

Abstract
Abnormal dendritic spine structure and function is one of the most prominent features associated with neurodevelopmental disorders including Down syndrome (DS). Defects in both spine morphology and spine density may underlie alterations in neuronal and synaptic plasticity, ultimately affecting cognitive ability. Here we briefly examine the role of astrocytes in spine alterations and more specifically the involvement of astrocyte-secreted thrombospondin 1 (TSP-1) deficits in spine and synaptic pathology in DS.
Keywords: Down syndrome, Alzheimer disease, dendritic spines, astrocytes, intellectual disability, dementia, thrombospondin, interferon
Graphical abstract
Down syndrome (DS) is characterized by various neurological abnormalities including reduced brain mass, impaired neuronal differentiation, altered dendritic spine morphology and reduced spine and synaptic density [1–6]. These structural abnormalities are directly linked to intellectual disability (ID), one of the invariable and most prominent features affecting individuals with DS. ID includes limitations in cognitive function such as lacking the ability to learn adaptive behaviors or problem solving skills. ID can also lead to deficits in self-supporting, interpersonal, and communication skills. The combination of these cognitive deficits in a person with DS often results in significant impairment in quality of life as well as in a compromised ability to live an independent lifestyle. Improvements in medical care in the last 20 years have dramatically increased the life expectancy of people with DS, who in developed countries now reach well into six decades of life. This notable advance in medical care is however dampened by the tremendous susceptibility of individuals with DS to develop Alzheimer’s disease (AD). In fact, approximately half of the DS population over 60 years of age develop AD dementia and nearly all subjects with DS develop AD neuropathology, including amyloid beta (Aβ) plaques and neurofibrillary tangles [7,8].
Advances in the understanding of the human genome have paved the way for the development of therapeutic interventions by allowing researchers to investigate candidate genes and molecular pathways leading to DS phenotypes. For example, as it relates to neurodevelopmental delay and cognitive dysfunction, DYRK1A (Dual-specificity Tyrosine Phosphorylation-Regulated Kinase 1A), RCAN1 (Regulation of Calcineurin 1), and DSCAM (Down Syndrome Cell Adhesion Molecule) have been identified as HSA21 candidate genes that regulate neuronal development and maturation in the brain. Studies in mice and humans have also implicated DYRK1A, RCAN1, and ETS2 (Avian Erythroblastosis Virus E26 Oncogene Homolog 2) in exacerbating AD pathology in DS individuals [9], highlighting the involvement of multiple genes in the DS-AD phenotype.
Gene expression analysis has shown dysregulation of trisomic and disomic genes in different tissues associated with DS phenotypes, suggesting that disomic genes are also important in understanding the genotype-phenotype relationship in DS [10]. Moreover, differences in gene expression have been documented in individuals with the same genotype, and not all trisomic genes are dysregulated at a 1.5 fold ratio. Some DS phenotypes may be more sensitive to expression of specific genes on HSA21, while other phenotypes arise from changes in disomic genes. This highlights the complexity of genetic interactions in the clinical manifestations of DS and the challenges in identifying therapeutic targets directed at attenuating specific phenotypes of the disease [9].
A large body of literature supports the notion that abnormalities in dendritic spine development and structure are associated with ID. In fact, reduced dendritic spine density and altered spine morphology have been widely reported in individuals with different degrees of ID [11,12]. Dendritic spines are neuronal specializations responsible for receiving input from excitatory synapses. Consequently, defects in spine density and structure are likely to lead to synaptic and circuit alterations. To date, no therapeutic approach exists to prevent or restore spine structure and function. This is a promising area of active research, which may pave the way for the discovery of new therapies to ameliorate functional connectivity at the cellular level in DS and other neurodevelopmental and neurodegenerative conditions.
Dendritic spine pathology in Down syndrome
Dendritic spines were originally described by neuroscientist Santiago Ramón y Cajal in 1888, as small protrusions emerging from dendrites of neurons having a potentially critical role in the connective function of the central nervous system (CNS). He postulated that these protrusions could be neuronal contact sites, which could change by neuronal activity [13]. Currently, dendritic spines are accepted as dynamic structures, whose morphology and lifespan are influenced by long-term potentiation (LTP) [14–16], and environmental stimulation [17]. Most dendritic spines represent the postsynaptic component of many excitatory and some inhibitory synapses in the CNS. They are involved in chemical compartmentalization of calcium homeostasis, protein synthesis, neuronal connectivity, and synaptic function [18–20].
Structurally, dendritic spines consist of a head anchored to the dendritic shaft through a thinner stem or neck. They are classified into three main categories depending on their size and shape: stubby, mushroom, and thin [21]. Stubby spines have a short neck and wide head with no constriction between the head and the dendrite. Mushroom spines have a large head and narrow neck, and thin spines are short and thin protrusions of uniform width. An additional type of spine, known as “filopodium-like spines,” are “hair-like” structures hypothesized to be precursors of mature spines which are present during development and can also be found in pathological states [22,23].
Dendritic spines are dynamic structures that are constantly undergoing changes in size, shape, and overall density throughout life. Dendritic growth is initially driven by genetic factors, but can be modified by levels and patterns of neuronal activity [14–16,19,20,24,25]. In fact, independent studies have shown that rats exposed to environmental enrichment [26,27], or trained on spatial learning tasks [28,29], exhibit increased dendritic spine number and density, demonstrating the close relationship between spine density and neuronal activity [30].
Morphological abnormalities in the architecture of dendritic spines have been linked to ID in DS and other neurological disorders, including Rett syndrome [31], Fragile X syndrome [32,33], and Williams syndrome [34,35]. Initial anatomopathological studies using Golgi staining and morphological analysis of cortical neurons in the brain of subjects with ID showed a high number of unusually long, thin spines and reduced short, thick spine numbers. Additionally, the degree of dendritic spine loss appears to correlate with the severity of the ID [36]. Analysis of dendrites in pyramidal neurons located in the motor cortex of an individual with DS showed reduced number of spines and abnormally short and long spines, consistent with an immature developmental state [1,37]. Later studies have expanded these early observations showing decreased number of dendritic spines in the hippocampus of young adults with DS and further reduction of spine density in individuals with DS who developed AD [38,39]. The decrease in dendritic spines in the brain of individuals with DS-AD may be associated with the deterioration in cognitive function observed in AD [40,41]. Although a progressive loss of spines has been reported as part of the natural aging process, incremental structural changes due to DS and later to the onset of AD can result in substantial dysfunction in neuronal networks and concurrent cognitive decline.
Animal models of DS have also shown a close correlation between dendritic spine abnormalities and deficits in learning and memory. For example, the Ts65Dn mouse, a segmental trisomic model of DS, exhibits dendritic spine pathology and behavioral abnormalities reminiscent of DS. Structural alterations in the brain of Ts65Dn mice include enlarged spines, irregular spine heads, and globular spine shapes [42]. Ts65Dn mice also exhibit degeneration of cholinergic basal forebrain neurons, as observed in DS adults and people with AD. Further studies have shown that Ts65Dn mice exhibit deficits in hippocampal-dependent memory, which correlate with structural abnormalities in dendritic spines and dendritic arborizations. Another segmental trisomic model commonly used to investigate the genotype-to-phenotype relationship is the Ts1Cje. Like the Ts65Dn model, Ts1Cje mice display poor performance in hippocampal-dependent tasks and altered dendritic spine morphology and density [43–45]. Because dendritic spines are the primary sites for excitatory input, it is hypothesized that these alterations may be structural correlates of the cognitive dysfunction seen in DS and other neurological disorders including a disbalance in the ratio of excitatory/inhibitory inputs [1,37,40,41,46].
Astrocyte-secreted thrombospondin 1 as a “spinotrophic” factor
Astrocytes constitute the major glial cell population in the CNS. At the genetic, physiological and functional level, astrocytes can be as heterogeneous as neurons [47,48]. Based on their morphology, anatomical location and antigenic phenotype, astrocytes can be classified in at least two categories: protoplasmic astrocytes and fibrous astrocytes [49], each of which differs from the other in some of its main functions. Astrocytes have an important role in CNS homeostasis and brain development. They participate directly in formation of the blood brain barrier, regulation of synaptogenesis, and modulation of neurotransmitters and ions. They influence neuronal path finding, neuronal metabolism, and synaptic transmission, by releasing trophic molecules such as thrombospondins (TSPs), cholesterol, nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophins 3 and 4 (NT3, NT4), and tumor necrosis factor-α (TNF-α) [50]. Additionally, astrocytes can become reactive during pathological states and produce different responses depending on the type of injury [51]. While most research on the topic has focused on rodent-derived astrocytes, recently Zhang and colleagues, developed protocols for the isolation and characterization of astrocytes from human brains and showed that, like their mouse counterparts, human astrocytes participate in multiple functions including neuronal survival, synapse formation and pruning and modulation of neuronal activity [52].
The interaction between astrocytes and neurons is critical for the formation and preservation of dendritic spines [20,53], furthermore, astrocytes modulate synaptic transmission through the secretion of diverse molecules such as cholesterol [54], and thrombospondins [55,56]. TSPs were originally identified in conditioned medium as astrocyte-secreted factors involved in the formation of excitatory synapses [55]. Addition of purified TSPs to retinal ganglion cells (RGCs), recapitulated the effect of astrocyte-conditioned medium on synapse number, implying an active role for TSPs in synaptogenesis [56]. Research in our laboratory is aimed at understanding the molecular mechanisms underlying dendritic spine pathology in DS. We identified thrombospondin-1 (TSP-1), an astrocyte-secreted protein, as a critical factor that modulates dendritic spine development. Additional experiments showed that TSP-1 secretion and the levels of TSP-1 in the conditioned medium of DS astrocyte cultures are markedly reduced. However, TSP-1 mRNA levels were not changed, suggesting a post-translational origin for TSP-1 deficiency in DS. Using a co-culture system of astrocytes and primary neurons, we demonstrated that reduced TSP-1 levels produce alterations in dendritic spine structure and reduce spine and synapse numbers [57]. These defects can be prevented by addition of recombinant TSP-1, and are reproduced in normal astrocytes by blocking TSP-1, suggesting an important role for TSP-1 in the formation and modulation of dendritic spines. More recently, we have investigated spine and synaptic structure in TSP-1 KO mice. The results show abnormal spine and synaptic densities in the hippocampal formation associated with behavioral deficits [58]. In this context, TSP-1 deficiency may be associated with defects in the number and morphology of spines. In DS, TSP-1 deficiency may also be associated with the development of AD later in life. For example, reduced TSP1 levels have been reported in sporadic AD brains [59], which may be linked to spine and synaptic pathology, neuronal dysfunction and cognitive impairment. These findings provide a mechanistic rationale for the exploration of TSP-1-based therapies to treat spine and synaptic pathology in DS. We explored the potential link between mitochondrial dysfunction and oxidative stress in DS cells as a possible cause of TSP-1 secretory deficits, given that a similar reduction in secreted amyloid precursor protein (APPs) is observed in the conditioned medium of DS astrocytes and neurons [60]. However, treatment with various antioxidants and mitochondrial cofactors were not able to increase TSP-1 secretion in DS astrocytes [57].
Contribution of Interferon in Down syndrome pathology
Alternatively, TSP-1 deficits may be associated with Interferon-γ (IFN-γ) hypersensitivity in DS [61,62]. IFN-γ is part of the interferon family, best known for its modulatory effects on the adaptive and innate immune response [63]. It is primarily secreted by T-cells, but low levels have also been reported in astrocytes [64], endothelial cells [65], and activated microglia [66]. In individuals with DS, there is increased sensitivity to interferon (IFN) [67], which is likely related to the presence of several genes encoding interferon ligands and four genes that encode IFN receptors in HSA21. In fact, a recent study showed that fibroblasts from DS individuals exhibit increased expression of interferon-stimulated genes [62]. Dysregulation of IFN-γ has been documented in DS mouse models [68–70], and maternal anti-IFN treatment has proven effective at improving embryonic development in DS mice [70]. Although IFN-γ has been shown to have protective properties, its overexpression can also cause negative effects on the brain. For example, in a mouse model of AD, IFN-γ suppressed AD pathology by decreasing hyperphosphorylated tau levels and increasing neurogenesis, all while enhancing accumulation of Aβ in the hippocampus [71]. Other studies have shown that the effect of IFN-γ is concentration dependent. At low concentrations, there seems to be a protective effect by supporting neurogenesis, while at high concentrations this protective effect is counteracted by TNF-α activity [72]. The connection between IFN hypersensitivity and TSP-1 low levels in DS relates to the fact that IFN has a potent inhibitory effect on protein glycosylation with a much less pronounced effect on protein translation [73]. Specifically, treatment of human cells with IFN inhibits TSP-1 secretion significantly without changing its mRNA levels [74], which is similar to our findings in DS astrocytes [57]. Together, these results suggest that the deficits in TSP-1 observed in DS astrocytes may be directly related to the upregulation of IFN-γ signaling. Ongoing studies in our laboratory are directed to examine this possibility and to develop pharmacological approaches to normalize TSP-1 levels.
In summary, the research reviewed here highlights the role of astrocytes in dendritic spine pathology in DS. Defects in spine structure and number may lead to synaptic and circuit alterations. To date, no effective treatment exists to prevent or restore dendritic spine structure and function. The exploration of astrocyte-secreted TSP-1 as a therapeutic target may prove useful to ameliorate spine pathology and cognitive impairment in DS and other neurological conditions associated with spine and synaptic loss.
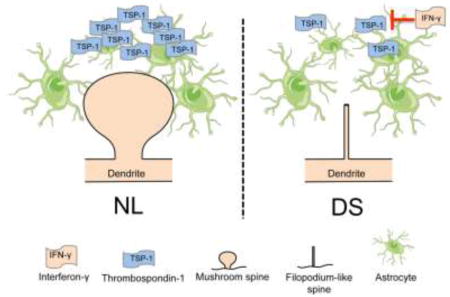
Figure 1. Modulation of dendritic spine morphology by TSP-1.

Under normal conditions (NL), TSP-1 supports the development of dendritic spine morphology and number (A). In DS, increased sensitivity to IFN-γ inhibits TSP-1 glycosylation and secretion resulting in increased number of thin and filopodium-like spines, reduced number of stubby and mushroom spines and an overall decrease in spine number (B). We propose that this maladaptive plasticity mechanism ultimately leads to changes in synaptic activity, neuronal network dysfunction and cognitive alterations.
Acknowledgments
We thank the T-32 “Training in the Neurobiology of Aging” Grant (AG000096) for providing financial support for some of the research discussed in this review. Part of this work was also supported by grants from National Institutes of Health (HD38466), and Alzheimer’s Disease Research Center grant AG16573), and. PAPIIT IN 304817 (O.G.).
ABBREVIATIONS
- Aβ
Beta-amyloid
- AD
Alzheimer’s disease
- APPs
Secreted amyloid precursor protein
- BDNF
Brain-derived neurotrophic factor
- CNS
Central Nervous System
- DS
Down syndrome
- DSCAM
Down syndrome Cell Adhesion Molecule
- DYRK1A
Dual-specificity Tyrosine Phosphorylation-Regulated Kinase 1A
- ETS2
Avian Erythroblastosis Virus E26 Oncogene Homolog 2
- HSA 21
Human Chromosome 21
- ID
Intellectual disability
- IFN
Interferon
- IFN-γ
Interferon- γ
- LTP
Long-term potentiation
- NGF
Nerve growth factor
- NT3
Neurotrophin-3
- NT4
Neurotrophin-4
- RCAN1
Regulation of Calcineurin 1
- TNFα
Tumor necrosis factor-α
- TSP-1
Thrombospondin-1
- TSPs
Thrombospondins
Footnotes
CONFLICT OF INTERESTS STATEMENT
The authors declare that they do not have financial or non-financial competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Marin-Padilla M. Short Communications Structural abnormalities of the cerebral cortex in human chromosomal aberrations: a Golgi study. Brain Res. 1972;44:625–629. doi: 10.1016/0006-8993(72)90324-1. [DOI] [PubMed] [Google Scholar]
- 2.Coyle JT, Oster-Granite ML, Gearhart JD. The neurobiologic consequences of Down syndrome. Brain Res Bull. 1986;16:773–787. doi: 10.1016/0361-9230(86)90074-2. [DOI] [PubMed] [Google Scholar]
- 3.Golden Ja, Hyman BT. Development of the superior temporal neocortex is anomalous in trisomy 21. J Neuropathol Exp Neurol. 1994;53:513–520. doi: 10.1097/00005072-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Nadel L. Down’s syndrome: a genetic disorder in biobehavioral perspective. Genes Brain Behav. 2003;2:156–166. doi: 10.1034/j.1601-183X.2003.00026.x. [DOI] [PubMed] [Google Scholar]
- 5.Benavides-Piccione R, Ballesteros-Yáñez I, Martínez De Lagrán M, Elston G, Estivill X, Fillat C, Defelipe J, Dierssen M. On dendrites in Down syndrome and DS murine models: A spiny way to learn. Prog Neurobiol. 2004;74:111–126. doi: 10.1016/j.pneurobio.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 6.Chakrabarti L, Scafidi J, Gallo V, Haydar TF. Environmental enrichment rescues postnatal neurogenesis defect in the male and female Ts65Dn mouse model of down syndrome. Dev Neurosci. 2011;33:428–441. doi: 10.1159/000329423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roizen NJ, Patterson D. Down’s syndrome. Lancet. 2003;361:1281–1289. doi: 10.1016/s0140-6736(03)12987-x. [DOI] [PubMed] [Google Scholar]
- 8.Ballard C, Mobley W, Hardy J, Williams G, Corbett A. Dementia in Down’s syndrome. Lancet Neurol. 2016;15:622–636. doi: 10.1016/S1474-4422(16)00063-6. [DOI] [PubMed] [Google Scholar]
- 9.Deitz SL, Blazek JD, Solzak JP, Roper RJ. Down Syndrome: A Complex and Interactive Genetic Disorder. In: Dey S, editor. Genet Etiol Down Syndr. InTech; 2011. pp. 65–96. [Google Scholar]
- 10.Antonarakis SE. Down syndrome and the complexity of genome dosage imbalance. Nat Rev Genet. 2016;18:147–163. doi: 10.1038/nrg.2016.154. [DOI] [PubMed] [Google Scholar]
- 11.Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1207/S15326942DN1603_18. [DOI] [PubMed] [Google Scholar]
- 12.Chechlacz M, Gleeson JG. Is mental retardation a defect of synapse structure and function? Pediatr Neurol. 2003;29:11–17. doi: 10.1016/S0887-8994(03)00152-8. [DOI] [PubMed] [Google Scholar]
- 13.García-López P, García-Marín V, Freire M. The discovery of dendritic spines by Cajal in 1888 and its relevance in the present neuroscience. Prog Neurobiol. 2007;83:110–130. doi: 10.1016/j.pneurobio.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Matsuzaki M, Honkura N, Ellis-Davies GCR, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1007/s11103-011-9767-z.Plastid. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44:759–767. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. TL - 44. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 17.Nithianantharajah J, Hannan AJ. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat Rev Neurosci. 2006;7:697–709. doi: 10.1038/nrn1970. [DOI] [PubMed] [Google Scholar]
- 18.DeFelipe J. Brain plasticity and mental processes: Cajal again. Nat Rev Neurosci. 2006;7:811–817. doi: 10.1038/nrn2005. [DOI] [PubMed] [Google Scholar]
- 19.Alvarez VA, Sabatini BL. Anatomical and Physiological Plasticity of Dendritic Spines. Annu Rev Neruoscience. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- 20.Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peters A, Kaiserman-Abramof IR. The small pyramidal neuron of the rat cerebral cortex. The perikaryon, dendrites and spines. Am J Anat. 1970;127:321–355. doi: 10.1002/aja.1001270402. [DOI] [PubMed] [Google Scholar]
- 22.Ziv NE, Smith SJ. Evidence for a role of dendritic filopodia in synaptogenesis and spine formation. Neuron. 1996;17:91–102. doi: 10.1016/S0896-6273(00)80283-4. [DOI] [PubMed] [Google Scholar]
- 23.Heike H, Morgan S. Dendritic Spines: Structure, Dynamics and Regulation. Nat Rev Neurosci. 2001;2:880–888. doi: 10.1038/35104061. [DOI] [PubMed] [Google Scholar]
- 24.Bartlett WP, Banker GA. An electron microscopic study of the development of axons and dendrites by hippocampal neurons in culture. I. Cells which develop without intercellular contacts. J Neurosci. 1984;4:1944–1953. doi: 10.1523/JNEUROSCI.04-08-01944.1984. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=6470762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buchs PA, Muller D. Induction of long-term potentiation is associated with major ultrastructural changes of activated synapses. Proc Natl Acad Sci U S A. 1996;93:8040–5. doi: 10.1073/pnas.93.15.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diamond MC, Lindner B, Johnson R, Bennett EL, Rosenzweig MR. Difference in occipital cortical synapses from environmentally enriched, impoverished, and standard colony rats. J Neurosci Res. 1975;1:109–119. doi: 10.1002/jnr.490010203. [DOI] [PubMed] [Google Scholar]
- 27.Leggio MG, Mandolesi L, Federico F, Spirito F, Ricci B, Gelfo F, Petrosini L. Environmental enrichment promotes improved spatial abilities and enhanced dendritic growth in the rat. Behav Brain Res. 2005;163:78–90. doi: 10.1016/j.bbr.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 28.Moser TAM-B, Moser APEI. Multiple substrates for spatial learning in the rat hippocampus. Soc Neurosci Abstr. 1997;23:621. [Google Scholar]
- 29.Eyre MD, Richter-Levin G, Avital A, Stewart MG. Morphological changes in hippocampal dentate gyrus synapses following spatial learning in rats are transient. Eur J Neurosci. 2003;17:1973–1980. doi: 10.1046/j.1460-9568.2003.02624.x. [DOI] [PubMed] [Google Scholar]
- 30.Harms KJ, Dunaevsky A. Dendritic spine plasticity: Looking beyond development. Brain Res. 2007;1184:65–71. doi: 10.1016/j.brainres.2006.02.094. [DOI] [PubMed] [Google Scholar]
- 31.Chapleau CA, Calfa GD, Lane MC, Albertson AJ, Larimore JL, Kudo S, Armstrong DL, Percy AK, Pozzo-Miller L. Dendritic Spine Pathologies in Hippocampal Pyramidal Neurons from Rett Syndrome Brain and after Expression of Rett-Associated MECP2 Mutations. Neurobiol Dis. 2009;35:219–233. doi: 10.1016/j.nbd.2009.05.001.DENDRITIC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comery Ta, Harris JB, Willems PJ, Oostra Ba, Irwin Sa, Weiler IJ, Greenough WT. Abnormal dendritic spines in fragile X knockout mice: Maturation and pruning deficits. Proc Natl Acad Sci U S A. 1997;94:5401–5404. doi: 10.1073/pnas.94.10.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruz-Martin A, Crespo M, Portera-Cailliau C. Delayed stabalization of dendritic spines in fragile x mice. J Neurosci. 2010;30:7793–7803. doi: 10.1523/JNEUROSCI.0577-10.2010.Delayed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galaburda AM, Wang PP, Bellugi U, Rossen M. Cytoarchitectonic anomalies in a genetically based disorder: Williams syndrome. Neuroreport. 1994;5:753–757. doi: 10.1097/00001756-199403000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/S0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 36.Purpura DP. Dendritic spine “dysgenesis” and mental retardation. Science (80-) 1974;186:1126–1128. doi: 10.1126/science.186.4169.1126. [DOI] [PubMed] [Google Scholar]
- 37.Marin-Padilla M. Pyramidal cell abnormalities in the motor cortex of a child with Down’s syndrome. A Golgi study. J Comp Neurol. 1976;167:63–81. doi: 10.1002/cne.901670105. [DOI] [PubMed] [Google Scholar]
- 38.Suetsugu M, Mehraein P. Spine distribution along the apical dendrites of the pyramidal neurons in Down’s syndrome - A quantitative golgi study. Acta Neuropathol. 1980;50:207–210. doi: 10.1007/BF00688755. [DOI] [PubMed] [Google Scholar]
- 39.Ferrer I, Gullotta F. Down’s syndrome and Alzheimer’s disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 1990;79:680–685. doi: 10.1007/BF00294247. [DOI] [PubMed] [Google Scholar]
- 40.Devenny DA, Silverman WP, Hill AL, Jenkins E, Sersen EA, Wisniewski KE. Normal ageing in adults with Down’s syndrome: A longitudinal study. J Intellect Disabil Res. 1996;40:208–221. doi: 10.1111/j.1365-2788.1996.tb00624.x. [DOI] [PubMed] [Google Scholar]
- 41.Lott IT, Head E. Alzheimer disease and Down syndrome: Factors in pathogenesis. Neurobiol Aging. 2005;26:383–389. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC. Synaptic structural abnormalities in the Ts65Dn mouse model of Down syndrome. J Comp Neurol. 2004;480:281–298. doi: 10.1002/cne.20337. [DOI] [PubMed] [Google Scholar]
- 43.Sago H, Carlson EJ, Smith DJ, Kilbridge J, Rubin EM, Mobley WC, Epstein CJ, Huang TT. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Genetics. 1998;95:6256–6261. doi: 10.1073/pnas.95.11.6256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belichenko PV, Kleschevnikov AM, Salehi A, Epstein CJ, Mobley WC. Synaptic and Cognitive Abnormalities inMouse Models of Down Syndrome:Exploring Genotype-PhenotypeRelationships. J Comp Neurol. 2007;504:329–345. doi: 10.1002/cne.21433. [DOI] [PubMed] [Google Scholar]
- 45.Gupta M, Dhanasekaran AR, Gardiner KJ. Mouse models of Down syndrome: gene content and consequences. Mamm Genome. 2016;27:538–555. doi: 10.1007/s00335-016-9661-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catuara-Solarz S, Espinosa-Carrasco J, Erb I, Langohr K, Gonzalez JR, Notredame C, Dierssen M. Combined Treatment With Environmental Enrichment and (-)-Epigallocatechin-3-Gallate Ameliorates Learning Deficits and Hippocampal Alterations in a Mouse Model of Down Syndrome. eNeuro. 2016;3 doi: 10.1523/ENEURO.0103-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hansson E. Regional heterogeneity among astrocytes in the central nervous system. Neurochem Int. 1990;16:237–245. doi: 10.1016/0197-0186(90)90097-D. [DOI] [PubMed] [Google Scholar]
- 48.Matyash V, Kettenmann H. Heterogeneity in astrocyte morphology and physiology. Brain Res Rev. 2010;63:2–10. doi: 10.1016/j.brainresrev.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Kettenmann H, Verkhratsky A. Neuroglia: the 150 years after. Trends Neurosci. 2008;31:653–659. doi: 10.1016/j.tins.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Verkhratsky A, Butt AM. Glial neurobiology. 2007. [Google Scholar]
- 51.Liddelow SA, Barres BA. Reactive astrocytes: Production, function, and therapeutic potential. Immunity. 2017;46:957–967. doi: 10.1016/j.immuni.2017.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, JAD, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MGH, Barres BA. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. 2016;89:37–53. doi: 10.1016/j.neuron.2015.11.013.Purification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haber M, Zhou L, Murai KK. Cooperative Astrocyte and Dendritic Spine Dynamics at Hippocampal Excitatory Synapses. J Neurosci. 2006;26:8881–8891. doi: 10.1523/JNEUROSCI.1302-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mauch DH. CNS Synaptogenesis Promoted by Glia-Derived Cholesterol. Science (80-) 2001;294:1354–1357. doi: 10.1126/science.294.5545.1354. [DOI] [PubMed] [Google Scholar]
- 55.Christopherson KS, Ullian EM, Stokes CCA, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 56.Eroglu Ç, Allen NJ, Susman MW, Rourke NAO, Young C, Özkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Christopher K, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres A. The Gabapentin Receptor α2δ-1 is the Neuronal Thrombospondin Receptor Responsible for Excitatory CNS Synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025.The. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garcia O, Torres M, Helguera P, Coskun P, Busciglio J. A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in down’s syndrome. PLoS One. 2010;5 doi: 10.1371/journal.pone.0014200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Torres MD, Bohannan RC, Tang C, Li A, Duong T, Zamponi E, Helguera P, Busciglio J. A role for Thrombospondin-1 in learning and memory and neuroplasticity. 2nd T21 Res. Soc. Int. Conf; Chicago, Illinois, USA. 2017. [Google Scholar]
- 59.Buée L, Hof PR, Roberts DD, Delacourte A, Morrison JH, Fillit HM. Immunohistochemical identification of thrombospondin in normal human brain and in Alzheimer’s disease. Am J Pathol. 1992;141:783–8. http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1886629&tool=pmcentrez&rendertype=abstract. [PMC free article] [PubMed] [Google Scholar]
- 60.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, et al. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–88. doi: 10.1016/S0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 61.Leonard M, Maroun ELE. Interferon Action and Chromosome 21 Trisomy (Down Syndrome): 15 Years Later. J Theor Biol. 1996;181:41–46. doi: 10.1006/jtbi.1996.0113. [DOI] [PubMed] [Google Scholar]
- 62.Sullivan KD, Lewis HC, Hill AA, Pandey A, Jackson LP, Cabral JM, Smith KP, Liggett LA, Gomez EB, Galbraith MD, Degregori J, Espinosa JM. Trisomy 21 consistently activates the interferon response. Elife. 2016;5 doi: 10.7554/eLife.16220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Weerd NA, Nguyen T. The interferons and their receptors;distribution and regulation. Immunol Cell Biol. 2012;90:483–491. doi: 10.1038/icb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Simone R, Levi G, Aloisi F. Interferon γ gene expression in rat central nervous system glial cells. Cytokine. 1998;10:418–22. doi: 10.1006/cyto.1997.0314. [DOI] [PubMed] [Google Scholar]
- 65.Wei YP, Kita M, Shinmura K, Yan XQ, Fukuyama R, Fushiki S, Imanishi J. Expression of IFN-γ in cerebrovascular endothelial cells from aged mice. J Interferon Cytokine Res. 2000;20:403–409. doi: 10.1089/107999000312342. [DOI] [PubMed] [Google Scholar]
- 66.Li HL, Kostulas N, Huang YM, Xiao BG, van der Meide P, Kostulas V, Giedraitas V, Link H. IL-17 and IFN-γ mRNA expression is increased in the brain and systemically after permanent middle cerebral artery occlusion in the rat. J Neuroimmunol. 2001;116:5–14. doi: 10.1016/S0165-5728(01)00264-8. [DOI] [PubMed] [Google Scholar]
- 67.Tan YH, Schneider EL, Tischfield J, Epstein CJ, Ruddle FH. Human chromosome 21 dosage: effect on the expression of the interferon induced antiviral state. Science. 1974;186:61–63. doi: 10.1126/science.186.4158.61. [DOI] [PubMed] [Google Scholar]
- 68.Hallam DM, Capps NL, Travelstead AL, Brewer GJ, Maroun LE. Evidence for an interferon-related inflammatory reaction in the trisomy 16 mouse brain leading to caspase-1-mediated neuronal apoptosis. J Neuroimmunol. 2000;110:66–75. doi: 10.1016/S0165-5728(00)00289-7. [DOI] [PubMed] [Google Scholar]
- 69.Ling KH, Hewitt CA, Tan KL, Cheah PS, Vidyadaran S, Lai MI, Lee HC, Simpson K, Hyde L, Pritchard MA, Smyth GK, Thomas T, Scott HS. Functional transcriptome analysis of the postnatal brain of the Ts1Cje mouse model for Down syndrome reveals global disruption of interferon-related molecular networks. BMC Genomics. 2014;15:624. doi: 10.1186/1471-2164-15-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maroun LE. Anti-interferon immunoglobulins can improve the trisomy 16 mouse phenotype. Teratology. 1995;51:329–335. doi: 10.1002/tera.1420510509. [DOI] [PubMed] [Google Scholar]
- 71.Mastrangelo MA, Sudol KL, Narrow WC, Bowers WJ. Interferon-γ differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am J Pathol. 2009;175:2076–2088. doi: 10.2353/ajpath.2009.090059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, Martino G, Schwartz M. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2005;31:149–160. doi: 10.1016/j.mcn.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 73.Maheshwari RK, Banerjee DK, Waechter CJ, Olden K, Friedman RM. Interferon treatment inhibits glycosylation of a viral protein. Nature. 1980;287:454–456. doi: 10.1038/287454a0. [DOI] [PubMed] [Google Scholar]
- 74.Nickoloff BJ, Riser BL, Mitra RS, Dixit VM, Varani J. Inhibitory effect of Gamma Interferon on Cultured Human Keratinocyte Thrombospondin Production, Distribution, and Biologic Activities. J Invest Dermatol. 1988;91:213–218. doi: 10.1111/1523-1747.ep12465005. [DOI] [PubMed] [Google Scholar]