

Summary
Nonapoptotic cell death is important for human health and disease. Here, we show how various tools and techniques drawn from the chemical biology field have played a central role in the discovery and characterization of nonapoptotic cell death pathways. Focusing on the example of ferroptosis, we describe how phenotypic screening, chemoproteomics, chemical genetic analysis, and other methods enabled the elucidation of this pathway. Synthetic small molecule inducers and inhibitors of ferroptosis identified in early studies have now been leveraged to identify an even broader set of compounds that impact ferroptosis and to validate new chemical methods and probes for various ferroptosis-associated processes. A number of limitations associated with specific chemical biology tools or techniques have also emerged and must be carefully considered. Nevertheless, the study of ferroptosis provides a roadmap for how chemical biology methods may be used to discover and characterize nonapoptotic cell death mechanisms.
Keywords: Phenotypic screening, apoptosis, necroptosis, ferroptosis, glutathione, GPX4, ROS, methuosis, ZDHHC5
eTOC:
Nonapoptotic cell death is important for normal and disease biology. In this review, Armenta and Dixon summarize how chemical biology approaches have been used to discover and characterize nonapoptotic cell death pathways such as ferroptosis and also consider some of the limitations of these approaches.
Graphical Abstract

Introduction
Cell death is a terminal fate resulting in elimination of the cell, either through physical disintegration or, more commonly in vivo, engulfment by a neighboring cell. Because cell death results in elimination of the cell itself, it is not trivial to capture dying cells and study the molecular mechanisms that regulate these processes. The acuteness of chemical tools (e.g. natural products, synthetic small molecules) therefore provides exceptional advantages in the study of cell death, as they can be used to initiate or inhibit cell death in a population of cells at a precise time. The inherent portability of these tools between systems is also invaluable to the study of cell death. A perfect illustration of these advantages was provided by J.R. Tata, who showed that the stereotypical regression of Xenopus tail explants induced by the addition of thyroid hormone was blocked by the acute addition of actinomycin D or cycloheximide, natural product inhibitors of transcription and protein synthesis, respectively, that had only recently been discovered (Kerridge, 1958; Reich et al., 1961; Tata, 1966). This classic study was one of the first to suggest that cell death could be a molecularly regulated process and helped establish chemical tools as key enablers of cell death research.
Discovery of Regulated Nonapoptotic Cell Death
Cells can perish in a number of ways that can be distinguished on the basis of morphological, genetic, and biochemical criteria. One fundamental distinction is between regulated and unregulated cell death, and a second distinction is between apoptosis and all forms of nonapoptotic or necrotic cell death. Regulation is evident if the cell death phenotype under observation is morphologically stereotypical and can be enhanced or suppressed by a specific genetic or chemical manipulation (Kerr et al., 1972; Wolpaw et al., 2011) (Figure 1). By contrast, unregulated cell death, as occurs in response to extreme physical stresses, nonspecific chemically-reactive compounds, or detergents, cannot be modified by any specific molecular intervention. At one time, regulated cell death was synonymous with apoptosis and unregulated cell death with necrosis. However, over the last twenty years this simple dichotomy has been shattered by the finding that nonapoptotic cell death can also occur in a regulated fashion (Galluzzi et al., 2018). Chemical biology approaches were central to establishing this new paradigm, as exemplified by the study of necroptosis.
Figure 1. Cell death pathways can be regulated by different genes and compounds.

Cell death can be triggered by various lethal stimuli and lead either to the activation of a regulated apoptotic or nonapoptotic cell death process, or to unregulated cell death. Regulation is evident if cell death involves stereotypical dead/dying cell morphology and can be modulated by specific genetic (e.g. gene deletion) or chemical perturbations. Here, two lethal compounds (a and b) induce regulated cell death, either apoptosis (a) or a form of nonapoptotic cell death (b). Cell death induced by a and b can be inhibited by deletion of specific genes (X, Y) or addition of specific inhibitors (1, 2, 3). Cell death induced by extreme physical stress, like heat, cold, or detergent treatment, cannot be inhibited by any specific genetic or chemical intervention (i.e. is unregulated).
The cytokine TNFα is a potent trigger for apoptosis, but over two decades ago it was observed that cells treated with the pro-apoptotic stimulus TNFα and a small molecule caspase inhibitor like zVAD-fmk still succumbed to cell death and, in fact, appeared to do so more readily (Vercammen et al., 1998). The thioxo-imidazolidinone necrostatin-1 (Nec-1) was identified from a small molecule screen as a specific inhibitor of cell death under these conditions (Degterev et al., 2005) (see Table 1, selected small molecule structures). This inhibitor blocked receptor interacting serine/threonine kinase 1 (RIPK1) function, implying that cell death was regulated by phosphorylation (Degterev et al., 2008). Nec-1 analogs also suppressed pathological cell death in the nervous system, demonstrating the in vivo relevance of this process (Degterev et al., 2005). This form of cell death was rechristened necroptosis, a portmanteau of necrosis and apoptosis, highlighting that it was a regulated form of cell death distinct from apoptosis. Small molecule screening subsequently helped identify additional important regulators of this process, including mixed lineage kinase domain-like pseudokinase (MLKL), which is inhibited by the small molecule necrosulfonamide (Sun et al., 2012). Thus, the necroptosis field provided a conceptual road map for how new nonapoptotic cell death pathways could be discovered using chemical biology approaches. The full power of chemical biology-driven approaches to pinpoint and characterize new forms of nonapoptotic cell death is, however, perhaps best demonstrated by the example of ferroptosis.
Table 1.
Chemical modulators of nonapoptotic cell death.
| Pathway | Effect of target inactivation on death | Target | Effect of small molecule on target activity | Small molecule name | Structure | References |
|---|---|---|---|---|---|---|
| Necroptosis | Suppress | RIPK1 | Inhibitor | Necrostatin-1 |  |
Degterev et al. (2005) |
| MLKL | Necro-sulfonamide |  |
Sun et al. (2012) | |||
| Ferroptosis | Induce | GPX4 | Inhibitor | RSL3 |  |
Yang & Stockwell (2008) |
| ML210 |  |
Weïwer et al. (2012) | ||||
| ML162 | 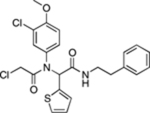 |
Weïwer et al. (2012), Yang et al. (2014) | ||||
| SQS | Activator | FIN56 |  |
Shimada et al. (2016b) | ||
| System xc− | Inhibitor# | Erastin | 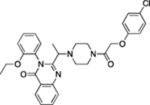 |
Dolma et al. (2003), Dixon et al. (2012) | ||
| Sorafenib | Dixon et al. (2014) | |||||
| Compound 4 |  |
Taylor et al. (2019) | ||||
| Suppress | Lipid peroxyl radicals | Inhibitor | Ferrostatin-1 |  |
Dixon et al. (2012) | |
| Liproxstatin-1 |  |
Friedmann Angeli et al. (2014) | ||||
| Iron | Chelator | Deferoxamine |  |
Yang and Stockwell (2008) | ||
| Methuosis | Induce | Unknown | Unknown | Compound 17 |  |
Sun et al. (2017) |
| Compound 13 |  |
Huang et al. (2018) | ||||
| PIKFYVE, other(s) | Inhibitor | MOMIPP |  |
Robinson et al. (2012), Cho et al. (2018) | ||
| CIL56- induced death | Induce | Unknown | Unknown | CIL56 | Shimada et al. (2016b) |
Examples of synthetic small molecule and natural product inducers and inhibitors of different nonapoptotic cell death pathways. For only some molecules have targets been established.
target inhibition is based on biochemical activity, not direct evidence of target engagement. RIPK1: receptor interacting serine/threonine kinase 1, MLKL: mixed lineage kinase domain like pseudokinase, GPX4: glutathione peroxidase 4, SQS: squalene synthase, PIKFYVE: phosphoinositide kinase, FYVE-type zinc finger containing.
Ferroptosis is one of several new nonapoptotic cell death processes to be recognized in the past two decades (Conrad and Pratt, 2019; Galluzzi et al., 2018). This process can be triggered by inactivation of the system xc− cystine/glutamate antiporter (leading to depletion of intracellular glutathione), direct inhibition of the glutathione-dependent lipid hydroperoxidase GPX4, and possibly other mechanisms. GPX4 inhibition ultimately leads to the iron-dependent accumulation of lipid peroxides which eventually kill the cell (Cao and Dixon, 2016; Stockwell et al., 2017). While ferroptosis was named in 2012, similar processes have been observed for decades in various contexts where glutathione metabolism is disrupted (Hirschhorn and Stockwell, 2018; Lewerenz et al., 2018). What crucially enabled ferroptosis to be recognized as a distinct mode of nonapoptotic cell death was the discovery and characterization of potent and specific small molecule inducers and inhibitors of this process (Table 1). These highly portable tools have been leveraged to investigate ferroptosis using a host of chemical biology approaches in a wide array of models.
Below, we describe how different chemical biology approaches enabled ferroptosis to be identified and studied (summarized in Figure 2). We provide examples of how early discoveries catalyzed a virtuous cycle of innovation leading to new methods and probes to study ferroptosis. This has not occurred without some bumps along the way, and we therefore point out some limitations of current methods. We conclude by considering how chemical biology approaches could help elucidate additional nonapoptotic cell death mechanisms.
Figure 2. Characterization of ferroptosis using chemical biology approaches.

Key molecules involved in ferroptosis are annotated to indicate how they were discovered using different chemical biology techniques. Proteins are denoted by rounded rectangles and metabolites are indicated by text only. Colored circles denote chemical biology techniques used to discover the role of key molecules in this pathway. A few important biochemical details are shown in simplified form, especially the process of lipid peroxidation (red highlight), which involves repeated cycles of initiation, propagation, and termination. The role of lipoxygenase (LOX) enzymes in ferroptosis initiation remains controversial, and initiation of lipid peroxidation can occur with or without these enzymes. Abbreviations: 4-HNE, 4-hydroxynonenal; CoQ10, coenzyme Q10; DHN, 1,4-dihydroxy-2-nonene; LOX, lipoxygenase enzyme; PL, phospholipid; PUFA, polyunsaturated fatty acid; TAG, triacylglycerol; TF, transferrin; TFRC1, transferrin receptor 1; TXN, thioredoxin; TxnR, thioredoxin reductase.
Chemical Biology and Ferroptosis
Phenotypic small molecule screening.
Small molecule phenotypic screening is a powerful means to discover compounds with novel mechanisms of action (Wagner and Schreiber, 2016). Here, libraries of small molecules are searched in an unbiased manner for those that can produce a given phenotype of interest. Advantageously, such screens have the potential to unveil new or unexpected biochemical mechanisms that modulate this cellular phenotype.
Erastin and 1S,3R-RSL3 (hereafter RSL3) are prototypic inducers of ferroptosis, originally discovered in large-scale (~25–50,000 compounds) phenotypic screens designed to identify compounds that were selectively lethal to engineered tumor cells expressing oncogenic mutant HRASV12 (Dolma et al., 2003; Yang and Stockwell, 2008). Serendipitously, the use of a generic metabolism-based cell viability readout made these screens agnostic to the nature of the cell death process that was activated by a given lethal small molecule. While not apparent initially, detailed characterization of the erastin and RSL3 mechanisms of action eventually led to the discovery that these compounds triggered ferroptosis (Dixon et al., 2012). Had the original phenotypic screens been designed to report specifically on the induction of apoptosis, these ferroptosis-inducing small molecules would likely have gone undiscovered. Subsequently, fourteen additional lethal molecules, including ML162 and ML210, were identified from over a million compounds tested for HRASV12-selective lethality and shown to trigger ferroptosis (Weiwer et al., 2012; Yang et al., 2014). Other phenotypic screens, looking for cell death without caspase activation, or enhanced cell death following glutathione depletion, identified additional inducers of ferroptosis, including sorafenib and FIN56 (Dixon et al., 2014; Lachaier et al., 2014; Shimada et al., 2016b).
Phenotypic chemical suppressor screens, where cell death induced by one molecule is blocked by the action of a second molecule, have helped dissect the ferroptosis mechanism and identify specific inhibitors of this process. From a library of over 2,000 annotated bioactive compounds, antioxidants (e.g. α-tocopherol, β-carotene, butylated hydroxytoluene) and iron chelators (e.g. deferoxamine) were found to block cell death induced by erastin (Yagoda et al., 2007). This study first illuminated the fundamental oxidative, iron-dependent nature of this process. Further chemical suppressor screening using large unannotated compound libraries tested in erastin-treated cancer cells or inducible Gpx4−/− mouse embryonic fibroblasts pinpointed the potent ferroptosis inhibitors ferrostatin-1 and liproxstatin-1 (Dixon et al., 2012; Friedmann Angeli et al., 2014). These two compounds have been essential for a range of mechanistic and animal model studies (Dixon et al., 2012; Fang et al., 2019; Friedmann Angeli et al., 2014; Li et al., 2017; Linkermann et al., 2014).
Chemoproteomics.
Identifying the target(s) of small molecules isolated in phenotypic screens is an important and often arduous task. A classic approach involves the use of affinity analogs, purification, and protein identification using mass spectrometry. An important success using this approach was the identification of GPX4 as a direct target of RSL3 (Yang et al., 2014). Two features of RSL3 aided the success of the chemoproteomic approach here. First, RSL3 contains a chloroacetamide ‘warhead’ that reacts covalently with GPX4. Second, only the 1S,3R diastereomer of RSL3 is active, making it possible to identify GPX4 as a protein uniquely bound to active (1S,3R) but not inactive (1R,3R) analogs. A similar chemoproteomic strategy has recently been successfully deployed to identify the small antioxidant protein thioredoxin (TXN) as a covalent target of the ferroptosis-inducing compound ferroptocide (Llabani et al., 2019). Mutagenesis studies indicate that ferroptocide binds to the active site cysteine residues of thioredoxin as well as to an adjacent cysteine residue. How thioredoxin inhibition triggers ferroptosis is not clear but could involve enhanced lipid peroxidation due to disruption of the thioredoxin/thioredoxin reductase cycle (Bjornstedt et al., 1995).
While powerful in principle, affinity chemoproteomics has not always yielded straightforward answers. These methods, used together with both lethal and non-lethal analogs of erastin, identified mitochondrial voltage anion channel (VDAC) 2 and 3 as targets of this molecule (Yagoda et al., 2007). Functionally, erastin reduces the permeability of VDAC1 and VDAC2 when these proteins are expressed in yeast (Yagoda et al., 2007), and can alter VDAC function, stability, and interaction with other proteins in mammalian cells (Maldonado et al., 2013; Yang et al., 2020). However, the ability of erastin to induce ferroptosis is best explained by inhibition of the cystine/glutamate antiporter system xc−, leading to depletion of glutathione and other metabolites (Dixon et al., 2014) (and see below). This link between erastin and system xc− was not discovered using unbiased methods, but rather using a small scale phenotypic similarity profiling analysis, driven by the hypothesis that erastin triggered oxidative cell death (Dixon et al., 2012). Chemoproteomic results are therefore not a panacea, perhaps especially when dealing with compounds that engage targets non-covalently.
Chemical genetic screens.
Lethal small molecules can be combined with genetic screening to identify genes that normally promote or suppress cell death. These methods have been applied successfully to the study of several nonapoptotic cell death pathways, including ferroptosis. Short hairpin RNAs (shRNAs), short interfering RNAs (siRNAs), insertional mutagenesis in human haploid cells, and CRISPR/Cas9-based technologies have all been used in ferroptosis loss of function suppressor screens (Table 2). Perhaps as could be anticipated from previous chemical suppressor screens, these genetic screens have identified genes that govern lipid metabolism (e.g. ACSL4, LPCAT3), iron homeostasis (e.g. IREB2, ATM), and redox metabolism (CARS, KEAP1) (Chen et al., 2019; Dixon et al., 2012; Dixon et al., 2015; Doll et al., 2017a; Hayano et al., 2015; Zou et al., 2019). Disruption of these genes limits ferroptosis by reducing the levels of oxidizable membrane lipids, lowering intracellular free iron, or increasing the antioxidant capacity of the cell.
Table 2.
Summary of chemical genetic screens for regulators of ferroptosis and related processes.
| Model | Focus/Scope | Method | Genes | Hits | Reference |
|---|---|---|---|---|---|
| HT-1080, Calu-1 + erastin | Mitochondrial genes | Arrayed shRNA | 1,087 | IREB2, RPL8, ACSF2, others | Dixon et al., 2012 |
| MEFs AA starvation + FBS | Signaling genes | Pooled shRNA | 4,625 | Tfrc, Aco1, Ulk1, Becn1, Atg4d, others | Gao et al., 2015 |
| HT-1080 + erastin | Genome-wide | siRNA | 21,687 | CARS, others | Hayano et al., 2016 |
| MDA-MB-231 + low cystine | Kinome | siRNA | 715 | ATM, ATR, TTK, SYK, others | Chen et al., 2019 |
| HAP1 haploid + RSL3 or ML162 | Genome-wide | Saturating retroviral insertional mutagenesis | ~20,000 | ACSL4, LPCAT3, AGPAT3, HSD17B11 | Dixon et al., 2015 |
| Pfa1 MEFs + RSL3, erastin | Genome-wide | CRISPR/Cas9 | ~20,000 | ACSL4 | Doll et al., 2017 |
| 786–O cells + ML210 | Genome-wide | CRISPR/Cas9 | ~18,000 | ACSL4, KEAP1, EPAS1, EP300, others | Zou et al., 2019 |
| U-2 OS + RSL3 | Apoptosis and cancer genes | CRISPR/Cas9 | 3,015 | FSP1 | Bersuker et al., 2019 |
| Pfa1 MEFs + MCF7 cDNA + tamoxifen (to induce Gpx4 deletion) | cDNA library | cDNA overexpression | Unknown | GPX4, FSP1 | Doll et al., 2019 |
| Pfa1 MEFs + IKE, RSL3, or tamoxifen (to induce Gpx4 deletion) | Genome-wide | CRISPR activation | ~20,000 | GCH1, others | Kraft et al., 2020 |
Important chemical genetic screens performed to isolate genes that regulate ferroptosis sensitivity in different contexts. Only the top or best validated hits from each screen are listed; in many cases there were other hits that were reported but not validated in detail. shRNA: short hairpin RNA; siRNA: short interfering RNA; MEF: mouse embryonic fibroblast.
Remarkably, with one exception (e.g. ACSL4), the hits recovered in these genetic suppressor screens have been largely non-overlapping. This could be explained by technical differences between the screens. For example, partial gene silencing using sh/siRNA may allow for the role of essential genes like the cysteinyl-tRNA synthetase CARS to be identified more readily than when using complete knockouts. Different screens have typically employed different cell line models. Cell type-specific differences in ferroptosis regulatory networks may therefore also contribute to differences between screens. It is possible that the execution of ferroptosis: (i) in mouse embryonic fibroblasts is especially dependent upon autophagy-related genes (e.g. Ulk1, Becn1, Atg4d) for the degradation of intracellular ferritin and the liberation of iron (Gao et al., 2016); (ii) in renal cell carcinoma cells is especially sensitive to modulation of the hypoxia inducible factor (HIF) pathway, which controls expression of the candidate lipid metabolic enzyme HILPDA (Zou et al., 2019); and, (iii) in triple negative breast cancer cells is dependent upon regulation of intracellular labile iron levels by an ATM kinase-dependent mechanism (Chen et al., 2019). Alternatively, differences in the design of each screen may have favored the detection of only a subset of genes from a larger shared regulatory network common to all cells.
Apart from suppressor screens, CRISPR/Cas9-mediated gene disruption has also been used in a loss of function sensitizer screen, resulting in the identification of ferroptosis suppressor protein 1 (FSP1, formerly AIFM2) which prevents ferroptosis (Bersuker et al., 2019). A cDNA overexpression screen likewise pinpointed FSP1 as a potent suppressor of ferroptosis (Doll et al., 2019). FSP1 acts as a reductase for the endogenous lipid electron carrier coenzyme Q10. This metabolite limits lipid ROS accumulation in the plasma membrane by acting as a radical trapping antioxidant, in parallel to GPX4. Another antioxidant mechanism was discovered using a CRISPR activation screen, finding that enhanced GTP cyclohydrolase-1 (GCH1) expression can suppress ferroptosis by boosting the synthesis of tetrahydrobiopterin/dihydrobiopterin, which are metabolites that can act as direct radical trapping antioxidants (Kraft et al., 2020).
Future genetic screens could yield additional regulators of ferroptosis, although it seems likely that most of these genes will ultimately impinge in some way upon iron, lipid, or antioxidant metabolism. In fact, ferroptosis screens could be a great way to identify new or unexpected genes involved in these three processes. Likewise, hits from other large-scale screens of metabolic genes can be tested for links to ferroptosis sensitivity, with a good chance of finding a connection to ferroptosis (Alvarez et al., 2017; Cao et al., 2019; Garcia-Bermudez et al., 2019).
Gene expression profiling.
Compound treatment can alter gene expression in informative ways, providing candidate molecular markers and potentially leading to new regulatory insights. Unbiased RNA sequencing identified CHAC1 as a highly upregulated gene in cells treated with erastin in vitro, an effect confirmed in tumors exposed to an erastin analog in vivo (Dixon et al., 2014; Zhang et al., 2019). PTGS2 is another gene expression marker of ferroptotic cells, discovered from a biased analysis of 83 candidate oxidative stress-sensitive genes, that can likewise be upregulated in cells undergoing ferroptosis (Yang et al., 2014; Zhang et al., 2019). Therefore, increased CHAC1 and PTGS2 expression can serve as molecular markers for ferroptosis-inducing drug exposure in vivo, although neither is likely specific enough to exclusively mark ferroptotic cells.
Gene expression profiling in response to compound treatment can also define mechanisms of cell death resistance. For example, resistance to erastin is correlated with overexpression of AKR1C1–3 (Dixon et al., 2014; Gagliardi et al., 2019). These genes encode aldo-keto reductases that may suppress ferroptosis by inactivating reactive carbonyls that form downstream of phospholipid oxidation, such as 4-hydroxynonenal (4-HNE) (Burczynski et al., 2001). In certain breast cancer cells, cell detachment from the extracellular matrix can induce ferroptosis (Brown et al., 2017; Brown et al., 2018). Cells that resist this process rapidly upregulate PROM2, encoding a transmembrane pentaspanin protein, that promotes exosomal release of iron-loaded ferritin from the cell (Brown et al., 2019). As demonstrated by the example of PROM2, which was not previously linked to iron metabolism, it is essential to couple gene expression profiling with functional studies to deduce the nature of the regulatory connection.
Differences in basal gene expression between cells can also yield important insights into the regulation of cell death. For example, correlating cell viability and basal gene expression data from the NCI-60 cell line panel identified an association between ferroptosis-inducing small molecules and genes regulating NAD(P)H metabolism (Shimada et al., 2016a). Mechanistically, high NAD(P)H levels correlate with ferroptosis resistance, perhaps due to enhanced activity of one or more NAD(P)H-dependent anti-ferroptotic proteins like TXN (Llabani et al., 2019) or FSP1 (Bersuker et al., 2019; Doll et al., 2019). In a similar vein, basal gene expression and dose-dependent compound sensitivity across > 700 cancer cell lines (Basu et al., 2013; Rees et al., 2016) demonstrate that ferroptosis sensitivity is linked to: (i) a mesenchymal gene expression state (Viswanathan et al., 2017); (ii) gene expression changes associated with melanoma dedifferentiation (Tsoi et al., 2018); and (iii) low expression of FSP1 or the lipid metabolic enzyme ACSL3 (Bersuker et al., 2019; Magtanong et al., 2019). It is possible that further analysis of this resource, available online through the Cancer Therapeutics Response Portal, will yield additional new regulators of ferroptosis.
Metabolomic analysis.
Ferroptosis is fundamentally the result of dysregulated iron, lipid, and antioxidant metabolism. Thus, combining ferroptosis-inducing small molecules with the direct measurement of individual metabolites using mass spectrometry-based techniques has been highly informative. Consistent with the notion that erastin induces ferroptosis by inhibiting cystine import and thereby preventing de novo GSH synthesis, polar metabolites including cysteine and GSH are among the most significantly depleted in erastin-treated HT-1080 cells (Skouta et al., 2014; Tarangelo et al., 2018; Yang et al., 2014). Intruigingly, depletion of the thiol-containing metabolite coenzyme A (CoA) contributes essentially to the induction of ferroptosis in response to cystine deprivation (i.e. system xc− inhibition) (Badgley et al., 2019; Leu et al., 2019). How CoA inhibits ferroptosis is unclear but could involve a direct antioxidant mechanism or effects on CoA-dependent lipid metabolism. The need for simultaneous depletion of GSH and CoA could help explain why a direct GSH biosynthetic inhibitor, buthionine sulfoximine, is a relatively poor inducer of ferroptosis (Cao and Dixon, 2016). The relative contribution of different antioxidant metabolites (e.g. GSH, CoA, tetrahydrobiopterin, NADPH) to the regulation of ferroptosis sensitivity will be important to clarify in future studies.
Ferroptosis involves oxidation of specific phospholipids and the analysis of this process has been greatly advanced by advances in the analysis of non-polar lipid metabolites. In cells undergoing ferroptosis numerous polyunsaturated fatty acid (PUFA) species (C20:4n6, C22:6n3, C20:5n3) are depleted, while various lyso-phospholipids accumulate (Skouta et al., 2014; Yang et al., 2014). Combining mass spectrometry with specific ferroptosis inhibitors has led to the suggestion that oxidation of PUFA-containing phosphatidylethanolamines (PUFA-PEs) is especially critical for the initiation of ferroptosis, both in vitro and in vivo (Anthonymuthu et al., 2018; Doll et al., 2017b; Kagan et al., 2017; Kenny et al., 2019). This may not be universal, however, as pronounced depletion of other lipid species including phosphatidylcholines (PCs) is observed in many ferroptosis-inducing conditions (Gaschler et al., 2018a; Kraft et al., 2020; Magtanong et al., 2019; Zhang et al., 2019). How the oxidation of these or other phospholipids may lead to membrane ‘blistering’ (Magtanong et al., 2019) and eventual lethal membrane permeabilization remains speculative.
Leveraging Chemical Biology for New Tools and Mechanistic Insights
Chemical biology-driven analyses have yielded numerous small molecule activators and inhibitors of nonapoptotic cell death. These initial reagents have subsequently been leveraged to discover additional chemical tools and further our understanding of lethal mechanisms. Below, we focus on examples from the ferroptosis field showing how this virtuous cycle can work in practice.
Expanding the constellation of small molecule ferroptosis modulators.
The discovery of specific inhibitors like ferrostatin-1 and liproxstatin-1 makes it straightforward to test whether any given lethal molecule of interest can induce ferroptosis. This has helped identify synthetic small molecules (e.g. FINO2), natural products or natural product derivatives (e.g. withaferin A, ironomycin, ferroptocide, open chain epothilone analogs), and other substances (e.g. ultrasmall silica nanoparticles) as novel ferroptosis-inducing molecules (Abrams et al., 2016; Hassannia et al., 2018; Kim et al., 2016; Llabani et al., 2019; Mai et al., 2017; Taylor et al., 2019).
First-generation ferroptosis inhibitors have also served as the basis for the development of improved analogs, typically with chemical properties more suitable for use in vivo, such as the improved ferrostatin-1 analogs SRS16–86 and UAMC-3203 (Devisscher et al., 2018; Linkermann et al., 2014; Skouta et al., 2014). Knowledge of the ferrostatin-1 and liproxstatin-1 mechanism of action has also facilitated the rational design of synthetic compounds with appropriate radical trapping properties, such as a new series of 1,8-tetrahydronaphthyridinols (Zilka et al., 2017). Further developments along these lines may yet yield potent ferroptosis modulators suitable for use in humans.
New chemical methods and chemical reporters.
Understanding that lipid peroxidation and lipid radical formation are important for ferroptosis has catalyzed efforts to develop new methods to study this process. The fluorescence-enabled inhibited autoxidation (FENIX) approach uses the reporter STY-BODIPY to assess the potency of putative radical trapping antioxidant inhibitors of ferroptosis, and represents a significant improvement over the existing 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay (Haidasz et al., 2016; Shah et al., 2019). Application of this method led to a new explanation for the potency of diarylamine-based ferroptosis inhibitors like ferrostatin-1 versus phenolic ferroptosis inhibitors; unlike diarylamine-based ferroptosis inhibitors, the phenolic inhibitors form hydrogen bonds with phospholipid headgroups that limit their reactivity with lipid radicals.
Small molecule inhibitors and inducers of ferroptosis have also aided the development of new chemical probes of the ferroptosis mechanism. For example, an alkyne-tagged ferrostatin-1 analog was localized to the endoplasmic reticulum, mitochondria, and lysosomes using stimulated Raman scattering microscopy, suggesting that inhibition of lipid peroxidation in one or more of these locations is required to block ferroptosis (Gaschler et al., 2018b). Ferroptosis inducers have also been useful to characterize a new specific fluorescent probe for glutathione (RealThiol, (Jiang et al., 2017)) and a FRET-based probe for labile iron (FIP-1, (Aron et al., 2016)). Interestingly, using FIP-1 it was found that the intracellular labile iron pool is increased by erastin analog treatment and that this may contribute directly to cell death, since the increase is prevented by cotreatment with a protective iron chelator. These new probes allow for key features of ferroptosis to be more easily examined in diverse settings.
Challenges with the Analysis of Nonapoptotic Cell Death Using Chemical Biology Approaches
Chemical biology methods and cell culture-based approaches are powerful but have limitations that can unintentionally introduce confusion or uncertainty into the analysis of nonapoptotic cell death. Consideration of these limitations may help avoid common pitfalls and, in the fullness of time, lead to useful reinterpretation of existing data. Below we illustrate some common limitations using examples from the ferroptosis field.
Limitations of cell-based assays.
Ferroptosis is a metabolic process that is expected to be more sensitive to cell growth conditions than other lethal processes (Stockwell et al., 2017). For example, differences in the concentration of individual metabolites in the culture medium (e.g. monounsaturated fatty acids, selenium) can significantly alter ferroptosis sensitivity (Magtanong et al., 2019; Vande Voorde et al., 2019). Ferroptosis sensitivity within a population of cells is also sensitive to cell density and cell-cell contact, likely in a cell type-specific manner (Brown et al., 2018; Wenz et al., 2019; Wu et al., 2019). Control over medium composition and cell seeding density is largely lacking between studies and, combined with genetic drift between cell stocks (Ben-David et al., 2018), could easily contribute to differences in results between studies. Moreover, the measurement of cell viability or death at a single arbitrary timepoint (e.g. 48 h), as remains common in the cell death field generally, may overestimate or fail to capture the contribution of a given biochemical process to ferroptosis. For example, overexpression of the glutathione efflux pump MRP1 or stabilization of the tumor suppressor p53 attenuates ferroptosis only over short timescales (~hours) (Cao et al., 2019; Tarangelo et al., 2018). These effects are readily apparent from time-lapse imaging but difficult to detect using traditional indirect end-point measures.
Small molecule off-target effects.
Small molecules are rarely entirely specific, and both erastin and RSL3 bind to more than one protein (Gao et al., 2018; Yagoda et al., 2007). The induction of ferroptosis by these classic agents could therefore conceivably involve the combined inhibition of multiple targets (Yang et al., 2020). This concern is somewhat mitigated by the fact that Gpx4 deletion alone is sufficient to induce cell death that is completely inhibited by liproxstatin-1 (Angeli et al., 2014; Seiler et al., 2008), while direct deprivation of the system xc− substrate cystine is sufficient to trigger ferroptosis that is suppressed by ferrostatin-1 (Magtanong et al., 2019; Tarangelo et al., 2018). Nonetheless, the development of more selective ferroptosis inducers, like the more selective GPX4 inhibitor ML210, will further help address this issue (Eaton et al., 2019).
More concerning are off-target effects associated with small molecule ferroptosis inhibitors. Early studies reported that the first-generation MEK1/2 inhibitor U0126 potently inhibited ferroptosis (Dixon et al., 2014; Yagoda et al., 2007). However, U0126 has a cryptic off-target effect as a radical trapping antioxidant that likely explains its ability to block ferroptosis (Gao et al., 2015). Likewise, several small molecule lipoxygenase (LOX) inhibitors in common use, including NDGA, zileuton, and PD146176, can suppress ferroptosis by acting as radical trapping antioxidants, independent of effects on LOX enzymes (Shah et al., 2018). While LOX enzymes do promote ferroptosis in some contexts (Dar et al., 2018; Wenzel et al., 2017), results obtained using putative small molecule LOX inhibitors must be interpreted cautiously. Of special note, the original Nec-1 molecule can inhibit ferroptosis at high doses, independent of effects on RIPK1 (Angeli et al., 2014). This is potentially problematic for mechanistic studies, but dual necroptosis/ferroptosis inhibitors could conceivably be highly desirable for the treatment of pathological cell death events that involve both processes.
Looking Ahead: Discovery and Characterization of Additional Nonapoptotic Cell Death Pathways
As illustrated by the history of necroptosis and ferroptosis, chemical biology-driven approaches have provided a fruitful means to discover and characterize nonapoptotic cell death mechanisms. Chemical biology-driven approaches are likewise providing evidence for several additional nonapoptotic lethal mechanisms, possibly distinct from known pathways. For example, a number of structurally distinct molecules including an ursolic acid derivative (Sun et al., 2017), a synthetic azaindole-based compound (Huang et al., 2018), and the indole-based chalcone MOMIPP (Robinson et al., 2012) (Table 1), can all induce a nonapoptotic cell death process in cultured cells termed methuosis. Methuosis is characterized by the perturbation of endomembrane trafficking and accumulation of cytoplasmic vacuoles derived from macropinosomes (Maltese and Overmeyer, 2015). Lethal compound treatment accelerates macropinosome and late endosome formation and inhibits the fusion of late endosomes and autophagosomes with lysosomes. This leads to altered protein trafficking, impaired glucose uptake, and activation of the c-Jun N-terminal kinase (JNK) pathway, which conspire together to trigger caspase-independent cell death, although the precise coup de grâce remains somewhat murky (Li et al., 2019; Mbah et al., 2017). The application of chemoproteomic and chemical genetic methods to identify key targets of the above lethal molecules and the pathways that regulate methuosis sensitivity is likely to be informative and currently in its infancy (Cho et al., 2018; Li et al., 2019).
Another distinct lethal mechanism is triggered by the synthetic oxime-containing small molecule caspase-independent lethal 56 (CIL56) (Shimada et al., 2016b) (Table 1). CIL56-induced cell death does not require key regulators of apoptosis, necroptosis, or ferroptosis, or involve immediate membrane permeabilization or bioenergetic disruption (Ko et al., 2019). Genome-wide shRNA chemical genetic screening identified two genes, zinc finger DHHC-type containing 5 (ZDHHC5) and Golgin A7 (GOLGA7), as being essential for CIL56-induced death. These two proteins together form a novel palmitoyl S-acyltransferase complex that appears to promote nonapoptotic cell death in CIL56-treated cells through effects on retrograde membrane trafficking from the plasma membrane. The use of chemoproteomic methods to identify the target(s) of CIL56 would be especially informative in the future.
Studies of methuosis and CIL56-induced cell death may help unveil new regulated cell death mechanisms and associated biochemical regulatory networks. Agents that induce these forms of cell death could be of use in the treatment of cancer, where the ultimate goal is cell death by any means necessary. Indeed, methuosis inducers and CIL56 (also known as CA3) can both attenuate xenograft tumor growth in vivo (Huang et al., 2018; Song et al., 2018). It is currently unclear whether methuosis-inducing small molecules or CIL56 mimics any physiological stimulus that a cell would normally experience in vivo. The identification of specific chemical suppressors for these pathways would enable roles for these pathways to be examined in vivo (e.g. during pathological cell death), as has been done sucessfully using necrostatins or ferrostatins/liproxstatins. Further application of chemical biology approaches will allow for a deeper exploration of these new cell death mechanisms and may enable to discovery of additional, as-yet-unknown nonapoptotic cell death mechanisms.
Significance
Chemical biology approaches have been central to the discovery and characterization of multiple nonapoptotic cell death pathways. The acuteness and portability of chemical tools make them mainstays of cell death research. These tools can be productively combined with genetic and biochemical methods to help characterize nonapoptotic cell death mechanisms and be further employed to discover additional chemical regulators of cell death. Small molecule off-target effects must be recognized, but if identified, would improve chemical understanding and represent potential chemotherapeutic repurposing opportunities. Broader application of chemical biology methods could also lead to the discovery of additional new forms of nonapoptotic cell death.
Acknowledgements
We thank L. Magtanong and C. Brown for comments. This work is supported by the NIH (T32GM007276 to D.A.A. and 1R01GM122923 to S.J.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.J.D. is on the scientific advisory board of Ferro Therapeutics, has consulted for Toray Industries and AbbVie Inc., and is an inventor on patents related to ferroptosis.
References
- Abrams RP, Carroll WL, and Woerpel KA (2016). Five-Membered Ring Peroxide Selectively Initiates Ferroptosis in Cancer Cells. ACS chemical biology 11, 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeli JPF, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthonymuthu TS, Kenny EM, Shrivastava IH, Tyurina YY, Hier ZE, Ting H-C, Dar H, Tyurin VA, Nesterova A, Amoscato AA, et al. (2018). Empowerment of 15-lipoxygenase catalytic competence in selective oxidation of membrane ETE-PE to ferroptotic death signals, HpETE-PE. Journal of the American Chemical Society 140, 17835–17839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aron AT, Loehr MO, Bogena J, and Chang CJ (2016). An Endoperoxide Reactivity-Based FRET Probe for Ratiometric Fluorescence Imaging of Labile Iron Pools in Living Cells. Journal of the American Chemical Society 138, 14338–14346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgley MA, Kremer D, Maurer HC, DelGiorno KE, Lee H-J, Purohit V, Sagalovskiy I, Ma A, Kapillian J, Firl CEM, et al. (2019). Induction of pancreatic tumor-selective ferroptosis through modulation of cystine import. BioRxiv doi: 10.1101/827972. [DOI] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, et al. (2018). Genetic and transcriptional evolution alters cancer cell line drug response. Nature 560, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornstedt M, Hamberg M, Kumar S, Xue J, and Holmgren A (1995). Human thioredoxin reductase directly reduces lipid hydroperoxides by NADPH and selenocystine strongly stimulates the reaction via catalytically generated selenols. J Biol Chem 270, 11761–11764. [DOI] [PubMed] [Google Scholar]
- Brown CW, Amante JJ, Chhoy P, Elaimy AL, Haibo L, Zhu LJ, Baer CE, Dixon SJ, and Mercurio AM (2019). Prominin2 Drives Ferroptosis Resistance by Stimulating Multivesicular Body/Exosome-Mediated Iron Export. SSRN Electronic Journal. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CW, Amante JJ, Goel HL, and Mercurio AM (2017). The α6β4 integrin promotes resistance to ferroptosis. The Journal of cell biology 216, 4287–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CW, Amante JJ, and Mercurio AM (2018). Cell clustering mediated by the adhesion protein PVRL4 is necessary for α6β4 integrin-promoted ferroptosis resistance in matrix-detached cells. Journal of Biological Chemistry 293, 12741–12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burczynski ME, Sridhar GR, Palackal NT, and Penning TM (2001). The reactive oxygen species--and Michael acceptor-inducible human aldo-keto reductase AKR1C1 reduces the alpha,beta-unsaturated aldehyde 4-hydroxy-2-nonenal to 1,4-dihydroxy-2-nonene. The Journal of biological chemistry 276, 2890–2897. [DOI] [PubMed] [Google Scholar]
- Cao JY, and Dixon SJ (2016). Mechanisms of ferroptosis. Cellular and molecular life sciences : CMLS 73, 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao JY, Poddar A, Magtanong L, Lumb JH, Mileur TR, Reid MA, Dovey CM, Wang J, Locasale JW, Stone E, et al. (2019). A Genome-wide Haploid Genetic Screen Identifies Regulators of Glutathione Abundance and Ferroptosis Sensitivity. Cell reports 26, 1544–1556.e1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen p.-H., Wu J, Ding C-KC, Lin C-C, Pan S, Bossa N, Xu Y, Yang W-H, Mathey-Prevot B, and Chi J-T (2019). Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell death and differentiation 23, 369–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Geno E, Patoor M, Reid A, McDonald R, Hild M, and Jenkins JL (2018). Indolyl-Pyridinyl-Propenone-Induced Methuosis through the Inhibition of PIKFYVE. ACS omega 3, 6097–6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad M, and Pratt DA (2019). The chemical basis of ferroptosis. Nat Chem Biol 15, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Dar HH, Tyurina YY, Mikulska-Ruminska K, Shrivastava I, Ting H-C, Tyurin VA, Krieger J, St Croix CM, Watkins S, Bayir E, et al. (2018). Pseudomonas aeruginosa utilizes host polyunsaturated phosphatidylethanolamines to trigger theft-ferroptosis in bronchial epithelium. The Journal of clinical investigation 128, 4639–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch'en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. (2008). Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature chemical biology 4, 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, and Yuan J (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature chemical biology 1, 112–119. [DOI] [PubMed] [Google Scholar]
- Devisscher L, Van Coillie S, Hofmans S, Van Rompaey D, Goossens K, Meul E, Maes L, De Winter H, Van Der Veken P, Vandenabeele P, et al. (2018). Discovery of Novel, Drug-Like Ferroptosis Inhibitors with in Vivo Efficacy. Journal of medicinal chemistry 61, 10126–10140. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, et al. (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 3, e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS chemical biology 10, 1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Grocin AG, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017a). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017b). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature chemical biology 13, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolma S, Lessnick SL, Hahn WC, and Stockwell BR (2003). Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer cell 3, 285–296. [DOI] [PubMed] [Google Scholar]
- Eaton JK, Ruberto RA, Kramm A, Viswanathan VS, and Schreiber SL (2019). Diacylfuroxans Are Masked Nitrile Oxides That Inhibit GPX4 Covalently. J Am Chem Soc 141, 20407–20415. [DOI] [PubMed] [Google Scholar]
- Fang X, Wang H, Han D, Xie E, Yang X, Wei J, Gu S, Gao F, Zhu N, Yin X, et al. (2019). Ferroptosis as a target for protection against cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America 116, 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nature cell biology 16, 1180–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi M, Cotella D, Santoro C, Cora D, Barlev NA, Piacentini M, and Corazzari M (2019). Aldo-keto reductases protect metastatic melanoma from ER stress-independent ferroptosis. Cell Death Dis 10, 902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell death and differentiation 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Yang F, Che J, Han Y, Wang Y, Chen N, Bak DW, Lai S, Xie X, Weerapana E, et al. (2018). Selenium-Encoded Isotopic Signature Targeted Profiling. ACS central science 4, 960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X (2016). Ferroptosis is an autophagic cell death process. Cell research 26, 1021–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Monian P, Quadri N, Ramasamy R, and Jiang X (2015). Glutaminolysis and Transferrin Regulate Ferroptosis. Molecular cell 59, 298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, et al. (2019). Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 343, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E, et al. (2018a). FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nature chemical biology 14, 507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018b). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS chemical biology 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haidasz EA, Van Kessel ATM, and Pratt DA (2016). A Continuous Visible Light Spectrophotometric Approach To Accurately Determine the Reactivity of Radical-Trapping Antioxidants. The Journal of organic chemistry 81, 737–744. [DOI] [PubMed] [Google Scholar]
- Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, Tyurina YY, Bayır H, Abhari BA, Angeli JPF, Choi SM, et al. (2018). Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. The Journal of clinical investigation 128, 3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayano M, Yang WS, Corn CK, Pagano NC, and Stockwell BR (2015). Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell death and differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn T, and Stockwell BR (2018). The development of the concept of ferroptosis. Free Radical Biology and Medicine 133, 130–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sun X, Li Y, He Z, Li L, Deng Z, Huang X, Han S, Zhang T, Zhong J, et al. (2018). Discovery and Identification of Small Molecules as Methuosis Inducers with in Vivo Antitumor Activities. Journal of medicinal chemistry 61, 5424–5434. [DOI] [PubMed] [Google Scholar]
- Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai Z-L, Tang M, Xue M, Cheng N, et al. (2017). Quantitative real-time imaging of glutathione. Nature communications 8, 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Mao G, Qu F, Angeli JPF, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature chemical biology 13, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny EM, Fidan E, Yang Q, Anthonymuthu TS, New LA, Meyer EA, Wang H, Kochanek PM, Dixon CE, Kagan VE, et al. (2019). Ferroptosis Contributes to Neuronal Death and Functional Outcome After Traumatic Brain Injury. Critical care medicine 47, 410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, and Currie AR (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer 26, 239–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerridge D (1958). The effect of actidione and other antifungal agents on nucleic acid and protein synthesis in Saccharomyces carlsbergensis. Journal of general microbiology 19, 497–506. [DOI] [PubMed] [Google Scholar]
- Kim SE, Zhang L, Ma K, Riegman M, Chen F, Ingold I, Conrad M, Turker MZ, Gao M, Jiang X, et al. (2016). Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nature nanotechnology 11, 977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, Merl-Pham J, Bao X, Anastasov N, Kossl J, et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaier E, Louandre C, Godin C, Saidak Z, Baert M, Diouf M, Chauffert B, and Galmiche A (2014). Sorafenib induces ferroptosis in human cancer cell lines originating from different solid tumors. Anticancer research 34, 6417–6422. [PubMed] [Google Scholar]
- Leu JI, Murphy ME, and George DL (2019). Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Ates G, Methner A, Conrad M, and Maher P (2018). Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Frontiers in neuroscience 12, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Han X, Lan X, Gao Y, Wan J, Durham F, Cheng T, Yang J, Wang Z, Jiang C, et al. (2017). Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI insight 2, e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Mbah NE, Overmeyer JH, Sarver JG, George S, Trabbic CJ, Erhardt PW, and Maltese WA (2019). The JNK signaling pathway plays a key role in methuosis (non-apoptotic cell death) induced by MOMIPP in glioblastoma. BMC cancer 19, 77–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz P-S, et al. (2014). Synchronized renal tubular cell death involves ferroptosis. Proceedings of the National Academy of Sciences of the United States of America 111, 16836–16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, and Hergenrother PJ (2019). Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nature chemistry 11, 521–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell chemical biology 26, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai TT, Hamaï A, Hienzsch A, Cañeque T, Müller S, Wicinski J, Cabaud O, Leroy C, David A, Acevedo V, et al. (2017). Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nature chemistry 9, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Sheldon KL, DeHart DN, Patnaik J, Manevich Y, Townsend DM, Bezrukov SM, Rostovtseva TK, and Lemasters JJ (2013). Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. Journal of Biological Chemistry 288, 11920–11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltese WA, and Overmeyer JH (2015). Non-apoptotic cell death associated with perturbations of macropinocytosis. Frontiers in physiology 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbah NE, Overmeyer JH, and Maltese WA (2017). Disruption of endolysosomal trafficking pathways in glioma cells by methuosis-inducing indole-based chalcones. Cell biology and toxicology 33, 263–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, Javaid S, Coletti ME, Jones VL, Bodycombe NE, et al. (2016). Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nature chemical biology 12, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich E, Franklin RM, Shatkin AJ, and Tatum EL (1961). Effect of actinomycin D on cellular nucleic acid synthesis and virus production. Science 134, 556–557. [DOI] [PubMed] [Google Scholar]
- Robinson MW, Overmeyer JH, Young AM, Erhardt PW, and Maltese WA (2012). Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. Journal of medicinal chemistry 55, 1940–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell metabolism 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Shah R, Farmer LA, Zilka O, Van Kessel ATM, and Pratt DA (2019). Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell chemical biology. [DOI] [PubMed] [Google Scholar]
- Shah R, Shchepinov MS, and Pratt DA (2018). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS central science 4, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Hayano M, Pagano NC, and Stockwell BR (2016a). Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell chemical biology 23, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016b). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nature chemical biology 12, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, et al. (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. Journal of the American Chemical Society 136, 4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, Skinner HD, Johnson RL, Ding S, and Ajani JA (2018). A Novel YAP1 Inhibitor Targets CSC-Enriched Radiation-Resistant Cells and Exerts Strong Antitumor Activity in Esophageal Adenocarcinoma. Molecular cancer therapeutics 17, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK, Kagan VE, et al. (2017). Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Li B, Su X, Chen G, Li Y, Yu L, Li L, and Wei W (2017). An Ursolic Acid Derived Small Molecule Triggers Cancer Cell Death through Hyperstimulation of Macropinocytosis. Journal of medicinal chemistry 60, 6638–6648. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. [DOI] [PubMed] [Google Scholar]
- Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell reports 22, 569–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tata JR (1966). Requirement for RNA and protein synthesis for induced regression of the tadpole tail in organ culture. Developmental biology 13, 77–94. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Fedorka SR, Gad I, Shah R, Alqahtani HD, Koranne R, Kuganesan N, Dlamini S, Rogers T, Al-Hamashi A, et al. (2019). Small-Molecule Ferroptotic Agents with Potential to Selectively Target Cancer Stem Cells. Scientific reports 9, 5926–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer cell 33, 890–904.e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Science advances 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, and Vandenabeele P (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. The Journal of experimental medicine 187, 1477–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BK, and Schreiber SL (2016). The Power of Sophisticated Phenotypic Screening and Modern Mechanism-of-Action Methods. Cell chemical biology 23, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiwer M, Bittker J, Lewis TA, Shimada K, Yang WS, MacPherson L, Dandapani S, Palmer M, Stockwell BR, Schreiber SL, et al. (2012). Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorganic & medicinal chemistry letters 22, 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenz C, Faust D, Linz B, Turmann C, Nikolova T, and Dietrich C (2019). Cell-cell contacts protect against t-BuOOH-induced cellular damage and ferroptosis in vitro. Archives of toxicology 93, 1265–1279. [DOI] [PubMed] [Google Scholar]
- Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641.e626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpaw AJ, Shimada K, Skouta R, Welsch ME, Akavia UD, Peer D, Shaik F, Bulinski JC, and Stockwell BR (2011). Modulatory profiling identifies mechanisms of small molecule-induced cell death. Proceedings of the National Academy of Sciences of the United States of America 108, E771–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, Chen Z-N, and Jiang X (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. (2007). RAS-RAF-MEK dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry and Biology 15, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Luo M, Zhang K, Zhang J, Gao T, Connell DO, Yao F, Mu C, Cai B, Shang Y, et al. (2020). Nedd4 ubiquitylates VDAC2/3 to suppress erastin-induced ferroptosis in melanoma. Nat Commun 11, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O'Connor OA, and Stockwell BR (2019). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell chemical biology 26, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, and Pratt DA (2017). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS central science 3, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, Tseng Y-Y, Deasy R, Kost-Alimova M, Dančík V, et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nature communications 10, 1617–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]