---|---|---|---|---|---|---|---|---|
| Chr12:110960044 | Case | NM_152442.3:g. 110960044 delA | p.Ser318Serfs*28 | Frameshift | / | / | / | 0 | 0 |
| Chr12:110968402 | Case | NM_152442.3:g. 110968402 insC | p.Ala401Argfs*4 | Frameshift | / | / | / | 0.000074 | 0.000072 |
| Chr12:110960151 | Case | NM_152442.3:c.1060A>G | p.Ser354Gly | nonsynonymous SNV | / | 0.36 | 0.010 | 0 | 0 |
| Chr12:110940170 | Case | NM_152442.3: c.28A>G | p.Ser10Gly | nonsynonymous SNV | rs372056091 | 0.13 | 0.000 | 0.000018 | 0.000016 |
| Chr12:110950631 | Case | NM_152442.3: c.436C>G | p.Gln146Glu | nonsynonymous SNV | / | 0.01 | 0.373 | 0 | 0 |
| Chr12:110956546 | Case | NM_152442.3:c.661G>A | p.Gly221Arg | nonsynonymous SNV | rs763079713 | 0.02 | 1.000 | 0.000008 | 0.000004 |
| Chr12:110944446 | Case | NM_152442.3: c.336A>G | p.Ile112Met | nonsynonymous SNV | / | 0.05 | 0.980 | 0 | 0 |
| Chr12:110956530 | Case | NM_152442.3: c.645T>A | p.Phe215Leu | nonsynonymous SNV | / | 0.13 | 0.946 | 0 | 0 |
| Chr12:110952867 | Control | NM_152442.3: c.501T>G | p.Asp167Glu | nonsynonymous SNV | rs142286458 | 1.00 | 0.009 | 0.001806 | 0.000965 |
| Chr12:110960041 | Control | NM_152442.3: c.950T>C | p. p.Ile317Thr | nonsynonymous SNV | rs143029596 | 0.03 | 0.557 | 0.002620 | 0.001489 |
| Chr12:110950665 | Control | NM_152442.3: c.470C>T | p.Thr157Met | nonsynonymous SNV | rs7969568 | 0.27 | 0.239 | 0.006222 | 0.002174 |
| Chr12:110952892 | Control | NM_152442.3: c.526C>G | p.Gln176Glu | nonsynonymous SNV | rs61758787 | 0.45 | 0.990 | 0.002586 | 0.002029 |
| Chr12:110941725 | Control | NM_152442.3:c.117+44A>T | / | intronic SNV | rs141349325 | – | 0.022950 | 0.023080 |
Note: The down panel showed the variants found in controls. The NCBI Reference Sequence: NP_689655.3.
PolyPhen and SIFT scores predicted whether the mutants are deleterious or not. The SIFT score: Ranges from 0 to 1. The amino acid substitution is predicted damaging is the score is <=0.05 and tolerated if the score is >0.05. PolyPhen Score: 0.957–1: probably damage; 0.453–0.956: possible damage; 0–0.452: benign.
FIGURE 1.
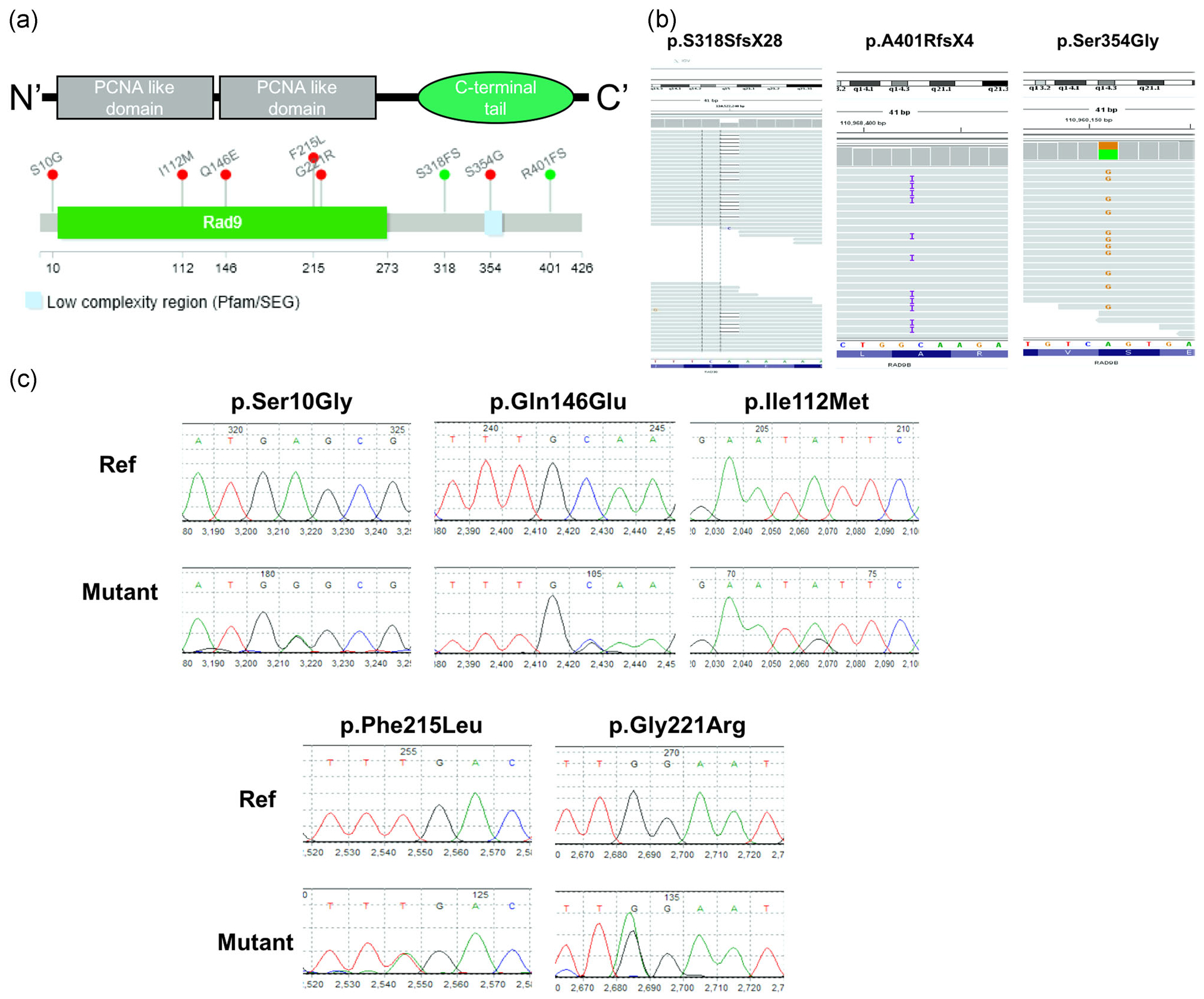
Identification of spina bifida-associated frameshift (FS) variants and missense variants in the Rad9B gene. (a). Conserved domains and total scheme of the Rad9B and positions of the detected novel mutations. Red mutations in the panel were identified as missense variants in the spina bifida cases, while green variants were determined to be frameshift mutations. (b). Examination of whole-genome bam-files of indicated mutations. Mutations are all covered with at least 60 reads in indicated regions. All detected mutations have Sequencing Quality Score Q >30. (c). Sanger Sequencing results for the 280-NTD-cohort, mutations were detected using a 3730 DNA analyzer and confirmed by Mutation Surveyor V 4.0.7. targeting NCBI protein (RAD9B, NT_009775, NC_000012.12). NTD, neural tube defect
Resequencing a larger cohort consisting of 280 SB cases as well as 190 bloodspot DNA samples from matched nonmalformed control infants was performed for validation of the observed association with RAD9B gene variants. Five rare deleterious missense variants (MAF < 1%, ExAC/gnomAD frequency (AF) <0.001, SIFT <0.1 or PolyPhen >0.9) were identified in SB patients (Table 1), while there were no LoF or predicted-to-be-damaging rare variants found in our control panel of 190 samples (Table 1). Notably, p.Gln146Glu, p.Ile112Met, and p.Phe215Leu variants were absent from the gnomAD database, suggesting that these are all novel variants. All five variants were found to be heterozygous in separate SB cases (Figure 1c). Combing the three WGS variants with the five variants identified by targeted sequencing, we have identified in total 8 variants in SB cases, indicating a significant enrichment of RAD9B rare deleterious variants in SB cases in our cohort (p = .0241). Together, the DNA sequencing results indicate that deleterious RAD9B variants are associated with an increased SB risk in humans. The information on the identified variants has been submitted to Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/). The Summary table of submission on ClinVar was shown as Table 2.
TABLE 2.
summary table of functional assay of the identified variants
| FS1 | FS2 | p.Ser354Gly | p.Ser10Gly | p.Gln146Glu | p.Gly221Arg | p.Ile112Met | p.Phe215Leu | |
|---|---|---|---|---|---|---|---|---|
| RAD9B nuclear localization | Abnormal | Abnormal | Normal | Normal | Abnormal | Normal | Normal | Normal |
| RAD9B protein level | ↓ | ↓ | NA | NA | NA | NA | NA | NA |
| Cell proliferation | ↓ | ↓ | NA | NA | ↓ | NA | NA | NA |
| Activating affection on JNK phosphorylation | – | ↓ | ↓ | ↓ | ↓ | NA | ↓ | NA |
Note: Abnormal means the mutants significantly affected RAD9B localization from the nucleus into spread over the cytoplasm and nucleus; ↓, downregulated.
Abbreviations: FS1, Frameshift1 (p.S318SfsX28); FS2, Frameshift2 (p.A401Rfs4X); NA, not affected.
3.2 |. Variants of RAD9B effect on its localization, DNA checkpoint function, and JNK activation
All SB-associated mutations identified in our sequencing data were annotated to be deleterious (Table 1). Previous studies showed that the RAD9B C-terminal tail domain is required for nuclear localization (Perez-Castro & Freire, 2012) and activates the DDR under UV or oxidative stress in cancer cell lines (Perez-Castro & Freire, 2012; Sierant et al., 2010). Therefore, the effects of these variants on RAD9B cellular localization were determined. N-eGFP-RAD9B wildtype and mutants were overexpressed in HeLa cell lines. The two frameshift mutants showed a lack or weak nuclear staining. Surprisingly, p.Gln146Glu mutation is not located in the C-terminal tail, but this variant still exhibits an expression pattern throughout both the cytoplasm and nucleus. The wildtype and other mutants maintained a proper nuclear-localization status. (Figure 2a). We next determined the impact of protein mislocalization on the DDR function of RAD9B. Both frameshift mutants and p.Gln146Glu mutants showed an inability to form DNA damage repair foci under conditions of DNA damage stress (e.g., Hydroxyurea [HU] treatment), while the wildtype protein produced a strong nucleus-located foci signal (Figure 2b). Furthermore, we noticed that the expression level of two frameshift mutants is much lower than others in the localization assay. Using western blotting, we confirmed that the bands from the two frameshifts were weaker than other RAD9B bands, which could be caused by decreased RAD9B gene expression or increased RAD9B protein instability. (Figure 2c).
FIGURE 2.
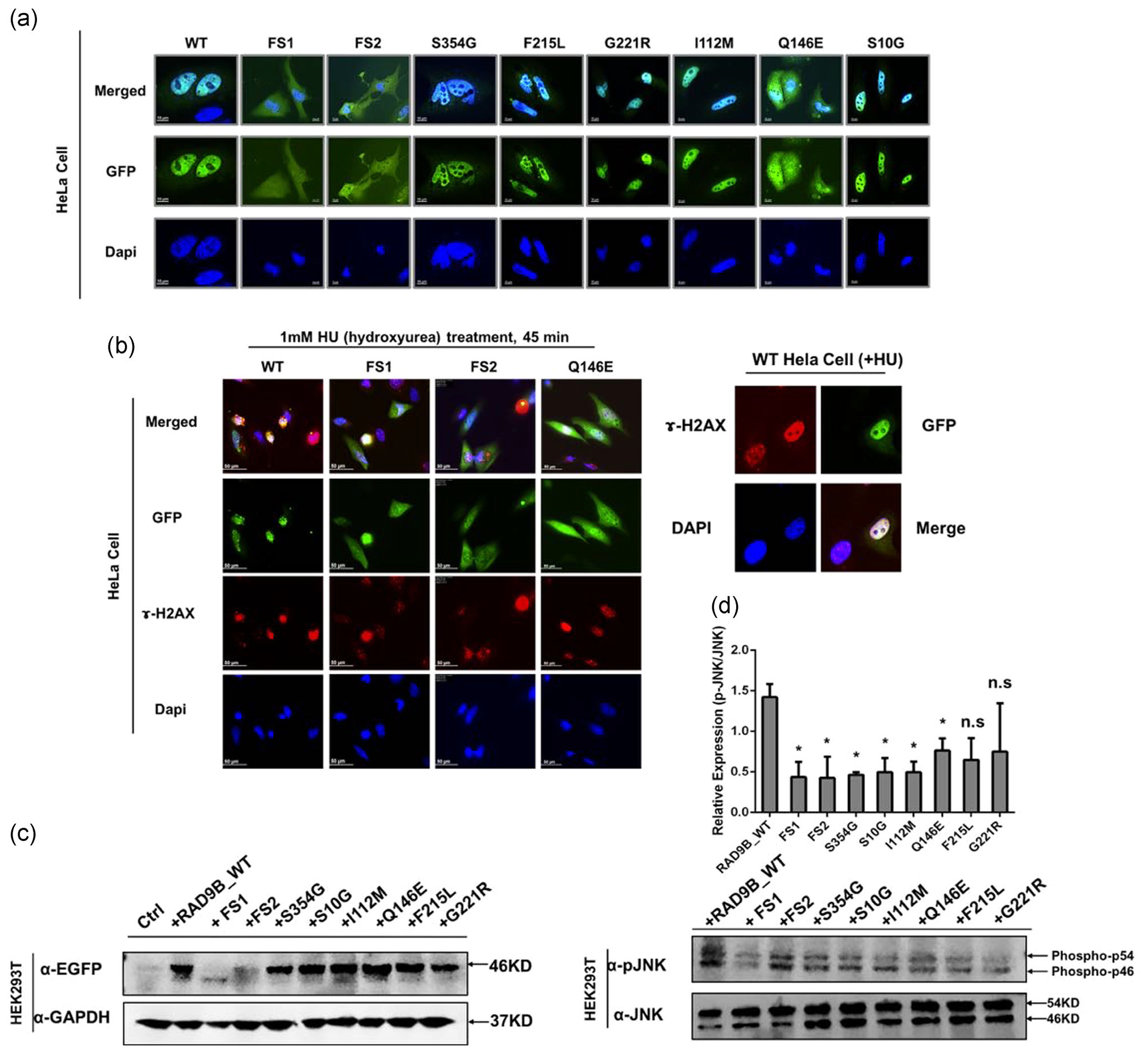
Mutated constructs impair Rad9B both subcellular localization, DDR checkpoint and JNK activation functions. (a) HeLa cells were transfected with mutated and wildtype constructs for 24-hr incubation and then were imaged under a deconvolution microscope. FS1: Ser318FS; FS2: Arg401FS. (b) HeLa cells were transfected for 24 hr and followed with 45 min hydroxyurea treatment and then imaged under a fluorescent microscope. The right panel shows a zoomed-in wildtype cell forming strong foci signal. ɤ-H2AX, a biomarker for DNA double-strand break (c) Western blot assay showed a decreased expression level of FS1 and FS2 mutants in HEK293T overexpression model. The predicted size of the wildtype and other mutants RAD9B are 46 Kd, while the FS1 is predicted to be 35 Kd and FS2 is 44 Kd. In (c), FS1 showed a band approximately 35 Kd, which is consistent with prediction. FS2 showed an unclear band at 44 Kd. We assumed that the protein may have a degradation. (d) HEK293T cells were transfected with wildtype and different RAD9B mutants. JNK and phosphorylated JNK level are both examined by western blot. RAD9B mutant overexpression except for p.Phe215Leu and P.Gly221Arg show a significant decreased level of p-JNK activation. *p < .05. DDR, DNA damage response; n.s, not significant; WT, wildtype
Overexpression of exogenous RAD9B has been shown to delay the G1/S progression in a cancer cell line and decrease the overall proliferation rate (Perez-Castro & Freire, 2012). An EdU assay was performed to determine the effects of RAD9B mutants on cell proliferation. KD of RAD9B (Figure S1A) significantly accelerated the proliferation rate in HeLa cells (Figure S1B). Moreover, replenishing exogenous wildtype RAD9B protein restored normal cellular kinetics; however, the proliferation rate could not be rescued by overexpressing the two frameshift mutants as well as the p.Ser354Gly mutant (Figure S1B). RAD9B was reported to be involved in regulating the JNK (c-Jun N-terminal kinase) signal (Perez-Castro & Freire, 2012). We further examined the effects of RAD9B mutants on regulating the p-JNK level under DNA damage stress. Notably, mutants save for Phe215Leu and p.Gly221Arg all show a decreased ability for activating JNK phosphorylation following HU treatment compared to the wildtype (p < .05; Figure 2d). These observations (Figure S1B) suggest that the variants identified in the SB infants affect the molecular functions (MFs) of RAD9B, including DDR foci formation and proliferation.
3.3 |. Loss of RAD9B influences cell fate specification in EBs derived from hESCs
Previous studies showed that Rad9b is highly expressed in the forebrain and ectodermal tissues of early gastrulation-stage mouse embryos (Leloup et al., 2010) and during embryonic Days E8.5–E9.5. In adult tissues, Rad9b protein is rather low in abundance, with the exception of several reproductive tissues (Dufault et al., 2003). These studies suggest that Rad9b plays an important functional role during early embryonic development.
To elucidate the potential roles of RAD9B during embryonic development, EBs were derived from hESCs using the protocol outlined in Figure 3a (Lippmann, Estevez-Silva, & Ashton, 2014). It is widely acknowledged that nonviral hESC KD experiments are difficult to perform; but by applying the protocol of Ma et al. (2010), a 60% reduction in RAD9B expression was achieved using small interfering RNAs (siRNAs) in an Oct4-GFP-hESC cell line (Figure S2A). Oct4 expression was found to be significantly downregulated in the RAD9B-KD EBs compared to controls (Figure 3b). Furthermore, flow cytometry experiments confirmed that the Oct4 positive cell population in KD EBs was decreased by approximately two-fold compared to control EBs (Figure 3c). Together, these data indicate that loss of RAD9B significantly impairs OCT4 expression in hESC derived EBs.
FIGURE 3.
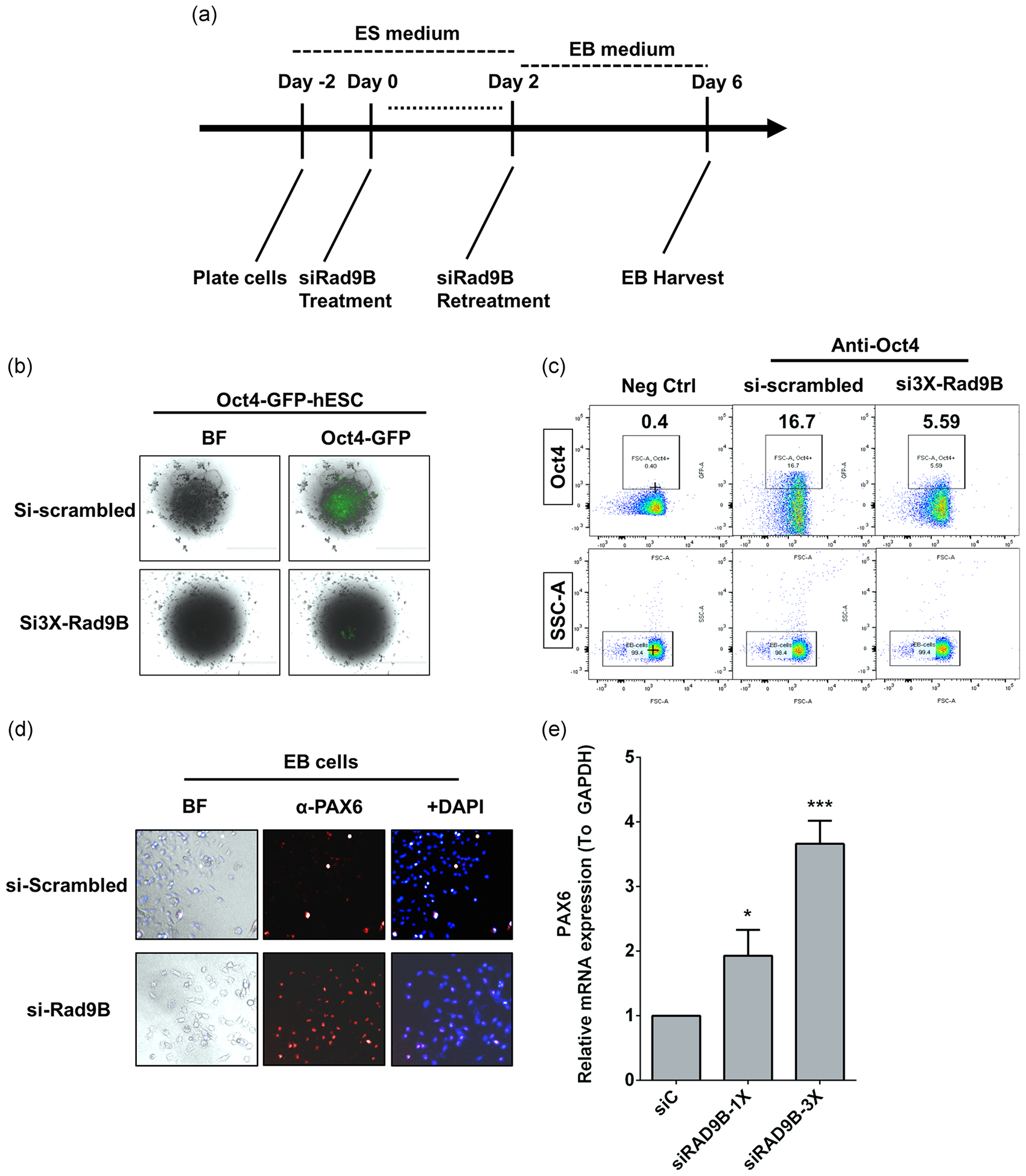
Loss of Rad9B compromised early human gastrulation processes by decreasing Oct4 expression but elevated Pax6 expression. (a) Scheme for the EB culture and staining experiments. (b) Flow data for Oct4 positive cell population detection. SSC-A: Side-scattered; Oct4: anti-POU5F1. Invents are locked for 10,000 cells. (c) Oct4-GFP cells were imaged under a fluorescent microscope under indicated treatments. (d) Immunofluorescent assay displayed the elevated expression of Pax6 in EB cells. (e) Elevated Pax6 mRNA level was detected in siRad9B treated human ES cells. Error bars represent ± SD for triplicate experiments. *p < .05; ***p < .001. EB, embryoid body; SD, standard deviation; siC, si-Scrambled
OCT4 expression is known to reflect the pluripotency of stem cells. Since OCT4 expression was impaired in RAD9B KD cells, we subsequently examined the role of RAD9B in cell fate specification. Pax6 (Paired box transcription factor 6), is a major driver of neuroectodermal cell fate determination and is known to restrict expression of Oct4 and other pluripotency factors (Zhang et al., 2010). As RAD9B is highly expressed in the ectoderm, we hypothesized that abnormal PAX6 activation may be involved in dysregulation of the OCT4. Therefore, both mRNA and endogenous protein levels of PAX6 were measured in RAD9B-KD cells. As predicted, both mRNA and protein levels of PAX6 were significantly elevated in the RAD9B-KD hESCs (Figure 3d,e). Pax6 mRNA concentrations were also observed to be dose-dependent with respect to RAD9B siRNA treatment.
Since PAX6 activation contributes to neural ectoderm specification (Zhang et al., 2010), we evaluated the expression of neural ectodermal markers in response to RAD9B KD. As expected, the TUBB3+ (aka Tuj1) cell population, which reflects pan-early-neural-cells, increased significantly in the RAD9B-KD cells (Figure S2B,C). Peripherin+ cells, reflecting the peripheral neuronal population, were also increased compared to the control cells. Together, these observations demonstrate that loss of functional RAD9B disrupts early cell fate specification in differentiating EBs through abnormal regulation of PAX6 and OCT4 circuitry.
3.4 |. Loss of RAD9B impairs neuronal differentiation and inhibits neural-organoid development
Human NTC starts at approximately 2 weeks postfertilization and is completed sometime around gestational Day 28. Although the loss of RAD9B resulted in the increased specification of neural ectoderm cells in EBs, whether this population can differentiate into functional neural stem cells (NSCs) or mature neurons remains unclear. We therefore tested the impact of RAD9B-KD on neural differentiation (Figure S3A) and established a long-term neural-organoid culture (Vichier-Guerre et al., 2017). After a second RAD9B-siRNA transfection on culture Day 10, the cells were harvested and stained on Day 21 to examine the formation of NSC populations. There were fewer mature neurons in the RAD9B-KD cells based expression of neural progenitor marker Nestin and pan-neuronal marker Tuj1 (Figure S3C). However, the AP2α expression did not differ significantly between the KD and control cells, suggesting that the neural crest cell population was not altered. PAX6 was also significantly decreased in the KD cells (Figure S3B), expressing a phenotype opposite to that observed in EBs. Taken together, these data suggest that RAD9B-KD can adversely affect the differentiation of neurons.
Cell colonies were cultured until Day 28 (Figure 4a), at which point a number of spherical organoids were observed in the control cultures (Figure 4B), that were not observed in the KD cultures. Immunohistochemistry revealed that these control organoids expressed high levels of SOX2, Nestin, and the pan-neuronal-marker Tuj1 (Figure 4C), suggesting that they were adopting a neural fate. In contrast, no neural organoids were observed at all in the RAD9B-KD cultures, even though the cell numbers were comparable between the two treatments. Indeed, Nestin expression remained low in RAD9B-KD cultures. These data indicate that the loss of RAD9B functionality in early embryogenesis may result in impaired neural development by compromising the differentiation of NSC populations.
FIGURE 4.
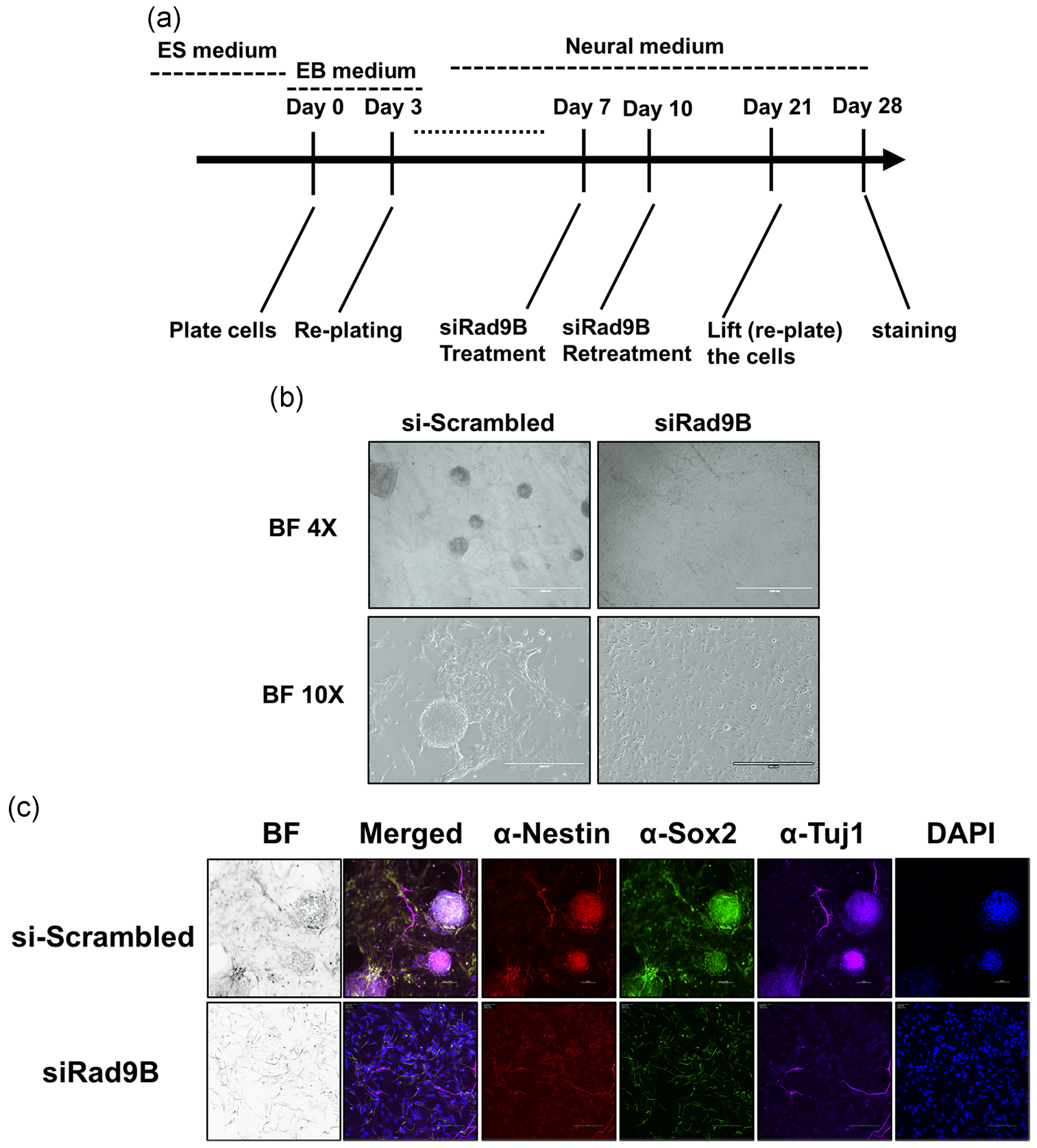
Neuro-organoid failed to form in siRad9B treated culture. (a) Culture scheme for establishing the neural-organoid (“mini-brain”). (b) Brightfield imaging landscape (×4; scale bar = 1 mm, ×10; scale bar = 400 μm) of the control and siRad9B treated neural-organoid culture. (c) Immunostaining on the neuro-organoid structure. Scale bar = 50 μm. Two parallel experiments are performed; each experiment has triplicate wells for staining
3.5 |. Gene expression analysis on RAD9B KD cells
To further identify the differential gene expression underlying the loss of organoid formation phenotype, we performed RNA-seq on both RAD9B-KD and wildtype hESC cells (Day 11). The RNA-seq analysis showed a nearly 60% decrease in RAD9B mRNA levels (Figure S4A) and revealed that over 2,000 genes were significantly differentially regulated (p-Adj-value < .05; Figure 5a). In the Gene Ontology term enrichment analysis (Yu, Wang, Han, & He, 2012), the axon development and axonogenesis pathways were specifically downregulated in terms of biological process (Figure 5a). Interestingly, we observed that a number of cells adhesion-related pathways were also highly enriched in terms of MF. GSEA confirmed that there is a robust upregulation of genes in the “KEGG Cell Adhesion Molecules CAM” and “KEGG ECM Receptor Interaction” gene set when comparing control cells versus RAD9B-depleted cells (Figure 5b). It was previously established that organoid formation relies on proper cell adhesion and mechanical controls (Dahl-Jensen & Grapin-Botton, 2017). Among these “leading edge” genes (Figure 5c), 7 of 16 play crucial roles in neuronal system adhesion (Reactome Database). For instance, NLGN and NRXN family proteins help connect neurons at the postsynaptic and presynaptic membranes, respectively. NCAM1 protein has been reported to be displaying a biphasic expression pattern contributing to the neural tube development in mice (Bally-Cuif, Goridis, & Santoni, 1993). These RNA-seq data strongly suggest that the loss of organoid formation in RAD9B-KD cells could be secondary to dysregulated cell adhesion pathways. Conversely, genes that are overrepresented in the gene sets of the cell cycle checkpoint and ATR response pathway were detected to be highly upregulated in RAD9B-KD cells (Figure S4B). This pattern implicates a cellular “rescue” response to the depletion of RAD9B.
FIGURE 5.
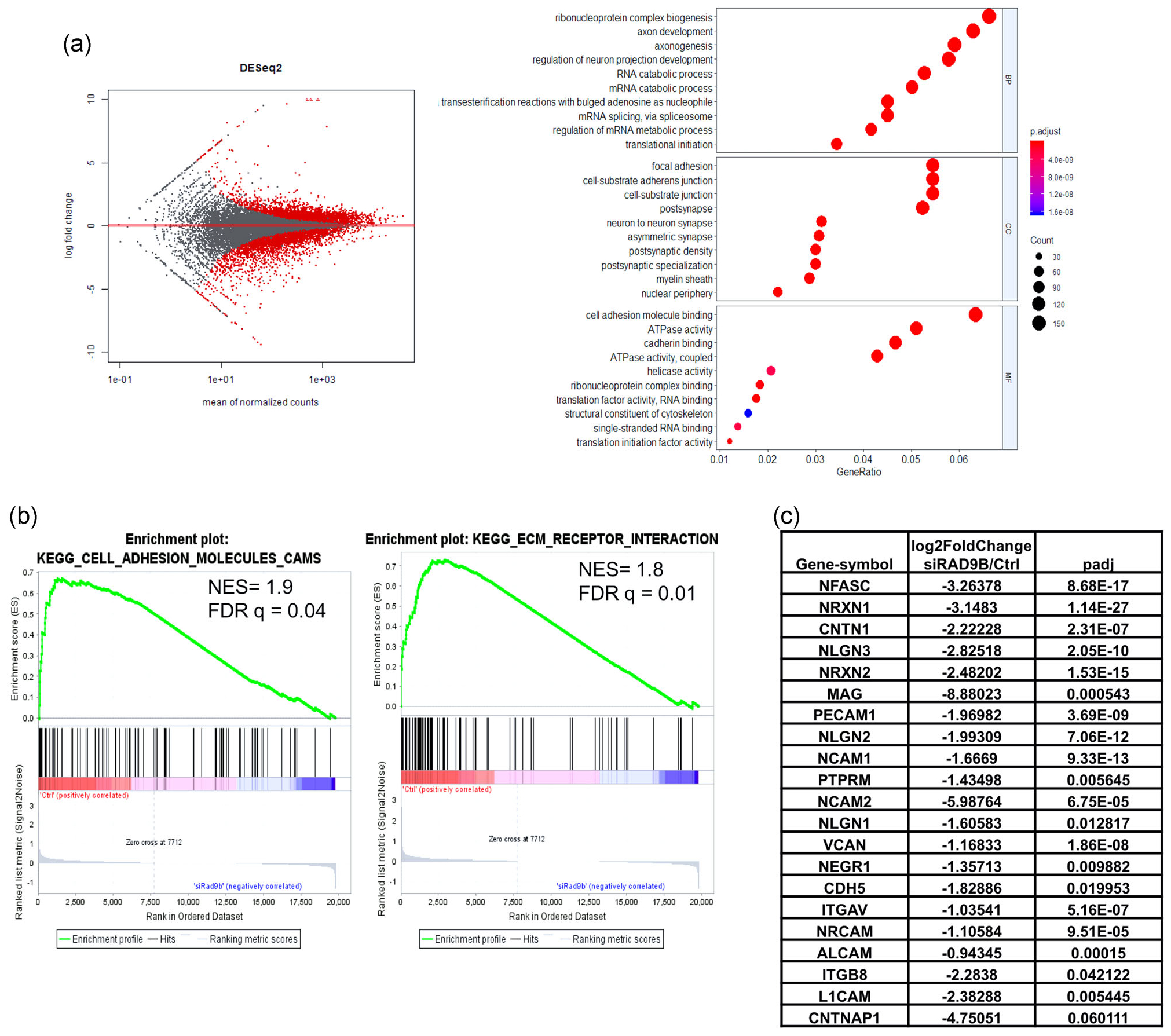
RNA-seq data analysis on RAD9B-KD hESC cells. Total RNAs of hESC cells are collected on Day 11 after second transfection of siRNA (Scheme in Figure S3A), then applied to sequencing protocols. (a) MA-plot of total expression pattern. The right panel shows the dotplot for GO term enrichment analysis. (b) Gene set enrichment analysis for mostly enriched gene sets plotted by enrichment of gene expression in si-Control-treated cells compared with siRAD9B transfected cells. (c) Gene lists of “leading edge” genes in GESA analysis, with expression foldchange from the “KEGG_CELL_ADHESION_MOLECULES_CAMS” gene sets. GESA, gene set enrichment analysis; hESC, human embryonic stem cell; siRNA, small interfering RNA
4 |. DISCUSSION
We identified eight rare and deleterious variants of RAD9B in two different human SB cohorts. This discovery is consistent with observations that Rad9b heterozygosity causes exencephaly in mouse embryos (Leloup et al., 2010). As Table 2 shows, functional analysis on three of these variants showed that they failed to fully translocate to the nucleus or form DNA checkpoint foci, while other variants did not activate the JNK signaling pathway properly in response to DNA damage stress. Taken together, these observations suggest that these variants fail to perform critical DNA damage checkpoint functions necessary to maintain genomic stability, which is likely a crucial component of proper NTC. Further support is derived from several mouse knockout models of DNA repair genes that similarly produced embryonic structural malformations, including NTDs. Brca1 and Brca2 nullizygous mice die between E5.5 and E10.5, and Brca1−/− embryos display neurodevelopmental defects (Gowen, Johnson, Latour, Sulik, & Koller, 1996). Furthermore, a subset of p53-deficient embryos present with exencephaly (Sah et al., 1995), while the loss of the DDR genes, Hus1 and Rad17 both result in NTDs (Weiss et al., 2000).
DNA repair mechanisms have also been linked to NTD etiology through several human studies. Our laboratory previously described polymorphisms in XPD (ERCC2), a gene involved in nucleotide excision repair, that were found to be associated with an increased risk for SB (Olshan, Shaw, Millikan, Laurent, & Finnell, 2005). In another study, hypomethylation of MGMT was a common factor identified in NTD-afflicted human embryos (Tran et al., 2012). To our knowledge, this is the first report of any human DDR genes upstream of the repair machinery being associated with an increased risk for clinical SB.
Folate one-carbon metabolism is the most extensively studied pathway influencing exogenous factors that impact NTC. One of the proposed mechanisms through which folate prevents NTDs is its role in nucleotide biosynthesis. Folate metabolism and folate status have previously been implicated in genome stability in several studies (Rosati, Ma, & Cabelof, 2012), and it is widely reported that if folate availability is limited, uracil can be mis-incorporated into DNA, resulting in DNA damage (Duthie, Narayanan, Brand, Pirie, & Grant, 2002). However, folate supplementation cannot overcome this situation if the DNA maintenance machinery itself is dysfunctional, suggesting that a compromised DDR, such as that which might occur with the LoF variants in RAD9B, may contribute to NTD risk burden that is not folate responsive.
Because of the low abundance of RAD9B in adult tissue, RAD9B is not well-studied compared to its paralog, RAD9A. In adults, RAD9B is expressed at only moderate levels in reproductive tissues, including the testes, ovaries, and cervix. However, in mouse embryos, Rad9b is expressed predominantly in the ectoderm at E7.5, and in the forebrain at E9.5. At E8.5, corresponding to early-stage NTC in the mouse, Rad9b is expressed in the anterior neuroepithelium. Furthermore, based on an analysis of a public mouse embryo RNA-seq data set (EXPression AloNg Development and Evolution (EXPANDE), NCBI_SRA_Accession: PRJDB3785), global embryonic expression of Rad9b diminishes after NTC (E8.5–E9.5). Other DDR genes, including Rad1, Hus1, and Atr, exhibit the same expression trends. Taken together, these data suggest that the DDR gene RAD9B, may play a critical role at this early stage of embryonic development.
Initially, cells within the embryo rapidly proliferate at these early timepoints, and must cope with replicative stress. It has been proposed that cells spending most of their cell cycle in S-phase, such as pluripotent stem cells, utilize replication-coupled DDR via ATR activation to protect genome integrity (Ahuja et al., 2016). To fulfill the downstream functions of ATR in certain high proliferation cell types including neural ectoderm or neuroepithelium, RAD9B may be the Rad9 homolog-of-choice. Furthermore, the rapidly proliferating cells of the neural plate and neural tube may be particularly susceptible to environmental stressors that can overload a compromised DDR response pathway through oxidative stress or other mechanisms. Examples include low folate status, arsenic exposure (Rao et al., 2017), valproic acid exposure (Tung & Winn, 2011), or exposure to polycyclic aromatic hydrocarbons via air pollution (Yuan et al., 2013), all of which are known human NTD risk factors. Furthermore, the mammalian embryo develops in an inherently hypoxic environment, which itself can act as a genomic stressor, given that hypoxia can drive genetic instability leading to altered cell cycle checkpoints and DDR through ATM and ATR activation. Therefore, it is not surprising that DDR genes should be so critical to embryonic development, given the sensitivity of rapidly proliferating embryonic cells to genotoxic influences.
As previously mentioned, Rad9b expression appears to be restricted to ectoderm and neural tissues in mouse embryos, leading us to evaluate how the loss of RAD9B would affect neural development in human cell culture models. In EBs, KD of RAD9B resulted in reduced expression of the pluripotency factor, OCT4, accompanied by an increased proportion of PAX6+ neural ectoderm cells. This suggests that RAD9B may contribute to pluripotency and control early cell fate determination. In addition, directed neural differentiation in RAD9B KD cells failed to produce mature neurons or neural organoids, suggesting that LoF variants for RAD9B may cause NTDs through impaired neural differentiation, a hypothesis supported by previous studies indicating a role for DDR pathways in neurogenesis.
The relationship between RAD9B and other DDR genes in neural development provides new insight to enhance our understanding of embryonic development, DNA integrity, and the pathology underlying the development of NTDs. The detailed elucidation of molecular interactions underlying the role of RAD9B in pluripotency and cell fate determination will be the subject of future investigations.
Supplementary Material
ACKNOWLEDGMENTS
We thank the families for their participation in this study. This project was supported by grants from NIH (HD081216 and HD083809 to Drs. Finnell and Lei, HD067244 to Drs. Finnell and Ross, HD074695 to Dr. Marini), and from CDC (CU01DD001033 to Dr. Shaw). We thank the California Department of Public Health, Maternal Child and Adolescent Health Division for providing surveillance data from California for this study. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention or the California Department of Public Health.
Funding information
NIH, Grant/Award Numbers: HD081216, HD083809, HD067244, HD074695; Centers for Disease Control and Prevention, Grant/Award Number: CU01DD001033
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
REFERENCES
- Ahuja AK, Jodkowska K, Teloni F, Bizard AH, Zellweger R, Herrador R, … Mendez J (2016). A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nature Communications, 7, 10660 10.1038/ncomms10660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avagliano L, Massa V, George TM, Qureshy S, Bulfamante GP, & Finnell RH (2018). Overview on neural tube defects: From development to physical characteristics. Birth Defects Research, 111, 1455–1467. 10.1002/bdr2.1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bally-Cuif L, Goridis C, & Santoni MJ (1993). The mouse NCAM gene displays a biphasic expression pattern during neural tube development. Development, 117(2), 543–552. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, & Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology, 34(5), 525–527. 10.1038/nbt.3519 [DOI] [PubMed] [Google Scholar]
- Chen Z, Lei Y, Cao X, Zheng Y, Wang F, Bao Y, … Wang H (2018). Genetic analysis of Wnt/PCP genes in neural tube defects. BMC Medical Genomics, 11(1), 38 10.1186/s12920-018-0355-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Lei Y, Zheng Y, Aguiar-Pulido V, Ross ME, Peng R, … Wang H (2018). Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Research, 28(10), 1039–1041. 10.1038/s41422-018-0061-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Stanier P, & Greene ND (2013). Neural tube defects: Recent advances, unsolved questions, and controversies. Lancet Neurology, 12(8), 799–810. 10.1016/S1474-4422(13)701108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl-Jensen S, & Grapin-Botton A (2017). The physics of organoids: A biophysical approach to understanding organogenesis. Development, 144(6), 946–951. 10.1242/dev.143693 [DOI] [PubMed] [Google Scholar]
- Dufault VM, Oestreich AJ, Vroman BT, & Karnitz LM (2003). Identification and characterization of RAD9B, a paralog of the RAD9 checkpoint gene. Genomics, 82(6), 644–651. 10.1016/s0888-7543(03)00200-3 [DOI] [PubMed] [Google Scholar]
- Duthie SJ, Narayanan S, Brand GM, Pirie L, & Grant G (2002). Impact of folate deficiency on DNA stability. Journal of Nutrition, 132(8 Suppl), 2444S–2449S. 10.1093/jn/132.8.2444S [DOI] [PubMed] [Google Scholar]
- Enriquez-Rios V, Dumitrache LC, Downing SM, Li Y, Brown EJ, Russell HR, & McKinnon PJ (2017). DNA-PKcs, ATM, and ATR interplay maintains genome integrity during neurogenesis. Journal of Neuroscience, 37(4), 893–905. 10.1523/JNEUROSCI.4213-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen LC, Johnson BL, Latour AM, Sulik KK, & Koller BH (1996). Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nature Genetics, 12(2), 191–194. 10.1038/ng0296-191. [DOI] [PubMed] [Google Scholar]
- Greene ND, & Copp AJ (2014). Neural tube defects. Annual Review of Neuroscience, 37, 221–242. 10.1146/annurev-neuro-062012-170354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay JJ, & Brouwer C (2016). Lollipops in the clinic: Information dense mutation plots for precision medicine. PLoS One, 11(8):e0160519 10.1371/journal.pone.0160519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharfallah F, Guyot MC, El Hassan AR, Allache R, Merello E, De Marco P, … Kibar Z (2017). Scribble1 plays an important role in the pathogenesis of neural tube defects through its mediating effect of Par-3 and Vangl1/2 localization. Human Molecular Genetics, 26(12), 2307–2320. 10.1093/hmg/ddx122 [DOI] [PubMed] [Google Scholar]
- Kibar Z, Bosoi CM, Kooistra M, Salem S, Finnell RH, De Marco P, … Gros P (2009). Novel mutations in VANGL1 in neural tube defects. Human Mutation, 30(7), E706–E715. 10.1002/humu.21026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lei Y, Guo J, Kim SE, Wlodarczyk BJ, Cabrera RM, … Finnell RH (2018). Formate rescues neural tube defects caused by mutations in Slc25a32. Proceedings of the National Academy of Sciences of the United States of America, 115(18), 4690–4695. 10.1073/pnas.180013811523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Kim SE, Chen Z, Cao X, Zhu H, Yang W, … Finnell RH (2019). Variants identified in PTK7 associated with neural tube defects. Molecular Genetics & Genomic Medicine, 7(4):e00584 10.1002/mgg3.584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei YP, Zhang T, Li H, Wu BL, Jin L, & Wang HY (2010). VANGL2 mutations in human cranial neural-tube defects. New England Journal of Medicine, 362(23), 2232–2235. 10.1056/NEJMc0910820 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, … Fennell T Exome Aggregation Consortium (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leloup C, Hopkins KM, Wang X, Zhu A, Wolgemuth DJ, & Lieberman HB (2010). Mouse Rad9b is essential for embryonic development and promotes resistance to DNA damage. Developmental Dynamics, 239(11), 2837–2850. 10.1002/dvdy.22415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippmann ES, Estevez-Silva MC, & Ashton RS (2014). Defined human pluripotent stem cell culture enables highly efficient neuroepithelium derivation without small molecule inhibitors. Stem Cells, 32(4), 1032–1042. 10.1002/stem.1622 [DOI] [PubMed] [Google Scholar]
- Ma Y, Jin J, Dong C, Cheng EC, Lin H, Huang Y, & Qiu C (2010). High-efficiency siRNA-based gene knockdown in human embryonic stem cells. RNA, 16(12), 2564–2569. 10.1261/rna.2350710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch JN, & Copp AJ (2010). The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Research. Part A, Clinical and Molecular Teratology, 88(8), 633–652. 10.1002/bdra.20686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshan AF, Shaw GM, Millikan RC, Laurent C, & Finnell RH (2005). Polymorphisms in DNA repair genes as risk factors for spina bifida and orofacial clefts. American Journal of Medical Genetics. Part A, 135(3), 268–273. 10.1002/ajmg.a.30713 [DOI] [PubMed] [Google Scholar]
- Perez-Castro AJ, & Freire R (2012). Rad9B responds to nucleolar stress through ATR and JNK signalling, and delays the G1-S transition. Journal of Cell Science, 125(Pt 5), 1152–1164. 10.1242/jcs.091124 [DOI] [PubMed] [Google Scholar]
- Rao CV, Pal S, Mohammed A, Farooqui M, Doescher MP, Asch AS, & Yamada HY (2017). Biological effects and epidemiological consequences of arsenic exposure, and reagents that can ameliorate arsenic damage in vivo. Oncotarget, 8(34), 57605 10.18632/oncotarget.17745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati R, Ma H, & Cabelof DC (2012). Folate and colorectal cancer in rodents: A model of DNA repair deficiency. Journal of Oncology, 2012, 105949 10.1155/2012/105949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, & Jacks T (1995). A subset of p53-deficient embryos exhibit exencephaly. Nature Genetics, 10(2), 175–180. [DOI] [PubMed] [Google Scholar]
- Sierant ML, Archer NE, & Davey SK (2010). The Rad9A checkpoint protein is required for nuclear localization of the claspin adaptor protein. Cell Cycle, 9(3), 548–556. 10.4161/cc.9.3.10553 [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, … Mesirov JP (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran S, Wang L, Le J, Guan J, Wu L, Zou J, … Zhang T (2012). Altered methylation of the DNA repair gene MGMT is associated with neural tube defects. Journal of Molecular Neuroscience, 47(1), 42–51. 10.1007/s12031-011-9676-2 [DOI] [PubMed] [Google Scholar]
- Tung EW, & Winn LM (2011). Valproic acid-induced DNA damage increases embryonic p27KIP1 and caspase-3 expression: A mechanism for valproic-acid induced neural tube defects. Reproductive Toxicology, 32(3), 255–260. 10.1016/j.reprotox.2011.05.020 [DOI] [PubMed] [Google Scholar]
- Vichier-Guerre C, Parker M, Pomerantz Y, Finnell RH, & Cabrera RM (2017). Impact of selective serotonin reuptake inhibitors on neural crest stem cell formation. Toxicology Letters, 281, 20–25. 10.1016/j.toxlet.2017.08.012 [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Niswander LA, Shaw GM, & Finnell RH (2013). The continuing challenge of understanding, preventing, and treating neural tube defects. Science, 339(6123), 1222002 10.1126/science.122200224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RS, Enoch T, & Leder P (2000). Inactivation of mouse Hus1 results in genomic instability and impaired responses to genotoxic stress. Genes & Development, 14(15), 1886–1898. [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, & He QY (2012). clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS, 16(5), 284–287. 10.1089/omi.2011.0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Jin L, Wang L, Li Z, Zhang L, Zhu H, … Ren A (2013). Levels of PAH–DNA adducts in placental tissue and the risk of fetal neural tube defects in a Chinese population. Reproductive Toxicology, 37, 70–75. 10.1016/j.reprotox.2013.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaganjor I, Sekkarie A, Tsang BL, Williams J, Razzaghi H, Mulinare J, … Rosenthal J (2016). Describing the Prevalence of Neural Tube Defects Worldwide: A Systematic Literature Review. PLoS One, 11(4): e0151586 10.1371/journal.pone.0151586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Huang CT, Chen J, Pankratz MT, Xi J, Li J, … Zhang SC (2010). Pax6 is a human neuroectoderm cell fate determinant. Cell Stem Cell, 7(1), 90–100. 10.1016/j.stem.2010.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.