

Summary
Excessive host inflammatory responses negatively impact disease outcomes in respiratory infection. Host-pathogen interactions during the infective phase of influenza are well studied, but little is known about the host’s response during the repair stage. Here, we show that influenza infection stimulated the expression of angiopoietin-like 4 (ANGPTL4) via a direct IL6-STAT3-mediated mechanism. ANGPTL4 enhanced pulmonary tissue leakiness and exacerbated inflammation-induced lung damage. Treatment of infected mice with neutralizing anti-ANGPTL4 antibodies significantly accelerated lung recovery and improved lung tissue integrity. ANGPTL4-deficient mice also showed reduced lung damage and recovered faster from influenza infection when compared to their wild-type counterparts. Retrospective examination of human lung biopsy specimens from infection-induced pneumonia with tissue damage showed elevated expression of ANGPTL4 when compared to normal lung samples. These observations underscore the important role that ANGPTL4 plays in lung infection and damage and may facilitate future therapeutic strategies for the treatment of influenza pneumonia.
Graphical Abstract

Highlights
-
•
ANGPTL4 is upregulated by a STAT3-mediated mechanism during influenza pneumonia
-
•
ANGPTL4-deficient mice show reduced lung damage and accelerated lung recovery
-
•
Antibodies targeting ANGPTL4 reduce pulmonary tissue leakiness and damage
-
•
ANGPTL4 is a potential biomarker for respiratory infection and pneumonia
Li et al. show that influenza infection stimulates the expression of ANGPTL4 through a STAT3-mediated mechanism. Host ANGPTL4 enhances pulmonary tissue leakiness and exacerbates inflammation-induced lung damage. ANGPTL4 deficiency improves pulmonary tissue integrity and accelerates lung tissue recovery.
Introduction
The occurrence of annual epidemics and random global pandemics of influenza exerts a large public health burden worldwide (Mizgerd, 2006, Armstrong et al., 1999). However, designing effective vaccines and treatment options has proven challenging in view of the rapid evolution of the virus. While many aspects of host-pathogen interactions during the course of an influenza infection have been studied, there is less information on the host response during the repair stage of an infection (Mizgerd, 2008). A better understanding of the host response during the pulmonary repair phase may facilitate innovative treatment strategies. Host-specific biomarkers, indicative of the severity of lung tissue damage, could be exploited to delineate opportunities for therapeutic intervention.
Host immune responses are extremely important for containing influenza infections (Julkunen et al., 2000). Through the combined action of innate and adaptive immune responses, the infectious pathogen becomes inactivated and cleared from the body, repair processes start to resolve the tissue damage, and long-term immunity is ultimately established. However, excessive and prolonged inflammation may be detrimental to the host and contribute to the greater morbidity and mortality associated with influenza-induced inflammatory injury (Akaike et al., 1996, Monsalvo, 2010, Nicholls and Peiris, 2005, Buchweitz et al., 2007). Exaggerated inflammatory responses in the lung parenchyma can destroy alveoli, induce excessive edema, precipitate hypoxia, and cause pulmonary impairment (Narasaraju et al., 2011). Studies have documented that inflammatory injury to the lungs represents a major factor for the fatalities associated with pandemic H1N1-2009, highly pathogenic avian influenza viruses, and severe acute respiratory syndrome (SARS) coronavirus (Monsalvo, 2010, Nicholls and Peiris, 2005). Although inflammatory processes represent important therapeutic targets, anti-inflammatory therapies may also inhibit critical immune functions that mediate pathogen clearance, and they run the risk of enhancing pathogen replication and secondary infection (Uchide and Toyoda, 2011, Snelgrove et al., 2006, Aldridge et al., 2009, Ballinger and Standiford, 2010). An ideal treatment regimen should minimize the tissue damage caused by inflammation and facilitate recovery without interfering with the host’s antiviral and antibacterial responses.
Angiopoietin-like 4 (ANGPTL4) belongs to a family of angiogenic-regulating, secreted proteins that bear a high similarity to members of the angiopoietin (ANG) family. However, ANGPTL4 does not bind to ANG receptor TIE1/2, indicating that ANGPTL4 exerts its distinct functions via a different mechanism from ANG proteins (Zhu et al., 2012, Grootaert et al., 2012). Native full-length ANGPTL4 (flANGPTL4) contains a secretory signal peptide, an N-terminal coiled-coil structure, and a C-terminal fibrinogen-like domain. ANGPTL4 undergoes proteolytic processing by proprotein convertases at the linker region, thereby releasing the N-terminal region (nANGPTL4) and the monomeric C-terminal portion (cANGPTL4) (Zhu et al., 2012, Grootaert et al., 2012). The nANGPTL4 assembles into oligomeric structures, which is important for its function as a lipoprotein lipase inhibitor (Lei et al., 2011, Dijk and Kersten, 2014). The cANGPTL4 interacts with integrin β1/5, vascular endothelial (VE)-cadherin, or claudin-5 to trigger intracellular pathways that aid wound healing and support tumor growth and metastasis (Goh et al., 2010a, Goh et al., 2010b, Huang et al., 2011, Zhu et al., 2011). The expression of ANGPTL4 is elevated by numerous stimuli that are also involved in influenza pneumonia, including glucocorticoids, transforming growth factor β, and hypoxia-inducible factor 1-α (HIF1-α) (Zhu et al., 2012, Grootaert et al., 2012). Furthermore, ANGPTL4 compromises the integrity of endothelial vascular junction by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 cluster (Huang et al., 2011).
Interestingly, pulmonary edema due to vascular leakiness is a component of the fully developed viral lesion in the mouse (Harford et al., 1950). However, to our knowledge, the role of ANGPTL4 has not been studied in detail in influenza pneumonia, and study on this host response factor may open door to future intervention strategies. Thus in this study, we elucidate the role of host response protein ANGPTL4 during influenza pneumonia.
Results
ANGPTL4 Expression Is Elevated in Influenza Virus-Infected Lungs
To investigate if ANGPTL4 is involved in the host response to influenza, we examined ANGPTL4 expression in mice infected with the PR8 influenza A H1N1 virus that is related to the 1918 pandemic influenza virus. The infection was performed via intratracheal inoculation with a nonlethal PR8 viral challenge that was sufficient to cause serious pulmonary damage. Viral replication was detected on bronchial structures at 3 days postinfection (dpi) and peaked at 5 dpi, as indicated by the presence of NS1 viral protein (Figure 1 A). Virus was also detected in alveolar type II epithelial cells throughout the lungs by coimmunofluorescence staining with surfactant protein C (Figure 1B). At day 7, the viral protein expression began to decrease, and it became undetectable at 9 dpi (Figure 1A). The viral replication profile during the disease progression was further confirmed by qPCR of viral nucleoprotein RNA from infected lungs (Figure 1C). In addition, the expression of interferon-γ (IFN-γ) and interleukin-6 (IL-6), cytokines critical for innate and adaptive immunity against viral infections, peaked at 5 dpi (Figure 1C).
Figure 1.

Elevated cANGPTL4 mRNA and Protein Expression during Influenza Virus-Induced Pneumonia
C57BL/6J mice were infected with the PR8 virus, and lungs were harvested at the indicated days postinfection (dpi).
(A) Representative immunofluorescence staining for viral protein NS1 (green) and cell nuclei (blue) of influenza-infected lungs. Scale bar, 50 μm.
(B) Representative dual immunofluorescence staining of alveolar epithelial type II cells (SPC, red) and viral protein NS1 (green) showing viral infection in the alveolar space is limited to alveolar epithelial type II cells. Scale bar, 60 μm.
(C) Relative mRNA expression of viral NP mRNA (means ± SEM, n = 5) and protein levels (means ± SEM, n = 4) of cytokine interleukin-6 (IL-6, blue) and interferon-γ (IFN-γ, red) in BALF as determined by Bioplex as described in Experimental Procedures.
(D and E) Relative expression of ANGPTL4 mRNA (D) and protein (E) in lungs harvested at the indicated dpi. mRNA expression was normalized to that of the housekeeping gene ribosomal protein L27, which did not change under any of the studied experimental conditions (means ± SEM, n = 5). β-Tubulin served as a loading and transfer control for immunoblotting.
(F) Representative H&E images and immunofluorescence-stained images of ANGPTL4 (green) counterstained with DAPI (blue) of infected lung sections. Scale bar, 1,000 μm.
All the staining pictures shown in this figure are representative images from 15 mice for each time point. See also Figure S1.
H&E staining of infected lung sections revealed that bronchial cells were damaged at 5 dpi, corresponding to the peak of viral load (Figure S1A). We observed extensive lung tissue damage that was marked by large regions of pulmonary hemorrhage and infiltration of host immune cells at 13 dpi (Figure S1A). Inflammatory cells, such as macrophages and neutrophils, infiltrated the alveolar spaces of infected lungs, causing tissue damage and bleeding (Figure S1B). Together with damaged tissue debris, the infiltrated cells formed dense cell clusters and filled up the alveolar spaces. In our model, the mice recovered from the infection, which allowed us to investigate the events that occurred during the recovery phase. Indeed, overall recovery of tissue integrity was observed at 19 dpi (Figure S1A).
Next, we determined the kinetics of ANGPTL4 mRNA and protein levels in the infected lungs by qPCR and immunoblot analyses, respectively. ANGPTL4 mRNA was significantly upregulated at day 5 and remained elevated until 9 dpi. Thereafter, its expression decreased, reaching the basal level by 17 dpi (Figure 1D). Elevated levels of ANGPTL4 protein, specifically cANGPTL4, were detected in lung tissue homogenates at day 7 and remained elevated until 19 dpi, compared to day 0 controls (Figure 1E). We also examined the spatiotemporal expression of cANGPTL4 during influenza infection by immunofluorescence staining (Figure 1F). At day 0, cANGPTL4 protein was restricted to tubular structures such as the blood vessels and bronchioles (Figure 1F). At 5 and 13 dpi, corresponding to the peaks of ANGPTL4 mRNA and protein respectively, we observed stronger cANGPTL4 staining within the inflamed regions infiltrated by immune cells (Figure 1F). To eliminate the possible interference from autofluorescence, we provided technical controls for the staining using isotype immunoglobulin G (IgG) control and Alexa Fluor 647 fluorescence antibody. Both the blood vessels and infiltrated alveolar space showed positive staining with anti-cANGPTL4 mouse monoclonal antibody as the primary antibody and anti-mouse Alexa Fluor 647 as the secondary antibody. In contrast, no staining of the blood vessel and infiltrated alveolar space at the corresponding regions in neighboring slide was observed when control IgG and anti-mouse Alexa Fluor 647 antibody were used (Figure S1C). The result confirmed the ANGPTL4 signal was specific. To confirm that the elevated cANGPTL4 expression was not virus-strain specific, we examined its expression in the lungs of mice challenged with a sublethal dose of a mouse-adapted H3N2 strain. H3N2 infection of BALB/c mice exhibited increased levels of cANGPTL4 as well as full-length ANGPTL4. The cANGPTL4 protein peaked at 10 dpi, while the expression of native ANGPTL4 protein remained elevated at 15 dpi during the recovery of lung tissues (Figures S1D and S1E). This confirmed that ANGPTL4 upregulation was not restricted to a specific strain of influenza virus or mouse. Notably, different strains of virus and mouse showed distinct ANGPTL4 mRNA and protein expression profiles.
ANGPTL4 Expression Is Regulated by a STAT3-Mediated Mechanism
As the ANGPTL4 mRNA expression profile mirrored the pattern of viral replication, we asked if viral infection could influence the expression of ANGPTL4. Dual immunofluorescence staining for NS1 viral protein and cANGPTL4 revealed that infected alveolar type II epithelial cells displayed cANGPTL4 staining, whereas the uninfected alveolar type II epithelial cells within the same lung did not exhibit cANGPTL4 staining (Figure 2 A). Although Clara cells were also stained positive for ANGPTL4 (Figure S2A), we did not detect any significant ANGPTL4 mRNA level in Clara cells isolated by laser capture microdissection (Figure S2B), suggesting that the ANGPTL4 staining most likely derived from secretion from heterotypic cell types. Next, we examined ANGPTL4 expression during influenza infection of mice with a lethal dose of PR8 virus (1,000 plaque-forming units [pfu]). We detected an earlier upregulation of ANGPTL4 mRNA and protein at 3 dpi, while the expression of native ANGPTL4 and cANGPTL4 remained elevated until the mice were euthanized at 7 dpi (Figure 2B).
Figure 2.
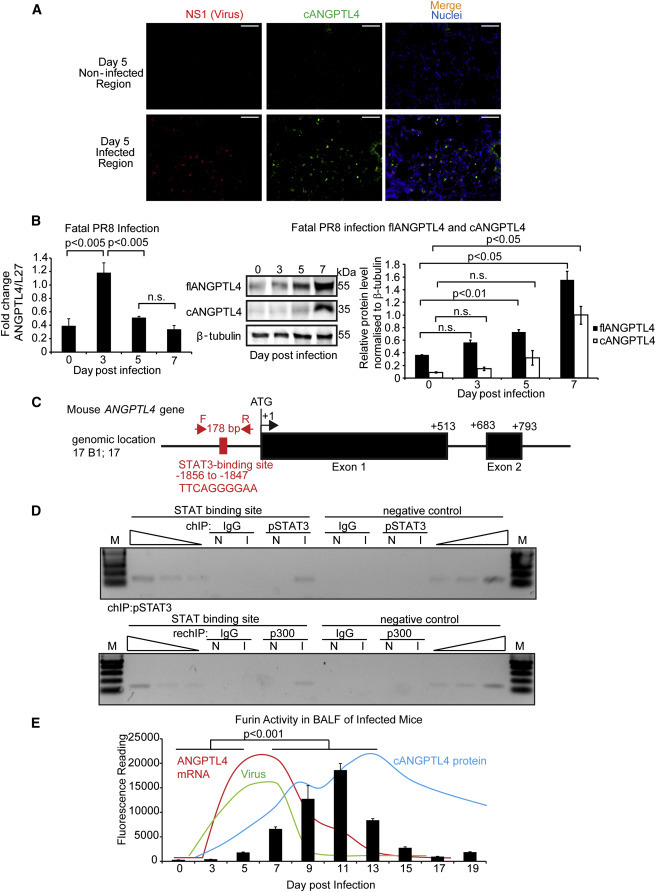
ANGPTL4 Expression Is Regulated by a STAT3-Mediated Mechanism
(A) Dual immunofluorescence staining of cANGPTL4 (green) and viral protein NS1 (red). Staining images are representative of five mice. Scale bar, 50 μm.
(B) Relative mRNA (left panel) and protein (middle and right panels) levels of ANGPTL4 in lungs of mice infected with a lethal dose of PR8 virus (1,000 pfu). mRNA expression was normalized to that of the housekeeping gene ribosomal protein L27, which did not change under any of the studied experimental conditions (means ± SEM, n = 3). β-Tubulin served as a loading and transfer control for immunoblotting.
(C) Schematic diagram showing the relative position of a putative STAT-binding site in the mouse ANGPTL4 gene promoter.
(D) ChIP assay was conducted using preimmune IgG or antibodies against pSTAT3 (top panel) and re-ChIP with anti-p300 (bottom panel) in infected (I) and mock-infected (N) lungs. The specific region spanning STAT3 binding site of ANGPTL4 gene was amplified using appropriate primers. A control region served as a negative control.
(E) Bar graph shows furin activity in the BALF extracts at the indicated time points after PR8 infection (means ± SEM, n = 10). Expression profile of viral NP mRNA (dark blue), ANGPTL4 mRNA (red), and protein (light blue) were plotted.
See also Figure S2.
We next sought to elucidate the underlying mechanism of this upregulation. HIF1-α has been shown to increase the expression of ANGPTL4 in endothelial cells (Zhu et al., 2012, Grootaert et al., 2012). To investigate if HIF1-α could be responsible for the expression of ANGPTL4 mRNA, we examined the HIF1-α protein expression profile in our animal model. The peak HIF1-α protein expression was detected at 13 dpi, which did not coincide with the peak mRNA expression of ANGPTL4 (Figure S2C), suggesting HIF1-α was unlikely to be a major regulator of ANGPTL4 at the early stages of infection. However, we cannot exclude that HIF1-α may maintain or sustain ANGPTL4 expression at later stages of infection. Interrogation of GEO data sets revealed that STAT3 deficiency in pulmonary alveolar type II epithelial cells was related to lower ANGPTL4 levels (Xu et al., 2007). Interestingly, viral infection triggered host responses through IFN-γ and IL-6 pathways, which activate STAT, and displayed overlapping profiles to the ANGPTL4 mRNA expression pattern (Figure 1C). In silico analysis of the promoter of the mouse ANGPTL4 gene also revealed a putative STAT-binding site (Figure 2C). Thus, we performed chromatin immunoprecipitation (ChIP) using phospho-STAT3 (pSTAT3) antibody on the ANGPTL4 promoter in uninfected and infected lung tissues. The sequences spanning the STAT-binding site were enriched in the immunoprecipitates obtained from the virus-infected lung tissues compared to uninfected tissues (Figure 2D). pSTAT3 ChIP experiments followed by re-ChIP with p300, a STAT3 coactivator, further confirmed the existence of direct regulation of ANGPTL4 via a STAT3-mediated mechanism (Figure 2D). To further strengthen our in vivo findings, we suppressed endogenous STAT3 expression by small interfering RNA (siRNA) and examined the expression of ANGPTL4 in a human small airway epithelial cell culture exposed to IL-6. We observed a significant increase in ANGPTL4 mRNA in response to an IL-6 challenge, which was abrogated when endogenous STAT3 was knocked down by ON-TARGETPlus siRNA (Figure S2D). We confirmed that the STAT3 protein was also reduced by western blot analysis (Figure S2D).
The flANGPTL4 undergoes proteolytic processing to release cANGPTL4. Furin proprotein convertase is known to cleave flANGPTL4 (Lei et al., 2011). To determine whether furin-like activity is present and consistent with the peak of cANGPTL4 protein expression in influenza-infected lungs, we measured furin activity in the bronchoalveolar lavage fluid (BALF) of mice that were sublethally infected with PR8 virus. Furin activity in the BALF started to increase at 5 dpi, coinciding with the peak in virus titer (Figure 2E). Importantly, the furin activity profile overlapped significantly with that of cANGPTL4 protein (Figure 2E). To strengthen the role of furin in ANGPTL4 cleavage, we performed similar experiments in the presence or absence of a furin inhibitor. Furin inhibitor was added to BALF taken from 11 dpi, a time point when the furin activity was elevated. Recombinant flANGPTL4 protein with FLAG tag at the C terminus was used as exogenous substrate. We detected a reduced cANGPTL4:flANGPTL4 ratio in BALF containing furin inhibitor when compared to BALF alone (Figure S2E), confirming that furin was a major contributor of posttranslational cleavage of flANGPTL4 in our animal model. Taken together, our data demonstrated that ANGPTL4 mRNA was upregulated via a STAT3-dependent pathway during influenza pneumonia. The concomitant increase in furin activity subsequently cleaved flANGPTL4 to generate cANGPTL4, which peaked at 13 dpi, corresponding to extensive lung injury marked by large regions of pulmonary hemorrhage and infiltration of host immune cells.
Immunoneutralization of cANGPTL4 Significantly Reduces Tissue Leakiness to Accelerate Lung Recovery
To understand the role of cANGPTL4 in influenza pathogenesis, we investigated the in vivo effect of a neutralizing cANGPTL4 monoclonal antibody (mAb; clone 3F4F5) on the host response to influenza viral pneumonia. We employed two treatment strategies based on the stage of disease progression (Figure 3 A). Virus-induced inflammation caused severe lung damage, which was observed until 13 dpi. Thereafter, tissue regeneration began to restore lung structural integrity and function. Thus, we defined the period before 13 dpi as the “damage window,” and days after 13 dpi as the “recovery window.” Daily intravenous injections of anti-ANGPTL4 mAb (10 mg/kg body weight) were administered for 5 days starting either at 6 dpi during the damage window or day 13 during the recovery period (Figure 3A). Negative control groups included mock-infected mice that received either isotype-matched control mouse IgG or anti-cANGPTL4 mAb alone. Mice infected with influenza virus and treated with control mouse IgG served as another control group. Lung tissues were harvested 24 hr after the last injection.
Figure 3.

Immunoneutralization of cANGPTL4 Improves Lung Tissue Recovery after Influenza Infection
(A) Schematic diagram showing administration protocols during “damage” (yellow arrows) and “recovery” (blue arrows) windows. Graph shows a summary of various events of mouse influenza infection model plotted based on earlier observations.
(B) Representative H&E images of lung sections from mice either mock-infected or PR8-infected and treated with control isotype IgG or anti-cANGPTL4 monoclonal antibody. Images show alveolar space, bronchiole, and pulmonary edema. Scale bar, 100 μm.
(C) Representative immunofluorescence staining of PDPN (red) as marker of alveolar type I epithelial cells and nuclei (blue) show tissue regeneration in the lungs. Scale bar, 50 μm. Representative images from five mice for each group.
(D) Representative immunofluorescence staining of ZO-1 (green) of confluent human microvascular endothelial cell and human small airway epithelial cell cultures treated with either vehicle PBS or recombinant cANGPTL4 protein (6 μg/mL). Scale bar, 40 μm, n = 3. Dotted scale bar, 10 μm.
(E) Infrared imaging of lungs in vivo (bottom panel) and ex vivo (top panel) using PEG-800 contrast agent to reveal pulmonary leakiness. Mice were treated with antibodies from 13 to 17 dpi, and the imaging was performed at 18 dpi. Scale bar, 1 cm. Representative images of three mice for each group were shown. Intensity of fluorescence signal from the lungs was measured ex vivo by Li-Cor MousePOD (means ± SEM, n = 3).
(F) H&E images of lungs from ANGPTL4−/− and ANGPTL4+/+ mice infected with sublethal PR8 at the indicated dpi. Scale bar, 100 μm.
(G) Immunofluorescence staining of PDPN (red) and nuclei (blue) showed tissue regeneration in the lungs. Scale bar, 50 μm. Representative images from five mice for each group are shown.
See also Figure S3.
The anti-cANGPTL4 mAb treatment during the “damage window” did not significantly alleviate the early inflammation-induced lung damage (Figure S3A). In contrast, anti-cANGPTL4 mAb treatment during the recovery stage resulted in reduced lung damage and a significant improvement in tissue recovery compared to control groups (Figure 3B). The alveolar spaces showed a remarkable increase of noninfiltrated areas with reduced pulmonary bleeding and accelerated regeneration of alveolar type I epithelial cells (Figure 3C). To pursue this further, we examined the in vivo integrity of the pulmonary vasculature by in vivo imaging of the mice receiving anti-cANGPTL4 mAb during the recovery period (Figure 3E). Mice were injected intravenously with IRDye 800CW PEG contrast agent (50 nmol/kg body weight) 24 hr after the last mAb injection. The presence of infrared (IR) signal indicates the accumulation of IRDye 800CW PEG in tissue due to increased leakiness of the local tissue. As expected, infected mice treated with control IgG showed a very high IR signal in the lung compared to mock-infected mice (Figure 3E). Infected mice treated with anti-cANGPTL4 mAb showed a significantly subdued IR signal in the lungs compared with control IgG treatment, indicating that immunoneutralization of cANGPTL4 markedly abrogated tissue leakiness. Ex vivo imaging of resected mouse lungs confirmed the reduced accumulation of IRDye 800CW PEG agent following anti-cANGPTL4 mAb therapy. To more precisely define the cell types responsible for the increased tissue leakiness, we examined the cell-cell junction of human small airway epithelial cells and microvascular endothelial cells after treatment with recombinant cANGPTL4 by immunofluorescence staining with tight junction protein zonula occludens-1 (ZO-1). The cANGPTL4 protein perturbed the cell-cell boundary of primary human endothelial and epithelial cells (Figure 3D), indicating that cANGPTL4 exacerbated pulmonary bleeding and edema and could accelerate the infiltration of immune cells inside the lung tissue to aggravate tissue damage.
To further strengthen our above observation, we examined the effect of influenza infection on wild-type (ANGPTL4+/+) and ANGPTL4 mutant mice (knockout ANGPTL4−/− and heterozygous ANGPTL4+/−). We observed lung damage in ANGPTL4−/− at 11 dpi, albeit at reduced severity when compared to influenza-infected ANGPTL4+/+ (Figure S3B). Consistent with above observation with neutralizing anti-cANGPTL4, infected ANGPTL4−/− mice showed rapid lung tissue recovery at 18 dpi compared with ANGPTL4+/+ (Figure 3F) and accelerated regeneration of alveolar type I epithelial cells (Figure 3G). We also studied the effect of ANGPTL4 gene dosage on lung tissue damage and recovery using ANGPTL4+/− mice. Similar to cANGPTL4 antibody-treated ANGPTL4+/+ mice, significant improvement in tissue recovery was observed in PR8-infected ANGPTL4+/− mice when compared with ANGPTL4+/+ mice at 18 dpi, but no significant difference in tissue damage was observed at 11 dpi (Figures S3C and S3D). We also confirmed the observations by immunoblotting of podoplanin (PDPN) as a marker of lung damage as well as scoring of lung tissue damage by trained pathologist (Figures S3E and S3F).
To explore the biological impact of anti-cANGPTL4 mAb therapy on infected lungs, we performed microarray gene expression analyses of lungs of mice treated with either anti-cANGPTL4 antibody or control IgG from 13 to 17 dpi. Data analyses revealed that mAb therapy resulted in differences in numerous major physiological activities, including angiogenesis, lung tissue development, inflammatory responses, and extracellular matrix and endopeptidase activities (Figures 4 A and S4). Genes involved in the development of lung alveoli, respiratory tubes, and blood vessels were identified in the anti-cANGPTL4 mAb-treated infected mice, indicating earlier tissue regeneration. The mAb treatment also dampened the inflammation-related tissue damage, consistent with the pathological phenotype. Taken together, our findings indicate that the functional neutralization of cANGPTL4 during the tissue recovery stage promoted lung tissue recovery and was associated with improved tissue integrity.
Figure 4.

ANGPTL4 Deficiency Reduces Infection-Associated Pulmonary Damage
(A) Heatmaps show gene expression profiles of influenza-infected lungs left untreated or treated with anti-cANGPTL4 antibody (I versus A). Genes are clustered according to their biological functions. The color spectrum from green to red depicts the fold change in LOG scale. See also Figure S4.
(B) Representative immunofluorescence images of cANGPTL4 expression in human lung biopsy specimens of patients with or without pneumonia. Scale bar, 100 μm. See also Table S2 for a complete list of patient information and immunofluorescence staining observations.
cANGPTL4 Expression Is Enhanced in Human Lung Biopsy Specimens of Patients with Infection-Induced Pneumonia
To underscore the clinical relevance of ANGPTL4 in lung infection and lung damage, we performed a retrospective immunofluorescence examination of ANGPTL4 expression in 40 archived human lung biopsy specimens from patients either without pneumonia or with pneumonia induced by various infections as approved by the institutional review board of the National University Hospital (reference number 2012/00661). The staining and microscopic imaging techniques were performed simultaneously under the same conditions, allowing the signal intensity to serve as a semiquantitative measure of the cANGPTL4 expression level. In the ten pneumonia samples that were symptomatic or microbe positive, we found brightly stained structures (Figure 4B; Table S2). These structures included the thickened layers around tubular structures as well as intensively stained structures in the damaged alveolar regions that appeared to be collapsed membrane-like structures (Figure 4B; Table S2). These structures were abundant in regions with dense infiltration or collapsed alveolar spaces. The nonpneumonia samples showed either very weak staining or staining restricted to a thin layer along the tubular structures, consistent with our observations in healthy mouse lung tissues (Figures 4B and 1F; Table S2). The obstructive pneumonia sample that was not caused by infection did not show positive staining of cANGPTL4, which is consistent with our proposed mechanism whereby an infection-induced STAT pathway contributes to ANGPTL4 upregulation. These findings suggest that cANGPTL4 may be a potential biomarker for respiratory infection and pneumonia. Clearly, further validation with a larger patient cohort that includes human virally induced ARDS cases and other bodily fluids such as blood and sputum will be necessary.
Discussion
One of the hallmarks of influenza pneumonia is the aggravated inflammatory host response accompanied by pulmonary edema and associated acute lung injury (Mizgerd, 2008). In this study, we showed that influenza infection elevated the expression of the host protein ANGPTL4 via direct transcriptional regulation by STAT3. The spatiotemporal expression of ANGPTL4 coincided with the inflammation phase of influenza infection, localized to lung regions with elevated immune cell infiltration and tissue damage. Similarly, our analysis of human clinical infection-associated pneumonia samples showed higher levels of ANGPTL4 compared to uncomplicated human lung sections. Notably, influenza infection of ANGPTL4-knockout mice and immunoneutralization of ANGPTL4 in wild-type mice showed significantly improved pulmonary tissue integrity and accelerated recovery from inflammation-induced tissue damage.
Recent high-throughput RNA sequencing of formalin-fixed, paraffin-embedded autopsy lung tissue samples from the 1918 and 2009 influenza pandemics revealed that ANGPTL4 mRNA was one of the most significantly upregulated genes in both samples (Xiao et al., 2013). Our interrogation of microarray data from influenza-infected mouse lungs also consistently detected elevated ANGPTL4 expression (Pommerenke et al., 2012). These observations underscore the importance of ANGPTL4 in response to pneumonia. However, its role in infected lungs remained unclear. From our influenza mouse model, we observed that the ANGPTL4 mRNA expression profile mirrored the pattern of viral replication. Probing further, we showed that IL-6-activated STAT3 directly regulated the expression of ANGPTL4. This is consistent with microarray analysis showing that pulmonary alveolar type II epithelial cells deficient in STAT3 have lower ANGPTL4 mRNA levels compared to their wild-type counterparts (Xu et al., 2007).
Furin or furin-like proprotein convertases (PCs) play multiple roles in host response to influenza infection. Toll-like receptor 7 (TLR7) triggers antiviral immune responses by recognizing viral single-stranded RNA in endosomes. hTLR7 is proteolytically processed by furin-like PCs, and the C-terminal fragment of hTLR7 selectively accumulates in endocytic compartments. TLR7 processing was required for its functional response to TLR7 agonists such as R837 or influenza virus (Hipp et al., 2013). Interestingly, we observed that the peak expression of cANGPTL4 protein at 13 dpi was likely attributed to the increase in activity of furin PCs that preceded and overlapped with cANGPTL4 protein expression. Recent work showed that two novel peptidomimetic furin inhibitors inhibit hemagglutinin cleavage and viral propagation of a highly pathogenic avian H7N1 influenza virus strain in vitro (Becker et al., 2012). The in vivo effect of such furin inhibitors, which conceivably prevent the proteolytic cleavage of flANGPTL4 to release cANGPTL4, on lung tissue recovery remains to be explored.
Previous work has shown that cANGPTL4 can affect the paracellular permeability of blood vessels in cancer (Huang et al., 2011, Guo et al., 2014). In vivo and ex vivo imaging of infected mice treated with neutralizing anti-cANGPTL4 mAb revealed diminished pulmonary tissue leakiness compared to isotype control IgG treatment. The direct consequence was reduced pulmonary edema and immune cell-infiltrated lung regions. The overall lung tissue integrity was improved and the alveolar space appeared to be well recovered for normal function as supported by microarray analysis. Administration of anti-cANGPTL4 mAb during the damage window, when cANGPTL4 protein levels were low, showed no observable difference compared to control. Thus, targeting cANGPTL4 to modulate tissue leakiness along with its collateral benefits is a promising approach toward the development of therapy for influenza treatment, specifically lung recovery. Our finding that host protein ANGPTL4 participates in pulmonary leakiness and lung injury responses during influenza pneumonia is an important step toward a better understanding of influenza pathogenesis and how it can be manipulated to reduce the burden of pneumonia.
Experimental Procedures
Mice, Viruses, and Infections
Female 8- to 12-week-old C57BL/6J mice and BALB/c mice were purchased from the Biological Resource Centre, Biopolis, Singapore. Wild-type mice and mice heterozygous for ANGPTL4 (C57/B6 background) were obtained from the Mutant Mouse Regional Resource Center (MMRRC), an NIH-funded strain repository, and were donated to the MMRRC by Genentech. ANGPTL4 KO mice were identified using RT-PCR quantification of ANGPTL4 expression compared to wild-type mice. Influenza H1N1virus A/PR/8/34 strain (PR8) was purchased from the American Type Culture Collection. PR8 was propagated in embryonated eggs at 37°C for 72 hr, and the allantoic fluid was harvested as a viral stock. Virus titers were determined by the plaque assay via infection of Madin-Darby canine kidney (MDCK) cells. Mice were housed in BSL2 facilities and infected with a sublethal dose of PR8 (30 pfu in 75 μl of PBS [pH 7.4] per mouse) or with a lethal dose (1,000 pfu) by intratracheal inhalation under anesthesia. Mice were anesthetized using a ketamine/xylazine cocktail. When fully anesthetized, the mouse was held up and 30 pfu of PR8 virus in 75 μl of PBS was introduced into the back of its mouth to be breathed into the trachea, with its tongue held tightly to prevent swallowing of the liquid pipetted in. Lungs were harvested from anesthetized mice at indicated time points and stored at −80°C until further use. Female 8- to 12-week-old BALB/c mice were infected with a sublethal dose of mouse-adapted influenza A/Aichi/2/68 H3N2 virus (1,500 pfu). Female 8- to 12-week-old ANGPTL4−/− mice were infected also using 30 pfu of PR8 virus for comparison with ANGPTL4+/+ mice. Lung tissues were harvested for further analysis at 11 and 18 dpi. All animal protocols were approved by the respective institutional animal care and use committees at National University of Singapore (050/11) and Nanyang Technological University (ARF SBS/NIE-A0200AZ).
Antibody Treatment of Mice
Anti-cANGPTL4 mAb (clone 3F4F5) was produced using hybridoma as described previously (Goh et al., 2010a, Goh et al., 2010b, Zhu et al., 2011). Mice were intraperitoneally injected daily with the antibody in 200 μl saline at a dose of 10 mg/kg body weight on 6–10 dpi (harvested on day 11) or on 13–17 dpi (harvested on day 18). More details on the experimental procedures can be found in Supplemental Experimental Procedures.
In Vivo Imaging of Mice
Mice were mock infected with heat-inactivated influenza virus, infected with influenza virus and injected with control IgG, or infected with influenza virus and treated with anti-cANGPTL4 antibody on 13–17 dpi as described above. At the day of lung harvesting, mice were injected through the tail vein with IRDye 800CW PEG contrast agent (Li-Cor 926-50401) and subjected to in vivo imaging under anesthesia, using the Li-Cor MousePOD in vivo imaging facility. The IR fluorescent-tagged PEG800 was used to detect tissue leakiness when the tissue exhibited abnormally high paracellular permeability. Following imaging, the lungs of the mice were harvested and reimaged, and quantification of fluorescence signal was done according to the manufacturer’s instructions.
Author Contributions
L.L., V.T.K.C., and N.S.T. conceived and designed the experiments. L.L., H.C.C., E.H.P.T., C.C.C., S.Y.N., K.W.K., and Z.T. performed the experiments. L.L., J.E.S., H.W.C., V.T.K.C., and N.S.T. analyzed the data. L.L. and N.S.T. wrote the manuscript. V.T.K.C., M.L.B., and N.S.T. supervised the work. J.E.S., M.L.B., V.T.K.C., and N.S.T. helped edit the paper.
Acknowledgments
This research is supported by the Singapore National Research Foundation under its Cooperative Basic Research Grant (NMRC/CBRG/0030/2013) and administered by the Singapore Ministry of Health’s National Medical Research Council. The mouse strain used for this research project, C57/B6;129S5-Angptl4Gt(OST352973)Lex/Mmucd, identification number 032147-UCD, was obtained from the Mutant Mouse Regional Resource Center, an NIH-funded strain repository, and was donated to the MMRRC by Genentech, Inc.
Published: February 5, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information includes Supplemental Experimental Procedures, four figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.01.011.
Accession Numbers
Microarray data have been submitted to the GEO database under accession number GSE58647.
Supplemental Information
References
- Akaike T., Noguchi Y., Ijiri S., Setoguchi K., Suga M., Zheng Y.M., Dietzschold B., Maeda H. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc. Natl. Acad. Sci. USA. 1996;93:2448–2453. doi: 10.1073/pnas.93.6.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge J.R., Jr., Moseley C.E., Boltz D.A., Negovetich N.J., Reynolds C., Franks J., Brown S.A., Doherty P.C., Webster R.G., Thomas P.G. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl. Acad. Sci. USA. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong G.L., Conn L.A., Pinner R.W. Trends in infectious disease mortality in the United States during the 20th century. JAMA. 1999;281:61–66. doi: 10.1001/jama.281.1.61. [DOI] [PubMed] [Google Scholar]
- Ballinger M.N., Standiford T.J. Postinfluenza bacterial pneumonia: host defenses gone awry. J. Interferon Cytokine Res. 2010;30:643–652. doi: 10.1089/jir.2010.0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker G.L., Lu Y., Hardes K., Strehlow B., Levesque C., Lindberg I., Sandvig K., Bakowsky U., Day R., Garten W., Steinmetzer T. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem. 2012;287:21992–22003. doi: 10.1074/jbc.M111.332643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchweitz J.P., Harkema J.R., Kaminski N.E. Time-dependent airway epithelial and inflammatory cell responses induced by influenza virus A/PR/8/34 in C57BL/6 mice. Toxicol. Pathol. 2007;35:424–435. doi: 10.1080/01926230701302558. [DOI] [PubMed] [Google Scholar]
- Dijk W., Kersten S. Regulation of lipoprotein lipase by Angptl4. Trends Endocrinol. Metab. 2014;25:146–155. doi: 10.1016/j.tem.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Goh Y.Y., Pal M., Chong H.C., Zhu P., Tan M.J., Punugu L., Lam C.R.I., Yau Y.H., Tan C.K., Huang R.L. Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration. Am. J. Pathol. 2010;177:2791–2803. doi: 10.2353/ajpath.2010.100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh Y.Y., Pal M., Chong H.C., Zhu P., Tan M.J., Punugu L., Tan C.K., Huang R.L., Sze S.K., Tang M.B.Y. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J. Biol. Chem. 2010;285:32999–33009. doi: 10.1074/jbc.M110.108175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grootaert C., Van de Wiele T., Verstraete W., Bracke M., Vanhoecke B. Angiopoietin-like protein 4: health effects, modulating agents and structure-function relationships. Expert Rev. Proteomics. 2012;9:181–199. doi: 10.1586/epr.12.12. [DOI] [PubMed] [Google Scholar]
- Guo L., Li S.Y., Ji F.Y., Zhao Y.F., Zhong Y., Lv X.J., Wu X.L., Qian G.S. Role of Angptl4 in vascular permeability and inflammation. Inflamm. Res. 2014;63:13–22. doi: 10.1007/s00011-013-0678-0. [DOI] [PubMed] [Google Scholar]
- Harford C.G., Hara M., Technical Assistance of Alice Hamlin Pulmonary edema on influenzal pneumonia of the mouse and the relation of fluid in the lung to the inception of pneumococcal pneumonia. J. Exp. Med. 1950;91:245–260. doi: 10.1084/jem.91.3.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp M.M., Shepherd D., Gileadi U., Aichinger M.C., Kessler B.M., Edelmann M.J., Essalmani R., Seidah N.G., Reis e Sousa C., Cerundolo V. Processing of human toll-like receptor 7 by furin-like proprotein convertases is required for its accumulation and activity in endosomes. Immunity. 2013;39:711–721. doi: 10.1016/j.immuni.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.L., Teo Z., Chong H.C., Zhu P., Tan M.J., Tan C.K., Lam C.R.I., Sng M.K., Leong D.T.W., Tan S.M. ANGPTL4 modulates vascular junction integrity by integrin signaling and disruption of intercellular VE-cadherin and claudin-5 clusters. Blood. 2011;118:3990–4002. doi: 10.1182/blood-2011-01-328716. [DOI] [PubMed] [Google Scholar]
- Julkunen I., Melén K., Nyqvist M., Pirhonen J., Sareneva T., Matikainen S. Inflammatory responses in influenza A virus infection. Vaccine. 2000;19(Suppl 1):S32–S37. doi: 10.1016/s0264-410x(00)00275-9. [DOI] [PubMed] [Google Scholar]
- Lei X., Shi F., Basu D., Huq A., Routhier S., Day R., Jin W. Proteolytic processing of angiopoietin-like protein 4 by proprotein convertases modulates its inhibitory effects on lipoprotein lipase activity. J. Biol. Chem. 2011;286:15747–15756. doi: 10.1074/jbc.M110.217638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd J.P. Lung infection—a public health priority. PLoS Med. 2006;3:e76. doi: 10.1371/journal.pmed.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizgerd J.P. Acute lower respiratory tract infection. N. Engl. J. Med. 2008;358:716–727. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsalvo A. Severe pandemic H1N1 influenza disease due to pathogenic immune complexes. Nat. Med. Journal of controlled release. 2010;17:195–199. doi: 10.1038/nm.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasaraju T., Yang E., Samy R.P., Ng H.H., Poh W.P., Liew A.A., Phoon M.C., van Rooijen N., Chow V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011;179:199–210. doi: 10.1016/j.ajpath.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J., Peiris M. Good ACE, bad ACE do battle in lung injury, SARS. Nat. Med. 2005;11:821–822. doi: 10.1038/nm0805-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommerenke C., Wilk E., Srivastava B., Schulze A., Novoselova N., Geffers R., Schughart K. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS ONE. 2012;7:e41169. doi: 10.1371/journal.pone.0041169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snelgrove R.J., Edwards L., Rae A.J., Hussell T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006;36:1364–1373. doi: 10.1002/eji.200635977. [DOI] [PubMed] [Google Scholar]
- Uchide N., Toyoda H. Antioxidant therapy as a potential approach to severe influenza-associated complications. Molecules. 2011;16:2032–2052. doi: 10.3390/molecules23100000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.L., Kash J.C., Beres S.B., Sheng Z.M., Musser J.M., Taubenberger J.K. High-throughput RNA sequencing of a formalin-fixed, paraffin-embedded autopsy lung tissue sample from the 1918 influenza pandemic. J. Pathol. 2013;229:535–545. doi: 10.1002/path.4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y., Ikegami M., Wang Y., Matsuzaki Y., Whitsett J.A. Gene expression and biological processes influenced by deletion of Stat3 in pulmonary type II epithelial cells. BMC Genomics. 2007;8:455. doi: 10.1186/1471-2164-8-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P., Tan M.J., Huang R.L., Tan C.K., Chong H.C., Pal M., Lam C.R.I., Boukamp P., Pan J.Y., Tan S.H. Angiopoietin-like 4 protein elevates the prosurvival intracellular O2(-):H2O2 ratio and confers anoikis resistance to tumors. Cancer Cell. 2011;19:401–415. doi: 10.1016/j.ccr.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Zhu P., Goh Y.Y., Chin H.F.A., Kersten S., Tan N.S. Angiopoietin-like 4: a decade of research. Biosci. Rep. 2012;32:211–219. doi: 10.1042/BSR20110102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.