

Abstract
We have generated polyclonal antibodies against the amino-terminal third of the Menkes protein (ATP7A; MNK) by immunizing rabbits with a histidine-tagged MNK fusion construct containing metal-binding domains 1–4. The purified antibodies were used in Western analysis of cell lysates and in indirect immunofluorescence experiments on cultured cells. On Western blots, the antibodies recognized the ∼165 kDa MNK protein in CHO cells and human fibroblasts. No MNK signal could be detected in fibroblasts from a patient with Menkes disease or in Hep3B hepatocellular carcinoma cells, confirming the specificity of the antibodies. Immunocytochemical analysis of CHO cells and human fibroblasts showed a distinct perinuclear signal corresponding to the pattern of the Golgi complex. This staining pattern was similar to that of α-mannosidase II which is a known resident enzyme of the Golgi complex. Using brefeldin A, a fungal inhibitor of protein secretion, we further demonstrated that the MNK protein is localized to the trans-Golgi network. This data provides direct evidence for a subcellular localization of the MNK protein which is similar to the proposed vacuolar localization of Ccc2p, the yeast homolog of MNK and WND (ATP7B), the Wilson disease gene product. In light of the proposed role of MNK both in subcellular copper trafficking and in copper efflux, these data suggest a model for how these two processes are linked and represent an important step in the functional analysis of the MNK protein.
Introduction
Menkes disease is a rare X-linked recessive disorder of copper transport that is usually fatal in the first few years of life. The link between the disease and copper metabolism was first made by Danks et al. (1), who noted that the brittle hair and the neurological features in Menkes disease are remarkably similar to the effects of copper deprivation seen in sheep raised on copper-deficient soil. Most of the phenotypic features of the disease can be understood as malfunctioning of several copperdependent enzymes caused by a general copper deficiency. The molecular basis of this deficiency has long been proposed to be a copper transport problem. Accumulation (2) and decreased export (3) of copper in cultured fibroblasts from patients with Menkes disease are in agreement with a hypothetical transport defect. The characteristics of the cloned Menkes disease gene (4–6) further supported this hypothesis. Analysis of the MNK cDNA sequence revealed the characteristic features of a P-type ATPase. The members of this family of ion-motive ATPases generally translocate positively charged ions across plasma or intracellular membranes in a cycle involving ATP hydrolysis coupled to phosphorylation of a highly conserved aspartic acid residue (7). Other features of MNK include six putative metal-binding domains, and a CPC (Cys-Pro-Cys) motif in a hydrophobic (transmembrane) domain thought to be involved in the translocation of the metal ion. Both these elements are present specifically in the subfamily of heavy metal-transporting P-type ATPases, several members of which have been described in bacteria and shown to play a role in heavy metal transport (8).
In mammals, the closest known family member of the Menkes disease gene is the Wilson disease gene (ATP7B; WND) which is expressed predominantly in the liver (9,10). WND is necessary for biliary excretion of copper and incorporation into ceruloplasmin (CP). Both these functions are impaired in Wilson's disease, leading to accumulation of copper in the liver and other tissues. The cellular localization of both the MNK and WND proteins is unknown. Recently a homolog of MNK and WND was cloned in Saccharomyces cerevisiae (11). This yeast gene, CCC2, encodes a P-type ATPase that is proposed to function in the subcellular transport of copper to a yeast ferroxidase, Fet3p (12). Fet3p has homology to human ceruloplasmin and, like CP, its function is dependent on proper cellular copper distribution and incorporation (13). The parallel between the yeast and mammalian systems, together with the sequence homology shared by MNK, WND and Ccc2p suggest that these three copper P-type ATPases may function similarly on the cellular level. The exact nature of their roles in cellular copper trafficking, however, remains unknown.
We report here immunocytochemical evidence for the subcellular perinuclear localization of MNK similar to α-mannosidase II, a known resident enzyme of the Golgi complex (14). Using brefeldin A, we further delineated the localization of MNK to the trans-compartment of the Golgi apparatus.
Figure 1.

Western blot analysis of whole cell lysates with 1/5000 dilution of anti-MNK polyclonal antibodies. The protein sources are shown above each lane. Lane 1 was loaded with 30 ng of purified MNK fusion product and shows a strong band of 57 kDa. A band of ∼165 kDa, corresponding to the MNK protein (large arrow), is visible in normal fibroblasts and in CHO cells. Larger non-specific bands are present above the MNK signal. These bands disappeared in a competition experiment and appear to be of no effect in the immunofluorescence experiments. No MNK band is visible in fibroblasts from a Menkes disease patient with no MNK RNA (Pt Fibr) or in Hep3B cells that have abundant WND RNA (data not shown). The small arrow shows the unidentified specific 35 kDa band in both normal and Menkes fibroblasts, which is clearly not present in the CHO or Hep3B cells.
Results
Western blot analysis of the anti-MNK polyclonal antibodies
Fourth bleed antisera diluted to 1/5000 reacted strongly with 30 ng of purified MNK fusion product which is 57 kDa in size (Fig. 1, lane 1). To analyze the specificity of the antibodies, we performed Western blot analysis on cell lysates from different cell lines. In normal human fibroblasts (Fig. 1, lane 3, large arrow), a band of the expected size (∼165 kDa) was present, while fibroblasts from a patient with no detectable MNK RNA (4,6) showed no signal (Fig. 1, lane 2). The antibodies did not detect a protein in liver-derived Hep3B cells (Fig. 1, lane 4). These results demonstrate that the polyclonal antibodies are indeed specific to MNK and do not cross-react with WND. MNK and WND expression in the Hep3B cells was also examined by Northern analysis. While WND RNA was abundantly present, MNK RNA was barely detectable (data not shown). The difference between the Western and Northern data in the liver cells is likely to be due to a lower sensitivity of our Western blot analysis. Lane 5 of Figure 1 shows a band in CHO cells in the same size range as in the human fibroblasts, demonstrating that the antibodies can be used in cross-species detection of MNK. A similar Western blot result was previously shown by Camakaris et al. (15) with antibodies that were also generated against the metal-binding domains of human MNK. These results are not surprising since the hamster cDNA is almost 90% identical to the human MNK cDNA (15), while the human WND cDNA is <60% identical to MNK (9,10). Faint larger bands can be seen in the human fibroblasts as well as the CHO cells, and represent non-specific signals due to binding to over-abundant high molecular weight proteins as analyzed by amido black staining of the membranes after Western analysis. Pre-adsorption of the antibodies with the MNK fusion protein did not have an effect on these non-specific bands, while the band representing the Menkes protein was eliminated completely (data not shown).
Figure 2.
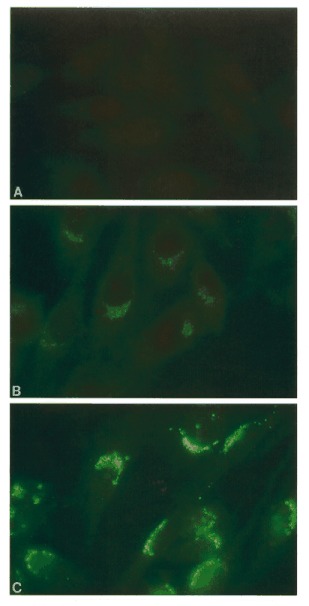
Indirect immunofluorescence of fixed CHO cells with a 1/200 dilution of pre-immune rabbit serum (A), a 1/200 dilution of anti-MNK polyclonal antibodies (B) and a 1/2000 dilution of anti-Man II antibodies (C). Both (B) and (C) show a similar distinct pattern of perinuclear fluorescence. The signal intensity in (B) is much lower than in (C) and reflects the lower abundance of MNK relative to mannosidase.
Both normal and Menkes syndrome fibroblasts also show an additional signal of ∼35 kDa (Fig. 1, lanes 2 and 3, small arrow). Because this band is present in both normal cells and cells from a patient with no MNK RNA, it cannot be a processed form of MNK. One explanation could be that the smaller band represents a protein with significant homology to the Menkes metal-binding region. This homology would have to be higher than the homology with WND which is not recognized by our antibody preparation. Alternatively, this band could represent an artifact caused by non-specific antibody reaction in the fibroblast lysates. However, in the above-mentioned competition experiment, in which the antibodies were pre-adsorbed with the MNK fusion protein, both the MNK signal and the smaller 35 kDa signal disappeared (data not shown), suggesting homology of this smaller protein to MNK. Further investigations will be necessary to clarify the nature of this signal.
Figure 3.
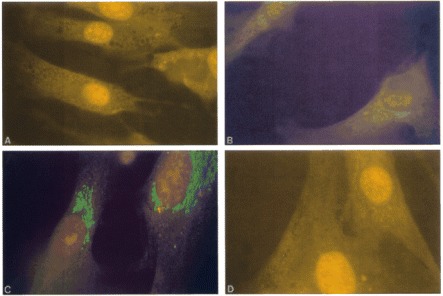
Indirect immunofluorescence of fixed normal fibroblasts with a 1/200 dilution of pre-immune rabbit serum (A), a 1/200 dilution of anti-MNK polyclonal antibodies (B) and a 1/2000 dilution of anti-Man II antibodies (C). (D) shows no signal in fixed fibroblasts from a Menkes disease patient treated with a 1/200 dilution of the anti-MNK antibodies. The results in (A-C) are similar to Figure 2A-C.
Immunolocalization of the MNK protein compared with-α-mannosidase II
We first investigated the localization of MNK by indirect immunofluorescence on fixed CHO cells, because in these cells there is no cross-reaction of the antibodies to the smaller 35 kDa protein band seen in human fibroblasts. The signal in these cells therefore reflects localization of the Menkes protein only. As shown in Figure 2B, a 1/200 dilution of the anti-MNK antibodies shows distinct fluorescent staining in the form of a perinuclear cap following the pattern of the Golgi apparatus. No staining was observed in cells treated with pre-immune serum (Fig. 2A). To confirm the Golgi localization, we compared the staining pattern with that of a known Golgi resident enzyme, α-mannosidase II (Man II). Panel C in Figure 2 shows staining with a 1/2000 dilution of polyclonal anti-Man II antibodies. The pattern is very similar to that of MNK (Fig. 2A), albeit more intense, reflecting the difference in expression of the two genes. Because the Golgi staining of MNK could reflect the transient passage of MNK through this cellular compartment, we blocked protein synthesis with cycloheximide (10 µg/ml) and observed no difference in the localization (data not shown).
We also analyzed normal and Menkes fibroblasts by indirect immunofluorescence. As expected, in normal fibroblasts a signal similar to that in the CHO cells can be seen with both anti-MNK (Fig. 3B) and anti-Man II (Fig. 3C) antibodies. Fibroblasts from the Menkes patient showed no signal with the anti-MNK antibodies (Fig. 3D). Although additional non-specific bands were seen on Western blots, this data further demonstrates the specificity of the antibodies to MNK in immunofluorescence experiments.
Brefeldin A treatment localizes MNK to the trans-Golgi network
The Golgi complex is made up of three functionally different subcompartments, the cis-Golgi network (CGN), the Golgi stack (or medial Golgi) and the trans-Golgi network (TGN) (16). The fungal macrocyclic lactone, brefeldin A (BFA), is known to affect these compartments differently and can be used to identify the sublocalization of proteins within the Golgi complex (17,18). We investigated the effects of BFA on the localization of MNK in comparison with Man II, which is known to localize predominantly to the medial cisternae of the Golgi stack in CHO cells based on electron microscopy data (14). The panels on the left (A-E) in Figure 4 show staining for MNK in CHO cells exposed to BFA for 0, 5, 10 and 30, and for 30 min followed by a 1 h recovery in BFA-free medium. The panels on the right (a-e) in Figure 4 show staining for Man II in CHO cells under the same treatment conditions. After 2 min of BFA treatment (not shown), the MNK perinuclear signal was essentially the same as before treatment (Fig. 4A). After 5 min (Fig. 4B), the signal started to show slightly less perinuclear capping. After 10 min (Fig. 4C), a more diffuse signal throughout the cytoplasm was visible with a strong circular staining next to the nucleus. BFA treatment for 30 min (Fig. 4D) stabilized this juxtanuclear staining. These BFA-induced changes are characteristic for proteins that reside in the TGN (17,19). The TGN is functionally connected to the cellular network of microtubuli that originates in the microtubule organizing center (MTOC). BFA treatment causes the TGN to collapse around the MTOC and this is responsible for the juxtanuclear signal described above. In contrast to these BFA effects on MNK localization, the Man II signal is affected very differently by BFA. Fluorescence starts to diffuse in the cytoplasm in a reticular pattern as early as 2 min after BFA induction (not shown). Upon longer exposures to BFA, the perinuclear cap seen in untreated cells completely disappears and the reticular cytoplasmic pattern that emerges after 2 min of BFA treatment becomes stronger (Fig. 4b-d). This pattern of BFA-induced endoplasmic reticulum redistribution of Man II has been reported previously (20) and is consistent with its localization to the medial cisternae of the Golgi stack in CHO cells. Recovery of the cells in BFA-free medium for 1 h resulted in nearly complete restoration of the normal signal in both anti-MNK (Fig. 4E) and anti-Man II (Fig. 4e) stained cells.

Indirect immunofluorescence of fixed CHO cells with a 1/200 dilution anti-MNK polyclonal antibodies (A–E) and with a 1/2000 dilution of anti-Man II antibodies (a–e) before and after BFA treatment. Normal perinuclear MNK staining (A) diffuses slightly after 5 min of BFA treatment (B). After 10 min, a more diffuse cytoplasmic staining occurs as well as a distinct circular signal next to the nucleus (C). After 30 min, the juxtanuclear signal has stabilized (D). In contrast to the BFA effects on MNK localization, BFA causes the normal perinuclear Man II signal (a) to diffuse into the cytoplasm in a reticular pattern typical of ER staining. This ER redistribution starts as early as 5 min (b) after BFA and increases upon longer exposures to BFA (c and d). Removal of BFA and recovery in normal media for 1 h restores both the MNK (E) and Man II (e) signals to near normal.
Discussion
We report here direct immunocytochemical evidence for the subcellular localization of MNK. The pattern of perinuclear staining obtained with our polyclonal anti-MNK antibodies was identical to that of the known Golgi resident enzyme α-mannosidase II. The effect of BFA treatment on this localization pattern shows that MNK resides within the TGN, one of the three functionally distinct compartments of the Golgi complex (16). Inhibition of protein synthesis with cycloheximide does not influence the Golgi localization of MNK, indicating that the Menkes protein does not just transiently pass through this subcellular compartment. A similar Golgi localization result has been obtained on a mutant CHO cell line with an amplification of the MNK gene resulting in the overexpression of the MNK protein (21), thus supporting these findings.
The MNK protein is proposed to function in two major aspects of cellular copper homeostasis. A role in intracellular copper transport has been suggested by the fact that several of the copper-dependent enzymes deficient in Menkes disease are located in subcellular organelles. It is also clear that MNK plays a role in copper efflux from the cell, based on the reduced efflux kinetics seen in cells from patients with Menkes disease (3) and the increased efflux kinetics in CHO cells overexpressing the MNK protein (15). The TGN is known to play a dynamic role in cellular protein trafficking and is involved in the transport to and from both the plasma membrane and the lysosomal compartment (22,23). Thus, a trans-Golgi localization is consistent with a functional role of MNK both in subcellular transport and in copper efflux.
MNK localization to the TGN is presumably directed by one or more as yet unidentified targeting signals within the MNK protein. For several proteins, a variety of targeting signals in cytoplasmic and transmembrane domains have been shown to be essential for Golgi retention (24,25), lysosomal targeting (26,27) and Golgi retrieval (28). Modification of these signals by directed mutagenesis has been shown to alter the cellular localization of such proteins significantly (28–31). Some proteins, such as furin (30) and TGN38 (28), are known to recycle from the plasma membrane to the TGN, based on the presence of an internalization or retrieval signal within their amino acid sequence (32,33). The involvement of MNK in copper efflux could be explained by targeting from the TGN to the plasma membrane, followed by return to the intracellular compartment through endocytosis, mediated by clathrin-coated vesicles, and based on the presence of a putative retrieval signal in MNK, as suggested by the data of Petris et al. (21). Alternatively, MNK could function directly in copper efflux due to specific targeting of the different MNK isoforms. We and others have shown previously that there is low level alternative splicing in the transmembrane region of the MNK cDNA removing either transmembrane domains three and four, or five and/or six (34,35 and H.A.D., unpublished data). Petruhkin et al. (36) showed that the same regions in the WND cDNA are also alternatively spliced. It is possible that skipping of the transmembrane domains could remove targeting signals necessary for MNK to localize to the TGN, and allowing these MNK isoforms to target to different cellular regions including the plasma membrane and the subcellular organelles. The low levels of the minor isoforms would make it difficult to visualize this phenomenon by indirect immunofluorescence. Studies using constructs of the different MNK isoforms will be necessary to prove this hypothesis. As an alternative to functioning directly in copper export, MNK could be involved indirectly in the efflux process. In this scenario, MNK would transport copper into the TGN vesicles, where it might be incorporated into another protein or small peptide which could leave the cell through the secretory pathway.
The Wilson disease gene product, WND (ATP7B), has a similar dual role in hepatocyte copper homeostasis. Decreased copper excretion into the bile in Wilson disease points to a role for WND in copper efflux from the hepatocyte, while decreased incorporation of copper into CP also suggests a role in subcellular transport. Recently, Dijkstra et al. (37) identified an active copper transport system in the plasma membrane of hepatocytes that they proposed represents the Wilson copper transporter. If the identity of this transport system is confirmed to be WND, this would prove a plasma membrane localization. These authors also presented the possibility that the observed copper transport activity could be due, at least in part, to contamination of the plasma membrane preparations with subcellular organelles. Subcellular localization of WND is therefore not excluded. Given the known similarities between MNK and WND, and considering the data presented here on MNK localization, it seems reasonable to predict that WND will have a similar localization pattern to MNK. Because of its high expression levels in comparison with MNK, localization studies for the Wilson gene product may reveal directly how WND functions in the efflux process. In yeast, a homolog of MNK and WND was cloned recently (11), and is proposed to have a subcellular localization. This yeast copper P-type ATPase, Ccc2p, was shown to provide copper to a yeast ferroxidase, Fet3p (12), that has homology to human CP and, like CP, is dependent on proper copper distribution and incorporation to form the mature ferroxidase. This process of copper-induced maturation of the yeast CP homolog occurs in a post-Golgi compartment (38). There is at present no evidence for a role of Ccc2p in copper efflux. A dynamic interaction between subcellular transport and efflux of copper in yeast based on Ccc2p, as suggested here for MNK and WND, therefore awaits further study.
In conclusion, we have shown by indirect immunofluorescence that MNK localizes to the TGN and have compared it with the known medial Golgi localization of α-mannosidase II in CHO cells (14). This localization is consistent with the proposed role of MNK in intracellular transport of copper for subsequent incorporation into the different cupro-enzymes. Based on the dynamic role of the TGN in cellular protein trafficking, it also helps to understand the role of MNK in copper efflux from the cell, either assuming that MNK participates directly in the dynamic interaction between the intracellular compartment and the plasma membrane, or indirectly through a mediating protein that leaves the cell through the secretory pathway.
Materials and Methods
Preparation of an MNK fusion construct and immunization of rabbits
An MNK fusion protein was constructed in the region of lowest homology between MNK and WND covering metal-binding domains 1–4. Using RT-PCR with primers at positions 159 and 1604 of the MNK cDNA (4), a fragment of 1456 bp was amplified. The upstream primer (5′-TGGGTGTGAATTCTGTTACCA-3′) contained the EcoRI site at position 166, while the downstream primer (5′-GGAAGCTTAATTCTTTCCTTCCTCCTTGTC-3′) was engineered with a HindIII restriction site generating an in-frame stop codon. The fragment was directionally cloned downstream of and in-frame to the six histidine residues in the pTRCHisC vector (Invitrogen), and verified by sequence analysis. An amino-terminal histidine-tagged MNK truncated protein of the expected size (57 kDa) was generated in Escherichia coli and purified on a nickel resin column according to the manufacturer's protocol (Invitrogen). Initial immunization of New Zealand white rabbits was performed using 1 mg of the purified fusion protein in 1 ml of Freund's complete adjuvant. Subsequent immunizations were performed with Freund's incomplete adjuvant at monthly intervals, and rabbits were bled 7–10 days after each immunization.
Antibody preparation and Western blot analysis
Antisera were prepared from whole blood after 1 h incubation at 37°C followed by overnight incubation at 4°C. The serum was isolated following 30 min centrifugation at 5000× g. The IgG fraction of the antisera was isolated by saturated ammonium sulfate (SAS) precipitation (39) and resuspended in one-tenth volume of phosphate-buffered saline (PBS). The remaining ammonium sulfate was removed by two successive dialyses against PBS. To remove cross-reacting antibodies to bacterial proteins, the SAS-purified IgG fraction was pre-adsorbed twice overnight at 4°C against a mixture of formalin-fixed and autoclaved protein preparations from E.coli (40).
Western blots were prepared from whole cell lysates of cultured cells. Approximately 107 cells were lysed with 1 ml of lysis buffer [50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.02% NaAz, 1 mM phenylmetylsulfonyl fluoride (PMSF), 1 µg/ml aprotinin, 1% Triton X-100] for 30 min at 4°C. After centrifugation, proteins were precipitated with 9 vol of absolute ethanol. The protein pellet was resuspended in 20 µl of Laemmli sample buffer. Samples were boiled for 2 min and loaded onto an 10% SDS-polyacrylamide gel. The separated proteins were electro-transferred to nitrocellulose and, after 1 h blocking [Tris-buffered saline/Tween-20; 137 mM NaCl, 20 mM Tris, 0.1% Tween-20, pH 7.6 (TBST) + 3% dry milk], the membrane was incubated for 1 h at room temperature with a 1/5000 dilution of the anti-MNK polyclonal antibodies. Following several washes in TBST, the membrane was incubated with a 1/2000 dilution of horseradish peroxidase (HRPO)-conjugated donkey anti-rabbit antibodies. The final detection was performed using ECL reagents according to the manufacturer's protocol (Amersham).
Cell culture and brefeldin A treatment
Human fibroblasts, Hep3B and CHO cells were cultured in monolayers with 5% CO2 at 37°C in Dulbecco's modified Eagle's medium (DMEM; Gibco-BRL) supplemented with 10% fetal calf serum (FCS; HyClone), 100 U/ml penicillin and 100 µg/ml streptomycin (Irvine Scientific). CHO cells are proline auxotrophs and were supplemented with 0.2 mM L-proline (Sigma). Cells were treated with 5 µg/ml BFA (Sigma) for various time points (2, 5, 10 and 30 min, and 30 min followed by 1 h recovery in DMEM) and washed twice in PBS prior to fixation and antibody incubation as described below. Cycloheximide treatments (10 µg/ml) were carried out for 4 h and then processed as described for the BFA-treated cells.
Immunocytochemistry
CHO cells were cultured on glass coverslips in DMEM for 15–36 h. The cells were rinsed twice in cold PBS and fixed for 20 min in 2% paraformaldehyde (Sigma) at 4°C. Following three washes in PBS, the cells were blocked for 1 h at room temperature in blocking buffer (0.25% dry milk, 0.1% Tween-20 in PBS). The coverslips were then incubated with 200 µl of a 1/200 dilution of the anti-MNK antibodies (diluted in blocking buffer) or a 1/2000 dilution of the anti-α-mannosidase II antibodies (kindly provided by Dr M.G. Farquhar, UC San Diego) for 2 h at room temperature. After three washes in PBS, the coverslips were overlaid with 200 µl of a 1/1000 dilution of affinity-purified fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (H+L) (GAR-FITC; Vector Laboratories) and incubated for 1 h in the dark at room temperature. Following three washes in PBS, the coverslip were mounted using 10 µl of Vectashield (Vector Laboratories) and examined by fluorescence microscopy using a Zeiss Axioskop epifluorescence microscope. Photographs were taken directly on Kodak ASA 400 Gold film.
Acknowledgements
We would like to thank Dr M.G. Farquhar for kindly providing polyclonal antibodies against the α-mannosidase II protein, Dr Dennis Thiele for helpful suggestions, Dr Charles M. Wilke for critically reading the manuscript and Drs Julian Mercer and Jim Camakaris for communicating unpublished data. This work was supported by grant R01 DK44130 from the National Institute of Diabetes and Digestive and Kidney Diseases, NIH (T.W.G.).
Note Added in Proof
While this manuscript was in press, Yamaguchi et al. (Proc. Natl. Acad. Sci. USA, 93, 14030–14035, 1996) demonstrated a similar localization of the Menkes protein in HeLa cells.
References
- 1.Danks D.M., Campbell P.E., Stevens B.J., Mayne V., Cartwright E. Menkes' kinky hair syndrome: an inherited defect in copper absorption with widespread effects. Pediatrics. 1972;50:188–201. [PubMed] [Google Scholar]
- 2.Goka T.J., Stevenson R.E., Hefferan P.M., Howell R.R. Menkes disease: a biochemical abnormality in cultured human fibroblasts. Proc. Natl Acad. Sci. USA. 1976;73:604–606. doi: 10.1073/pnas.73.2.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herd S.M., Camakaris J., Christofferson R., Wookey P., Danks D.M. Uptake and efflux of copper-64 in Menkes'-disease and normal continuous lymphoid cell lines. Biochem. J. 1987;247:341–347. doi: 10.1042/bj2470341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vulpe C., Levinson B., Whitney S., Packman S., Gitschier J. Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nature Genet. 1993;3:7–13. doi: 10.1038/ng0193-7. [DOI] [PubMed] [Google Scholar]
- 5.Chelly J., Tumer Z., Tonnesen T., Petterson A., Ishikawa-Brush Y., Tommerup N., Horn N., Monaco A.P. Isolation of a candidate gene for Menkes disease which encodes for a potential heavy metal binding protein. Nature Genet. 1993;3:14–19. doi: 10.1038/ng0193-14. [DOI] [PubMed] [Google Scholar]
- 6.Mercer J.F.B., Livingston J., Hall B., Paynter J.A., Begy C., Chandrasekharappa S., Lockhart P., Grimes A., Bhave M., Siemieniak D., Glover T.W. Isolation of a partial candidate for Menkes disease by positional cloning. Nature Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 7.Pedersen P.L., Carafoli E. Ion motive ATPases I. Ubiquity, properties, and significance to cell function. Trends Biochem. Sci. 1987;12:146–150. [Google Scholar]
- 8.Cooksey D.A. Molecular mechanisms of copper resistance and accumulation in bacteria. FEMS Microbiol. Rev. 1994;14:381–386. doi: 10.1111/j.1574-6976.1994.tb00112.x. [DOI] [PubMed] [Google Scholar]
- 9.Bull P.C., Thomas G.R., Rommens J.M., Forbes J.R., Cox D.W. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nature Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 10.Tanzi R.E., Petrukhin K., Chernov I., Pellequer J.L., Wasco W., Ross B., Romano D.M., Parano E., Pavone L., Brzustowicz L.M., Devoto M., Peppercorn J., Bush A.I., Sternlieb I., Pirastu M., Gusella J.F., Evgrafov O., Penchaszadeh G.K., Honig B., Edelman I.S., Soares M.B., Scheinberg I.H., Gilliam T.C. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nature Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 11.Fu D., Beeler T.J., Dunn T.M. Sequence, mapping and disruption of CCC2, a gene that cross-complements the Ca2+-sensitive phenotype of csg1 mutants and encodes a P-type ATPase belonging to the Cu2+-ATPase subfamily. Yeast. 1995;11:283–292. doi: 10.1002/yea.320110310. [DOI] [PubMed] [Google Scholar]
- 12.Yuan D.S., Stearman R., Dancis A., Dunn T., Beeler T., Klausner R. The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc. Natl Acad. Sci. USA. 1995;92:2632–2636. doi: 10.1073/pnas.92.7.2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Askwith C., Eide D., Van Ho A., Bernard P.S., Li L., Davis-Kaplan S., Sipe D.M., Kaplan J. The FET3 gene of S.cerevisiae encodes a multicopper oxidase required for ferrous iron uptake. Cell. 1994;76:403–410. doi: 10.1016/0092-8674(94)90346-8. [DOI] [PubMed] [Google Scholar]
- 14.Velasco A., Hendricks L., Moremen K.W., Tulsiani D.R.P., Touster O., Farquhar M.G. Cell type-dependent variations in the subcellular distribution of α-mannosidase I and II. J. Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Camakaris J., Petris M.J., Bailey L., Shen P., Lockhart P., Glover T.W., Barcroft C.L., Patton J., Mercer J.F.B. Gene amplification of the Menkes (MNK; ATP7A) P-type ATPase gene of CHO cells is associated with copper resistance and enhanced copper efflux. Hum. Mol. Genet. 1995;4:2117–2123. doi: 10.1093/hmg/4.11.2117. [DOI] [PubMed] [Google Scholar]
- 16.Mellman I., Simons K. The Golgi complex: in vitro veritas? Cell. 1992;68:829–840. doi: 10.1016/0092-8674(92)90027-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reaves B., Banting G. Perturbation of the morphology of the trans-Golgi network following brefeldin A treatment: redistribution of a TGN-specific integral membrane protein TGN38. J. Cell Biol. 1992;116:85–94. doi: 10.1083/jcb.116.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klausner R.D., Donaldson J.G., Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chege N.W., Pfeffer S.R. Compartmentation of the Golgi complex: brefeldin-A distinguishes trans-Golgi cisternae from the trans-Golgi network. J. Cell Biol. 1990;111:893–899. doi: 10.1083/jcb.111.3.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe S.L., Wong S.H., Hong W. The mammalian ARF-like protein 1 (Arl1) is associated with the Golgi complex. J. Cell Sci. 1996;109:209–220. doi: 10.1242/jcs.109.1.209. [DOI] [PubMed] [Google Scholar]
- 21.Petris M., Mercer J.F.B., Culvenor J.G., Lockhart P., Gleeson P.A., Camakaris J. Ligand-regulated transport of the Menkes copper P-type ATPase from the Golgi apparatus to the plasma membrane; a novel mechanism of regulated trafficking. EMBO J. 1996;15:6084–6095. [PMC free article] [PubMed] [Google Scholar]
- 22.Griffith G., Simons K. The trans Golgi network: sorting at the exit site of the Golgi complex. Science. 1986;234:438–443. doi: 10.1126/science.2945253. [DOI] [PubMed] [Google Scholar]
- 23.Schwartz A.L. Cell biology of intracellular protein trafficking. Annu. Rev. Immunol. 1990;8:195–229. doi: 10.1146/annurev.iy.08.040190.001211. [DOI] [PubMed] [Google Scholar]
- 24.Machamer C.E., Mentone S.A., Rose J.K., Farquhar M.G. The E1 glycoprotein of an avian coronavirus is targeted to the cis Golgi complex. Proc. Natl Acad. Sci. USA. 1990;87:6944–6948. doi: 10.1073/pnas.87.18.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teasdale R.D., D'Agostaro G., Gleeson P.A. The signal for Golgi retention of bovine β1,4-galctosyltransferase is the transmembrane domain. J. Biol. Chem. 1992;267:4084–4096. [PubMed] [Google Scholar]
- 26.Letourneur F., Klausner R.D. A novel di-leucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell. 1992;69:1143–1157. doi: 10.1016/0092-8674(92)90636-q. [DOI] [PubMed] [Google Scholar]
- 27.Hunziker W., Geuze H.J. Intracellular trafficking of lysosomal membrane proteins. BioEssays. 1996;18:379–389. doi: 10.1002/bies.950180508. [DOI] [PubMed] [Google Scholar]
- 28.Bos K., Wraight C., Stanley K.K. TGN38 is maintained in the trans-Golgi network by a tyrosine-containing motif in the cytoplasmic domain. EMBO J. 1993;12:2219–2228. doi: 10.1002/j.1460-2075.1993.tb05870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aoki D., Lee N., Yamaguchi N., Dubois C., Fukuda M.N. Golgi retention of a trans-Golgi membrane protein, galactosyl transferase, requires cysteine and histidine residues within the membrane-anchoring domain. Proc. Natl Acad. Sci. USA. 1992;89:4319–4323. doi: 10.1073/pnas.89.10.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molloy S.S., Thomas L., Van Slyke J.K., Stenberg P.E., Thomas G. Intracellular trafficking and activation of the furin proprotein convertase: localization to the TGN and recycling from the cell surface. EMBO J. 1994;13:18–33. doi: 10.1002/j.1460-2075.1994.tb06231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schafer W., Stroh A., Berghofer S., Seiler J., Vey M., Kruse M.-L., Kern H.F., Klenk H.-D., Garten W. Two independent targeting signals in the cytoplasmic domain determine trans-Golgi network localization and endosomal trafficking of the proprotein convertase furin. EMBO J. 1995;14:2424–2435. doi: 10.1002/j.1460-2075.1995.tb07240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohrer J., Schweizer A., Johnson K.F., Kornfeld S. A determinant in the cytoplasmic tail of the cation-dependent mannose 6-phosphate receptor prevents trafficking to lysosomes. J. Cell Biol. 1995;130:1297–1306. doi: 10.1083/jcb.130.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valiquette M., Bonin H., Hnatowich M., Caron M.G., Lefkowitz R.J., Bouvier M. Involvement of tyrosine residues located in the carboxyl tail of the human β2-adrenergic receptor in agonist-mediated down-regulation of the receptor. Proc. Natl. Acad. Sci. USA. 1990;87:5089–5093. doi: 10.1073/pnas.87.13.5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dierick H.A., Ambrosini L., Spencer J., Glover T., Mercer J.F.B. Molecular structure of the Menkes disease gene (ATP7A) Genomics. 1995;28:462–469. doi: 10.1006/geno.1995.1175. [DOI] [PubMed] [Google Scholar]
- 35.Das S., Levinson B., Vulpe C., Whitney S., Gitschier J., Packman S. Similar splicing mutations of the Menkes/mottled copper transporting ATPase in occipital horn syndrome and the blotchy mouse. Am. J. Hum. Genet. 1995;56:570–576. [PMC free article] [PubMed] [Google Scholar]
- 36.Petrukhin K., Lutsenko S., Chernov I., Ross B. M., Kaplan J.H., Gilliam T.C. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum. Mol. Genet. 1994;3:1647–1656. doi: 10.1093/hmg/3.9.1647. [DOI] [PubMed] [Google Scholar]
- 37.Dijkstra M., In't Veld G., van den Berg G.J., Muller M., Kuipers F., Vonk R.J. Adenosine triphosphate-dependent copper transport in isolated rat liver plasma membranes. J. Clin. Invest. 1995;95:412–416. doi: 10.1172/JCI117670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stearman R., Yuan D.S., Yamaguchi-Iwai Y., Klausner R.D., Dancis A. A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science. 1996;271:1552–1557. doi: 10.1126/science.271.5255.1552. [DOI] [PubMed] [Google Scholar]
- 39.Cooper H. M, Paterson Y. In: Current Protocols in Molecular Biology. Ausubel F.M., Brent R., Kingston R.E., Moore D.D., Smith J.A., Struhl K., editors. J. Wiley & Sons, Unit 11.13; 1994. [Google Scholar]
- 40.Gruber A., Bianca Z. Alternative method to remove antibacterial antibodies from antisera used for screening of expression libraries. Biotechniques. 1995;19:28–30. [PubMed] [Google Scholar]