

Abstract
The antiandrogen therapeutics apalutamide and darolutamide entered the clinic in 2018 and 2019, respectively, for the treatment of castration-resistant prostate cancer (CRPC). Increased expression of the enzyme aldo-keto reductase 1C3 (AKR1C3) is phenotypic of CRPC. The enzyme acts to circumvent castration by producing potent androgens that drive proliferation. Furthermore, AKR1C3 mediates chemotherapeutic resistance to the standard of care, enzalutamide, a structural analogue of apalutamide. Resistance develops in almost all CRPC patients within three months of beginning treatment. Herein, we report that both apalutamide and the structurally distinct darolutamide induce AKR1C3 expression in in vitro models of prostate cancer and are susceptible to AKR1C3-mediated resistance. This effect is countered by pretreatment with a potent and highly selective AKR1C3 inhibitor, sensitizing high AKR1C3 expressing prostate cancer cell lines to the action of both chemotherapeutics with a concomitant reduction in expression of AKR1C3 and the biomarker prostate-specific antigen.
Graphical Abstract

The standard treatment for advanced prostate cancer (PCa), androgen deprivation therapy, results in reduced production of androgens,1 leading to initial improvement with concomitant suppression of prostate-specific antigen (PSA).2 However, metastatic PCa almost always progresses to a fatal and aggressive form termed castration-resistant prostate cancer (CRPC) within three years of diagnosis.3 The exact biochemical mechanism of CRPC is poorly understood, but the androgen receptor (AR) is known to remain activated, despite low levels of circulating androgens.4 The standard of care for CRPC combines the CYP17A inhibitor abiraterone acetate (AA)5 with an AR antagonist, which until recently was limited to enzalutamide (ENZ; Figure 1). Invariably, resistance develops to ENZ within three months. To provide alternatives, the AR antagonists apalutamide (ARN; Figure 1) and darolutamide (ODM; Figure 1) were introduced to the clinic in 2018 and 2019, respectively.6
Figure 1.
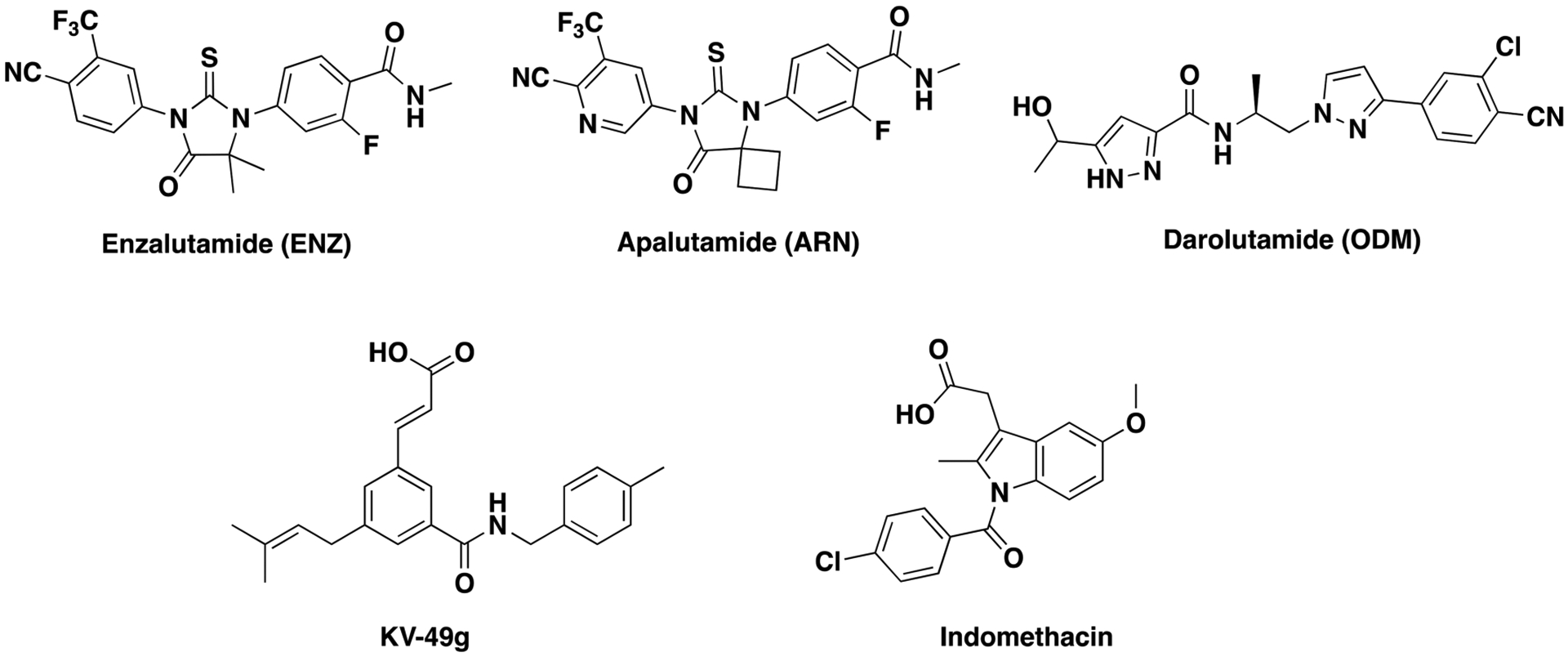
Structures of enzalutamide, apalutamide, and darolutamide and the AKR1C3 inhibitors KV-49g and indomethacin.
Several mechanisms have been proposed to explain ENZ resistance largely focusing on alterations of the AR gene through AR mRNA splice variants such as AR-V77 and point mutations in the AR ligand-binding domain, which have been associated with resistance to ARN, ENZ, or AA.8 The intratumoral androgen biosynthetic pathway has also been associated with resistance,9 as intracellular androgen biosynthesis is regulated by enzymes that are elevated in CRPC.10 One such enzyme is aldo-keto reductase 1C3 (AKR1C3).11 The enzyme produces dihydrotestosterone, the predominate intracellular transcriptional signal to androgen-responsive genes in intact human prostate cells.10 Overexpression of AKR1C3 has been shown to enhance human PCa resistance to a variety of chemotherapeutics.12,13 Hence, the delivery of a pharmacological AKR1C3 inhibitor represents a promising therapeutic strategy to counter CRPC drug resistance.14 We have previously disclosed AKR1C3 inhibitors15 that counter AKR1C3-mediated resistance to ENZ.12 Recently, the most potent and selective AKR1C3 inhibitor known, KV-49g (IC50 = 70 nM, >2800-fold selectivity over the highly homologous isoforms AKR1C1 and C2; Figure 1), was reported from our lab.16 This inhibitor demonstrated activity as a chemotherapeutic potentiator across a range of therapeutics in hematological malignancies that overexpress AKR1C3.17,18
Herein, we report that elevated AKR1C3 expression in PCa cell lines confers resistance to the recently approved AR antagonists, ARN and ODM. Gratifyingly, 24 h pretreatment of high AKR1C3 expressing PCa cells with our highly potent and selective AKR1C3 inhibitor, KV-49g, abolishes ARN and ODM resistance, leading to increased rates of PCa cell death and a significant decrease in PSA expression, a biomarker directly correlated to the severity of the disease. Additionally, KV-49g provides a greater potentiation effect than indomethacin (INDO; Figure 1), a compound that possesses relatively low AKR1C3 activity (IC50 = 2.3 μM, 22-fold selectivity over other isoforms)19 but is currently in a clinical trial to evaluate its effect in overcoming ENZ resistance in patients with recurrent or metastatic CRPC.20
Resistance to ARN was noted shortly after its approval and associated with AR gene mutation.8 However, the overall frequency of detected mutations in resistant subjects suggests this is not the most common contributor to resistance development.21 To the best of our knowledge, no resistance to ODM has been reported to date. To determine the baseline effect of ARN and ODM, we employed three PCa cell lines possessing varied AKR1C3 expression levels, LNCaP (AKR1C3 null), 22Rv1 (AKR1C3 expressing),12 and LNCaP cells overexpressing AKR1C3 (LNCaP1C3), generated by stable transfection with AKR1C3 plasmid as previously described.22 Each cell line was cultured either in RPMI media or in androgen-deprived conditions using charcoal-stripped serum (CSS). We have previously reported that cells cultured in CSS media overexpress AKR1C3 as a mechanism to counter the androgen-deprived conditions of CSS.12 The IC50 values of ARN and ODM in the three PCa cell lines were directly compared (Supplementary Table S1). Increasing IC50 values of ARN were observed that directly correlated to the increased expression of AKR1C3: 11 ± 3 μM in LNCaP cells, 77 ± 17 μM in 22Rv1 cells, and 101 ± 10 μM in LNCaP1C3 cells. Moreover, when AKR1C3 expression was induced further using CSS media, the IC50 value of ARN further increased to 42 ± 15 μM, 203 ± 30 μM, and 134 ± 25 μM, respectively. A similar correlation to AKR1C3 expression was observed for ODM treatment: IC50 values of 16 ± 3 μM in LNCaP cells, 46 ± 10 μM in 22Rv1, and 100 ± 20 μM in LNCaP1C3 cells. Upon culture in CSS media, increased IC50 values were noted: 31 ± 10 μM, 74 ± 16 μM, and 175 ± 35 μM in LNCaP, 22Rv1, and LNCaP1C3 cells, respectively. These data strongly correlate the expression of AKR1C3 to the development of resistance to both ARN and ODM.
To evaluate the ability of our AKR1C3 inhibitor KV-49g to resensitize these cell lines to the action of ARN and ODM, we first screened for inherent antineoplastic effect. Consistent with our previous findings,16 KV-49g did not induce cytotoxicity up to 100 μM in any of the cell lines (Supplementary Figures S1–3). We previously demonstrated that 24 h pretreatment with AKR1C3 inhibitor in PCa cell lines, followed by chemotherapy, resulted in significant synergistic effects,12,16 and this same strategy was applied herein. In LNCaP cells, no significant difference was observed between pretreatment with AKR1C3 inhibitor and either ARN or ODM alone due to the low expression of AKR1C3 (Figure 2A,C). In LNCaP1C3 cells with extremely high expression of AKR1C3, a significant potentiation effect was observed upon 24 h pretreatment with AKR1C3 inhibitor compared to ARN or ODM alone (Figure 2B,D). Concentrations of KV-49g as low as 0.1 μM potentiated 1 μM ARN, resulting in a 35% decrease in cell viability. A similar synergistic effect was seen with 1 μM KV-49g and 10 μM ODM. A dose–response relationship was observed. These data clearly support inhibiting AKR1C3 as a therapeutic strategy to counter resistance to both ARN and ODM.
Figure 2.

Combination effects of AKR1C3 inhibitor (KV-49g) and ARN or ODM in LNCaP and LNCaP1C3 cell lines. KV-49g and ARN in (A) LNCaP and (B) LNCaP1C3 cells. KV-49g and ODM in (C) LNCaP and (D) LNCaP1C3 cells. One-way ANOVA; 95% confidence interval; *p < 0.05; **p < 0.001; ***p < 0.0002; ****p < 0.0001; ns = no significance.
To further evaluate the synergistic effect, we screened the same combination treatment in 22Rv1 cells grown in CSS. This results in AKR1C3 expression levels similar to clinical samples. A remarkable synergistic effect was observed with both ARN or ODM (Figure 3A). Concentrations of KV-49g as low as 0.1 μM were capable of potentiating ARN effect in 22Rv1 cells. Pretreatment with 1 μM KV-49g and just 10 μM ARN resulted in >60% loss of cell viability. The IC50 of ARN in this cell line is 203 μM. Immunoblotting revealed that the treatment of 22Rv1 cells with 50 μM ARN alone resulted in the induction of AKR1C3 by 2-fold, further correlating the expression of the enzyme to resistance. Almost no change in PSA expression was observed (Figure 3B). However, 24 h pretreatment with just 1 μM KV-49g resulted in reduced AKR1C3 expression, with a remarkable 4-fold decrease in PSA expression.
Figure 3.

Combination of 24 h pretreatment of KV-49g followed by 72 h treatment with ARN or ODM in 22Rv1 cells (CSS): (A, C) cell viability; (B, D) immunoblots showing AKR1C3 and PSA expression. One-way ANOVA; 95% confidence interval; **p < 0.001; ***p < 0.0002; ****p < 0.0001; ns = no significance.
Likewise, the same combination of KV-49g and ODM provided a significant potentiation effect: 0.1 μM KV-49g was sufficient to sensitive 22Rv1 cells to a 10 μM concentration of ODM, with 1 μM KV-49g sufficient to sensitize the cell line to just 1 μM ODM (Figure 3C). Immunoblotting revealed a 3-fold increase in AKR1C3 expression upon exposure to 50 μM ODM, which was accompanied by overexpression of PSA (Figure 3D). Pretreatment of the cell line with KV-49g for 24 h at concentrations as low as 0.1 μM induced an approximate 3-fold reduction in AKR1C3 expression, which was accompanied by a marked decrease in PSA expression. Collectively, this data demonstrated that KV-49g, through inhibition of AKR1C3 activity, resensitizes 22Rv1 PCa cells to both ARN and ODM treatment, induces a marked loss of cell viability, and induces reduction of PSA expression.
The cyclooxygenase (COX) inhibitor INDO elicits AKR1C3 inhibition. However, it has lower potency and selectivity (AKR1C3 IC50 = 2.3 μM, 22-fold selectivity toward the C3 isoform vs KV-49g, AKR1C3 IC50 = 70 nM, >2800-fold selectivity toward the C3 isoform).16 To directly compare the effect of both compounds to resensitize 22Rv1 cells (CSS) to the effects of ARN or ODM, the effect of INDO alone was determined. No significant toxicity was observed until 100 μM (Supplementary Figure S4). In subsequent combination experiments, the clinically determined maximum blood concentration of ARN or ODM (12 μM) was employed.23,24 Gratifyingly, 24 h pretreatment with KV-49g at just 0.1 μM showed significant effect to sensitize 22Rv1 cells (CSS) to the action of both ARN and ODM (Figure 4A,C). A much lower potentiation effect was observed upon 24 h pretreatment with INDO followed by ARN or ODM; 1 μM KV-49g was sufficient to induce 50% cell death in combination with 12 μM ARN, while 1 μM INDO produced no statistically significant effect. Indeed, 10 μM INDO resulted in just 40% cell death in combination with 12 μM ARN. A similar effect was observed on AKR1C3 and PSA expression (Figure 4B,D). Pretreatment with 1 μM KV-49g and exposure to ARN resulted in the same reduction of PSA expression (50%) as 10 μM INDO. Combination of 24 h pretreatment with KV-49g followed by exposure to ODM resulted in an even more significant difference. A concentration of 1 μM KV-49g was more effective than 10 μM INDO, decreasing PSA expression by 50% and 30%, respectively. This data shows that the more potent and selective AKR1C3 inhibitor, KV-49g, is more effective than the weaker, less selective INDO in resensitizing 22Rv1 cells (CSS) to the cytotoxic action of both ARN and ODM.
Figure 4.

Combination of 24 h pretreatment with KV-49g or INDO prior to 72 h treatment with 12 μM ARN or ODM in 22Rv1 cells (CSS): (A, C) cell viability; (B, D) immunoblots showing AKR1C3 and PSA expression. One-way ANOVA; 95% confidence interval; *p < 0.05; **p < 0.001; ***p < 0.0002; ****p < 0.0001; ns = no significance.
Quantification of the degree of synergism in 22Rv1 cell lines (CSS) was performed using the Chou–Talalay method (Supplementary Table S2).25 Combination of KV-49g with either ARN or ODM resulted in high synergism and 126-fold or 75-fold dose-reduction indices (DRI), respectively. Combination treatment with INDO resulted in only 8-fold and 21-fold DRI with ARN and ODM, respectively. Notably, combination with KV-49g delivered IC50 values for both ARN and ODM at or below the maximum concentration levels attainable in patients (12 μM).
In conclusion, we report for the first time, to the best of our knowledge, that AKR1C3 overexpression is a critical regulator of resistance to the newly approved AR antagonists ARN and ODM in CRPC. Inhibition of AKR1C3 via a potent and highly selective compound, KV-49g, resensitized PCa cells to the chemotherapeutic effect of both clinical agents, completely countering resistance. Pharmacological inhibition of AKR1C3 is thus a promising target to counter drug-resistance in prostate cancer treatment, and KV-49g represents an advanced lead compound for preclinical evaluation.
METHODS
Synthesis.
The ARK1C3 inhibitor, KV-49g, was synthesized as previously described.16 Structural characterization matched that of the reported compound and was determined to be of >97% purity by HPLC (performed on a Agilent 1260 Infinity HPLC system with ELSD detector using Agilent ChemStation software with an Eclipse plus (C8, 3.5 μM, 150 mm × 4.6 mm) column, with gradient elution using 5–95% acetonitrile/water).
1H NMR (400 MHz; CD3OD): δ 1.75 (3H, s, CH3), 1.76 (3H, s, CH3), 2.31 (3H, s, CH3), 4.53 (2H, d, J = 5.4 Hz, CH2), 6.51 (1H, d, J = 15.9 Hz, CH), 7.15 (2H, d, J = 7.8 Hz, ArCH), 7.23 (2H, d, J = 8.0 Hz, ArCH), 7.53 (1H, s, ArCH), 7.70 (1H, d, J = 16.0 Hz, CH), 7.88 (1H, s, ArCH), 7.93 (1H, s, ArCH).
13C NMR (100 MHz; CD3OD): δ 16.5, 19.7, 24.5, 33.5, 42.9, 110.75, 122.1, 123.8, 127.1, 128.7, 128.8, 130.6, 133.0, 134.8, 135.6, 136.5, 141.9, 142.6, 143.1, 166.0, 166.9.
Cell Culture and Reagents.
The 22Rv1 and LNCaP cells were purchased from the American Type Culture Collection (ATCC in 2016) and cultured in RPMI 1640 supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg mL−1 streptomycin. LNCaP1C3 cells overexpressing AKR1C3 were generated by stable transfection with AKR1C3 plasmid as previously described.22 All cell lines were authenticated via short tandem repeat analysis and tested for mycoplasma using the MycoAlert mycoplasma detection kit as per the manufacturer’s instructions (Texas cancer cell repository) in May 2017, showing no contamination. Cell line use was limited to passage nine or lower.26 Where indicated, cells were cultured in charcoal-stripped (CSS) media prepared by supplementing RPMI 1640 without phenol red with charcoal-stripped FBS. All cells were maintained at 37 °C in a humidified incubator with 5% carbon dioxide. Apalutamide (ARN; catalog no. 75837–196) was purchased from VWR. Darolutamide (ODM; catalog no. 50-187-3532) and indomethacin (INDO; catalog no. AAA1991006) were purchased from Fisher Scientific. Stock solutions (200 μM) of ARN, ODM, KV-49g, and INDO were prepared in DMSO and were serially diluted for cell culture treatments to a final concentration range of 0.01 to 200 μM, maintaining the final DMSO concentration at less than 1%.
Cell Viability Assays.
Cells were seeded at a density of 10 000 cells/well in 96-well plates and were incubated in either normal media or CSS media. For pretreatment experiments, cells were treated with KV-49g or INDO for 24 h followed by the addition of ARN or ODM and incubated for a further 72 h. Cell viability was determined by the MTS tetrazolium dye assay as described previously.14 No intrinsic absorbance was noted with ARN, ODM, KV-49g, or INDO in the MTS assay.
Western Blotting. Cells were washed once with PBS and lysed with RIPA buffer (ThermoFisher). Proteins were quantified via BCA assay (ThermoFisher no. PI-23221) and then were resolved by SDS-PAGE on 4–20% Tris-Glycine gradient gels (BioRad). After transfer to nitrocellulose membranes, blocking was conducted for 1 h in Tris-buffered saline (10 mM Tris-HCl, 100 mM NaCl, pH 7.5) containing 0.1% Tween-20 (TBST) and 1% BSA. Samples were probed overnight at 4 °C with anti-aldo-keto reductase 1C3 enzyme (AKR1C3; Sigma; no. A6229, mouse mAb, 1:500), anti-prostate-specific antigen (PSA; Cell Signaling Technology; no. 5877S, rabbit mAb, 1:1000), or anti-β-actin (Sigma; no. A5441, mouse mAb, 1:1000), followed by incubation with anti-mouse (Sigma; no. SAB4600224) or anti-rabbit (Perkin-Elmer; no. NEF812001EA) secondary antibody (1:2000) horseradish peroxidase conjugate for 2 h, and bands were quantified by densitometry using ImageJ software.
Supplementary Material
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number R01CA226436. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.0c00069.
IC50 values of ARN and ODM in PCa cell lines, percentage cell viability of LNCaP, 22Rv1, and LNCaP1C3 cells grown in CSS media upon treatment with KV-49g, percentage cell viability of 22Rv1 cells grown in CSS media upon treatment with INDO, and calculated combination index (CI) and dose-reduction index (DRI) of treatment combinations in 22Rv1 cells grown in CSS media (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschembio.0c00069
The authors declare the following competing financial interest(s): P.C.T. is a named inventor on a patent application describing KV-49g (PCT Int. Appl. (2018), WO2018148721 A1).
Contributor Information
Ahmed Morsy, Department of Pharmaceutical Sciences and Fred and Pamela Buffet Cancer Center, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States.
Paul C. Trippier, Department of Pharmaceutical Sciences, Fred and Pamela Buffet Cancer Center, and UNMC Center for Drug Discovery, University of Nebraska Medical Center, Omaha, Nebraska 68198, United States;.
REFERENCES
- (1).Sharifi N, Gulley JL, and Dahut WL (2005) Androgen deprivation therapy for prostate cancer. JAMA 294, 238–244. [DOI] [PubMed] [Google Scholar]
- (2).Hotte SJ, and Saad F (2010) Current management of castrate-resistant prostate cancer. Curr. Oncol 17 (Suppl 2), S72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Attard G, and de Bono JS (2011) Translating scientific advancement into clinical benefit for castration-resistant prostate cancer patients. Clin. Cancer Res 17, 3867–3875. [DOI] [PubMed] [Google Scholar]
- (4).Bambury RM, and Rathkopf DE (2016) Novel and next-generation androgen receptor-directed therapies for prostate cancer: Beyond abiraterone and enzalutamide. Urol. Oncol 34, 348–355. [DOI] [PubMed] [Google Scholar]
- (5).Thakur A, Roy A, Ghosh A, Chhabra M, and Banerjee S (2018) Abiraterone acetate in the treatment of prostate cancer. Biomed. Pharmacother 101, 211–218. [DOI] [PubMed] [Google Scholar]
- (6).Dellis AE, and Papatsoris AG (2019) Perspectives on the current and emerging chemical androgen receptor antagonists for the treatment of prostate cancer. Expert Opin. Pharmacother 20, 163–172. [DOI] [PubMed] [Google Scholar]
- (7).Nakazawa M, Paller C, and Kyprianou N (2017) Mechanisms of therapeutic resistance in prostate cancer. Curr. Oncol. Rep 19, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Fizazi K, Smith MR, and Tombal B (2018) Clinical development of darolutamide: A novel androgen receptor antagonist for the treatment of prostate cancer. Clin. Genitourin. Cancer 16, 332–340. [DOI] [PubMed] [Google Scholar]
- (9).Cai C, and Balk SP (2011) Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr.-Relat. Cancer 18, R175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Jernberg E, Thysell E, Bovinder Ylitalo E, Rudolfsson S, Crnalic S, Widmark A, Bergh A, and Wikstrom P (2013) Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS One 8, No. e77407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Adeniji AO, Chen M, and Penning TM (2013) AKR1C3 as a target in castrate resistant prostate cancer. J. Steroid Biochem. Mol. Biol 137, 136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Verma K, Gupta N, Zang T, Wangtrakluldee P, Srivastava SK, Penning TM, and Trippier PC (2018) AKR1C3 inhibitor KV-37 exhibits antineoplastic effects and potentiates enzalutamide in combination therapy in prostate adenocarcinoma cells. Mol. Cancer Ther 17, 1833–1845. [DOI] [PubMed] [Google Scholar]
- (13).Liu C, Armstrong CM, Lou W, Lombard A, Evans CP, and Gao AC (2017) Inhibition of AKR1C3 activation overcomes resistance to abiraterone in advanced prostate cancer. Mol. Cancer Ther 16, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Verma K, Zang T, Gupta N, Penning TM, and Trippier PC (2016) Selective AKR1C3 Inhibitors Potentiate Chemotherapeutic Activity in Multiple Acute Myeloid Leukemia (AML) Cell Lines. ACS Med. Chem. Lett 7, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zang T, Verma K, Chen M, Jin Y, Trippier PC, and Penning TM (2015) Screening baccharin analogs as selective inhibitors against type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3). Chem.-Biol. Interact 234, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Verma K, Zang T, Penning TM, and Trippier PC (2019) Potent and highly selective aldo-keto reductase 1C3 (AKR1C3) inhibitors act as chemotherapeutic potentiators in acute myeloid leukemia and T-Cell acute lymphoblastic leukemia. J. Med. Chem 62, 3590–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Desmond JC, Mountford JC, Drayson MT, Walker EA, Hewison M, Ride JP, Luong QT, Hayden RE, Vanin EF, and Bunce CM (2003) The aldo-keto reductase AKR1C3 is a novel suppressor of cell differentiation that provides a plausible target for the non-cyclooxygenase-dependent antineoplastic actions of non-steroidal anti-inflammatory drugs. Cancer Res. 63, 505–512. [PubMed] [Google Scholar]
- (18).Moradi Manesh D, El-Hoss J, Evans K, Richmond J, Toscan CE, Bracken LS, Hedrick A, Sutton R, Marshall GM, Wilson WR, Kurmasheva RT, Billups C, Houghton PJ, Smith MA, Carol H, and Lock RB (2015) AKR1C3 is a biomarker of sensitivity to PR-104 in preclinical models of T-cell acute lymphoblastic leukemia. Blood 126, 1193–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Byrns MC, Steckelbroeck S, and Penning TM (2008) An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem. Pharmacol 75, 484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pan C. x., Lara P, Evans CP, Parikh M, White R d V, Dall’era M, Liu C, Robles D, and Gao A (2018) A phase Ib/II trial of indomethacin and enzalutamide to treat castration-resistant prostate cancer (CRPC). J. Clin. Oncol 36, TPS394. [Google Scholar]
- (21).Rathkopf DE, Smith MR, Ryan CJ, Berry WR, Shore ND, Liu G, Higano CS, Alumkal JJ, Hauke R, Tutrone RF, Saleh M, Chow Maneval E, Thomas S, Ricci DS, Yu MK, de Boer CJ, Trinh A, Kheoh T, Bandekar R, Scher HI, and Antonarakis ES (2017) Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol 28, 2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Byrns MC, Mindnich R, Duan L, and Penning TM (2012) Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5alpha-reductase inhibitor finasteride. J. Steroid Biochem. Mol. Biol 130, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pharma Janssen. 2018. ERLEADA: Highlights of prescribing information. Retrieved from https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210951s000lbl.pdf. Revised February 2018. Accessed December 20, 2019.
- (24).Bayer and Orion Pharma NUBEQA: Highlights of prescribing information. Retrieved from https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212099Orig1s000lbl.pdf. Revised July 2019. Accessed December 20, 2019.
- (25).Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446. [DOI] [PubMed] [Google Scholar]
- (26).Kinarivala N, Shah K, Abbruscato TJ, and Trippier PC (2017) Passage variation in PC12 cells results in inconsistent susceptibility to externally induced apoptosis. ACS Chem. Neurosci 8, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.