

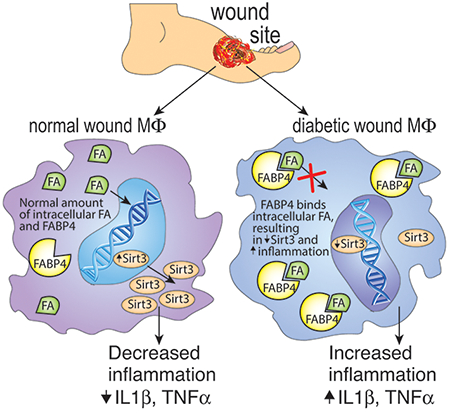
Abstract
Control of inflammation is critical for the treatment of non-healing wounds, but a delicate balance exists between early inflammation that is essential for normal tissue repair and the pathologic inflammation that can occur later in the repair process. This necessitates the development of novel therapies that can target inflammation at the appropriate time during repair. Here, we found that SIRT3 is essential for normal healing and regulates inflammation in wound macrophages post-injury. Under ‘pre-diabetic’ conditions, SIRT3 was decreased in wound macrophages and resulted in dysregulated inflammation. Further, we found that FABP4 regulates SIRT3 in human blood monocytes and inhibiting FABP4 in wound macrophages decreases inflammatory cytokine expression making FABP4 a viable target for the regulation of excess inflammation and wound repair in diabetes. Using a series of ex vivo and in vivo studies with genetically engineered mouse models, as well as diabetic human monocytes, we demonstrate that FABP4 expression is epigenetically upregulated in diabetic wound macrophages and, in turn, diminishes SIRT3 expression thereby promoting inflammation. These findings have significant implications for controlling inflammation and promoting tissue repair in diabetic wounds.
Keywords: Inflammation, Wound Healing, Macrophages, Type 2 Diabetes
Graphical abstract
INTRODUCTION
Impaired wound healing associated with chronic inflammation is a major complication of type 2 diabetes (T2D) and is the leading cause of non-traumatic amputation in the United States (Boulton et al., 2005). Wound healing is a tightly regulated process with distinct phases occurring in programmed succession (Diegelmann and Evans, 2004, Reinke and Sorg, 2012). Failure of progression through these discrete phases results in impaired healing. Normally, following the initial coagulation phase, neutrophils and monocyte/macrophages are recruited to the wound bed and the inflammatory phase ensues. As the injured tissue progresses through the inflammatory phase, the predominant macrophage phenotype in the wound bed changes when the wound transitions to the proliferative phase (Galli et al., 2011, Landen et al., 2016). During the initial part of the inflammatory phase, infiltrating monocytes recruited to the site of injury secrete proinflammatory mediators (Barrientos et al., 2008, Hubner et al., 1996). Then, towards the end of the inflammatory phase, they shift towards a reparative phenotype and secrete anti-inflammatory mediators that promote tissue repair (Barrientos et al., 2008, Galli et al., 2011, Landen et al., 2016, Porcheray et al., 2005). However, in T2D, the macrophages remain in a proinflammatory state and fail to shift to the reparative phenotype, resulting in chronic inflammation (Boniakowski et al., 2018, Falanga, 2005, Kimball et al., 2018, Kimball et al., 2017a, Kimball et al., 2017b, Wetzler et al., 2000).
While it is unknown what drives inflammatory macrophage phenotype transition during wound healing, recent studies have suggested a strong link between inflammation and metabolism (Liu et al., 2012a). The coupling of these two entities appears to be, in part, due to sirtuins (Liu et al., 2015). Sirtuins are a family of nicotinamide adenine dinucleotide (NAD+)- dependent protein deacetylases that regulate important metabolic homeostatic mechanisms that are known to influence inflammatory pathways (Buechler et al., 2017, Imai et al., 2000, Liu et al., 2012a, Liu et al., 2012b). Sirtuin 3 (SIRT3), the primary mitochondrial deacetylase (Hirschey et al., 2010, Lombard et al., 2007, Onyango et al., 2002), has been shown to be of particular importance to inflammation in adipose tissue (Xu et al., 2016). Importantly, SIRT3 has been implicated in the regulation of inflammation in human tissues and was recently found to be decreased in pancreatic islet cells of T2D patients (Caton et al., 2013). Although SIRT3 has been associated with adipocyte inflammation (Xu et al., 2016), its role in tissue repair, and specifically macrophage-mediated inflammation, in normal and diabetic wound repair has not been examined.
While the exact regulation of SIRT3 is unknown, free monounsaturated fatty acids have been shown to upregulate SIRT3. Further, a fatty acid binding protein, FABP4, has been shown to inversely correlate with Sirt3 expression (Xu et al., 2016). The suggested mechanism is based upon findings that high levels of FABP4 result in fewer free monounsaturated fatty acids, thus decreasing their availability to increase SIRT3 (Xu et al., 2015). Additionally, recent evidence suggests that epigenetic modifications, such as histone methylation, play a critical role in the regulation of macrophage phenotype in both normal and diabetic wounds (Gallagher et al., 2015, Kimball et al., 2017a). Although direct epigenetic regulation of inflammatory cytokine expression in wound macrophages has been examined, the epigenetic regulation of upstream proteins, such as FABP4, during wound repair is unknown. To date, the role of SIRT3/FABP4 in vivo in macrophages and wound repair is unknown.
Here, we demonstrate that SIRT3 is essential for normal wound repair and is impaired in SIRT3-deficient mice. We show that wound macrophages from diabetic mice display reduced SIRT3 during the transition from an inflammatory to reparative phenotype and this results in increased inflammatory cytokine production. Further, we demonstrate that FABP4 is increased in diabetic wound macrophages and regulates SIRT3 in human blood monocytes. Further, diabetic wound macrophages and human T2D monocytes treated ex vivo with an FABP4 inhibitor display increased Sirt3 and decreased inflammatory cytokine production. We demonstrate that FABP4 is epigenetically regulated, with increased activating histone 3 lysine 4 trimethylation (H3K4me3) on the Fabp4 promoter in diabetic wound macrophages. These findings suggest that FABP4 may be a viable therapeutic target to regulate macrophage phenotype and inflammation in diabetic wounds.
RESULTS
Sirt3 is essential for wound inflammation and tissue repair.
Recent studies have shown that SIRT3 plays a key role in the regulation of adipose tissue inflammation (Xu et al., 2016). Given that a regulated, macrophage-mediated inflammatory response is essential for tissue repair, we examined Sirt3 in wound macrophages following injury. Macrophages (CD11b+Ly6G−CD3−CD19−) were isolated by cell sorting from full thickness biopsy wounds in C57BL/6 mice on days 1, 3, 5, and 7 post-injury and Sirt3 gene expression was examined. We identified a significant increase in Sirt3 expression on days 3 and 5, during the transition in wound macrophages from an inflammatory to a reparative phenotype (Figure 1A). To determine whether this early increase in Sirt3 expression in macrophages is critical for wound healing, we wounded Sirt3−/− mice (Lombard et al., 2007) and their littermate controls (Sirt3+/+) and analyzed healing rates. We found significantly delayed wound healing in the Sirt3−/− mice compared with littermate controls, particularly during the early stages of healing (Figure 1B). Representative images of the delayed healing in the Sirt3−/− mice on day 3 compared with controls are shown. To further validate our findings we performed histological analysis of wounds at days 3 and 5. Histologic assessment confirmed that Sirt3−/− wound diameter was significantly greater and had less re-epithelialization on the wound edge as compared to littermate controls (Figure 1C). Further, trichrome analysis revealed that collagen deposition was reduced in Sirt3−/− wounds as compared to littermate controls. We performed Ki67 staining to evaluate whether cellular proliferation was impacted by Sirt3 deletion. Microscopically, there appears to be a reduction in Ki67 staining at the wound edge in Sirt3−/−compared to controls but this did not reach significance when quantified (Figure S1). Despite no significant changes in Ki67 staining, these data show that Sirt3 is necessary for normal wound healing.
Figure 1. SIRT3 is important for normal wound healing.
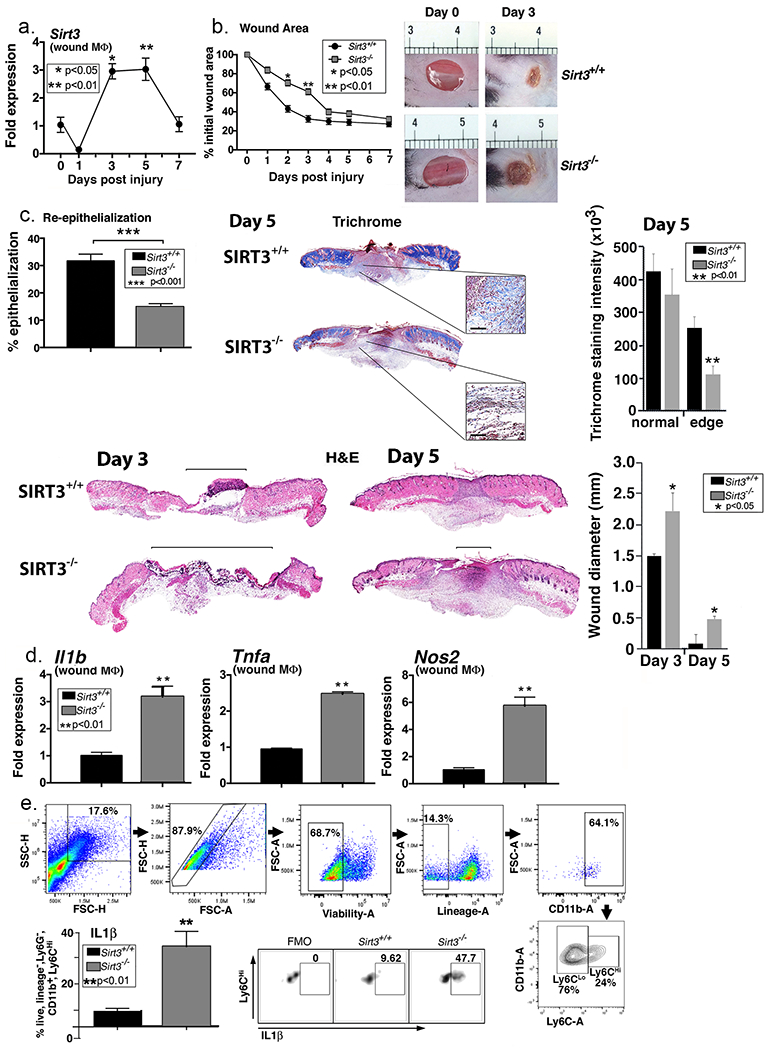
A. Two wounds were created using a 4mm punch on the backs of wild type C57BL/6 mice. Wound macrophages (CD11b+CD3− CD19−Ly6G−) were isolated at baseline (day 0) and various time points post-injury and Sirt3 gene expression was assessed (n=20 mice; repeated 2x). B: 4mm punch biopsies were created in Sirt3−/− mice and littermate controls (Sirt3+/+) and analyzed daily for changes in wound area using ImageJ software (n=10 mice; repeated 1x). Representative photographs of the wounds of on days 0 and 3 post-injury are shown. C: Wounds from Sirt3−/− and Sirt3+/+ controls were harvested on day 5, paraffin embedded and sectioned. Sections were selected at the wound center at the maximum wound diameter. 5 μm sections were stained with hematoxylin-eosin (H&E) and Masson’s Trichrome. Representative sections are shown. Percent re-epithelialization was calculated by measuring distance traveled by epithelial tongues on both sides of wound divided by total distance for full re-epithelialization. Wound diameter was calculated from and to the next leading epithelial wound edge across the maximum diameter of the wound. Black bar over H&E sections denotes wound diameter. Trichrome staining was calculated using ImageJ software (n=3 mice). Black scale bar in magnified blowout denotes 100 µm scale. D: Wound macrophages (CD11b+CD3− CD19−Ly6G−) were isolated from Sirt3−/− mice and Sirt3+/+ controls on day 3 post-injury. Relative gene expression of Il1b, Tnfa, and Nos2 was measured (n=8 mice; repeated 2x). E: Wounds were harvested from Sirt3−/− and Sirt3+/+ controls on day 3 and processed for ex vivo intracellular flow cytometry. The gating strategy used for intracellular flow cytometry selecting live, lineage− (CD3−CD19−Ter119−NK1.1−), non-neutrophil (Ly6G−), CD11b+ cells is shown. Conservative gates were chosen to limit including cells in early apoptosis. Flow cytometry quantification of Ly6C revealed two populations of cells in both Sirt3−/− and Sirt3+/+ wounds, translating to Ly6Chi and Ly6Clo monocyte/macrophages. Intracellular cytokine quantification by flow cytometry of IL1β in Ly6Chi wound monocyte macrophages is shown (n=10 mice, repeated 1x). Data are presented as mean ± SEM. Data were analyzed for normality and 2-test Student t-test was performed. For data with multiple comparisons, ANOVA followed by Newman-Keuls multiple comparisons test was performed.
Since SIRT3 is an important regulator of inflammation in adipose tissue, we examined if the absence of SIRT3 in macrophages alters wound healing by disrupting early inflammation that is necessary for tissue repair. To define the effects of SIRT3 on macrophage-mediated inflammation in wound repair, we examined the in vivo expression of key inflammatory cytokines important for healing in wound macrophages isolated from SIRT3-deficient mice. Wound macrophages (CD11b+Ly6G−CD3−CD19−) sorted from Sirt3−/− mice and littermate controls on day 3 were analyzed for inflammatory gene expression and Il1b, Tnfa, and Nos2 expression were significantly increased in wound macrophages from Sirt3−/− wounds compared to controls (Figure 1D). To examine if the changes in gene expression correlated with cytokine production, intracellular flow cytometry of wound macrophages was performed. Using our previously described gating strategy for in vivo wound flow cytometry, we examined live, lineage- (CD3−CD19−Ter119−NK1.1−), non-neutrophil (Ly6G−CD11b+Ly6Chi) cells (Kimball et al., 2018, Kimball et al., 2017a, Kimball et al., 2017b) from wounds of Sirt3−/− and controls and found that there was significantly increased IL1β in the Sirt3−/− recruited wound macrophages compared with controls (Figure 1E). Taken together, this data suggests that SIRT3 is important in vivo for macrophage-mediated inflammation in wound repair.
Adoptive transfer of Sirt3-competent macrophages to Sirt3-deficient wounds reduces inflammation and improves healing.
Since SIRT3 was upregulated in normal wound macrophages and mice lacking SIRT3 demonstrated impaired healing, we sought to examine whether replenishment of Sirt3+/+ macrophages into SIRT3-deficient mice would restore wound healing. To investigate this, we performed an adoptive transfer with macrophages (CD11b+CD3−CD19−CD11c−Ly6G−NK1.1−) isolated from Sirt3+/+ and Sirt3−/− spleens and adoptively transferred 1×106 cells into peripheral blood of Sirt3−/− mice. We found that the impairment in early wound healing was restored in SIRT3-deficient mice that received Sirt3+/+ macrophages. Representative mouse wound images on days 0 and 3 are shown (Figure 2A). To ensure that the transferred cells effectively home to the wound site, we adoptively transferred splenic bone marrow macrophages (CD11b+CD3−CD19−CD11c−Ly6G−NK1.1−) from B6.SJL-PtprcaPepcb/BoyJ mice expressing CD45.1 into peripheral blood of wounded Sirt3−/− mice. We found that 3 days following transfer, CD45.1+ cells were present in wounds from SIRT3-deficient mice (Figure 2B).
Figure 2. Adoptive transfer of Sirt3-expressing macrophages decreases inflammation and improves wound healing in Sirt3-deficient mice.
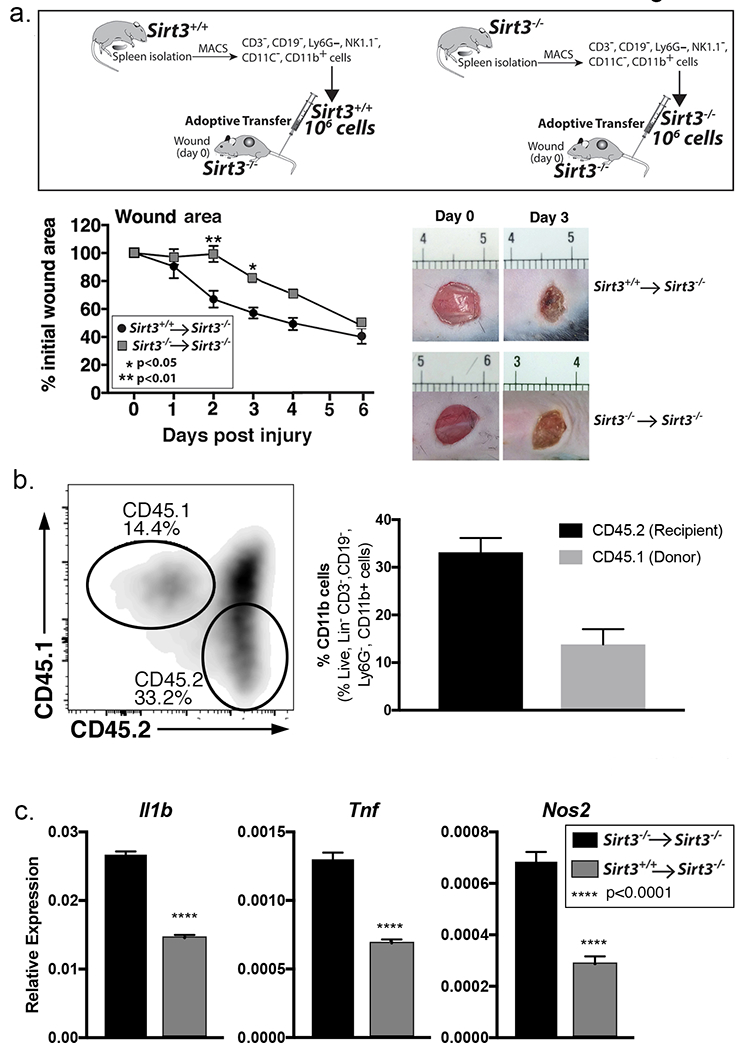
A: Monocyte/macrophage (CD11b+CD3−CD11c−CD19−Ly6G−NK1.1−) single cell suspensions were isolated from Sirt3−/− and Sirt3+/+ spleens by cell sorting. 1 × 106 cells were injected intravenously into wounded Sirt3−/− mice. Wound closure was measured using ImageJ software (n=5 mice, repeated 1x). B: Monocyte/macrophage (CD11b+CD3−CD11c−CD19−Ly6G−NK1.1−) single cell suspensions were isolated from B6.SJL-PtprcaPepcb/BoyJ spleens by cell sorting. 1 × 106 cells were injected intravenously into wounded Sirt3−/− mice. Wounds were harvested and processed for CD11b+CD3− CD19−Ly6G− 3 days post-injury and analyzed for CD45.1 and CD45.2 surface marker expression by flow cytometry. Bar chart shows relative levels of CD45.1 and CD45.2 as a percent of CD11b+CD3− CD19−Ly6G− cells (n=3 mice). C: Monocyte/macrophage (CD11b+CD3−CD11c−CD19−Ly6G−NK1.1−) single cell suspensions were isolated from Sirt3+/+ or Sirt3−/− spleens by cell sorting. 1 × 106 cells were injected intravenously into Sirt3−/− mice and wounded. Wounds were harvested and processed for CD11b+CD3− CD19−Ly6G− 3 days post-injury. Relative gene expression of Il1b, Tnfa, and Nos2 was measured (n=6 mice). Data are presented as mean ± SEM. Data were analyzed for normality and 2-test Student t-test was performed. For data with multiple comparisons, ANOVA followed by Newman-Keuls multiple comparisons test was performed.
To examine if the improvement in wound healing seen in Sirt3−/− mice receiving Sirt3-expressing cells was due to reduced expression of pro-inflammatory cytokines, we performed an adoptive transfer with macrophages (CD11b+CD3−CD19−CD11c−Ly6G−NK1.1−) isolated from Sirt3+/+ and Sirt3−/− spleens and injected 1×106 cells into tail vein of Sirt3−/− mice. On day 3 we isolated macrophages directly from the wounds of recipient mice and analyzed for expression of Il1b, Tnfa, and Nos2. Wound macrophages isolated from mice that received Sirt3+/+ cells showed reduced expression of these inflammatory cytokines compared to those that received the Sirt3−/− cells (Figure 2C). Taken together, these results suggest that SIRT3 is essential for normal wound repair and that myeloid-specific SIRT3 expression regulates inflammatory signaling leading to improved healing in SIRT3-deficient mice.
Sirt3 is significantly reduced in diabetic wound macrophages.
Impaired wound healing in T2D is secondary to failure to progress through the normal healing cascade and impaired resolution of inflammation. Further, previous studies have shown that mice lacking SIRT3 demonstrate significant metabolic abnormalities, rendering them functionally diabetic (Caton et al., 2013, Lantier et al., 2015). Given that SIRT3-deficient mice demonstrate similar metabolic derangements to that seen in T2D and the established importance of regulated inflammation in diabetic wound repair, we examined the role of SIRT3 in diabetic wound healing. We used the diet-induced obese (DIO) mouse as a model of ‘prediabetes’ because it physiologically mirrors the development of T2D in humans and does not have leptin alterations that can alter innate immunity (Francisco et al., 2018). We first examined wound healing in a cohort of Sirt3−/− mice, DIO mice, and normal diet littermate controls, and found that SIRT3-deficient mice and DIO mice demonstrated similarly impaired wound healing following acute injury, particularly in early healing. Representative images of mouse wounds on days 0 and 3 are shown for comparison (Figure 3A). Histological analysis confirmed that for both Sirt3−/− and Sirt3+/+ DIO wounds, wound diameter was increased and collagen deposition decreased similarly compared to Sirt3+/+ controls (Figure 3A). Comparably, SIRT3 protein was significantly decreased in DIO whole wounds and wound macrophages compared with controls at day 3 (Figure 3B and Figure S2). Similar to Sirt3−/− wounds, although we visualized a decrease in Ki67 staining in Sirt3+/+ DIO wounds, this did not show to be significant when quantified (Figure S3). To verify that the difference between control and DIO was intrinsic to macrophages, we measured Sirt3 expression in wound macrophages (CD11b+CD3−CD19−Ly6G−) and non-myeloid cells (CD11b−CD3+CD19+Ly6G+) isolated from the wound. We found that Sirt3 expression was significantly lower in the non-myeloid cells compared to wound macrophages (Figure 3C). Next, to examine Sirt3 expression in DIO wound macrophages over the course of wound healing, we isolated DIO and control wound macrophages (CD11b+CD3−CD19−Ly6G−) on days 1, 3, 5, and 7 post-injury and assessed Sirt3 gene expression. Sirt3 expression was significantly elevated in normal wound macrophages on days 3 and 5, while it remained low in the DIO macrophages (Figure 3D). Taken together, these findings suggest that SIRT3 is altered in DIO wound macrophages during healing.
Figure 3. Sirt3 expression is significantly reduced in diabetic mice compared with controls.
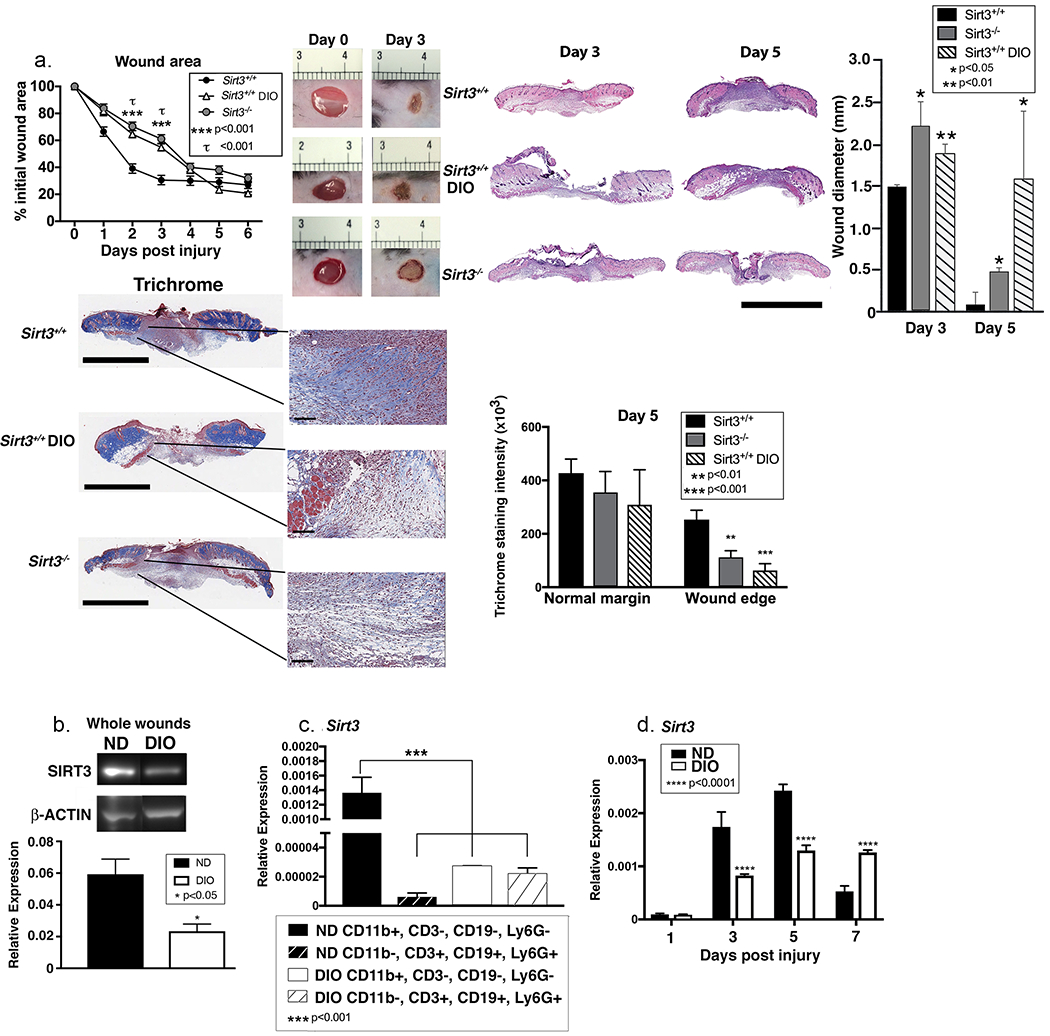
A: Wounds were created using 4 mm punch biopsies on the backs of DIO, Sirt3−/− mice and littermate controls (Sirt3+/+). Change in wound area was analyzed daily using ImageJ software (n=10 mice; repeated 2x). Wounds from DIO, Sirt3−/− mice and wild-type littermate controls (Sirt3+/+) were harvested on days 3 and 5, paraffin embedded and sectioned. Sections were selected at the wound center at the maximum wound diameter. 5 μm sections were stained with hematoxylin-eosin (H&E) and Masson’s Trichrome. Representative sections are shown. Scale bars represent 2mm (trichrome) or 3mm (H&E) for low magnification images and 100µm for blowouts (trichrome). Wound diameter was calculated from and to the next leading epithelial wound edge across the maximum diameter of the wound. Trichrome staining was calculated using ImageJ software (n=3 mice). B: Whole wounds were harvested from DIO mice and normal diet controls on day 3 post-injury. SIRT3 protein expression was assessed using Western Blot (n=8 mice; repeated 4x). C: Wound macrophages (CD11b+CD3−CD19−Ly6G−) or CD11b−CD3+CD19+Ly6G+ cells were isolated from DIO mice and controls on day 3 post-injury by cell sorting. Sirt3 gene expression was measured. (n=10 mice, repeated 2x). D: Wound macrophages (CD11b+CD3−CD19−Ly6G−) were isolated from DIO mice and controls on days 1, 3, 5, and 7 post-injury by cell sorting. Sirt3 gene expression was measured. (n=10 wounds, repeated 2x). Data are presented as mean ± SEM. Data were analyzed for normality and 2-test Student t-test was performed. For data with multiple comparisons, ANOVA followed by Newman-Keuls multiple comparisons test was performed.
FABP4 epigenetically regulates Sirt3 in diabetic wound macrophages and controls wound inflammation.
Recent studies have found that in vitro macrophages lacking FABP4 demonstrate decreased inflammation via a SIRT3-dependent mechanism (Xu et al., 2016). Given the connection between FABP4, SIRT3, and inflammation, we first examined if inhibition of FABP4 could alter Sirt3 expression in human monocytes. Human peripheral blood CD14+ monocytes were sorted and incubated ex vivo with an FABP4 inhibitor (BMS-309403) or vehicle control. We identified a significant increase in SIRT3 expression in normal human monocytes incubated with the FABP4 inhibitor (Figure 4A), suggesting that FABP4 may regulate SIRT3 in human monocytes. Given that recent literature suggests that FABP4 is increased in a variety of tissues in T2D and our findings show that FABP4 regulates SIRT3 in human monocytes, we examined the role of FABP4 in vivo in wound macrophages from our DIO mice and controls. We identified that there was a significant increase in Fabp4 expression in early DIO wound macrophages, starting at day 3, compared with controls (Figure 4B). Protein levels were also significantly increased in DIO wound macrophages compared with controls (Figure 4C). Since FABP4 levels were increased in DIO wound macrophages, we examined the effect of FABP4 inhibition on Sirt3 and inflammatory cytokine gene expression. When DIO wound macrophages were cultured ex vivo with the FABP4 inhibitor (BMS-309403) or vehicle control for 6 hours, we found a significant reduction in the expression of inflammatory cytokines Il1b and Tnfa in FABP4 inhibitor-treated DIO wound macrophages as compared to controls (Figure 4D).
Figure 4. FABP4 regulates SIRT3 in human monocytes.
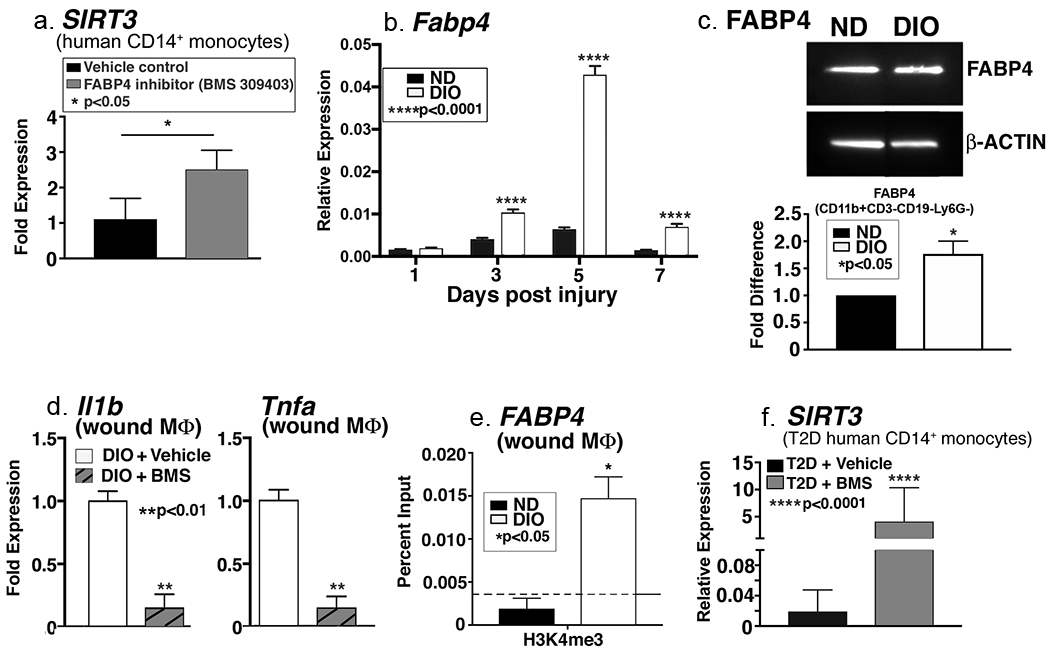
A: Human peripheral blood monocytes (CD14+) were isolated and incubated ex vivo with FABP4 inhibitor (BMS309403) or vehicle control for 6 hours. Related SIRT3 gene expression was quantified and is shown as fold expression compared to vehicle-treated (n=3, repeated 1x in triplicate). B: Wound macrophages (CD11b+CD3−CD19−Ly6G−) were isolated from DIO mice and controls on days 1, 3, 5, and 7 post-injury by cell sorting. Relative Fabp4 gene expression is shown (n=4 mice/time point, repeated 2x). C: FABP4 protein expression was assessed using Western Blot analysis (n=4 mice, repeated 2x). D: DIO wound macrophages (CD11b+CD3−CD19−Ly6G−) were isolated and incubated ex vivo for 6h with an FABP4 inhibitor (BMS309403) or vehicle control. Il1b and Tnfa gene expression is shown as fold change compared to vehicle-treated (n=14 mice, repeated 2x). E: DIO and control wound macrophages (CD11b+CD3−CD19−Ly6G−) were isolated by cell sorting on day 3. ChIP analysis was performed for H3K4me3 at the Fabp4 promoter (n=12 mice, repeated 2x). F. Human peripheral blood monocytes (CD14+) were isolated from diabetic patients and incubated ex vivo with FABP4 inhibitor (BMS309403) or vehicle control for 6 hours. Sirt3 gene expression was quantified (n=3, repeated 1x). Data are presented as mean ± SEM. Data were analyzed for normality and 2-test Student t-test was performed. For data with multiple comparisons, ANOVA followed by Newman-Keuls multiple comparisons test was performed.
Epigenetic regulation via histone methylation has been found to play a role in macrophage inflammation in wounds and T2D (Gallagher et al., 2015, Jaenisch and Bird, 2003). We and others have identified that trimethylation of histone 3, lysine 4 (H3K4me3) regulates the expression of inflammatory cytokine genes in wound macrophages (Carson et al., 2017, Kimball et al., 2017a, Kittan et al., 2013). Since there was a dramatic difference in FABP4 in diabetic wound macrophages compared to controls, we examined whether Fabp4 gene expression was epigenetically regulated by H3K4me3 in DIO wound macrophages. DIO and control wound macrophages were isolated and ChIP analysis was performed on the Fabp4 promoter for H3K4me3 as previously described (Kimball et al., 2017a). DIO wound macrophages demonstrated significantly increased H3K4me3 on the Fabp4 promoter as compared to control wound macrophages (Figure 4E). This correlates with the increased FABP4 seen in the DIO wound macrophages and suggests that FABP4 may be epigenetically regulated. To translate these findings, we tested whether SIRT3 expression could be reversed by FABP4 inhibition in human diabetic monocytes. We isolated diabetic CD14+ peripheral blood monocytes and cultured them ex vivo with FABP4 inhibitor or vehicle. Consistent with our previous results, while SIRT3 expression was extremely low in the presence of the vehicle, FABP4 inhibition resulted in a greater than 100-fold increase in SIRT3 expression in the human diabetic monocytes (Figure 4F). Taken together, these findings suggest that FABP4, likely through SIRT3 regulation, may be a viable target to control inflammation in DIO wounds. Further, our data suggest that FABP4 is epigenetically regulated, providing another therapeutic target to control macrophage-mediated inflammation in wounds through inhibition of Fabp4 gene expression.
DISCUSSION
It is well known that dysregulated inflammation is responsible for significant non-healing in T2D wounds (Boniakowski et al., 2017, Davis et al., 2018, Dinh et al., 2012); however, the molecular mechanisms responsible for this remain unclear. In this study, we found that SIRT3 is important for regulation of macrophage-mediated inflammation in normal wound healing, and that myeloid-specific SIRT3 was sufficient to rescue wound healing in SIRT3-deficient mice. Given the role of SIRT3 in wound repair and inflammation, we evaluated SIRT3 in DIO wound macrophages and found these had decreased SIRT3 expression. We further identified that FABP4 is increased in DIO wound macrophages. Finally, we interrogated FABP4, an upstream regulator of SIRT3, and were able to increase SIRT3 expression and decrease inflammatory cytokine production in wound macrophages and human monocytes. Taken together, our results suggest that SIRT3 plays a crucial role in the regulation of macrophage-mediated inflammation in wound healing, and that the manipulation of its upstream regulator, FABP4, may provide a therapeutic target to improve wound healing by controlling inflammation in diabetes.
Although our study displays insight into the mechanism behind dysregulated inflammation in wounds, there are a few limitations that should be addressed. While our study specifically examined the role of macrophages in inflammation in wound healing, it is well known that the wound healing process is complex, and that other cells and compensatory mechanisms are playing a role (Diegelmann and Evans, 2004, Reinke and Sorg, 2012). For example, since neutrophils play a key role in the early stages of wound healing (Theilgaard-Monch et al., 2004), it is possible that some of the healing deficits seen in our global Sirt3−/− mice are mediated by neutrophils, although the role of SIRT3 in neutrophils has not yet been investigated. Given the ubiquitous role SIRT3 plays in regulating inflammation and metabolism in multiple tissues (Dittenhafer-Reed et al., 2015, Giralt and Villarroya, 2012), it is possible that some of the effects mediated by SIRT3 may be impacting wound healing via an indirect influence on other tissues. For example, murine wounds heal by a combination of re-epithelialization and contraction (Chen et al., 2015) and studies have suggested that macrophages may regulate contraction in the wound (Goren et al., 2009, Newton et al., 2004). Thus, contraction-related effects could be contributing to the delayed healing we observe in Sirt3−/− mice. Further, SIRT3 likely has other end target effects within wound macrophages that influence macrophage behavior. Since the macrophage response to injury is multifactorial, further studies assessing the role of SIRT3 in wound macrophages using more objective sequencing approaches is warranted.
Additionally, in our adoptive transfer studies, healing in Sirt3−/− mice receiving Sirt3−/− macrophages was delayed more than in naïve Sirt3−/− mice. This is likely due to manipulation of these mice and non-resident cells being introduced to them since the allogeneic transfer (Sirt3+/+ into Sirt3−/−) also showed a shift in the healing curve. Although this is a side effect associated with this technique, adoptive transfer is one of the most commonly used methods for determining cell autonomy. Regardless of the difference between transferred and naïve wound curves, the allogenic transfer conferred an improvement in wound healing compared to the syngeneic transfer suggesting that restoring Sirt3 expression in circulating macrophages could improve wound repair.
To our knowledge, this study is the first to examine the role of SIRT3 on macrophage inflammation in both normal and diabetic wound healing. We have established that SIRT3 is dynamic during wound repair and is crucial during the early stages of healing. Loss of SIRT3 in macrophages results in an impaired inflammatory response and alters wound resolution via increased wound inflammation. These findings suggest that SIRT3 plays a significant role in dictating wound macrophage phenotype, which may have significance for many other secondary complications of T2D dictated by macrophage-mediated inflammation. Importantly, we identified that modulation of FABP4 can regulate SIRT3 in human blood monocytes and effectively decrease inflammation in diabetic wound macrophages. Thus, FABP4 may serve as a therapeutic target to improve wound healing in T2D. Although the exact molecular mechanisms that regulate FABP4 expression remain vague, understanding how FABP4/SIRT3 contribute to macrophage function in wounds will enhance knowledge regarding chronic inflammation in diabetic wounds.
MATERIAL & METHODS
Mice.
Male C57BL/6 and B6.SJL-PtprcaPepcb/BoyJ mice (Jackson Laboratory, Bar Harbor, ME) were obtained and maintained in breeding pairs by the University of Michigan Unit for Laboratory and Animal Medicine (ULAM). Mice were maintained on normal diet chow (ND) (13.5% kcal fat; lab diet) or high fat diet chow (HFD) (60% kcal fat, Research Diets, Inc.) for 10-12 weeks to generate the diet induced obese (DIO) model of glucose intolerance/insulin resistance. Sirtuin3−/− (Sirt3−/−) mice on a C57BL/6 background were obtained from Dr. David Lombard (University of Michigan). All animal procedures were approved under the University of Michigan Institutional Animal Care and Use Committee.
Wound Healing Assessment and Tissue Harvest.
Before wounding, mice were anesthetized, dorsal hair was removed with Veet, and skin was cleaned with sterile water. Full thickness back wounds were created by 4-mm punch biopsy (two wounds for wound monitoring, and four wounds for cell isolation, RNA, and protein experiments). An 8-megapixel iPad camera with an internal scale was used to monitor wound size. Wound area was calculated using ImageJ Software (Schneider et al., 2012) and was performed as a double-blind analysis to minimize bias. Initial wound size was calculated immediately after wounding, and wound closure was assessed over time as a percent of initial wound area. Wound tissue was harvested post-injury by 6-mm punch biopsy. Wounds were digested at 37°C for 30 min with Liberase (50 μg/mL; Roche) and DNaseI (20 units/mL; Sigma-Aldrich). Samples were filtered over a 100-mm cell strainer to produce a single-cell suspension.
Wound histology.
Wounds were harvested and fixed in 10% formalin overnight before embedding in paraffin. Sections were selected at the wound center at the maximum wound diameter. 5 μm sections were stained with hematoxylin-eosin (H&E) and with Masson’s trichrome stain. For immunohistochemistry staining, slides were deparaffinized followed by antigen retrieval in citrate buffer. Slides were subsequently stained with Ki67 antibody (Abcam) as previously described (Bagchi et al., 2017). Images were captured using Olympus BX43 microscope and Olympus cellSens Dimension software. Percent re-epithelialization was calculated by measuring distance traveled by epithelial tongues on both sides of wound divided by total distance needed for full re-epithelialization. Wound diameter was calculated from and to the next leading epithelial wound edge across the maximum diameter of the wound. Trichrome and Ki67 staining were quantified using ImageJ software (Schneider et al., 2012). Double-blind analyses were performed to minimize bias.
Wound Macrophage Isolation and Magnetic-Activated Cell Sorting (MACS).
Wounds were digested as described (Mirza et al., 2013). Single-cell suspensions were incubated with fluorescein isothiocyanate (FITC)-labeled anti-CD19, anti-CD3, and anti-Ly6G magnetic beads (Biolegend) followed by anti-FITC microbeads. The eluent was then collected and incubated with anti-CD11b microbeads (Miltenyi Biotec) to isolate the non-neutrophil, non-lymphocyte, CD11b+ cells. Cells were saved in Trizol® (Invitrogen) for quantitative PCR or fixed with 10% formalin for ChIP.
Quantitative Polymerase Chain Reaction (qPCR).
After being placed in Trizol® RNA was isolated following the standard protocol using chloroform, isopropyl alcohol, and ethyl alcohol. cDNA was then created using either iScript® (BioRad) or Superscript III® Reverse Transcriptase (ThermoFisher Scientific). Quantitative PCR was performed using primers specific for Il1b, Tnfa, Nos2, Sirt3, and Fabp4 (Thermo Fisher Scientic). All genes were normalized to 18s rRNA (Thermo Fisher Scientific) and their expression was analyzed using the 2∆Ct method. Data was compiled in Microsoft Excel (Microsoft) and presented using Prism software (GraphPad).
Western Blot.
Cells were lysed and centrifuged at 12,000g for 15 minutes. Equal amounts of cell extracts were fractionated by SDS-PAGE and transferred to PVDF membrane (ThermoFisher Scientific). Membranes were incubated in primary antibodies anti-SIRT3 (Cell Signaling), anti-FABP4 (Cell Signaling), and anti-β-actin (Sigma) overnight followed by secondary antibodies conjugated to horseradish peroxidase (Cell Signaling) and visualized using enhanced chemiluminescent substrate (Thermo-Fisher).
Flow Cytometry.
Before flow cytometry, single cell suspensions placed in ex vivo culture for 2 hours with GolgiStop 1:2,000 dilution (BD Biosciences). Gating strategy used selected live, lineage− (CD3−CD19−Ter119−NK1.1−), non-neutrophil (Ly6G−), CD11b+ cells. Cells were then fixed with 2% formaldehyde and then washed/permeabilized with BD perm/wash buffer (BD Biosciences) for intra-cellular flow cytometry. After permeabilization, intra-cellular stain for anti-IL1β (BD Biosciences) was added. Cells were acquired on a 3-Laser Novocyte Flow Cytometer (Acea Biosciences, Inc.) and data was analyzed using FlowJo software version 10.0 (Treestar, Inc.). To verify gating and purity, all populations were routinely back-gated.
Adoptive transfer.
Spleens from wild type Sirt3+/+ and Sirt3−/− mice were harvested and CD11b+CD3−CD11c−CD19−Ly6G−NK1.1− cells were isolated as described above. 1×106 cells in 200 uL total volume were injected via tail vein into each recipient Sirt3−/− mouse. Recipient mice were wounded as described above and monitored for wound closure or euthanized, and wounds harvested for CD11b+CD3−CD19−Ly6G− macrophage isolation. Alternatively, spleens from B6.SJL-PtprcaPepcb/BoyJ mice were harvested and CD11b+CD3−CD11c−CD19−Ly6G−NK1.1− cells were isolated and 1×106 cells in 200 uL total volume were injected into the tail vein of Sirt3−/− mice as described. Recipient mice were wounded, and wound macrophages were processed for flow cytometry for CD45.1/CD45.2 surface expression.
Ex vivo Macrophage Culture with FABP4 Inhibitor.
Cells isolated from wounds were cultured in the absence or presence of 50 μM FABP4 inhibitor, BMS309403 (Tocris Bioscience), in complete medium. After 2h, Trizol was added to cells and RNA was extracted for analysis.
Chromatin Immunoprecipitation (ChIP) Assay.
ChIP was performed as previously described by our group (Ishii et al., 2009, Wen et al., 2008). Briefly, cell lysates from CD11b+CD3−CD19−Ly6G− wound cells from control and DIO mice were incubated with anti-H3K4me3 antibody (Abcam) overnight followed by incubation with Protein A agarose beads (ThermoFisher Scientific). DNA eluted from beads was further purified and used in a PCR reaction with oligonucleotide primers to the promoter region of Fabp4 (5’-TGATCATTGCCAGGGAGAAC-3’, 5’- GGGCCAGATCATTTCCTTCA-3’).
Isolation of Human Monocytes from Peripheral Blood.
Thirty milliliters of peripheral blood were obtained from diabetic or non-diabetic subjects under the direction of institutional review board protocol (HUM#00060733). After red blood cell lysis and Ficoll separation, monocytes were isolated by using anti-CD14 magnetic beads.
Statistical Analysis.
Data were analyzed with GraphPad Prism software version 6.0. The statistical significance of differences between two groups was assessed by two-tailed Student t test. For experiments comparing more than 2 groups, a 2-way ANOVA followed by Newman-Keuls test for multiple comparisons was used. Unless otherwise specified, data are expressed as the mean +/− the standard error of the mean (SEM). A p-value of 0.05 or less was considered statistically significant.
IRB Exemption.
Patient consent for experiments was not required because this study was deemed EXEMPT (IRB HUM# 00060733) since it uses human tissue left over from surgery as discarded material.
DATA AVAILABILITY.
There are no datasets included in this manuscript. No sequencing or microarray data is part of this manuscript.
Supplementary Material
ACKNOWLEDGEMENTS.
The authors thank Robin Kunkel, research associate in the Pathology Department, University of Michigan, for the artwork.
FUNDING. This work was supported in part by National Institutes of Health grant R01 HL137919.
Footnotes
Conflict of interest statement: The authors have no conflicts of interest to declare.
CONFLICT OF INTEREST STATEMENT. The authors have no conflicts of interest to disclose.
REFERENCES
- Bagchi S, He Y, Zhang H, Cao L, Van Rhijn I, Moody DB, et al. CD1b-autoreactive T cells contribute to hyperlipidemia-induced skin inflammation in mice. J Clin Invest 2017;127(6):2339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen 2008;16(5):585–601. [DOI] [PubMed] [Google Scholar]
- Boniakowski AE, Kimball AS, Jacobs BN, Kunkel SL, Gallagher KA. Macrophage-Mediated Inflammation in Normal and Diabetic Wound Healing. J Immunol 2017;199(1):17–24. [DOI] [PubMed] [Google Scholar]
- Boniakowski AE, Kimball AS, Joshi A, Schaller M, Davis FM, denDekker A, et al. Murine macrophage chemokine receptor CCR2 plays a crucial role in macrophage recruitment and regulated inflammation in wound healing. Eur J Immunol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet 2005;366(9498):1719–24. [DOI] [PubMed] [Google Scholar]
- Buechler N, Wang X, Yoza BK, McCall CE, Vachharajani V. Sirtuin 2 Regulates Microvascular Inflammation during Sepsis. J Immunol Res 2017;2017:2648946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WFt, Cavassani KA, Soares EM, Hirai S, Kittan NA, Schaller MA, et al. The STAT4/MLL1 Epigenetic Axis Regulates the Antimicrobial Functions of Murine Macrophages. J Immunol 2017;199(5):1865–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caton PW, Richardson SJ, Kieswich J, Bugliani M, Holland ML, Marchetti P, et al. Sirtuin 3 regulates mouse pancreatic beta cell function and is suppressed in pancreatic islets isolated from human type 2 diabetic patients. Diabetologia 2013;56(5):1068–77. [DOI] [PubMed] [Google Scholar]
- Chen L, Mirza R, Kwon Y, DiPietro LA, Koh TJ. The murine excisional wound model: Contraction revisited. Wound Repair Regen 2015;23(6):874–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis FM, Kimball A, Boniakowski A, Gallagher K. Dysfunctional Wound Healing in Diabetic Foot Ulcers: New Crossroads. Curr Diab Rep 2018;18(1):2. [DOI] [PubMed] [Google Scholar]
- Diegelmann RF, Evans MC. Wound healing: an overview of acute, fibrotic and delayed healing. Front Biosci 2004;9:283–9. [DOI] [PubMed] [Google Scholar]
- Dinh T, Tecilazich F, Kafanas A, Doupis J, Gnardellis C, Leal E, et al. Mechanisms involved in the development and healing of diabetic foot ulceration. Diabetes 2012;61(11):2937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, et al. SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell Metab 2015;21(4):637–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falanga V Wound healing and its impairment in the diabetic foot. Lancet 2005;366(9498):1736–43. [DOI] [PubMed] [Google Scholar]
- Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernandez C, Mera A, Gonzalez-Gay MA, et al. Obesity, Fat Mass and Immune System: Role for Leptin. Front Physiol 2018;9:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher KA, Joshi A, Carson WF, Schaller M, Allen R, Mukerjee S, et al. Epigenetic changes in bone marrow progenitor cells influence the inflammatory phenotype and alter wound healing in type 2 diabetes. Diabetes 2015;64(4):1420–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol 2011;12(11):1035–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giralt A, Villarroya F. SIRT3, a pivotal actor in mitochondrial functions: metabolism, cell death and aging. Biochem J 2012;444(1):1–10. [DOI] [PubMed] [Google Scholar]
- Goren I, Allmann N, Yogev N, Schurmann C, Linke A, Holdener M, et al. A transgenic mouse model of inducible macrophage depletion: effects of diphtheria toxin-driven lysozyme M-specific cell lineage ablation on wound inflammatory, angiogenic, and contractive processes. Am J Pathol 2009;175(1):132–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464(7285):121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner G, Brauchle M, Smola H, Madlener M, Fassler R, Werner S. Differential regulation of pro-inflammatory cytokines during wound healing in normal and glucocorticoid-treated mice. Cytokine 1996;8(7):548–56. [DOI] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000;403(6771):795–800. [DOI] [PubMed] [Google Scholar]
- Ishii M, Wen H, Corsa CA, Liu T, Coelho AL, Allen RM, et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009;114(15):3244–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003;33 Suppl:245–54. [DOI] [PubMed] [Google Scholar]
- Kimball A, Schaller M, Joshi A, Davis FM, denDekker A, Boniakowski A, et al. Ly6C(Hi) Blood Monocyte/Macrophage Drive Chronic Inflammation and Impair Wound Healing in Diabetes Mellitus. Arterioscler Thromb Vasc Biol 2018;38(5):1102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball AS, Joshi A, Carson WFt, Boniakowski AE, Schaller M, Allen Rv, et al. The Histone Methyltransferase MLL1 Directs Macrophage-Mediated Inflammation in Wound Healing and Is Altered in a Murine Model of Obesity and Type 2 Diabetes. Diabetes 2017a;66(9):2459–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimball AS, Joshi AD, Boniakowski AE, Schaller M, Chung J, Allen R, et al. Notch Regulates Macrophage-Mediated Inflammation in Diabetic Wound Healing. Front Immunol 2017b;8:635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittan NA, Allen RM, Dhaliwal A, Cavassani KA, Schaller M, Gallagher KA, et al. Cytokine induced phenotypic and epigenetic signatures are key to establishing specific macrophage phenotypes. PLoS One 2013;8(10):e78045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landen NX, Li D, Stahle M. Transition from inflammation to proliferation: a critical step during wound healing. Cell Mol Life Sci 2016;73(20):3861–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, et al. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects Against Severe Insulin Resistance in High-Fat-Fed Mice. Diabetes 2015;64(9):3081–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Brown CM, El Gazzar M, McPhail L, Millet P, Rao A, et al. Fueling the flame: bioenergy couples metabolism and inflammation. J Leukoc Biol 2012a;92(3):499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem 2015;290(1):396–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TF, Vachharajani VT, Yoza BK, McCall CE. NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J Biol Chem 2012b;287(31):25758–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007;27(24):8807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza RE, Fang MM, Ennis WJ, Koh TJ. Blocking interleukin-1beta induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes 2013;62(7):2579–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton PM, Watson JA, Wolowacz RG, Wood EJ. Macrophages restrain contraction of an in vitro wound healing model. Inflammation 2004;28(4):207–14. [DOI] [PubMed] [Google Scholar]
- Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A 2002;99(21):13653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, Dereuddre-Bosquet N, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol 2005;142(3):481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res 2012;49(1):35–43. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9(7):671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theilgaard-Monch K, Knudsen S, Follin P, Borregaard N. The transcriptional activation program of human neutrophils in skin lesions supports their important role in wound healing. J Immunol 2004;172(12):7684–93. [DOI] [PubMed] [Google Scholar]
- Wen H, Dou Y, Hogaboam CM, Kunkel SL. Epigenetic regulation of dendritic cell-derived interleukin-12 facilitates immunosuppression after a severe innate immune response. Blood 2008;111(4):1797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol 2000;115(2):245–53. [DOI] [PubMed] [Google Scholar]
- Xu H, Hertzel AV, Steen KA, Bernlohr DA. Loss of Fatty Acid Binding Protein 4/aP2 Reduces Macrophage Inflammation Through Activation of SIRT3. Mol Endocrinol 2016;30(3):325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Hertzel AV, Steen KA, Wang Q, Suttles J, Bernlohr DA. Uncoupling lipid metabolism from inflammation through fatty acid binding protein-dependent expression of UCP2. Mol Cell Biol 2015;35(6):1055–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There are no datasets included in this manuscript. No sequencing or microarray data is part of this manuscript.